

Abstract
Pathophysiological conditions that lead to the release of the prototypic damage-associated molecular pattern molecule high mobility group box 1 (HMGB1) also result in activation of poly(ADP-ribose) polymerase 1 (PARP1; now known as ADP-ribosyl transferase 1 [ARTD1]). Persistent activation of PARP1 promotes energy failure and cell death. The role of poly(ADP-ribosyl)ation in HMGB1 release has been explored previously; however, PARP1 is a versatile enzyme and performs several other functions including cross-talk with another nicotinamide adenine dinucleotide- (NAD+) dependent member of the Class III histone deacetylases (HDACs), sirtuin-1 (SIRT1). Previously, it has been shown that the hyperacetylation of HMGB1 is a seminal event prior to its secretion, a process that also is dependent on HDACs. Therefore, in this study, we seek to determine if PARP1 inhibition alters LPS-mediated HMGB1 hyperacetylation and subsequent secretion due to its effect on SIRT1. We demonstrate in an in vitro model that LPS treatment leads to hyperacetylated HMGB1 with concomitant reduction in nuclear HDAC activity. Treatment with PARP1 inhibitors mitigates the LPS-mediated reduction in nuclear HDAC activity and decreases HMGB1 acetylation. By utilizing an NAD+-based mechanism, PARP1 inhibition increases the activity of SIRT1. Consequently, there is an increased nuclear retention and decreased extracellular secretion of HMGB1. We also demonstrate that PARP1 physically interacts with SIRT1. Further confirmation of this data was obtained in a murine model of sepsis, that is, administration of PJ-34, a specific PARP1 inhibitor, led to decreased serum HMGB1 concentrations in mice subjected to cecal ligation and puncture (CLP) as compared with untreated mice. In conclusion, our study provides new insights in understanding the molecular mechanisms of HMGB1 secretion in sepsis.
INTRODUCTION
High mobility group box 1 (HMGB1) is a DNA-binding protein that has been well characterized as a prototypical damage-associated molecular pattern molecule (1–3). The initial characterization of HMGB1 as a late-acting mediator of lipopolysaccharide (LPS)- (4) or sepsis-induced (5) lethality in mice was further affirmed in patients with sepsis and septic shock (6,7). Extracellular HMGB1 activates inflammatory responses in immune cells, endothelial cells and enterocytes (4,8,9). While much attention has been focused on the function of extracellular HMGB1, the mechanisms of HMGB1 release in sepsis have received little consideration (10).
Poly(ADP-ribose) polymerases (PARPs; also known as ADP-ribosyl transferases [ARTDs]) are a family of enzymes found in eukaryotes and prokaryotes that generate ADP-ribose modifications onto acceptor proteins. PARP1 is the most abundant isoform of the PARP enzyme family and, upon activation by genotoxic stimuli, cleaves nicotinamide adenine dinucleotide (NAD+) into nicotinamide (NAM), resulting in the formation of ADP-ribose moieties; these moieties covalently attach to various acceptor proteins, including PARP itself. The continued activation of PARP leads to depletion of its substrate, NAD+ and, consequently, adenosine-5′-triphosphate (ATP), energy failure and cell death (11–20). The benefits conferred by pharmacological inhibitors of poly(ADP-ribosyl)ation in several experimental disease models, including sepsis, further emphasize the potential importance of PARP1 as a pharmacological target (21–44). In addition to posttranslational poly(ADP-ribosyl)ation of nuclear proteins, PARP1 also regulates transcription (45–53). Ditsworth et al., in an in vitro DNA-alkylating damage model, suggested that PARP1 activity may play a role in the nuclear-to-cytosolic translocation of HMGB1 (54). Similarly, Davis et al., demonstrated that the release of HMGB1 to the extracellular milieu requires not only direct protein–protein interaction with PARP1, but also PARP1 enzymatic activity, which results in poly(ADP-ribosyl)ation of HMGB1 (55). However, PARP1 is a versatile enzyme and performs several other functions including cross-talk with another NAD+-dependent member of the Class III histone deacetylases (HDACs), sirtuin-1 (SIRT1) (56–58). In this study, we aim to define the role of PARP1 in the modulation of LPS-mediated HMGB1 expression, more specifically, the effect of PARP1-SIRT1 cross-talk on HMGB1 acetylation. We show in an in vitro model that LPS treatment leads to hyperacetylated HMGB1with concomitant reduction in nuclear HDAC activity. Treatment with PARP1 inhibitor mitigates the LPS-mediated reduction in nuclear HDAC activity and preserves the HDAC activity by increasing SIRT1 activity.
MATERIALS AND METHODS
Cell Culture
The human acute monocytic leukemia cell line THP-1 was purchased from American Type Culture Collection (ATCC #TIB-202; Manassas, VA, USA). For all experiments, cells first underwent a differentiation step by treatment with interferon (IFN)-γ (100 U/mL; Pierce Biotechnology, Rockford, IL, USA) for 18 h. THP-1 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 0.35% β-mercaptoethanol (2-ME), and 2% glutamine, 10 mmol/L 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) (pH 7.2).
Mouse fibroblasts from a strain genetically deficient in PARP1 and fibroblasts from the corresponding wild-type (WT) controls were created by immortalization by a standard 3T3 protocol (59). Cell monolayers were grown at 37°C in 5% CO2 air in Dulbecco’s modified Eagle medium (DMEM) (Gibco [Thermo Fisher Scientific Inc., Waltham, MA, USA) containing 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL).
Studies were performed in tissue culture plates (Becton, Dickinson and Company [BD], Franklin Lakes, NJ, USA) at a density of 1.0 × 106 cells. Where indicated, THP-1 cells were stimulated with 10 μg/mL LPS (Escherichia coli, serotype O55:B5, Sigma-Aldrich, St. Louis, MO, USA). Where indicated, cells were pretreated with PARP1 inhibitor, 5 dihydroxyisoquinoline (DIQ, 100 μmol/L; Sigma-Aldrich), EB-47 (50 and 100 nmol/L; Calbiochem, Billerica, MA, USA), SRT1720 (0.01 and 0.1 μmol/L; Calbiochem) or NAM (5 and 10 mmol/L; Sigma-Aldrich)
Bone Marrow–Derived Macrophage Cultures
Isolation of murine bone marrow–derived macrophages has been described previously (60). Briefly, bone marrow cells were obtained from 129S1/SvImJ, PARP1−/− (Jackson Laboratory, Bar Harbor, ME, USA), mice and were centrifuged in lymphocyte separation medium (Mediatech Inc., Herndon, VA, USA). Cells harvested from the interface were cultured in 75-cm2 flasks in Eagle MEM (EMEM; Mediatech Inc.) supplemented with 10% FBS and 10 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (Thermo Scientific [Thermo Fisher Scientific]). After a 24-h adherence step (Day 2), which allowed for the removal of mature monocytes and fibroblasts from the bone marrow, nonadherent cells from each flask were transferred to a second flask and were supplemented with medium containing GM-CSF (10 ng/mL). The cells were again supplemented with GM-CSF (10 ng/mL) on d 4. After a total of 7 d in culture, macrophages were removed enzymatically with the neutral protease, dispase (Worthington Biochemical Corp., Lakewood, NJ, USA), and collected by gentle scraping. The cells were resuspended in complete EMEM and exposed to experimental conditions. Where indicated, stimulation was performed with 10 μg/mL LPS (Escherichia coli, serotype O55:B5, Sigma-Aldrich) for 18 h.
Nuclear Protein Extraction
All nuclear protein extraction procedures were performed on ice with ice-cold reagents. Cells were washed twice with phosphate-buffered saline (PBS) and harvested by scraping. Cells were pelleted in 1.5 mL of PBS at 268 × g for 5 min. The pellet was washed twice with PBS and resuspended in lysis buffer (10 mmol/L Tris-HCl, pH 7.8, 10 mmol/L KCl, 1 mmol/L ethylenegly-coltetraacetic acid [EGTA], 5 mmol/L MgCl2, 1 mmol/L dithiothreitol (DTT), and 0.5 mmol/L phenylmethylsulfonyl fluoride [PMSF]). The suspension was incubated on ice for 15 min and 10% Non-idet P-40 was added. This was followed by centrifugation at 4°C at 268 × g for 5 min. The supernatant was discarded and the cell pellet was dissolved in extraction buffer (20 mmol/L Tris-HCl, pH 7.8, 32 mmol/L KCl, 0.2 mmol/L EGTA, 5 mmol/L MgCl2, 1 mmol/L DTT, 0.5 mmol/L PMSF and 25% v/v glycerol) was added to the nuclear pellet and incubated on ice for 15 min. Nuclear proteins were isolated by centrifugation at 13,148 × g at 4°C for 10 min. Protein concentrations of the resultant supernatants were determined using the bicinchoninic acid assay (Pierce Biotechnology [Thermo Fisher Scientific]).
Real-Time Reverse Transcriptase-PCR Analysis
Cells were harvested in 1.0 ml of TRIzol Reagent as directed by the manufacturer (Thermo Fisher Scientific). Bromochloro-propane was used for the extraction. The final RNA pellet was dissolved in 50.0 uL of nuclease-free water and quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Extracted RNA (1.0 μg/reaction) was converted to single-stranded cDNA in a 20 μL reaction using the RetroScript Reverse Transcriptase System Kit (Ambion [Thermo Fisher Scientific]) as directed by the manufacturer. The mixture was used in a TaqMan qPCR reaction, according to the manufacturer’s instructions: 10 μL TaqMan Universal PCR Master Mix, 1 μL of each Gene Expression Assay mix, 1 μL cDNA template and 7 μL of water (Applied Biosystems [Thermo Fisher Scientific]). HDAC qPCR primers used were as follows: HDACI-Hs02621185_s1; HDAC2- Hs00231032_m1; HDAC3- Hs00187320_m1 (Life Technologies [Thermo Fisher Scientific]). Human ACTB was used as the endogenous control. PCR reactions were performed in an Applied Biosystems thermocycler 7300 Real-Time PCR System (Thermo Fisher Scientific). After an initial denaturing step of 95°C for 2 min, the following steps were repeated for 40 cycles: a denaturing step of 95°C for 30 s, annealing step of 54°C for 30 s, elongation step of 68°C for 45 s. Each sample was assayed in duplicate and the Ct values were averaged. Lastly, the standard ΔΔCt relative quantification method was used to calculate mRNA levels for all HDACs, normalized to ACTB.
SiRNA-Mediated Inhibition of PARP Expression
In this experiment, cells were incubated for 6 h and transfected with 20 nm siRNA duplexes using Lipofectamine2000 (Invitrogen [Thermo Fisher Scientific]) according to the manufacturer’s instructions. Commercially available stealth small interference RNA (siRNA) sequence for PARP1 (accession number NM_001618) and control sequences were obtained (Life Technologies [Thermo Fisher Scientific]; catalog numbers 4390824 and 4390846). For these experiments, we used differentiated THP-1 cells. Studies were performed in 12-well plates (BD) at a density of 5 × 106 cells. All the experiments were performed 18 h after transfection. The efficiency and specificity of siRNA gene knockdown of PARP1 was determined by real time PCR for PARP1 mRNA and Western blotting for PARP1 expression.
Western Blot Analysis
Concentrated supernatant, whole cell lysates or nuclear extracts were mixed with Laemmli buffer (20% glycerol, 10% β-mercaptoethanol, 5% sodium dodecyl sulfate (SDS), 0.2 mol/L Tris-HCl, pH 6.8, and 0.4% bromphenol blue). After boiling for 10 min, the samples were subjected to 10% SDS-polyacrylamide gel electrophoresis. Proteins were separated on 85% to 16% polyacrylamide gels and subsequently transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Buckinghamshire, UK). For immunoblotting, membranes were blocked with 5% nonfat dried milk in PBS for 1 h. Primary polyclonal rabbit antibody to SIRT1 (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA), primary polyclonal mouse HMGB1 antibody (1:1000; R&D Systems, Minneapolis, MN, USA), β-actin (1:5000, Sigma-Aldrich), polyclonal mouse PAR and PARP1antibody (1:1000, Trevigen, Gaithersburg, MD, USA) were used for Western blot. After washing twice with PBS containing 0.5% Tween 20 (PBST), secondary antibody (horseradish peroxidase-conjugated rabbit anti-mouse immunoglobulin G) (Sigma-Aldrich) was applied at 1:10,000 dilution for 1 h. Blots were washed in PBST thrice for 10 min, incubated in Enhanced Chemiluminescence Reagent (GE Healthcare, Buckinghamshire, UK), and used to expose X-ray film (GE Healthcare).
HDAC and SIRT1 Activity
Histone deacetylase, histone acetyltransferase and SIRT1 activity were measured with the Fluor-de-Lys-green fluorimetric HDAC assay kit, HAT and SIRT1 activity kit (Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer’s instructions.
Serum HMGB1 Concentrations
We measured serum HMGB1 using a commercially available enzyme-linked immunoassay (ELISA) according to the manufacturer’s instructions (ST51011, Shino-Test Corporation, Kanagawa, Japan). We used the normal range standard curve: the limit of detection was 1.0 ng/mL.
Proximity Ligation Assay
PLAs were carried out using a Duolink II detection kit (Olink Bioscience, Uppsala, Sweden) as described previously. The cells are prepared as cytospin slides and two primary antibodies selected from two different host species, rabbit monoclonal SIRT1 antibody (Santa Cruz Biotechnology) and PARP1mouse monoclonal antibody (Enzo Life Sciences), are used together with the Duolink species-specific secondary antibodies. These secondary antibodies contain unique DNA strands that template the hybridization of added oligonucleotides. When in close proximity (<40 nm), the oligonucleotides are ligated by a ligase to form a circular template. This template, still anchored to the antibody, is subsequently amplified and detected using complementary labeled oligonucleotide probes. Detection was performed with a fluorescent microscopy for bright-field detection. The resulting distinct spots are derived from single-molecule protein interaction events, which were visualized.
Immunofluorescence Confocal Microscopy
THP-1cells were grown on 12-mm glass coverslips. After incubation for 20 h in the absence or presence of LPS (in the absence or presence of other compounds), the cells were fixed in 2% paraformaldehyde in PBS for 1 h. The cells were washed three times in PBS and then three times (5 min each) with PBS supplemented with 0.5% bovine serum albumin (PBB). The cells were permeabilized in 0.1% Triton X-100 in PBB for 30 min and then washed once with PBB. Cells were blocked with 2% bovine serum albumin in PBS for 40 min and then washed once with PBB. Rabbit anti-HMGB1 antibodies (1:2000) were added to cells in PBB incubated at room temperature for 1 h. Cells were washed four times in PBB, and then secondary antibodies (goat anti-rabbit Alexa 488; 1:500 dilution; Molecular Probes [Thermo Fisher Scientific]), rhodamine-phalloidin (1:250 dilution; Molecular Probes [Thermo Fisher Scientific]), and the nuclear dye DRAQ5 (1:1000 dilution; Biostatus, Shepshed, UK) were added for 1 h at room temperature. The cells were washed three times in PBB and then three times in PBS. The cells were mounted using gelvatol [23 g of poly(vinyl alcohol) 2000, 50 mL glycerol and 0.1% sodium azide to 100 mL of PBS] and then viewed on a confocal scanning fluorescence microscope (Fluoview 1000; Olympus, Malvern, NY, USA).
Immunoprecipitation
Whole cell lysates (300 μg) that had been harvested in RIPA buffer (10 mmol/L Tris-HCl, pH 8.0, 1.0 mmol/L ethylenedi-aminetetraacetic acid (EDTA), 0.5 mmol/L EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mmol/L NaCl, with 1.0 mmol/L fresh PMSF), were precleared for 1 h with Protein G DynaBeads (Thermo Fisher Scientific). Samples were then incubated with normal, non-specific mouse IgG (20 μL, Santa Cruz Biotechnology; sc-2343), mouse monoclonal anti-HMGB1 antibody (R&D Systems; catalog # MAB1690), rabbit monoclonal SIRT1 antibody (Santa Cruz Biotechnology), or rabbit polyclonal anti-acetylated-lysine antibody (Cell Signaling Technology, Danvers, MA, USA; catalog # 9441S), overnight at 4°C. The next day, the samples were applied to a Protein G DynaBead reaction, and incubated at 4°C to bind the primary antibody. The effluent was reserved, and the Protein G beads were washed three times with sample wash buffer. Samples were then eluted in denatured conditions in 100 μL of 2× SDS sample buffers and analyzed by Western blot as previously described in this manuscript, using a 10% polyacrylamide gel.
Cecal Ligation and Puncture (CLP) Model
Twenty WT mice, 4–6 wks old were subjected to CLP. Mice were anesthetized with a mixture of buprenorphine (0.1 mg/kg) and pentobarbital (90 mg/kg) by subcutaneous and intraperitoneal (IP) injections. Mice were treated with N-(6-oxo-5, 6-dihydro-phenanthridin-2-yl)-N, N-dimethyacetamide HCl (PJ34) (Sigma-Aldrich) 10 mg/kg 3h prior to CLP. Under aseptic conditions, cecal ligation was performed via a short (1 cm) midline incision and cecum is delivered into the wound and ligated 1.0 cm from the tip using a single 4-0 silk suture. The cecum is punctured through and through (that is, front wall and back wall) with a single pass of a 20-gauge hypodermic needle. The cecum was then gently squeezed to extrude a small amount of feces from the perforation sites. The cecum was returned to the peritoneal cavity and the laparotomy was closed with 4.0 silk sutures. The animals were returned to their cages with free access to food and water. Mice were euthanized at 24 h after CLP and blood samples were collected for measuring HMGB1 concentrations.
RESULTS
PARP1 Inhibition (Chemical or Genetic) Decreases LPS-Mediated HMGB1 Secretion
We first investigated the effect of PARP1 inhibition on LPS-mediated HMGB1 secretion and determined dose response inhibition of PARP1 in THP-1 cells. We treated THP-1 cells with a competitive inhibitor of PARP1, 10 to 300 μmol/L (1,5-dihydroxyisoquinoline [DIQ]) and achieved ~80% inhibition of PARP1 activity with 300 μmol/L of DIQ (data not shown). Next, we examined the effect of PARP1 inhibition on HMGB1 production; THP-1 cells were treated with varying concentrations of DIQ for 45 min. Subsequently, they were exposed to 10 μg/mL LPS for 18 h and supernatant HMGB1 levels were assessed by immunoblotting. As expected, cells incubated with LPS demonstrated increased HMGB1 secretion as compared with naïve cells (Figure 1A). However, when the cells were pretreated with DIQ, LPS-induced HMGB1 secretion was significantly attenuated (see Figure 1A). To confirm that PARP1 is responsible for the decrease in HMGB1 supernatant levels, we repeated the experiment with a new potent PARP1 inhibitor, EB-47. Similar to DIQ, pretreatment with EB-47 (50 and 100 nmol/L) led to a significant decrease in LPS-mediated HMGB1 secretion (Figure 1B). Together, these results indicate that chemical inhibition of PARP1 inhibits LPS-induced HMGB1 secretion.
Figure 1.

(A,B) Representative autoradiograph of Western blot analysis for supernatant HMGB1 levels in THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L), a PARP1 inhibitor (Figure 1A). Experiment was repeated by treating THP-1 cells with EB-47, a new potent PARP1 inhibitor and supernatant HMGB1 concentrations were analyzed (Figure 1B). THP-1 cells were treated with LPS (10 μg/mL) for 18 h. The gel is representative of three experiments with similar results. (C) Representative autoradiograph of Western blot analysis for supernatant HMGB1 levels in bone marrow–derived macrophages from WT and PARP1−/− treated with LPS. The gel is representative of three experiments with similar results. (D) Representative Western blot analysis for supernatant HMGB1 concentrations in naïve, nontarget siRNA and PARP1 siRNA transfected THP-1 cells treated with LPS. Cells were treated with LPS (10 μg/mL) for 18 h. The gel is representative of three experiments with similar results. (E) PJ-34 (10 mg/kg, IP, n = 10), or saline (IP, n = 10) was administered 3 h prior to CLP. CLP was performed and blood was collected 24 h later. Serum HMGB1 concentrations were measured by ELISA.
To validate the specificity of PARP1 protein in LPS-mediated HMGB1 secretion, we examined this process in bone marrow–derived macrophages obtained from PARP1−/− and WT mice. Bone marrow–derived macrophages were harvested from WT and PARP1−/− mice and were pretreated with LPS for 18 h and supernatant HMGB1 levels were assessed by immunoblotting. Following LPS stimulation, WT macrophages demonstrated a significant increase in supernatant HMGB1 levels as compared with untreated macrophages. In contrast, PARP1−/− macrophages exposed to LPS demonstrated no detectable HMGB1 levels in the supernatant (Figure 1C). Taken together, the results of our experiments suggest that either chemical inhibition or genetic deletion of PARP1 results in a decrease in LPS-mediated HMGB1 secretion.
To further verify that the decrease in LPS-mediated HMGB1 secretion is not a compensatory response to the missing PARP1 gene, we employed posttranscriptional gene silencing technology by RNA interference. Cells were transfected with siRNA for 12 h, exposed to LPS, and supernatant was collected at 18 h for measuring HMGB1 concentrations. After LPS exposure, naïve cells demonstrated a significant increase in supernatant HMGB1 concentrations. The supernatant HMGB1 concentration in cells transfected with nontarget siRNA was comparable to naïve cells after LPS exposure (Figure 1D). PARP1 siRNA-transfected cells that were exposed to LPS demonstrated a significant decrease in supernatant HMGB1 concentrations as compared with naïve or nontarget siRNA transfected cells (see Figure 1D). Taken together, these data support the view that both genetic and chemical inhibition of PARP1 gene inhibits LPS-induced HMGB1 secretion.
Serum HMGB1 Levels Are Decreased with PARP1 Inhibition in a Murine Model of Sepsis
To investigate the significance of our findings, we tested our hypothesis in an in vivo model of sepsis. CLP is a reliable and clinically relevant model of sepsis that is widely used. The median serum HMGB1 concentrations were significantly higher in the CLP mice as compared with the sham group (160.0 ± 26 ng/mL versus 11.8 ± 3.9 ng/mL; P ≤ 0.05) respectively (Figure 1E). Mice treated with PJ34 demonstrated a significant decrease in serum HMGB1 levels as compared with untreated mice (62.4 ± 22.7 versus 160.0 ± 26 ng/mL; P ≤ 0.05) respectively. Together, these data support the view that inhibition of PARP1 decreases HMGB1 both in vitro and in vivo models of sepsis.
PARP1 Inhibition Has No Significant Effect on LPS-Mediated HMGB1 Transcription
Considering the significant decrease in HMGB1 secretion after PARP1 inhibition, we next sought to define the mechanisms involved in greater detail. Using real time RT-PCR we examined the effect of PARP1 inhibition on LPS-mediated HMGB1 mRNA expression at 3 and 6 h after exposing THP-1 cells to LPS. HMGB1 mRNA levels in THP-1 demonstrated no significant change at 3 and 6 h after stimulation with LPS as compared with naïve cells (Figure 2). DIQ-treated cells demonstrated a slight decrease in HMGB1 mRNA as compared with LPS-stimulated cells, but this was not significant either. These data suggest that the pharmacological inhibition of PARP1 activity has no appreciable effect on HMGB1 gene expression as evidenced by steady-state HMGB1 mRNA levels.
Figure 2.

RT-PCR of HMGB1 expression in THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L), a PARP1 inhibitor. THP-1 cells were treated with LPS (10 μg/mL) for 3 or 6 h. Error bars indicate mean ± SEM
Effect of PARP1 Inhibition on the Compartmentalization of HMGB1 after LPS Stimulation in THP-1 Cells
After LPS exposure, activation of monocytes leads to relocalization of HMGB1 from the nucleus into cytoplasmic organelles that belong to the endolysosomal compartment (61), this is a seminal event prior to HMGB1 release into the extracellular compartment. We investigated the intracellular localization of HMGB1 in LPS-treated THP-1 cells with immunofluorescence. In the basal state, HMGB1 staining was present both in the nuclear and cytoplasmic compartments (Figure 3C), however, following LPS stimulation, the cells demonstrated morphological changes with decreased nuclear and cytoplasmic staining for HMGB1 as compared with naïve cells. In contrast, cells that were pretreated with a PARP1 inhibitor demonstrated a significant increase in nuclear staining for HMGB1 (Figure 3K). Taken together, these findings are consistent with the view that DIQ pretreatment leads to accumulation of HMGB1 in the nucleus and prevents the relocation of HMGB1 from nucleus to the extracellular compartment with LPS treatment.
Figure 3.
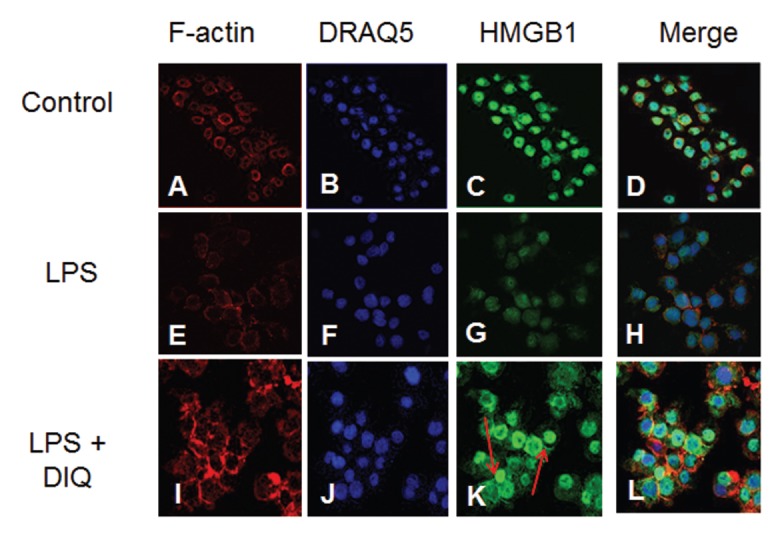
Representative confocal images of THP-1 cells demonstrate the nuclear and cytoplasmic distribution of HMGB1 after LPS exposure in the absence (E–H) or presence of DIQ (300 μmol/L) (I–L). The cells were exposed to 10 μg/mL LPS in the absence or presence of 300 μmol/L DIQ. In these panels, the green staining indicates the presence of HMGB1 and the red staining indicates the presence of F-actin. The nuclear stain DRAQ5 is blue. Red arrows point to an increase in nuclear staining for HMGB1 with PARP1 inhibition.
PARP1 Inhibition Decreases LPS-Mediated HMGB1 Acetylation
The secretion of HMGB1 occurs via a nonclassic secretory pathway, and hyperacetylation of several lysine residues distributed throughout the molecule is a vital step prior to its release. As a next step, we sought to determine whether DIQ pretreatment alters the hyperacetylation status of HMGB1, thereby leading to a decrease in LPS-mediated HMGB1 secretion. In this experiment, we immunoprecipitated nuclear lysates with antibodies against HMGB1, and these immunoprecipitated proteins were immunoblotted with acetyl lysine antibody. As shown in Figure 4A, acetylated HMGB1 was immunoprecipitated efficiently with HMGB1 antibody, and no signal was observed when IgG was used as a control for immunoprecipitation (Figure 4, panel 1, lane 1 versus lane 5). In addition, DIQ pretreatment alone had no appreciable effect (Figure 4, panel 1, lane 3 versus lane 2). Cells incubated with LPS demonstrated an increase in acetylated HMGB1 as compared with unstimulated cells. Coincubation with DIQ demonstrated a significant inhibition of LPS-mediated acetylation of HMGB1 (Figure 4, panel 1, lane 4 versus lane 2). The blot was then stripped and reprobed for HMGB1to demonstrate equal HMGB1 loading in lanes 1–4 (Figure 4, panel 2).
Figure 4.

Coimmunoprecipitation analysis from THP-1 cell lysates. (Panel 1) Samples were immunoprecipitated with anti-HMGB1 and immunoblotted with anti-acetyl-lysine. The blot was then stripped and reprobed for HMGB1 (panel 2). (Panel 3) Samples were immunoprecipitated with an anti-acetyl-lysine antibody and immunoblotted for HMGB1. The blot was then stripped and reprobed for acetyl-lysine (panel 4). The blot shown is representative of three experiments with similar results.
To confirm the results obtained by HMGB1 immunoprecipitation, we conducted the reverse experiment by immunoprecipitating whole cell lysates with an acetyl lysine antibody and subsequent analysis of the immunoprecipitate for HMGB1 by immunoblotting. Whole cell lysates of cells incubated with LPS demonstrated a reduction in acetylated HMGB1 as compared with controls, as some acetylated HMGB1 may have been secreted at 8 h (Figure 4, panel 3, lane 2). Cells pretreated with a PARP1 inhibitor alone demonstrated a slight increase in acetylated HMGB1 content as compared with LPS treated cells (Figure 4, panel 3, lane 3 versus lane 2). Analogous to our finding above, whole cell lysates pretreated with a PARP1 inhibitor prior to LPS exposure demonstrated a decrease in acetylated HMGB1 as compared with untreated cells. The blot was then stripped and reprobed for acetyl lysine to demonstrate equal acetyl lysine loading in lanes 1–4 (Figure 4, panel 4).
Taken together, these results suggest that the decrease in LPS-mediated HMGB1 secretion with PARP1 inhibition is related to a decrease in HMGB1 acetylation leading to nuclear retention of HMGB1 and decreased secretion.
DIQ Pretreatment Abrogates LPS-Mediated Reduction in HDAC Activity
On the basis of the results in Figure 4, we next sought to determine the enzymes that play a role in acetylation of HMGB1. We examined the effect of PARP1 inhibition on the HAT activity of CBP/p300, transcriptional coactivators that can acetylate intranuclear histones and were unable to discern any effect (data not shown). Next, we investigated histone deacetylases, a family of enzymes that can efficiently deacetylate nuclear proteins by removing acetyl groups from lysine residues. Accordingly, for these experiments, we examined the nuclear HDAC activity in THP-1 cells pretreated with DIQ in the presence or absence of LPS by using a colorimetric assay. As shown in Figure 5, LPS-treated THP-1 cells demonstrated a 20% and 30% reduction in nuclear HDAC activity as compared with naïve cells at 4 and 8 h respectively, and this reduction in HDAC activity persisted at 16 h after exposure to LPS. DIQ pretreatment abrogates LPS-mediated reduction in HDAC activity and, furthermore, maintains it at 100% to 120% as compared with naïve cells. Taken together, our results suggest that LPS-mediated suppression in HDAC activity leads to hyperacetylation and subsequent HMGB1 release; in contrast, DIQ pretreatment preserves the HDAC activity leading to decreased supernatant HMGB1 concentrations.
Figure 5.

PARP1 inhibition abrogates LPS-mediated reduction in HDAC activity. Nuclear proteins were extracted from THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L), a PARP1 inhibitor. THP-1 cells were treated with LPS (10 μg/mL) for 4, 8 or 16 h. HDAC activity was determined by colorimetric assay. (*Represents P ≤ 0.05 versus LPS treated cells at the same time point.) Assay shown is representative of three experiments with similar results.
Evankovich et al. had demonstrated previously that HDACI was critical in regulating HMGB1 release in mouse hepatocytes (62). Gene expression for HDACI was examined by performing RT-PCR on mRNA isolated from THP-1 cells at 4 and 8 h after LPS exposure. There was no change in HDAC transcripts for HDACI, II, and III in LPS exposed THP-1 cells with or without pretreatment with DIQ (Figure 6 demonstrates results for HDACI only). Therefore, these data suggest that the PARP1 regulates LPS-mediated HMGB1 hyperacetylation by altering the nuclear acetylation/deacetylation balance, leading to HMGB1 retention in the nucleus.
Figure 6.

RT-PCR of HDACI expression in THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L), a PARP1 inhibitor. THP-1 cells were treated with LPS (10 μg/mL) for 4 or 8 h. Error bars indicate mean ± SEM.
Although basal activation of PARP1 is needed for maintenance of cell function, overactivation of PARP1 consumes NAD+ and results in cell death due to depletion of intracellular NAD+ stores. One class of HDACs that are affected by changes in intracellular levels of NAD+ is the class III HDACs, also called as SIRT1. We next examined the effect of PARP1 inhibition on the nuclear SIRT1 activity in THP-1 cells pretreated with DIQ in the presence or absence of LPS by using a colorimetric assay. As shown in Figure 7A, nuclear SIRT1 activity was reduced to <40% 8 h after LPS treatment as compared with naïve cells. This reduction in SIRT1 activity persisted at 12 h after exposure to LPS. DIQ pretreatment significantly abrogates LPS-mediated reduction in SIRT1 activity and furthermore, maintains it at 66% as compared with untreated cells at 8 h (P ≤ 0.05). At 12 h, the SIRT1 activity is comparable in DIQ untreated and treated cells that were exposed to LPS (P ≥ 0.05) respectively. SIRT1 protein levels were unchanged in LPS-exposed cells treated with DIQ as compared with naïve and untreated cells (Figure 7B), data as shown for 4 and 8 h only. Consistent with this possibility, PARP1−/− cells demonstrated significantly higher SIRT1 activity as compared with WT cells (2.05 ± 0.1 versus 1 ± 0.09 arbitrary units (P ≤ 0.05)) (Figure 7C).
Figure 7.


(A) Nuclear proteins were extracted from THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L) and NAM (5 and 10 mmol/L). THP-1 cells were treated with LPS (10 μg/mL) for 4, 8, 12 h. SIRT1 activity was determined by colorimetric assay. (*Represents P ≤ 0.05 LPS exposed cells in the absence or presence of DIQ; #represents P ≤ 0.05 LPS exposed cells treated with DIQ or DIQ and NAM.) Assay shown is representative of three experiments with similar results. (B) Representative autoradiograph of Western blot analysis for nuclear SIRT1 concentration in THP-1 cells treated with LPS in the presence or absence of DIQ (100 and 300 μmol/L). THP-1 cells were treated with LPS (10 μg/mL) for 4 and 8 h. The gel is representative of three experiments with similar results. (C) Nuclear proteins were extracted from WT and PARP1−/− cells and SIRT1 activity was determined by colorimetric assay. (*Represents P ≤ 0.05 versus WT cells.) (D) Representative autoradiograph of Western blot analysis for supernatant HMGB1 levels in THP-1 cells treated with LPS in the presence or absence of SRT1720 (0.01 and 0.1 μmol/L), a SIRT1 activator. THP-1 cells were treated with LPS (10 μg/mL) for 18 h. The gel is representative of three experiments with similar results.
To better understand the effect of PARP1 inhibition on SIRT1 activity and its dependence on NAD+ concentration, we repeated the experiment in the presence of NAM. NAM is an end product of ADP-ribosylation reactions, a noncompetitive inhibitor of SIRT1, which blocks NAD+ hydrolysis by binding to a conserved pocket adjacent to NAD+ binding site on SIRT1. Importantly, while DIQ pretreated cells exposed to LPS demonstrated an increase in SIRT1 activity, cells pretreated with DIQ and NAM (10 mmol/L) failed to exhibit an increase in SIRT1 activity (66% versus 44%; P > 0.05) (Figure 7A). These findings underscore the importance of NAD+ in mediating the increase in SIRT1 activity with PARP1 inhibition as the addition of NAM leads to an inhibition of NAD+ consumption, thereby blocking the increase in SIRT1 activity.
To further validate these findings, we examined the effect of SIRT1 activation on HMGB1 production. THP-1 cells treated with the SIRT1 activator, SRT1720 for 45 min and were subsequently exposed to 10 μg/mL LPS for 18 h. Supernatant HMGB1 levels were assessed by immunoblotting. As expected, cells incubated with LPS demonstrated increased HMGB1 secretion as compared with naïve cells (Figure 7D). However, when the cells were pretreated with SRT1720, LPS-induced HMGB1 secretion was significantly attenuated (Figure 7D). Taken together, these findings indicate that PARP1 inhibition mitigates LPS-mediated HMGB1 acetylation and subsequent secretion by increasing the deacetylase activity of SIRT1, and along with the results above, provide insight that this mechanism is NAD+ dependent.
PARP1 and SIRT1 Interaction
To examine whether we could detect endogenous complexes between PARP1 and SIRT1 proteins in THP-1 cells in situ, we performed proximity ligation assays (PLA) (63), whereby each red signal is indicative of one detected interaction event. As compared with naïve cells, a slight increase in the red signal, indicating an increase in nuclear complexes of PARP1 and SIRT1 was observed with LPS exposure. Pretreatment with DIQ leads to a significant increase in the fluorescent signal, indicating a stronger interaction between PARP1 and SIRT1. Specificity of the assay was confirmed by testing this interaction in the absence of specific antibodies (Figure 8A).
Figure 8.

(A) Representative confocal images of proximity ligation assay (PLA). Nuclear proteins were extracted from THP-1 cells treated with LPS in the presence or absence of DIQ (300 μmol/L), a PARP1 inhibitor. THP-1 cells were treated with LPS (10 μg/mL) for 6 h. PLA amplification corresponds with the interaction of PARP1 with SIRT1 and is visualized as red-pink spots localized mainly in the nucleus. (B) Coimmunoprecipitation analysis from THP-1 cell lysates. Panel 1: Samples were immunoprecipitated with anti-SIRT1 and immunoblotted with anti-poly(ADP-ribose). The blot was then stripped and reprobed for SIRT1 (panel 2).
To test whether SIRT1 is ADP-ribosylated, we immunoprecipitated nuclear lysates with antibodies against HMGB1, and these immunoprecipitated proteins were immunoblotted with anti-poly(ADP-ribose) antibody. Cells incubated with LPS demonstrated a slight increase in ribosylated SIRT1 as compared with naive cells (Figure 8B, panel 1, lane 2 versus lane 1). Cells treated with PARP1 inhibitor alone demonstrated a slight decrease in the ribosylation of SIRT as compared with LPS treated cells (Figure 8B, panel 1, lane 3 versus lane 2). However, ribosylation of SIRT1 in nuclear extracts of cells that were pretreated with DIQ and subsequently exposed to LPS was similar to untreated cells (Figure 8B, panel 1, lane 4 versus lane 2). Although we did not find any modulation in SIRT1 ribosylation with DIQ pretreatment, the following two mechanisms underlie PARP1 inhibition of LPS-mediated HMGB1 secretion: (1) using an NAD+ dependent mechanism, PARP1 inhibition increases the deacetylase activity of SIRT1; (2) PARP1 interacts with SIRT1 as demonstrated by PLA.
DISCUSSION
Wang et al. first demonstrated the cytokine-like properties of HMGB1 and established HMGB1 as a prototype for endogenous danger signals or the so-called “alarmins” (1–3). Alarmins are cellular proteins rapidly released from cells in response to infection or tissue damage that serve to activate the immune system (64). Active secretion of HMGB1 from monocytes/macrophages begins 8 to 12 h after exposure to the inflammatory stimulus and represents a delayed onset of release as compared with the early proinflammatory mediators. In a murine model of sepsis, circulating HMGB1 levels increase 18 h after induction of peritonitis and remain elevated for 3 d (65). The delayed kinetics of HMGB1 release parallels the onset of animal lethality in animal models of sepsis. Furthermore, treatment with neutralizing anti-HMGB1 antibodies can rescue mice from LPS- or sepsis-induced lethality (4), thereby, solidifying its role as a potential therapeutic target. Collectively, these in vivo and in vitro studies support a paradigm in which HMGB1 plays a pathogenic role in sepsis, and is a late mediator of systemic inflammation. Therefore, fully understanding the molecular mechanisms involved in HMGB1 release will expand our therapeutic armamentarium in sepsis.
In this study, we examine the role of acetylation in HMGB1 release after THP-1cells were treated with LPS. HMGB1 protein lacks a leader signal sequence; therefore, it cannot be secreted via the classical endoplasmic reticulum-Golgi secretory pathway. In resting cells, there is a continuous HMGB1 shuttle between the nucleus and cytoplasm. HMGB1 protein undergoes several posttranslational modifications, for example, acetylation and phosphorylation prior to its nuclear export (66,67). Hassa et al. have previously demonstrated that PARP1-dependent gene expression not only requires the enzymatic activity of p300/CBP but also that PARP1 itself is acetylated in vivo (68). Therefore, we hypothesized that the acetylation of HMGB1 may require the intrinsic HAT activity of CBP/p300 proteins, but we were unable to detect any evidence of alteration of HAT activity in LPS-treated. The coactivator activity of CBP/p300 for gene expression may be dependent on the stimuli and cell type, therefore, we were unable to detect any change in HAT activity.
HMGB1 has two main clusters of lysines that are crucial for acetylation and nuclear localization (69). Bonaldi et al. have previously demonstrated that when the balance between HATs and HDACs is tilted in favor of acetylation, nuclear import of HMGB1 is reduced and it accumulates in vesicles or secretory lysosomes (69). Gardella et al. first noted that HMGB1 accumulates in the cytoplasm of activated monocytes, and is subsequently sequestered into lysosomes prior to its release into the extracellular space (61). We provide evidence that LPS-treated cells demonstrate a significant reduction in nuclear HDAC activity as compared with naïve cells, and this reduction persists even 16 h after exposure to LPS, leading to an increase in HMGB1 secretion. DIQ pretreatment significantly abrogates LPS-mediated reduction in HDAC activity, thereby tilting the balance to deacetylation, consequently leading to reduced LPS-mediated HMGB1 secretion.
In addition to its influence on vital cellular processes, recent studies indicate PARP1 plays a role in gene-specific transcription (49,53,70,71). PARP1 regulates transcription by modifying chromatin-associated proteins, and also functions as a cofactor for transcription factors, most notably, nuclear factor-κB (NF-κB) and activating protein (AP)-1 (46,72). PARP1 is frequently associated with transcriptionally active regions of chromatin suggesting a possible role of poly(ADP-ribosyl)ation in the regulation of transcription (73,74). To this end, we found that pharmacological inhibition of PARP1 activity has no appreciable effect on HMGB1 gene expression as evidenced by steady-state HMGB1 mRNA levels.
It has been demonstrated previously that LPS stimulation induces PARP1 enzymatic activity and the ADP-ribosylation of histones in macrophages (75). Furthermore, the targets of ADP-ribosylation in LPS-stimulated macrophages are PARP1, core histones (76), nucleosome-occupied promoters of IL-1β, MIP-2 and CSF2, facilitating NF-κB recruitment and transcription (76). Similarly, Davis et al., (55) have shown that HMGB1 release to the extracellular milieu requires not only directs protein–protein interaction with PARP1, but also PARP1 enzymatic activity, which results in HMGB1 poly(ADP-ribosyl)ation. Therefore, LPS-mediated ADP-ribosylation of PARP1, HMGB1 and other inflammatory mediators is well published. However, we focus on another function of PARP1, its ability to cross-talk with another NAD+-dependent member of the Class III HDACs, SIRT1 (55–58). The effects of PARP1 inhibition on LPS-mediated HMGB1 secretion were associated with the modulation of HDAC activity, specifically SIRT1. SIRT1 has been implicated in transcriptional silencing, genetic control of aging, cell metabolism and calorie restriction-mediated longevity of the organism.
Both PARP1 and SIRT1 use NAD+ for their activity and are capable of performing several common functions, therefore, cross-talk between these proteins has been suggested (77). PARP and SIRT proteins operate as the “yin and yang” of the cellular response to stress that determines cell survival or death (57,78). Using SIRT1 deficient mouse cardiomyocytes, Rajamohan et al. noted increased levels of PARP1 acetylation in response to mechanical stress (58). However, no similar posttranslational modification has been seen on SIRT1 by PARP1 in response to DNA damage (79). Our results are in agreement with this observation as we were unable to find any changes in ADP-ribosylation of SIRT1 after PARP1 inhibition in LPS-exposed THP-1 cells.
We believe that PARP1 modulates SIRT1 activity by two mechanisms: (a) Cellular NAD+ levels: Since NAD+ serves as a substrate for poly(ADP-ribosyl)ation and NAD+ dependent deacetylases, cellular NAD+ concentrations form a framework for the basis of cross-talk between these proteins. The Km of PARP1 and SIRT1 toward NAD+ is 20–60 (80,81) and 100–200 μmol/L (82) respectively, indicating that PARP1 has a 5- to 10-fold higher affinity for NAD+. Consequently, overactivation of PARP1 with a precipitous drop in cellular NAD+ levels leads to reduced SIRT1 activity (82). Therefore, while PARP1 will continue to consume NAD+, SIRT1 activity will decrease. Conversely, PARP1 chemical or genetic inhibition will increase cellular SIRT1 activity. In our study, we demonstrate that DIQ pretreatment significantly abrogates LPS-mediated reduction in SIRT1 activity. However, cells pretreated with DIQ and NAM failed to show a significant increase in SIRT1 activity. These findings indicate that PARP1 inhibition mitigates LPS-mediated HMGB1 acetylation by increasing the deacetylase activity of SIRT1, and along with the results above, provide insight that this mechanism is NAD+-dependent. (b) PARP1 and SIRT1 interaction: PARP1 and SIRT1 interact with each other in the presence of LPS stimulation; furthermore, this interaction is efficiently enhanced upon inhibition of PARP1.
Finally, there are remarkably few therapeutic inhibitors of HMGB1 release discovered so far. One such inhibitor of HMGB1 release is ethyl pyruvate (EP). Mice subjected to cecal perforation can be rescued from lethal sepsis by treatment with anti-HMGB1 antibodies, HMG A box or EP, in a dose-dependent manner, even when the first dose is administered 24 h after the onset of sepsis (67,81). With such a limited number of clinically efficacious inhibitors available, we wanted to focus on finding new targets. Further understanding of these mechanisms will enable us to design new drugs to neutralize HMGB1, a crucial late mediator of sepsis.
CONCLUSION
To summarize, two distinct mechanisms support our conclusion that PARP1 inhibition decreases LPS-mediated HMGB1 acetylation and its subsequent secretion. These include: (1) Using an NAD+-dependent mechanism, PARP1 inhibition mitigates LPS-mediated HMGB1 acetylation by increasing the deacetylase activity of SIRT1. (2) PARP1 and SIRT1 interact with each other in the presence of LPS stimulation and this interaction is efficiently enhanced upon inhibition of PARP1. These novel findings further elucidate signaling pathways that govern cellular responses to sepsis and open up new avenues to develop targeted therapies to treat sepsis.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant R01GM098474 to RK Aneja).
Footnotes
Online address: http://www.molmed.org
DISCLOSURES
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–73. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Castiglioni A, Canti V, Rovere-Querini P, Manfredi AA. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 343:189–99. doi: 10.1007/s00441-010-1033-1. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 5.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angus DC, et al. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med. 2007;35:1061–7. doi: 10.1097/01.CCM.0000259534.68873.2A. [DOI] [PubMed] [Google Scholar]
- 7.Sunden-Cullberg J, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–73. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- 8.Yang R, et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–14. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S, et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J Physiol Cell Physiol. 2006;290:C990–9. doi: 10.1152/ajpcell.00308.2005. [DOI] [PubMed] [Google Scholar]
- 10.Aneja RK, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;84:1326–34. doi: 10.1189/jlb.0108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–93. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 12.Bakondi E, et al. Detection of poly(ADP-ribose) polymerase activation in oxidatively stressed cells and tissues using biotinylated NAD substrate. J Histochem Cytochem. 2002;50:91–8. doi: 10.1177/002215540205000110. [DOI] [PubMed] [Google Scholar]
- 13.Butler AJ, Ordahl CP. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Mol Cell Biol. 1999;19:296–306. doi: 10.1128/mcb.19.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci. 2002;23:122–9. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- 15.de Murcia G, Menissier de Murcia J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem Sci. 1994;19:172–6. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 16.de Murcia G, et al. Structure and function of poly(ADP-ribose) polymerase. Mol Cell Biochem. 1994;138:15–24. doi: 10.1007/BF00928438. [DOI] [PubMed] [Google Scholar]
- 17.Desmarais Y, Menard L, Lagueux J, Poirier GG. Enzymological properties of poly(ADP-ribose)polymerase: characterization of automodification sites and NADase activity. Biochim Biophys Acta. 1991;1078:179–86. doi: 10.1016/0167-4838(91)99007-f. [DOI] [PubMed] [Google Scholar]
- 18.Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol Life Sci. 2002;59:1534–53. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogata N, Ueda K, Kawaichi M, Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J Biol Chem. 1981;256:4135–7. [PubMed] [Google Scholar]
- 20.Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet. 1999;64:1282–8. doi: 10.1086/302389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zingarelli B, Szabo C, Salzman AL. Blockade of poly(ADP-ribose) synthetase inhibits neutrophil recruitment, oxidant generation, and mucosal injury in murine colitis. Gastroenterology. 1999;116:335–45. doi: 10.1016/s0016-5085(99)70130-7. [DOI] [PubMed] [Google Scholar]
- 22.Zingarelli B, Salzman AL, Szabo C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]
- 23.Zingarelli B, O’Connor M, Wong H, Salzman AL, Szabo C. Peroxynitrite-mediated DNA strand breakage activates poly-adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol. 1996;156:350–8. [PubMed] [Google Scholar]
- 24.Eliasson MJ, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 25.Liaudet L, et al. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc Natl Acad Sci U S A. 2000;97:10203–8. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pieper AA, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:3059–64. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 28.Mota RA, et al. Poly(ADP-ribose) polymerase-1 inhibition increases expression of heat shock proteins and attenuates heat stroke-induced liver injury. Crit Care Med. 2008;36:526–34. doi: 10.1097/01.CCM.0000299735.43699.E9. [DOI] [PubMed] [Google Scholar]
- 29.Jagtap P, et al. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med. 2002;30:1071–82. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 30.Liaudet L, et al. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: long-term morphological and functional consequences. Br J Pharmacol. 2001;133:1424–30. doi: 10.1038/sj.bjp.0704185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliver FJ, et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. Embo J. 1999;18:4446–54. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano FG, et al. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17:286–92. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 33.Stern Y, Salzman A, Cotton RT, Zingarelli B. Protective effect of 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase, against laryngeal injury in rats. Am J Respir Crit Care Med. 1999;160:1743–9. doi: 10.1164/ajrccm.160.5.9902024. [DOI] [PubMed] [Google Scholar]
- 34.Szabo C, et al. Inhibition of poly (ADP-ribose) synthetase attenuates neutrophil recruitment and exerts antiinflammatory effects. J Exp Med. 1997;186:1041–9. doi: 10.1084/jem.186.7.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabo E, et al. Peroxynitrite production, DNA breakage, and poly(ADP-ribose) polymerase activation in a mouse model of oxazolone-induced contact hypersensitivity. J Invest Dermatol. 2001;117:74–80. doi: 10.1046/j.0022-202x.2001.01388.x. [DOI] [PubMed] [Google Scholar]
- 36.Valenzuela MT, et al. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene. 2002;21:1108–16. doi: 10.1038/sj.onc.1205169. [DOI] [PubMed] [Google Scholar]
- 37.Veres B, et al. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem Pharmacol. 2003;65:1373–82. doi: 10.1016/s0006-2952(03)00077-7. [DOI] [PubMed] [Google Scholar]
- 38.Virag L, Salzman AL, Szabo C. Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J Immunol. 1998;161:3753–9. [PubMed] [Google Scholar]
- 39.Virag L, Szabo C. Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotoxicity by zinc chelation. Br J Pharmacol. 1999;126:769–77. doi: 10.1038/sj.bjp.0702332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Virag L, Szabo C. BCL-2 protects peroxynitrite-treated thymocytes from poly(ADP-ribose) synthase (PARS)-independent apoptotic but not from PARS-mediated necrotic cell death. Free Radic Biol Med. 2000;29:704–13. doi: 10.1016/s0891-5849(00)00359-2. [DOI] [PubMed] [Google Scholar]
- 41.Virag L, Szabo C. Purines inhibit poly(ADP-ribose) polymerase activation and modulate oxidant-induced cell death. FASEB J. 2001;15:99–107. doi: 10.1096/fj.00-0299com. [DOI] [PubMed] [Google Scholar]
- 42.Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabo C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc Res. 1997;36:205–15. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- 43.Zingarelli B, et al. Differential regulation of activator protein-1 and heat shock factor-1 in myocardial ischemia and reperfusion injury: role of poly(ADP-ribose) polymerase-1. Am J Physiol Heart Circ Physiol. 2004;286:H1408–15. doi: 10.1152/ajpheart.00953.2003. [DOI] [PubMed] [Google Scholar]
- 44.Zingarelli B, O’Connor M, Hake PW. Inhibitors of poly (ADP-ribose) polymerase modulate signal transduction pathways in colitis. Eur J Pharmacol. 2003;469:183–94. doi: 10.1016/s0014-2999(03)01726-6. [DOI] [PubMed] [Google Scholar]
- 45.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–68. [PMC free article] [PubMed] [Google Scholar]
- 46.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem. 2001;276:45588–97. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- 47.Oei SL, Griesenbeck J, Ziegler M, Schweiger M. A novel function of poly(ADP-ribosyl)ation: silencing of RNA polymerase II-dependent transcription. Biochemistry. 1998;37:1465–9. doi: 10.1021/bi9727390. [DOI] [PubMed] [Google Scholar]
- 48.Andreone TL, O’Connor M, Denenberg A, Hake PW, Zingarelli B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170:2113–20. doi: 10.4049/jimmunol.170.4.2113. [DOI] [PubMed] [Google Scholar]
- 49.Cervellera MN, Sala A. Poly(ADP-ribose) Polymerase Is a B-MYB Coactivator. J Biol Chem. 2000;275:10692–6. doi: 10.1074/jbc.275.14.10692. [DOI] [PubMed] [Google Scholar]
- 50.Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem. 2003;278:45145–53. doi: 10.1074/jbc.M307957200. [DOI] [PubMed] [Google Scholar]
- 51.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–9. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 52.Kannan P, Yu Y, Wankhade S, Tainsky MA. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res. 1999;27:866–74. doi: 10.1093/nar/27.3.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–83. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 54.Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the proinflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282:17845–54. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis K, et al. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med. 2012;18:359–69. doi: 10.2119/molmed.2011.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bai P, Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–5. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 57.Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5:873–7. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- 58.Rajamohan SB, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–29. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang ZQ, et al. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–20. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 60.Robert SM, Sjodin H, Fink MP, Aneja RK. Preconditioning with high mobility group box 1 (HMGB1) induces lipoteichoic acid (LTA) tolerance. J Immunother. 2010;33:663–71. doi: 10.1097/CJI.0b013e3181dcd111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardella S, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Evankovich J, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–97. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 64.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 65.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonaldi T, et al. Monocytic cells hyper-acetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–97. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 68.Hassa PO, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280:40450–64. doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 69.Bonaldi T, et al. Monocytic cells hyper-acetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson MG, Scoggin KE, Simbulan-Rosenthal CM, Steadman JA. Identification of poly(ADP-ribose) polymerase as a transcriptional coactivator of the human T-cell leukemia virus type 1 Tax protein. J Virol. 2000;74:2169–77. doi: 10.1128/jvi.74.5.2169-2177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pavri R, et al. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell. 2005;18:83–96. doi: 10.1016/j.molcel.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 72.Aguilar-Quesada R, et al. Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem. 2007;14:1179–87. doi: 10.2174/092986707780597998. [DOI] [PubMed] [Google Scholar]
- 73.De Lucia F, Mennella MR, Quesada P, Farina B. Poly(ADPribosyl)ation system in transcriptionally active rat testis chromatin fractions. J Cell Biochem. 1996;63:334–41. doi: 10.1002/(sici)1097-4644(19961201)63:3<334::aid-jcb8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 74.Rouleau M, Aubin RA, Poirier GG. Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci. 2004;117:815–25. doi: 10.1242/jcs.01080. [DOI] [PubMed] [Google Scholar]
- 75.Martinez-Zamudio R, Ha HC. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol. 2012;32:2490–502. doi: 10.1128/MCB.06667-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petrilli V, et al. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J Clin Invest. 2004;114:1072–81. doi: 10.1172/JCI21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J. Are poly(ADP-ribosyl)ation by PARP-1 and deacetylation by Sir2 linked? Bioessays. 2003;25:808–14. doi: 10.1002/bies.10317. [DOI] [PubMed] [Google Scholar]
- 78.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–30. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 79.Luna A, Aladjem MI, Kohn KW. SIRT1/ PARP1 crosstalk: connecting DNA damage and metabolism. Genome Integr. 2013;4:6. doi: 10.1186/2041-9414-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ame JC, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–8. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 81.Alvarez-Gonzalez R, Mendoza-Alvarez H. Dissection of ADP-ribose polymer synthesis into individual steps of initiation, elongation, and branching. Biochimie. 1995;77:403–7. doi: 10.1016/0300-9084(96)88153-3. [DOI] [PubMed] [Google Scholar]
- 82.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]