

Abstract
Emergence of antibiotic-resistant infections highlights the need for novel antibiotic leads, perhaps with a broader spectrum of activity. Herein, we disclose a semisynthetic, catalytic approach for structure diversification of vancomycin. We have identified three unique peptide catalysts that exhibit site-selectivity for the lipidation of the aliphatic hydroxyls on vancomycin, generating three new derivatives 9a, 9b and 9c. Incorporation of lipid chains into vancomycin scaffold provides promising improvement of its bioactivity against vancomycin-resistant enterococci (Van A and Van B phenotypes of VRE). The MICs for 9a, 9b, and 9c against MRSA and VRE (Van B phenotype) range from 0.12 to 0.25 μg/mL. We have also performed a structure activity relationship (SAR) study to investigate the effect of lipid chain length at the newly accessible G4-OH derivatization site.
Keywords: vancomycin, glycopeptides, infectious diseases, antibiotics, peptide catalysis, site-selective chemistry, vancomycin-resistant bacteria
INTRODUCTION
Natural products and their structural derivatives continue to be the lead therapeutics for the treatment of various diseases.1-3 In particular, numerous antibiotics approved for clinical applications have been either natural products or their derivatives.4 Emerging problem of drug resistance highlights the need for new drug leads to combat infectious diseases, perhaps via development of novel synthetic analogues of existing bioactive molecules.4-6 Synthetic endeavors to modify natural products can be a challenging task due to their structural complexity and the presence of a large array of reactive functional groups.2,7 As such, efficient and highly selective approaches that employ minimal protecting group manipulations are desirable. In recent years, catalyst dependent diversification of complex drug molecules has gained considerable interest.8-11 Many important approaches, including biocatalysis, may be brought to bear on the problem.12
Our research group (Figure 1)13-18 and others19,20 have demonstrated that synthetic oligopeptide-based catalysis enables a wide range of chemical transformations to derivatize polyol natural products of medicinal interest.10,21,22 For example (Figure 1a), we have developed peptide-based catalyst wherein the critical catalytic residue p(methyl)-histidine (Pmh) is central to the selective transfer of acyl groups,13 phosphoryl groups,23 sulfur-based electrophiles24,25 to individual sites in structures containing multiple hydroxyl groups. As an example in the context of complex glycopeptides like vancomycin (1a), we also showed that catalytic thiocarbonylation – as a prelude to site-selective deoxygenation – could be enabled with the Pmh-based catalyst construct, allowing excision of either the G6-oxygen atom, or the Z6-oxygen atom, in a catalyst-dependent manner (Figure 1b).15
Figure 1.

(a) A schematic outline of various group transfer reactions catalyzed by Pmh-based peptides for polyol modifications. (b) A representative set of catalyst-dependent maneuvers leading to two unique deoxygenation vancomycin analogues.15
Catalytic, site-selective acyl transfer reactions of long-chain fatty acid functionality (i.e., lipidation) present a particularly intriguing avenue for the synthesis of positional isomers of lipidated analogues of glycopeptide-based polyols. Lipid groups in lipopeptide and lipoglycopeptide antibacterial natural products have been implicated in their potency26-29 and such modifications enable medicinal chemists to tune the bioactivity of antibacterial natural products.28,30,31 We describe in this paper several advances in the use of Pmh-based catalysts for the preparation of site-selectively lipidated vancomycins, along with data on their biological activity.
The glycopeptide vancomycin and the teicoplanins, lipoglyco-peptides (Figure 2, 1a and 2) are potent antibiotics for the treatment of several drug resistant bacterial infections, including methicillin-resistant Staphylococcus aureus (MRSA).26,32,33 Glycopeptides and lipoglycopeptides bind to the ‘Lys-DAla-DAla’ peptide region of peptidoglycan monomer, which leads to inhibition of bacterial cell wall biosynthesis, and cell death. Due to its unique mode of action and potency, vancomycin has been an antibiotic of interest for synthetic modifications in order to target drug-resistant pathogens.7,32,34-40 In particular, several analogues containing lipid motifs at the chemically most accessible positions on vancomycin (V3-NH2, X1-NHMe, or G6-OH, Figure 2) have been reported, which exhibit improved antibacterial activity against glycopeptide-resistant bacteria.34,35,37
Figure 2.

Structures of vancomycin (1a), a G6-modified vancomycin (1b), and teicoplanin A2-2 (2)
Differentiating the reactivity of six aliphatic hydroxyls, the phenolic OH-group and the resorcinol-like OH-groups on vancomycin is a challenging task. This has restricted comprehensive structural modifications on the peripheral hydroxyls of vancomycin to date. Moreover, limited knowledge is available regarding the importance of these functional groups on structure and bioactivity. Our recent study of the catalytic synthesis of two deoxy-vancomycin analogues noted above revealed biological consequences.15 Z -Deoxy-vancomycin exhibits conformational heterogeneity, and modified potency in MIC assays. We speculate that additional perturbations to the hydroxyl group array of vancomycin would also unveil additional modulation of biological activity. Related observations followed our discovery of catalysts for the site-selective phosphorylation of hydroxyls within the teicoplanin A2-2 structure.
Moreover, based on the heightened bioactivity of alkyl- and acyl-vancomycin analogues and teicoplanins, 41,42 we hypothesized that site-selective lipidation of vancomycin would yield structurally diverse and biologically promising analogues. In this vein, previous studies suggest that hydrophobic groups on vancomycin greatly improve its potency via at least two distinct proposed mechanisms: (i) enhancement of proximity induced binding of vancomycin analogues to peptidoglycan monomer via bacterial cell membrane association31,43,44; (ii) binding of lipidated vancomycin derivatives to transglycosylase and inhibition of cross-linking of disaccharides during peptidoglycan biosynthesis.45-47 The research described below culminates in the discovery of three unique peptide-based catalysts for site-selective incorporation of lipid motifs at the G6, G4, and Z6 positions on vancomycin. Each is portable for the transfer of acyl side chains of varying length, leading to a library of previously unknown vancomycin analogues. Their structural characterization and the antibacterial activity are also disclosed.
RESULTS AND DISCUSSION
Chemical catalysis
Based on previous studies, we began with the synthesis of a partially allyl-protected vancomycin (3) in order to gain desirable solubility in organic solvents, and prevent undesired reactions with amines, phenols or resorcinol-like OH-groups of 1a.48 Since vancomycin is readily available from commercial sources, we synthesized 3 on multi-gram scale (15 g) via a sequential formation of alloc-carbamates, ally-ester and allyl-ethers.15,48,49 From our initial screening, we found that symmetric anhydrides worked well for the acyl-transfer reactions; while acyl chlorides showed unselective reactivity and N-hydroxysuccinimide active esters were not reactive under the reaction conditions.
Under optimized reaction conditions, 3 exhibited no reactivity towards an anhydride in the absence of a nucleophilic catalyst, and negligible reactivity in the presence of N-methylimidazole (NMI) as an achiral control catalyst. Once we established a sense of the background reactivity, we investigated a library of peptide-based catalysts for the acyl-transfer reaction. All of these peptides contain the catalytic amino acid Pmh, which assists in the acyl transfer reaction. For this study, we investigated two parallel approaches to identify catalysts to achieve site-selective lipidation: (i) screening of catalysts from legacy peptide-based catalysts in our laboratory. (These catalysts and their analogues have been previously explored for regioselective or enatioselective reactions.50,51), and (ii) Screening of catalyst structures designed by placing the Pmh residue within the ‘Lys-DAla-DAla’ scaffold. Analysis of a X-ray crystal structure of vancomycin-ligand complex (Figure 3)52 and our previous findings15 suggest that placement of the Pmh residue proximal to disaccharides on vancomycin often yields selective modification of the hydroxyls based on this binding model.15-18
Figure 3.
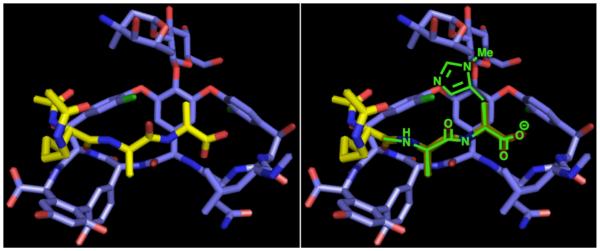
(a) a co-crystal structure of the ligand, N-Ac-Lys(Ac)-DAla-DAla (yellow) bound to vancomycin and (b) a drawing of a segment of a designed catalyst over the ligand (PDB 1FVM).52
Since numerous lipopeptides and lipoglycopeptides (i.e. daptomycin, teicoplanins) generally contain a decanoyl-group as the lipid motif, we began the lipidation chemistry using decanoic anhydride as the longest lipid chain. We first screened a collection of 25 catalysts from the legacy peptide library. Starting material 3 was reacted with decanoic anhydride (2 equiv), 1,2,2,6,6-pentamethylpiperidine (PEMP, 2 equiv), and catalyst (20 mol%) in 3:1 CH2Cl2/THF (0.007 M) for 24 h (Figure 4 and Scheme 1). Intriguingly, the peptide catalysts exhibited superior reactivity compared to NMI, and provided reasonable conversion to the decanoyl-vancomycin derivatives (See Supporting Information for details). In fact, we identified several catalysts that yielded a mixture of G6 and G4-products in very high conversion, including catalyst 5. Each product was characterized using a combination of LC-MS/MS fragmentation analysis, and 2D-NMR spectroscopy to elucidate the site of modification (see Supporting Information).
Figure 4.

RP-HPLC traces for uncatalyzed, NMI (0.2 equiv) catalyzed, and peptide 5 catalyzed reactions of 3 using decanoyl anhydride.
Scheme 1.

Exploration of catalysts for site-selective modification of 3.
Within the first set of legacy library, we were pleased to identify catalysts 6 and 7 for selective modification of the G4 (4a:4b:4c = 1:5:0, 87% conv. of 3) and Z6 (4a:4b:4c = 0:1:17, 94% conv. of 3) hydroxyls respectively.53 It is worth noting that 6 and 7 were historically relevant to us, having been previously identified as hit catalysts for the kinetic resolution of formamides,54 and for the asymmetric phosphorylation of myo-inositol,23 respectively. Moreover, 7 was also a key catalyst for the Z6-selective thiocarbonylation of vancomycin.15 Interestingly, no catalyst from the legacy peptide library delivered G6-selectivity. Thus, we turned our attention to the second approach, where rationally designed ‘Xaa-DAla-DPmh’ type catalysts were evaluated. As this approach had been successful previously, allowing observation of high selectivity for G6-position for thiocarbonylation, we hoped its selectivity would carry over to the transfer of a different reagent. Strikingly, catalyst 8, which was a hit catalyst for the G6-selective thiocarbonylation15 also yielded good selectivity for the acylation of the G -position (4a:4b:4c = 6:1:0, >99% conv. of 3).53 It is gratifying that we rediscovered new reactivity and selectivity for 6, 7 and 8, which suggests that these ‘legacy catalysts’ are very versatile for various group transfer reactions. This state of affairs is observed in some cases as different types of electrophiles, but not all, as we have observed previously.15 Nevertheless, a set of catalysts selective for three distinct hydroxyl groups within vancyomycin (G6-, Z6- and G4-, with G4-being previously unknown in our experience) represents an expansion of catalytic capabilities for derivatization of this particular structure.
Although the catalyst dependent acyl-transfer reaction enables modification of the various hydroxyls, we wanted to confirm that the selectivity observed is not due to an internal, intramolecular acyl group migration among the hydroxyls within a glucose ring. More specifically, we wanted to ensure that conversion of 4a into 4b is not occurring under the reaction condition. To investigate this, we prepared pure samples of 4a and 4b and subjected them to the reaction condition (Scheme 2). Since 2 equiv of PEMP was used for the optimized acylation reaction to modify vancomycin, we subjected the products (4a and 4b) to the same reaction condition to evaluate the acyl migration pathway. We were pleased to find that 4a remained stable in the presence of up to 2 equiv of PEMP for 20 h, suggesting that the G6-acyl group (4a) is not migrating to G4-position (4b). When 4b was treated with 2 equiv of PEMP, it was stable for up to 5 h; however, at 20 h, 4b progressively equilibrated to 4a (4a: 4b = 1:1, see supporting materials), indicating under strong basic conditions, the acyl group at G4-position migrated to the G6-position. Yet, when only 0.5 equiv of PEMP was used, there was no equilibration of 4b to 4a for up to 20 h. These findings suggest that the migration is not occurring under mild basic condition (with 0.5 equiv PEMP). In addition, when acylation reactions (Scheme 1) were monitored over 24 h, 30 h and 40 h intervals, no change in selectivity was observed, perhaps indicative of a buffering effect wherein the RCO2H byproduct of the anhydride inhibits migration. Taken together, we believe that both 4a and 4b are formed in a catalyst-dependent manner under kinetic control under our catalytic reaction conditions, and once these products are formed, they remain stable under the reaction condition.
Scheme 2.
Investigation of acyl-migration within a glucose ring.
We also performed a brief study to understand better the nature of any catalyst-substrate complex suggestive of the possible mode of binding of 6, 7, and 8 to the vancomycin-based substrate. From earlier work,15 we have compelling evidence that 8 binds to the ligand binding pocket of vancomycin through a series of hydrogen bonds with the backbone amides (Figure 3b).55 However, it is unclear if the same sets of amide bonds are also playing a role in recruiting 6 and 7. To probe this, we performed the acylation reactions with each catalyst in the presence of Boc-Leu-DAla-DAla-OH (1 equiv) as a competing ligand for the binding pocket (see Supporting Information). RP-HPLC analysis confirmed that 8 considerably lost its selectivity (4a:4b, from 6:1 to 1.6:1) in the presence of the ligand. Moreover, 6 also lost its selectivity to a noticeable extent (4a:4b, from 1:5 to 1:1.6). On the other hand, 7 had insignificant effect in the presence of the competing ligand. These results further support the hypothesis that ‘Xxa-DAla-DPmh’ type catalysts interact with the substrate in the canonical ligand binding pocket, and these interactions contribute to high selectivity. As for the catalysts from the legacy library (6 and 7), it is difficult to propose a binding model for the observed selectivity, as we have limited knowledge from these experiments.
With the discovery of catalysts 6, 7, and 8, desired mono-acylated products were synthesized in sufficient amounts (100 mg scale reaction) for structural characterization and biological studies. Since starting material 3 is available in multi-gram scale, the lipidation reactions can be done in gram-scale quantity to access these derivatives if necessary (See Supporting Information for details). Following selective acylation, the compounds were purified via RP-MPLC, then the allyl/alloc groups were removed reliably under standard conditions (Pd(PPh3)2Cl2 and PhSiH3; Scheme 3). A final RP-HPLC purification yielded the decanoyl-vancomycin analogues 9a (17 mg, 38% over two steps), 9b (32 mg, 24% over two steps) and 9c (11 mg, 24% over two steps) in greater than 95% purity based on RP-HPLC analysis (Please see Supporting Information).
Scheme 3.

Site-selective incorporation of decanoyl motifs into vancomycin scaffold.
The structure of the acyl-vancomycin derivatives 9a, 9b, and 9c were all characterized by 2D-NMR spectroscopy. Figure 7 shows an overlaid edited-HSQC spectrum of native vancomycin (green spots), and 9b (red and blue spots). All peaks have been assigned using a combination of HSQC, COSY, TOCSY and HMBC experiments and verified with literature reports.15,56 There are clear changes in the chemical shifts of glucose ring protons relative to that of native vancomycin. The G4 proton has shifted about 1.5 ppm downfield. The G3 and G5 protons also showed noticeable downfield shifts.
Figure 7.

An overlay of HSQC-DEPT spectra of vancomycin analogue 9b (methyls and methines shown in red, and methylenes shown in blue), and native vancomycin (green). DEPT: distortionless enhancement by polarization transfer.
The G1 and G2 protons, on the other hand, exhibit a very small downfield shift, while the G6 protons reflects a very small up-field shift (Figure 7). We also observed a minute perturbation of some of the amino acid α-protons, perhaps due to subtle conformational changes introduced by the decanoyl moiety. However, almost all of the remaining protons’ chemical shifts overlap well with that of native vancomycin. Similarly, 9a and 9c were also characterized, where the chemical shift of G6 proton on 9a and the Z6 proton on 9c had significant downfield shift, confirming the site of modification (see Supporting Information).
We then used the newly identified G4-selective catalyst to facilitate the synthesis of a library of acyl-vancomycin derivatives to perform a SAR study, wherein the acyl side chain was varied in length and composition at this newly modifiable site. Butanoyl, pentanoyl, hexanoyl, octanoyl, pentenoyl and methyl-adipoyl moieties were selectively incorporated at the G4 position (10 – 15, Figure 8) using catalyst 6. The different aliphatic chains enable an interrogation of the role of chain length for antibacterial activity. We envisioned that incorporation of a terminal alkene (14), or a carboxylate-handle (15) could provide a useful chemical handle for further derivatization of vancomycin scaffold as a biomarker or imaging probe.57 Catalyst 6 was highly faithful in delivering the anticipated selectivity for G4-OH site. Each new compound was synthesized using the established protocol with catalyst 6 (20 mol%), and isolated in pure form by RP-HPLC.
Figure 8.

Structures of G4-modified vancomycin derivatives.
The site of modification was confirmed by LC-MS/MS analysis and multidimensional NMR spectroscopy (see Supporting Information).
Antibacterial evaluation
We evaluated the set of nine new lipidated-vancomycin derivatives against a panel of both vancomycin sensitive and vancomycin resistant strains, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecalis (VRE). The activity is reported as minimum inhibitory concentration (MIC) where complete killing effect was observed. Vancomycin, teicoplanin A2-2, and linezolid were used as control compounds to compare the MIC values. All of the biologically tested compounds are greater than 95% pure as ascertained by RP-HPLC, as exhibited in the SI (9a, page S-8; 9b, page S-11; 9c, page S-14; 10, page S-16; 11, page S-16; 12, page S-17; 13, page S-17; 14, page S-18; 15, page S-18). All decanoyl-vancomycin derivatives (9a, 9b, and 9c) exhibited remarkable potency against all strains tested, even against a VRE (Van B phenotype) strain. The G4-modified analogue 9b is 10 times more active against vancomycin-sensitive strains and 64 times more active against a VRE strain (Van B phenotype). In fact, the values are comparable or marginally enhanced in some cases compared to teicoplanin A2-2. Remarkably, derivatives 9a, 9b, and 9c exhibit a promising activity of 16 μg/mL against Van A phenotype VRE strain as well. As a comparison, vancomycin and teicoplanin A2-2 exhibit an MIC value of 512 μg/mL and 128 μg/mL respectively against Van A phenotype VRE strain based on literature data.37
We also evaluated the bioactivity of the derivatives containing shorter acyl chains or functionalized acyl-moieties. The results indicate that analogues containing lipid chain that is smaller than an octanoyl moiety exhibited reduced bioactivity against VRE strains. A clear trend of loss of activity with reduced length of lipid chain is observed. However, the bioactivity is not completely lost compared to native vancomycin. This trend may support the previously proposed cell membrane binding model by which lipidated-vancomycin analogues exhibit potency against VRE strains.34,35,37 Compounds 14 and 15 retained respectable bioactivity against vancomycin-sensitive strains, suggesting that these sites are potential places for the incorporation of chemical probes for studying vancomycin and its biological fate.57
CONCLUSION
With the increased emergence of vancomycin resistance among clinically relevant pathogens, significant effort is being directed towards identifying new vancomycin analogues with improved bioactivity. Herein we report our discovery of three unique peptide-catalysts that allow us to incorporate lipid moieties into vancomycin scaffold in a highly site-selective manner. This semi-synthetic approach enables us to modify three different positions (G4, G6, and Z6) on vancomycin. Since enhancing the bioactivity of vancomycin via incorporating lipid motifs is of high interest in literature37,43,45,46, our approach provides new opportunities to efficiently modify three distinct positions on vancomycin. We also prepared a series of lipidated vancomycins derivatized at the site for which the newest catalyst capability was achieved, the G4-OH group. The antibacterial evaluation revealed that incorporation of a decanoyl group considerably improves vancomycin’s activity against MRSA and VRE (Van A and Van B phenotypes). That said, it seems that the added lipophilicity of the presently reported vancomycin derivatives might render them more teicoplanin-like. Since teicoplanin is known to be vulnerable to the development of resistance,58 it seems much further study would be required to for the presently reported compounds to contribute to useful therapies.
Ultimately, the site-selective catalysis approach may allow one not only to identify biologically interesting analogues, but perhaps also to study the function of the peripheral hydroxyl groups of vancomycin in a position-specific manner.57
EXPERIMENTAL PROCEDURES
Materials and instrumentation. All commercially available reagents were purchased and used without further purification, unless otherwise stated. Air and moisture sensitive reactions were done under nitrogen atmosphere employing flame-dried glassware. Commercially available solvents (> 99.0% purity) were used for column chromatography without any further purification. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), and toluene were purified by a Seca Solvent Purification System from Glass Contour (Nashua, NH). Proton and carbon NMR spectra were recorded on a 600 MHz or 500 MHz spectrometer. Proton chemical shifts are reported in ppm (δ) relative to residual CHCl3 (δ 7.26 ppm) or DMSO-d6 (2.50 ppm). Data are reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), triplet (t), quartet (q), quintet (p), sextet (h), multiplet (m), apparent singlet (app. s), apparent doublet (app. d)], coupling constants [Hz], integration). Carbon chemical shifts are reported in ppm with the respective solvent resonance as the internal standard (CDCl3, δ 77.1 ppm, DMSO-d6, δ 40.0). Unless otherwise noted, all NMR spectra were acquired at ambient temperature.
Infrared spectra (thin film) were recorded on a FT-IR, = (cm-1) and are partially reported. Analytical thin-layer chromatography (TLC) was performed using Silica Gel 60 Å F254 precoated plates (0.25 mm thickness). The following visualization methods were used for monitoring reactions and column chromatography: UV absorption by fluorescence quenching; Ninhydrin spray (Ninhydrin: acetic acid: n-butanol/ 0.6 g:6 mL:200 mL) or PMA stain (phosphomolybdic acid, H2SO4). Flash column chromatography was performed using Flash Silica Gel (32-63 micron). Mass spectrometry data were collected using a Ultra high performance LC/MS, which was was performed on a UPLC/MS instrument equipped with a reverse-phase C18 column (1.7 μm particle size, 2.1 × 50 mm), dual atmospheric pressure chemical ionization (API)/electrospray (ESI) mass spectrometry detector, and photodiode array detector. Minimum inhibitory concentrations (MICs) were determined by Micromyx, LLC (Kalamazoo, MI) in accordance with CLSI guidelines.
HR-LC/MS analyses were done on a Waters Acquity UPLC® instrument. Method 1: Column = Waters Acquity UPLC® BEH C18 1.7 μm, 2.1 Å~ 100 mm; Temperature = 25 °C; Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 0.8 mL/min; Gradient: Start at 20% B, ramped to 100% B over 3 min, held at 100% B for 1 min. Method 2: Column = Waters Acquity UPLC® BEH C8 1.7 μm, 2.1 Å~ 100 mm; Temperature = 25 °C; Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 0.3 mL/min; Gradient: Held at 5% B for 1 min, ramped to 95% B over 5 min and held for 1.5 min.
RP-HPLC analysis and purification were done using these methods. Method 3: Column = Waters SymmetryPrep C8 7 μm, 300 Å, 7.8 × 300 mm; Temperature = 25 °C; Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 4.0 mL/min; Gradient: Held at 5% B for 3 min, ramped to 57% B over 17 min, maintained at 57% B for 5 min, ramped to 99% B over 25 min, held at 99% B for 10 min, ramped to 5% B over 2 min and held for 5 min. Method 4: Column = Waters SymmetryPrep C8 7 μm, 300 Å, 19 × 300 mm; Temperature = 23 °C; Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 24 mL/min; Gradient: Held at 5% B for 3 min, ramped to 57% B over 17 min, held at 57% B for 3 min, ramped to 90% B over 20 min, held at 90% B for 3 min, ramped to 5% B over 2 min and held for 2 min. Method 5: Column = Waters SymmetryPrep C8 7 μm, 300 Å, 19 × 300 mm; Temperature = 23 °C; Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 24 mL/min; Gradient: Held at 2% B for 7 min, ramped to 60% B over 20 min, ramped to 95% B over 2 min, held at 95% B for 2 min, ramped to 2% B over 2 min and held for 2 min.
RP-MPLC purification was done on a Biotage® instrument. Method 6: Column = Biotage SNAP C18, 120 g column with 12-g samplet; Temperature = 23 °C; Solvent A = H2O; Solvent B = MeOH; Flow Rate = 50 mL/min; Gradient: Held at 75% B for 1 column volume (CV), ramped to 80% B over 4 CV, held at 80% B for 6 CV, ramped to 90% B over 1 CV, held at 90% B for 2 CV. Method 7: Column = Biotage SNAP C18 60 g column with 12-g samplet; Temperature = 23 °C; Solvent A = H2O ; Solvent B = MeCN; Flow Rate = 50 mL/min; Gradient: Held at 5% B for 4 column volume (CV), ramped to 40% B over 4 CV, ramped to 95% B over 18 CV, held at 95% B for 5 CV.
General procedure 1: Screening of peptide catalysts. In a flame-dried small glass vial containing a stir bar, 3 (5 mg, 2.81 μmol, 1 equiv) and peptide catalyst (0.2 equiv) were dissolved in THF (100 μL) to yield a clear, homogeneous solution. Then, CH2Cl2 (300 μL) and PEMP (1.1 μL, 6.07 μmol, 2 equiv) were added to the vial. Decanoic anhydride (2.1 μL, 5.62 μmol, 2 equiv) was added subsequently and the reaction mixture was allowed to stir at rt for 24 h. An aliquot (20 μL) was taken and quenched with 2-propanol (500 μL), and analyzed by HR-LC/MS (Method 2) and RP-HPLC (Method 3). These data were used for preliminary determination of conversion, product selectivity, and site of reaction (See Supporting Information for HPLC data).
General procedure 2: Synthesis of decanoyl-vancomycin derivatives 9a-9c. In a flame-dried 10-mL round bottom flask containing a stir bar, 3 (50 mg, 28.1 μmol, 1 equiv) and the peptide catalyst (5.69 μmol, 0.2 equiv) were dissolved in THF (1 mL) to yield a clear, homogeneous solution. Then, CH2Cl2 (2 mL) and PEMP (56.2 μmol, 2 equiv) were added sequentially to the flask, and stirred under N2 atmosphere. A solution of decanoic anhydride (56.2 μmol, 2 equiv) in CH2Cl2 (1 mL) was added over 10 h using a syringe pump, and the reaction was allowed to stir for an additional 14 h. An aliquot (20 μL) was taken and quenched with 2-propanol (500 μL), and analyzed by HR-LC/MS (Method 2) and RP-HPLC (Method 3) to determine if the reaction was complete. At this time, the reaction was quenched with 2-propanol (2 mL). The solution was then concentrated in vacuo to a volume of ~1 mL and diluted with another 2 mL of 2-propanol. The solution was loaded onto a C18 Biotage samplet and purified using RP-MPLC (Method 7). Fractions containing the desired mono-decanoyl vancomycin derivatives were combined and concentrated to yield the mixture of products as a white residue.
The protected decanoyl vancomycin derivative (15.0 μmol, 1 equiv) was dissolved in degassed 1,4-dioxane (2 mL) in a 10-mL round bottom flask. The flask was evacuated, and refilled with argon gas four times, and kept under argon atmosphere. To the flask, Pd(PPh3)2Cl2 (10.6 mg, 15.1 μmol, 1 equiv), and AcOH (0.5 mL) were added quickly, and the flask was sealed with a septum. The flask was again evacuated and refilled with argon gas four times, and kept under argon atmosphere. Then, PhSiH3 (84 μL, 0.68 mmol, 45 equiv) was added drop wise over 30 min. The reaction was stirred for another 2 h under argon atmosphere at rt. The reaction mixture turned from a cloudy yellow mixture into a darker brown mixture over the course of the reaction. After 2 h of stirring, the reaction mixture was poured into a separatory funnel containing 0.01 M aqueous HCl (100 mL), and washed with hexanes (3 × 100 mL). The aqueous layer was passed through a pad of celite. The filtrate was concentrated to ~20 mL, and purified using preparative RP-HPLC (Method 5). The fractions containing the desired product were concentrated in vacuo to remove the organic solvent, and the water was removed via freeze-drying to yield 9a-9c a white residue in greater than 95% purity based on RP-HPLC (Method 3), and 24 – 38% yield over 2 steps.
Synthesis of G6-decanoyl vancomycin derivative 9a using general procedure 2. Protected G6-decanoyl vancomycin derivative was isolated as a white residue (29 mg yield; 28.1 μmol scale reaction), which was carried forward for the next reaction. Analytical RP-HPLC retention time of protected G6-decanoyl vancomycin derivative (Method 3): 35.5 min; LC-MS (ESI+) for C96H117Cl2N9O29K [M+K]+: Calc’d = 1968.697; found = 1968.696.
The deprotected G6-decanoyl vancomycin derivative 9a was isolated as a white residue (17 mg, 38% over two steps). Analytical RP-HPLC retention time of 9a (Method 3): 13.4 min; LC-MS (ESI+) for C76H94Cl2N9O25 [M+H]+: Calc’d = 1602.573; found = 1602.579. (See Supporting Information for HPLC & NMR data)
Synthesis of G4-decanoyl vancomycin derivative 9b using general procedure 2. Protected G4-decanoyl vancomycin derivative was isolated as a white residue (47 mg yield; 56.2 μmol scale reaction), which was carried forward for the next reaction. Analytical RP-HPLC retention time of protected G4-decanoyl vancomycin derivative (Method 3): 38.0 min; LC-MS (ESI+) for C96H117Cl2N9O29K [M+K]+: Calc’d = 1968.696; found = 1968.695.
The deprotected G4-decanoyl vancomycin derivative 9b was isolated as a white residue (32 mg, 24% over two steps). Analytical RP-HPLC retention time of 9b (Method 3): 14.2 min; LC-MS (ESI+) for C76H94Cl2N9O25 [M+H]+: Calc’d = 1602.573; found = 1602.582. (See Supporting Information for HPLC & NMR data)
Synthesis of Z6-decanoyl vancomycin derivative 9c using general procedure 2. Protected Z6-decanoyl vancomycin derivative was isolated as a white residue (29 mg yield; 28.1 μmol scale reaction), which was carried forward for the next reaction. Analytical RP-HPLC retention time of protected Z6-decanoyl vancomycin derivative (Method 3): 39.6 min; LC-MS (ESI+) for C96H118Cl2N9O25 [M+H]+: Calc’d = 1930.741; found = 1930.742.
The deprotected Z6-decanoyl vancomycin derivative 9c was isolated as a white residue (11 mg, 24% over two steps). Analytical RP-HPLC retention time of 9c (Method 3): 12.3 min; LC-MS (ESI+) for C76H94Cl2N9O29 [M+H]+: Calc’d = 1602.573; found = 1602.588. (See Supporting Information for HPLC & NMR data)
General procedure 3: Synthesis of G4-acyl vancomycin derivatives 10 – 15. In a flame-dried 10-mL round bottom flask containing a stir bar, 3 (100 mg, 56.2 mmol, 1 equiv) and catalyst 6 (8 mg, 11.6 μmol, 0.2 equiv) were dissolved in THF (2 mL) to yield a clear, homogeneous solution. Then, CH2Cl2 (5 mL) and PEMP (20.3 μL, 112.4 μmol, 2 equiv) were added sequentially to the flask, and stirred under N2 atmosphere. A solution of anhydride (112.6 μmol, 2 equiv) in CH2Cl2 (1 mL) was added over 10 h using a syringe pump, and the reaction was allowed to stir for an additional 14 h. An aliquot (20 μL) was taken and quenched with 2-propanol (500 μL), and analyzed by HR-LC/MS (Method 2) and RP-HPLC (Method 3) to determine if the reaction was complete. At this time, the reaction was quenched with 2-propanol (2 mL). The solution was then concentrated in vacuo to a volume of ~1 mL and diluted with another 2 mL of 2-propanol. The solution was loaded onto a C18 Biotage samplet and purified using RP-MPLC (Method 7). Fractions containing the desired mono-decanoyl vancomycin derivatives were combined and concentrated to yield the mixture of products as a white residue (40 – 50% yield), which was carried forward for the next reaction.
The protected G4-acyl vancomycin derivative (28 μmol, 1 equiv) was dissolved in degassed 1,4-dioxane (2 mL) in a 10-mL round bottom flask. The flask was evacuated, and refilled with argon gas four times, and kept under argon atmosphere. To the flask, Pd(PPh3)2Cl2 (19.7 mg, 28 μmol, 1 equiv), and AcOH (0.5 mL) were added sequentially, and the flask was sealed with a septum. The flask was again evacuated and refilled with argon gas four times, and kept under argon atmosphere. Then, PhSiH3 (157 μL, 1.27 mmol, 45 equiv) was added drop wise over 30 min. The reaction was stirred for another 2 h under argon atmosphere at rt. The reaction mixture turned from a cloudy yellow mixture into a darker brown mixture over the course of the reaction. After 2 h of stirring, the reaction mixture was poured into a separatory funnel containing 0.01 M aqueous HCl (200 mL), and washed with hexanes (3 × 100 mL). The aqueous layer was passed through a pad of celite. The filtrate was concentrated to ~20 mL, and purified using preparative RP-HPLC (Method 5). The fractions containing the desired product were concentrated in vacuo to remove the organic solvent, and the water was removed via freeze-drying to yield a white residue (25 – 32% yield over two steps) in greater than 95% purity based on RP-HPLC (Method 3). (See Supporting Information for HPLC & NMR data)
G4-butyryl-vancomycin derivative (10): Analogue 10 was synthesized using the general procedure 3 and isolated as a white solid (14.5 mg, 17% over two steps). Analytical RP-HPLC retention time (Method 3): 9.7 min; LC-MS (ESI+) for C71H84Cl2N9O25 [M+H]+: Calc’d = 1518.479; found = 1518.469.
G4-pentanoyl-vancomycin derivative (11): Analogue 11 was synthesized using the general procedure 3 and isolated as a white solid (10 mg, 23% over two steps). Analytical RP-HPLC retention time (Method 3): 9.8 min; LC-MS (ESI+) for C71H86Cl2N9O25 [M+H]+: Calc’d = 1532.495; found = 1532.526.
G4-hexanoyl-vancomycin derivative (12): Analogue 12 was synthesized using the general procedure 3 and isolated as a white solid (11 mg, 25% over two steps). Analytical RP-HPLC retention time (Method 3): 11.2 min; LC-MS (ESI+) for C74H90Cl2N9O25 [M+H]+: Calc’d = 1546.511; found = 1546.510.
G4-octanoyl-vancomycin derivative (13): Analogue 13 was synthesized using the general procedure 3 and isolated as a white solid (14 mg, 32% over two steps). Analytical RP-HPLC retention time (Method 3): 13.2 min; LC-MS (ESI+) for C74H90Cl2N9O25 [M+H] : Calc’d = 1574.542; found = 1574.540.
G4-pentenoyl-vancomycin derivative (14): Analogue 14 was synthesized using the general procedure 3 and isolated as a white solid (18 mg, 21% over two steps). Analytical RP-HPLC retention time (Method 3): 10.2 min; LC-MS (ESI+) for C71H84Cl2N9O25 [M+H]+: Calc’d = 1530.479; found =1530.476.
G4-methyladipoyl-vancomycin derivative (15): Analogue 15 was synthesized using the general procedure 3 and isolated as a white solid. Analytical RP-HPLC retention time (Method 3): 10.1 min; LC-MS (ESI+) for C71H86Cl2N9O25 [M+H]+: Calc’d = 1590.500; found = 1590.529.
General procedure 4: Competition studies using Boc-Leu-DAla-DAla-OH, a tripeptide ligand for vancomycin. In a flame-dried small glass vial containing a stir bar, 3 (5 mg, 2.81 μmol, 1 equiv), Boc-Leu-DAla-DAla-OH (1.1 mg, 2.94 μmol, 1 equiv) and peptide catalyst (0.9 μmol, 0.3 equiv) were dissolved in THF (100 μL) to yield a clear, homogeneous solution. Then, CH2Cl2 (300 μL) and PEMP (1.1 μL, 6.07 μmol, 2 equiv) were added to the vial. Decanoic anhydride (2.1 μL, 5.62 μmol, 2 equiv) was added subsequently and the reaction mixture was allowed to stir for 24 h at rt. An aliquot (20 μL) was taken and quenched with 2-propanol (500 μL), and analyzed by HR-LC/MS (Method 2) and RP-HPLC (Method 3). These data were used for preliminary determination of the conversion, product selectivity, and site of reaction (See Supporting Information for HPLC data).
Supplementary Material
Figure 5.

Structures of catalysts identified for selective lipidation.
Figure 6.

RP-HPLC traces for catalyst dependent reactivity of 3 with decanoyl anhydride. (a) G4-selective catalysis with 6, (b) Z6-selective catalysis with 7, and (c) G6-selective catalysis with 8.
Table 1.
Antibacterial activity data for lipidated-vancomycin derivatives against vancomycin-sensitive and vancomycin-resistant bacteria.
| Compound |
S. aureus
(MSSA) ATCC 29213 |
S. aureus
(MRSA) ATCC 43300 |
E. .faecalis
(Van S) ATCC 29212 |
E. faecalis.
(Van B) ATCC 51299 |
E. faecalis
(Van A) MMX 486a |
|---|---|---|---|---|---|
| 9a | 0.25 | 0.25 | 0.5 | 1 | 16 |
| 9b | 0.12 | 0.12 | 0.25 | 0.25 | |
| 9c | 0.25 | 0.5 | 0.25 | 0.5 | 16 |
| 10 | 4 | 4 | 8 | 32 | >64 |
| 11 | 2 | 4 | 4 | 32 | >32 |
| 12 | 2 | 2 | 4 | 16 | >64 |
| 13 | 0.5 | 0.5 | 1 | 8 | >64 |
| 14 | 4 | 4 | 8 | 32 | >32 |
| 15 | 4 | 4 | 8 | 64 | >64 |
| Vancomycin | 1 | >64b | |||
| Teicoplanin | 1 | 0.5 | 0.25 | 0.25 | >64b |
| Linezolid | 4 | 4 | 2 | 2 | 2 |
MMX: Micromyx isolate number.
Vancomycin exhibits an MIC of 512 μg/mL and teicoplanin A2-2 exhibits an MIC of 128 μg/mL against Van A phenotype VRE strain, based on a literature report.37
ACKNOWLEDGMENT
We are grateful to the National Institute of General Medical Sciences of the United States National Institutes of Health (GM-068649) for support. We also wish to thank Dr. Brandon S. Fowler for providing peptide catalysts for the initial screening.
ABBREVIATIONS USED
- NMI
N-methylimidazole
- PEMP
1,2,2,6,6-pentamethylpiperidine
- pmh
p(methyl)- histidine
- DEPT
distortionless enhancement by polarization transfer
- MSSA
methicillin-sensitive Staphylococcus aureus
- methicillin-resistant
Staphylococcus aureus
- VRE
vancomycin-resistant Enterococcus faecalis
Footnotes
Supporting Information Availability:
Figures, schemes and characterization (RP-HPLC data and NMR data) of the compounds are provided. This material is available free of charge via the Internet at: http://pubs.acs.org
REFERENCES
- (1).Li JWH, Vederas JC. Drug discovery and natural products: End of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- (2).Clardy J, Walsh C. Lessons from natural molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- (3).Szychowski J, Truchon J-F, Bennani YL. Natural products in medicine: Transformational outcome of synthetic chemistry. J. Med. Chem. 2014;57:9292–9308. doi: 10.1021/jm500941m. [DOI] [PubMed] [Google Scholar]
- (4).Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pootoolal J, Neu J, Wright GD. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. 2002;42:381–408. doi: 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- (6).Wright GD. Antibiotics: A new hope. Chem. Biol. 2012;19:3–10. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- (7).James RC, Pierce JG, Okano A, Xie J, Boger DL. Redesign of glycopeptide antibiotics: Back to the future. ACS Chem. Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Robles O, Romo D. Chemo- and site-selective derivatizations of natural products enabling biological studies. Nat. Prod. Rep. 2014;31:318–334. doi: 10.1039/c3np70087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mahatthananchai J, Dumas AM, Bode JW. Catalytic selective synthesis. Angew. Chem. Int. Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- (10).Beale TM, Taylor MS. Synthesis of cardiac glycoside analogs by catalyst-controlled, regioselective glycosylation of digitoxin. Org. Lett. 2013;15:1358–1361. doi: 10.1021/ol4003042. [DOI] [PubMed] [Google Scholar]
- (11).Yoganathan S, Yin N, He Y, Mesleh MF, Gu YG, Miller SJ. An efficient chemical synthesis of carboxylate-isostere analogs of daptomycin. Org. Biomol. Chem. 2013;11:4680–4685. doi: 10.1039/c3ob40924d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Thaker MN, Wright GD. Opportunities for Synthetic Biology in Antibiotics: Expanding Glycopeptide Chemical Diversity. ACS Synth. Biol. 2014 doi: 10.1021/sb300092n. DOI: 10.1021/sb300092n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lewis CA, Miller SJ. Site-selective derivatization and remodeling of erythromycin A by using simple peptide-based chiral catalysts. Angew. Chem. Int. Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- (14).Lewis CA, Longcore KE, Miller SJ, Wender PA. An approach to the site-selective diversification of apoptolidin A with peptide-based catalysts. J. Nat. Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fowler BS, Laemmerhold KM, Miller SJ. Catalytic site-selective thiocarbonylations and deoxygenations of vancomycin reveal hydroxyl-dependent conformational effects. J. Am. Chem. Soc. 2012;134:9755–9761. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pathak TP, Miller SJ. Site-selective bromination of vancomycin. J. Am. Chem. Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pathak TP, Miller SJ. Chemical tailoring of teicoplanin with site-selective reactions. J. Am. Chem. Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Han S, Miller SJ. Asymmetric catalysis at a distance: Catalytic, site-selective phosphorylation of teicoplanin. J. Am. Chem. Soc. 2013;135:12414–12421. doi: 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ueda Y, Mishiro K, Yoshida K, Furuta T, Kawabata T. Regioselective diversification of a cardiac glycoside, lanatoside C, by organocatalysis. J. Org. Chem. 2012;77:7850–7857. doi: 10.1021/jo301007x. [DOI] [PubMed] [Google Scholar]
- (20).Yoshida K, Furuta T, Kawabata T. Perfectly regioselective acylation of a cardiac glycoside, digitoxin, via catalytic amplification of the intrinsic reactivity. Tetrahedron Lett. 2010;51:4830–4832. [Google Scholar]
- (21).Wilcock BC, Uno BE, Bromann GL, Clark MJ, Anderson TM, Burke MD. Electronic tuning of site-selectivity. Nat. Chem. 2012;4:996–1003. doi: 10.1038/nchem.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lee D, Williamson CL, Chan L, Taylor MS. Regioselective, borinic acid-catalyzed monoacylation, sulfonylation and alkylation of diols and carbohydrates: Expansion of substrate scope and mechanistic studies. J. Am. Chem. Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- (23).Sculimbrene BR, Morgan AJ, Miller SJ. Enantiodivergence in small-molecule catalysis of asymmetric phosphorylation: Concise total syntheses of the enantiomeric D-myo-inositol-1-phosphate and D-myo-inositol-3-phosphate. J. Am. Chem. Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- (24).Evans JW, Fierman MB, Miller SJ, Ellman JA. Catalytic enantioselective synthesis of sulfinate esters through the dynamic resolution of tert-butanesulfinyl chloride. J. Am. Chem. Soc. 2004;126:8134–8135. doi: 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- (25).Fiori KW, Puchlopek ALA, Miller SJ. Enantioselective sulfonylation reactions mediated by a tetrapeptide catalyst. Nat. Chem. 2009;1:630–634. doi: 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kahne D, Leimkuhler C, Wei L, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- (27).Baltz RH, Miao V, Wrigley SK. Natural products to drugs: Daptomycin and related lipopeptide antibiotics. Nat. Prod. Rep. 2005;22:717–741. doi: 10.1039/b416648p. [DOI] [PubMed] [Google Scholar]
- (28).Nakama Y, Yoshida O, Yoda M, Araki K, Sawada Y, Nakamura J, Xu S, Miura K, Maki H, Arimoto H. Discovery of a novel series of semisynthetic vancomycin derivatives effective against vancomycin-resistant bacteria. J. Med. Chem. 2010;53:2528–2533. doi: 10.1021/jm9017543. [DOI] [PubMed] [Google Scholar]
- (29).Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J. Membrane active vancomycin analogues: A strategy to combat bacterial resistance. J. Med. Chem. 2014;57:4558–4568. doi: 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- (30).Cochrane SA, Lohans CT, Brandelli JR, Mulvey G, Armstrong GD, Vederas JC. Synthesis and structure-activity relationship studies of N-terminal analogues of the antimicrobial peptide tridecaptin A(1) J. Med. Chem. 2014;57:1127–1131. doi: 10.1021/jm401779d. [DOI] [PubMed] [Google Scholar]
- (31).Lunde CS, Hartouni SR, Janc JW, Mammen M, Humphrey PP, Benton BM. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob. Agents Chemother. 2009;53:3375–3383. doi: 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nicolaou KC, Boddy CNC, Brase S, Winssinger N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- (33).Butler MS, Hansford KA, Blaskovich MAT, Halai R, Cooper MA. Glycopeptide antibiotics: Back to the future. J. Antibiot. 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- (34).Nagarajan R, Schabel AA, Occolowitz JL, Counter FT, Ott JL. Synthesis and antibacterial activity of N-acyl vancomycins. J. Antibiot. 1988;41:1430–1438. doi: 10.7164/antibiotics.41.1430. [DOI] [PubMed] [Google Scholar]
- (35).Nagarajan R, Schabel AA, Occolowitz JL, Counter FT, Ott JL, Feltyduckworth AM. Synthesis and antibacterial evaluation of N-alkyl vancomycins. J. Antibiot. 1989;42:63–72. doi: 10.7164/antibiotics.42.63. [DOI] [PubMed] [Google Scholar]
- (36).Williams DH, Bardsley B. The vancomycin group of antibiotics and the fight against resistant bacteria. Angew. Chem. Int. Ed. 1999;38:1173–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- (37).Kerns R, Dong SD, Fukuzawa S, Carbeck J, Kohler J, Silver L, Kahne D. The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc. 2000;122:12608–12609. [Google Scholar]
- (38).Okano A, Nakama Y, Schammel AW, Boger DL. Total synthesis of [Ψ[C(-NH)NH]Tpg4]vancomycin and its (4-chlorobiphenyl)methyl derivative: Impact of peripheral modifications on vancomycin analogues redesigned for dual d-Ala-d-Ala and d-Ala-d-Lac binding. J. Am. Chem. Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pinchman JR, Boger DL. Probing the role of the vancomycin E-ring aryl chloride: Selective divergent synthesis and evaluation of alternatively substituted E-ring analogues. J. Med. Chem. 2013;56:4116–4124. doi: 10.1021/jm4004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ashford P-A, Bew SP. Recent advances in the synthesis of new glycopeptide antibiotics. Chem. Soc. Rev. 2012;41:957–978. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- (41).Liu YC, Li YS, Lyu SY, Hsu LJ, Chen YH, Huang YT, Chan HC, Huang CJ, Chen GH, Chou CC, Tsai MD, Li TL. Interception of teicoplanin oxidation intermediates yields new antimicrobial scaffolds. Nat. Chem. Biol. 2011;7:304–309. doi: 10.1038/nchembio.556. [DOI] [PubMed] [Google Scholar]
- (42).Lyu SY, Liu YC, Chang CY, Huang CJ, Chiu YH, Huang CM, Hsu NS, Lin KH, Wu CJ, Tsai MD, Li TL. Multiple complexes of long aliphatic N-acyltransferases lead to synthesis of 2,6-diacylated/2-acyl-substituted glycopeptide antibiotics, effectively killing vancomycin-resistant Enterococcus. J. Am. Chem. Soc. 2014;136:10989–10995. doi: 10.1021/ja504125v. [DOI] [PubMed] [Google Scholar]
- (43).Sharman GJ, Try AC, Dancer RJ, Cho YR, Staroske T, Bardsley B, Maguire AJ, Cooper MA, O'Brien DP, Williams DH. The roles of dimerization and membrane anchoring in activity of glycopeptide antibiotics against vancomycin-resistant bacteria. J. Am. Chem. Soc. 1997;119:12041–12047. [Google Scholar]
- (44).Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KA, Sandvik E, Hubbard JM, Kaniga K, Schmidt DE, Gao QF, Cass RT, Karr DE, Benton BM, Humphrey PP. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- (46).Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc. Natl. Acad. Sci. USA. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Roy RS, Yang P, Kodali S, Xiong YS, Kim RM, Griffin PR, Onishi HR, Kohler J, Silver LL, Chapman K. Direct interaction of a vancomycin derivative with bacterial enzymes involved in cell wall biosynthesis. Chem. Biol. 2001;8:1095–1106. doi: 10.1016/s1074-5521(01)00075-8. [DOI] [PubMed] [Google Scholar]
- (48).Thompson C, Ge M, Kahne D. Synthesis of vancomycin from the aglycon. J. Am. Chem. Soc. 1999;121:1237–1244. [Google Scholar]
- (49).Griffith BR, Krepel C, Fu X, Blanchard S, Ahmed A, Edmiston CE, Thorson JS. Model for antibiotic optimization via neoglycosylation: Synthesis of liponeoglycopeptides active against VRE. J. Am. Chem. Soc. 2007;129:8150–8155. doi: 10.1021/ja068602r. [DOI] [PubMed] [Google Scholar]
- (50).Davie EAC, Mennen SM, Xu YJ, Miller SJ. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- (51).Miller SJ. In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- (52).Nitanai Y, Kikuchi T, Kakoi K, Hanmaki S, Fujisawa I, Aoki K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J. Mol. Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- (53).Conversion and selectivity data based on uncorrected RP-HPLC analysis at 220 nm.
- (54).Fowler BS, Mikochik PJ, Miller SJ. Peptide-catalyzed kinetic resolution of formamides and thioformamides as an entry to nonracemic amines. J. Am. Chem. Soc. 2010;132:2870–2871. doi: 10.1021/ja9107897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Han S, Le BV, Hajare HS, Baxter RHG, Miller SJ. X-ray crystal structure of teicoplanin A2-2 bound to a catalytic peptide sequence via the carrier protein strategy. J. Org. Chem. 2014;79:8550–8556. doi: 10.1021/jo501625f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pearce CM, Williams DH. Complete assignment of the C-13 nmr-spectrum of vancomycin. J. Chem. Soc. Perkin Trans. 2. 1995:153–157. [Google Scholar]
- (57).Tiyanont K, Doan T, Lazarus MB, Fang X, Rudner DZ, Walker S. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc. Natl. Acad. Sci. USA. 2006;103:11033–11038. doi: 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Holmes NE, Ballard SA, Lam MMC, Johnson PDR, Grayson ML, Stinear TP, Howden BP. Genomic analysis of teicoplanin resistance emerging during treatment of vanB vancomycin-resistant Enterococcus faecium infections in solid organ transplant recipients including donor-derived cases. J. Antimicrob. Chemother. 2013;68:2134–2139. doi: 10.1093/jac/dkt130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.