

Abstract
Nosocomial infections pose a significant threat to patient health; however, the gold standard laboratory method for determining bacterial relatedness (pulsed-field gel electrophoresis [PFGE]) remains essentially unchanged 20 years after its introduction. Here, we explored bacterial whole-genome sequencing (WGS) as an alternative approach for molecular strain typing. We compared WGS to PFGE for investigating presumptive outbreaks involving three important pathogens: vancomycin-resistant Enterococcus faecium (n = 19), methicillin-resistant Staphylococcus aureus (n = 17), and Acinetobacter baumannii (n = 15). WGS was highly reproducible (average ≤ 0.39 differences between technical replicates), which enabled a functional, quantitative definition for determining clonality. Strain relatedness data determined by PFGE and WGS roughly correlated, but the resolution of WGS was superior (P = 5.6 × 10−8 to 0.016). Several discordant results were noted between the methods. A total of 28.9% of isolates which were indistinguishable by PFGE were nonclonal by WGS. For A. baumannii, a species known to undergo rapid horizontal gene transfer, 16.2% of isolate pairs considered nonidentical by PFGE were clonal by WGS. Sequencing whole bacterial genomes with single-nucleotide resolution demonstrates that PFGE is prone to false-positive and false-negative results and suggests the need for a new gold standard approach for molecular epidemiological strain typing.
INTRODUCTION
Hospital-acquired infections cause significant patient morbidity and mortality around the world (1–3) and incur substantial burdens with respect to health care costs; current estimates exceed $9.7 billion yearly in the United States alone (3). In addition to preventative measures (2), effective infection control is achieved through active surveillance of infection and recognition of burgeoning disease outbreaks that can be interrupted through health care interventions (1). Molecular epidemiological investigations have become increasingly integral to these functions and rely on methods that can rapidly and effectively assess genetic relatedness among strains (1, 4, 5).
Pulsed-field gel electrophoresis (PFGE) has long been considered the gold standard approach for such investigations (6). Developed at a time when phenotypic strain typing approaches were common (7, 8), PFGE was among the first technologies to propel the era of molecular epidemiology (8). Broadly applicable to bacteria of many different genera, PFGE entails the liberation of intact genomic DNA by in situ lysis within an agarose plug, restriction enzyme digestion, and separation of fragments using pulsed-field gel electrophoresis (7). Pairwise comparisons are made among strains' banding patterns, which are interpreted according to the number of dissimilar restriction fragments that are observed: “indistinguishable” (no difference), “closely related,” “possibly related,” and “different” (signaling the greatest amount of strain divergence) (7). Although alternative methods have been developed to enable molecular typing of bacteria (1, 9, 10) or of particular species (11), none has displaced PFGE as a gold standard approach (8), in part because the approach provides a measurement of differences spanning the entire genome (1).
Despite its utility, there are legitimate criticisms of PFGE. PFGE is both time-consuming and labor-intensive and is technically challenging for many multipurpose clinical laboratories to implement (12, 13). Reproducibility among different laboratories can be difficult to achieve (8, 13–15), and disparities between predicted and achieved PFGE banding patterns have been reported (16). Processing isolates using different restriction enzymes can yield distinct PFGE patterns and, subsequently, discordant interpretations of strain relationships (16). Genetic diversity in closely related outbreak strains (1, 16) or clonally dominant endemic strains (17) may be insufficient to permit their correct interpretation, and some isolates with different phenotypes share the same PFGE pattern (18). PFGE results may correlate poorly with the true relatedness of isolates (16, 19, 20), especially among more distantly related strains (21), and at least one study reported that greater discriminatory power could be obtained through alternative molecular approaches (22). Due in part to these concerns, alternative, amplification-based molecular methods for strain typing of bacteria have been developed, including multilocus variable-number tandem-repeat analysis (MLVA) (23), multilocus sequence typing (MLST) (24), and randomly amplified polymorphic DNA (RAPD) typing (25).
Whole-genome sequencing (WGS) represents a relatively new and increasingly accessible means for tracking disease outbreaks that has garnered success in multiple applied contexts (26–34). Using massively parallel (or “next-generation”) DNA sequencing technologies, it is now possible to examine the complete or nearly complete genomes of bacterial isolates. WGS can theoretically distinguish strains which differ at only a single nucleotide and, in the limited number of studies where direct comparisons with PFGE have been performed, has provided greater resolution (28, 35, 36). However, previous work has focused on individual bacterial species, has employed nonuniform validation methodologies, and has utilized heterogeneous library preparation, sequencing, and data analysis methods, making it difficult to integrate and generalize results across studies or to evaluate its suitability for clinical use.
Here we explored the utility of WGS as a strain typing approach for the clinical laboratory using a single, universal protocol (encompassing library preparation, sequencing, and data analysis) for 3 biologically and genomically distinct organisms: methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), and multidrug-resistant Acinetobacter baumannii. We examined isolates sent to our laboratory for investigation of suspected clinical outbreaks and systematically evaluated WGS results with matched PFGE data.
MATERIALS AND METHODS
Strains.
Three different clinically relevant organisms with phenotypical and genomic properties were selected for this study (Table 1). All strain collections were part of outbreak investigations and were collected from patients with suspected epidemiological relationships. MRSA isolates were derived from sputum (15 isolates), stool (1 isolate), and urine (1 isolate). A. baumannii strains originated from sputum (7 isolates), bronchoalveolar lavage fluid (3 isolates), tissue (2 isolates), a wound (1 isolate), a chest tube drain (1 isolate), and urine (1 isolate). Information on the source of VRE strains was not available. For PFGE analyses, all strains were freshly harvested from primary, single-colony isolates at the time of the outbreak investigation. Strains were cryopreserved until the time of whole-genome sequencing. All strains were maintained on blood agar at 37°C when actively grown. Control strains not spatiotemporally related to the outbreak investigation were selected for each strain collection and were included in all analyses.
TABLE 1.
Characteristics of organisms studied
| Organism | No. of isolates examined | Reference genome size (bp) | GC% | Avg read depth per isolate [range] |
|---|---|---|---|---|
| Vancomycin-resistant Enterococcus (Enterococcus faecium) | 19a | 2,698,137 | 18.99 | 54.77 [43.1–64.1] |
| Methicillin-resistant Staphylococcus aureus | 17 | 2,872,769 | 32.75 | 45.01 [39.2–50.0] |
| Acinetobacter baumannii | 15 | 3,976,747 | 28.83 | 38.26 [30.3–47.3] |
One additional isolate was E. faecalis, not included here.
PFGE.
Agarose plugs were prepared using a rapid method as previously described (37). Genomic DNA was digested with apaI (New England BioLabs, Ipswich, MA) for VRE and A. baumannii or with smaI (New England BioLabs) for MRSA. Restriction fragments were resolved by PFGE using a temperature-controlled Chef-DR III system (Bio-Rad, Hercules, CA). Gels were photographed using a Bio-Rad Gel Doc 1000 system and were interpreted according to standard guidelines (7).
Whole-genome sequencing.
DNA was purified using an UltraClean microbial DNA isolation kit (Mo-Bio, Carlsbad, CA). Genomic DNA (30 to 100 ng) was digested for 60 to 90 min at 37°C in a 10-μl volume using 0.3 μl NEBNext double-stranded DNA (dsDNA) Fragmentase (New England BioLabs). DNA was end repaired and A-tailed in a 40-μl reaction mixture containing 1× Rapid ligation buffer (Enzymatics Inc., Beverly, MA), 0.1675 mM (each) deoxynucleoside triphosphate (dNTP) (New England BioLabs), 0.1 μl Escherichia coli DNA polymerase I (New England BioLabs), 0.5 μl T4 polynucleotide kinase (New England BioLabs), and 0.02 μl Taq DNA polymerase (New England BioLabs) and was incubated at 37°C for 30 min and 72°C for 20 min. Annealed Y-adaptors (0.2 μM) (5′-[PO4]GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG-3′ and 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) were added and ligated at 25°C for 20 min using T4 DNA ligase in Rapid ligation buffer (Enzymatics Inc.). Following purification with Agencourt AMPure XP beads (Beckman Coulter, Brea, CA), PCR amplification was performed with KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Wilmington, MA) using primer PRECAP_FWD_AMP_COMMON (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC-3′) and sample-specific indexed primers (5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXCGGTCTCGGCATTCCTGCTGAACCG-3′, where XXXXXXXX indicates the 8-bp index). Cycling conditions were 95°C for 3 min, 10 cycles of 98°C for 20 s, 65°C for 15 s, and 72°C for 1 min, and 1 cycle of 72°C for 5 min. PCR products were purified using AMPure beads, pooled in equimolar amounts, and sequenced on a MiSeq sequencer (Illumina, San Diego, California) using 150-bp paired-end reads with a custom index primer (5′-AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG-3′). Oligonucleotides were synthesized by IDT. Average read depths obtained are summarized in Table 1.
Data analysis.
Adaptors were trimmed and PCR duplicates removed using Fastq-Mcf (http://code.google.com/p/ea-utils/) with skew filtering disabled and other parameters at their defaults. Sequence reads were aligned to completed reference genomes for Enterococcus faecium DO (GenBank identifier [ID] 389867183), Acinetobacter baumannii strain AYE (GenBank ID 169147133), or Staphylococcus aureus USA300 FPR3757 (GenBank ID 87125858) using bwa (v0.6.2) (38) and samtools (v0.1.19) (39). Reads with a mapping quality value of less than 10 were discarded. Calling of single nucleotide variants and small insertions and deletions (indels) was performed using samtools with a haploid genome model and a minimum variant frequency value of 0.5. Variants supported by fewer than 15 reads or a likelihood score of less than 200 were masked as “unknown” data. All-by-all pairwise distance matrices were constructed by comparing sites of variation among isolates, masking sites at which one or both isolates displayed “unknown” data or less than 15× read coverage, and counting only variant sites for which both isolates in the comparison could be confidently genotyped. Pairwise distances were expressed as the absolute number of passing variant sites which distinguished such pairs, with indels weighted the same as single nucleotide variants. Technical replicates were separately considered in order to determine assay variability, and then paired replicates were merged prior to performing intraisolate comparisons. Approximately maximum-likelihood phylogenetic trees were constructed using FastTree 2.1 (40) (see Fig. S1 in the supplemental material).
Analysis of discrepant strains.
One VRE isolate evidencing poor mapping statistics despite adequate sequence reads was subjected to additional analysis. A de novo assembly was produced for this isolate using AbySS v1.3.5 (41), with parameters empirically optimized to maximize the N50 statistic (the length for which all contigs of equal or larger size contain half the sum of the entire assembly). To perform taxonomic classification of the isolate, contigs in the assembly were compared to the NCBI nonredundant nucleotide database using a BLAST (42) search. The top match for each contig was recorded, as well as the length of that contig in nucleotides. The organism for which the greatest cumulative numbers of nucleotides were identified as the top matches was examined, as were matches corresponding to 70% or more of the matched nucleotides associated with the top hit.
Nucleotide sequence accession number.
Sequence reads generated in this study are available from the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession no. SRP042634.
RESULTS
Reproducibility of WGS.
Inaccuracies in whole-genome sequence data may result from errors that occur during library preparation (43), sequencing (44), and bioinformatic analysis (45). We limited the numbers of artifacts through laboratory procedures, including the use of high-fidelity DNA polymerase and limiting PCR cycling during library preparation, and performed the sequencing analysis using the highly accurate Illumina platform (46). Bioinformatics artifacts were minimized by discarding reads with low mapping quality, using a model-based variant caller tuned to a haploid genome (39), and by imposing quality filtering on variant calls.
We assessed the reproducibility of WGS using technical replicates; duplicate sequencing libraries prepared from separate aliquots of the same genomic DNA were sequenced and analyzed in parallel (Table 2). Technical replicates were produced for all specimens utilized in this study (51 isolates), allowing robust assessment of the inherent variability of the assay. Of the 51 paired technical replicates, 35 were 100% identical at the genomic sequence level. In general, duplicate genome sequences of bacterial isolates differed by less than a single variant, with an average of 0.39 (standard deviation of 0.63) variants distinguishing among paired controls. We did not observe a statistically significant difference in the burden of technical errors in comparing organisms with the highest and lowest frequencies of discordant replicates (A. baumannii and VRE, respectively; P = 0.21 by a 2-tailed t test), indicating that the levels of reproducibility of the assay were similar across all three species. There was only a weak correlation between genome size and the rate of technical errors (R2 = 0.69). We conclude that our protocol is highly reproducible and generalizable across the three different organisms studied.
TABLE 2.
Reproducibility of WGS
| Organism | No. of technical replicates | No. of genomic differencesa |
|---|---|---|
| VRE | 19 | 0.467 ± 0.333 |
| MRSA | 17 | 0.333 ± 0.691 |
| Acinetobacter baumannii | 15 | 0.533 ± 0.533 |
| All organisms | 51 | 0.392 ± 0.629 |
Data represent averages ± standard deviations.
We consequently set a threshold for interpreting isolates as truly clonal, or genomically indistinguishable, corresponding to detection of 3 or fewer pairwise differences between them (equal to the average technical replicate error frequency + 4.15 standard deviations). Given a normal distribution for the number of sequencing errors encountered per isolate, this threshold is sufficiently sensitive that instances in which isolate differences were attributable to technical artifacts alone would be limited to less than <0.0017%.
Correlation of WGS and PFGE.
For each of the strain collections, we considered every possible pairwise comparison of isolates. We identified the PFGE category assignment for each pairwise comparison and tabulated the number of genomic variants identified by WGS which distinguished between the two isolates (Fig. 1 and Table 3).
FIG 1.
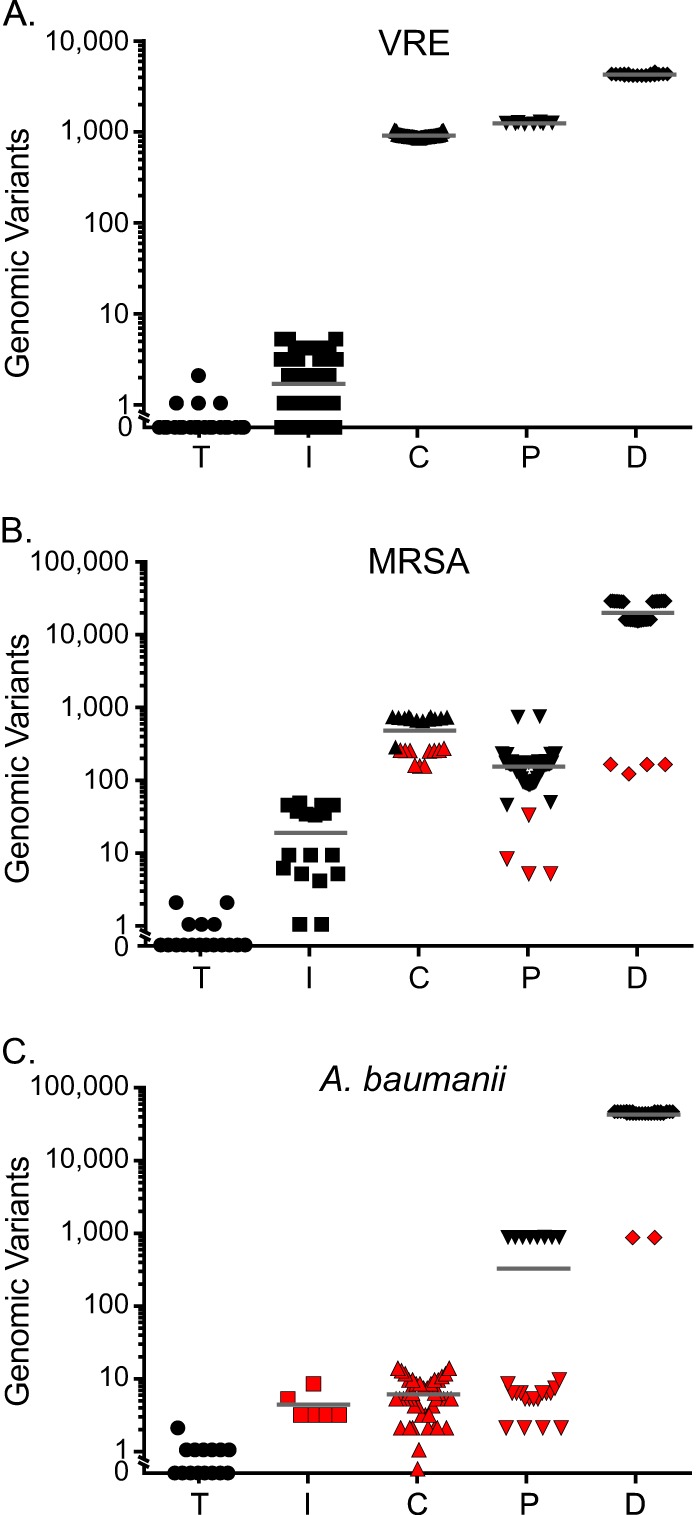
Numbers of disparate genomic polymorphisms detected among pairwise comparisons of technical replicates and different PFGE classification categories. Pairwise comparisons of isolates are stratified according to their status as whole-genome sequencing technical replicates (T) or classification by PFGE as “indistinguishable” (I), “closely related” (C), “possibly related” (P), or “different” (D). The numbers of genomic variants identified by whole-genome sequencing which distinguished isolate pairs are plotted along the y axis. Results are separately shown for VRE (A), MRSA (B), and A. baumannii (C). Red shading indicates a pairwise comparison with a count of genomic differences within 1 standard deviation of the mean observed in a different PFGE category for the same organism.
TABLE 3.
Numbers of genomic polymorphisms detected among PFGE categories
| Organism | No. of genomic polymorphisms by PFGE classificationa: |
|||
|---|---|---|---|---|
| Indistinguishable | Closely related | Possibly related | Different | |
| VRE | 1.625 ± 1.473 (n = 64) | 821.59 ± 41.658 (n = 81) | 1,118.625 ± 21.564 (n = 8) | 3,796.11 ± 87.658 (n = 18) |
| MRSA | 18.15 ± 17.805 (n = 20) | 467.087 ± 234.213 (n = 23) | 149.224 ± 119.201 (n = 58) | 19,446.51 ± 9,101.092 (n = 35) |
| Acinetobacter baumannii | 4.167 ± 1.863 (n = 6) | 5.773 ± 3.267 (n = 44) | 296.963 ± 380.451 (n = 27) | 37,277.180 ± 10,184.950 (n = 28) |
| All organisms | 5.467 ± 10.890 (n = 90) | 523.960 ± 371.877 (n = 148) | 275.505 ± 349.501 (n = 93) | 22,132.33 ± 15,125.14 (n = 81) |
Data represent average numbers of genomic polymorphisms ± standard deviations, with number of pairwise comparisons indicated in parentheses.
The number of genomic differences separating technical replicates was substantially lower than that observed among strains categorized as “indistinguishable” by PFGE for all organisms studied (VRE, P = 5.6 × 10−8; MRSA, P = 0.0004; A. baumannii, P = 0.007 [using 2-tailed t tests]). This finding indicates that WGS is significantly better able to resolve closely related isolates than PFGE.
Additionally, for all organisms, the number of variants identified by WGS correlated roughly with the degree of dissimilarity determined by PFGE. However, the relationship between the two measures was not directly proportional.
In other regards, we observed apparently species-specific differences.
VRE.
VRE demonstrated the most concordant results of comparisons between PFGE and WGS (Fig. 1A and Table 3). The average number of genomic variants distinguishing strains increased consistently with the degree of dissimilarity registered by PFGE. PFGE categories were discrete and well-defined by the number of genomic differences they displayed (“indistinguishable” versus “closely related,” P = 7.6 × 10−9;“closely related” versus “possibly related,” P = 1.9 × 10−6; “possibly related” versus “different,” P = 3.9 × 10−14), with no overlap observed among isolates in different PFGE categories (as defined by isolates demonstrating a number of polymorphisms falling within 1 standard deviation of the mean observed for a different PFGE category).
One VRE isolate demonstrated poor (<0.05%) alignment coverage of the reference genome despite adequate numbers of sequencing reads (>667,000) and was excluded from primary analysis. Unlike the other isolates, further investigation revealed that this organism best matched Enterococcus faecalis, rather than E. faecium. Although the PFGE pattern derived from the strain marked it as different from the others, identifying the erroneous taxonomic classification of this organism would not have been achievable by PFGE.
MRSA.
The average number of genomic polymorphisms that distinguished pairs of MRSA strains correlated less well with the PFGE typing category than was the case for VRE strains, and the distribution of variants observed in each category was greater than that observed for VRE strains (Fig. 1B and Table 3). The average number of pairwise differences among strains classified as “indistinguishable” was considerably higher than that observed for VRE isolates bearing the same PFGE designation (P = 6.9 × 10−4 [2-tailed t test]). Paired comparisons of “possibly related” isolate pairs had on average significantly fewer genomic differences than paired comparisons among strains classified as “closely related” by PFGE (149.2 variants versus 467.1 variants; P = 1.9 × 10−6 [2-tailed t test]), a category defined as demonstrating less divergence.
The numbers of genomic differences separating isolate pairs within specific PFGE groups showed significant overlap. A total of 10 pairwise comparisons in the “closely related” category (43.5% of pairwise comparisons in that category) had a count of genomic differences (range, 152 to 264 variants) that fell within 1 standard deviation of the mean observed for the “possibly related” category (149.22 ± 119.2 variants). Four comparisons in the “different” category (11.4% of such pairwise comparisons; range, 118 to 160 variants) fell within this range and, surprisingly, were also less than 1 standard deviation from the mean for the “closely related” category (232.87 variants). Similarly, 4 “possibly related” isolate pairs (6.9%; range, 5 to 32 variants) were within 1 standard deviation of the mean observed in the “indistinguishable” category (18.15 ± 17.81 variants).
The distributions of pairwise genomic distances observed for the “indistinguishable,” “closely related,” and “different” categories were bimodal (Fig. 1B), indicating an underlying population structure, not evident by PFGE, that is readily detected by WGS with single-nucleotide resolution (see Fig. S1 in the supplemental material).
A. baumannii.
Although summary statistics for the number of genomic differences defining A. baumannii PFGE categories were comparable to those calculated for the other organisms (Table 3), the PFGE results were discordant with the whole-genome data (Fig. 1C).
The numbers of genomic differences that differentiated PFGE categories were least discrete for A. baumannii, and there was substantial overlap in the numbers of polymorphisms demonstrated among separate PFGE categories. The “indistinguishable” and “closely related” PFGE classifications were not significantly different based on the numbers of genomic polymorphisms they exhibited (P = 0.206 [2-tailed t test]). For all 5 (100%) isolate pairs classified as “indistinguishable,” the counts of the distinguishing polymorphisms that they displayed (range, 3 to 8) were within 1 standard deviation of the mean for values exhibited by the “closely related” classification (5.77 ± 3.27 variants). Counts of distinguishing genomic variants for all 5 pairwise comparisons from the “indistinguishable” category and all 44 (100%) paired comparisons deemed in the “closely related” category (range, 0 to 13 variants) fell within 1 standard deviation of the mean value observed for the “possibly related” PFGE classification (296.96 ± 380.45 variants). The “different” category was the category best separated from other PFGE groups; however, two pairs of outliers (793 and 794 variants) fell within the range of genomic differences compatible with the “possibly related” group.
The pairwise comparisons in the “possibly related” category were distributed almost equally across two groups (Fig. 1C). One group was characterized by a higher number of distinguishing mutations and the other by a much lower number, similar to the number of differences observed among the “closely related” PFGE classification.
Evaluation of clonality by PFGE and WGS.
Finally, we compared the abilities of WGS and PFGE to resolve clonal isolates (Table 4), a critical determination in outbreak investigations (1).
TABLE 4.
Numbers of discrepancies between whole-genome sequencing and PFGE for paired-strain comparisons
| Organism | No. of strains |
|||||||
|---|---|---|---|---|---|---|---|---|
| Indistinguishable |
Closely related |
Possibly related |
Different |
|||||
| Clonal by WGS | Nonclonal by WGS | Clonal by WGS | Nonclonal by WGS | Clonal by WGS | Nonclonal by WGS | Clonal by WGS | Nonclonal by WGS | |
| VRE | 55 | 9 | 0 | 81 | 0 | 8 | 0 | 18 |
| MRSA | 5 | 15 | 0 | 23 | 0 | 58 | 0 | 35 |
| Acinetobacter baumannii | 4 | 2 | 12 | 32 | 4 | 23 | 0 | 28 |
| All organisms | 64 | 26 | 12 | 136 | 4 | 89 | 0 | 81 |
Given our empirical definition of clonality, a substantial fraction (28.9%) of the total pairwise comparisons interpreted as “indistinguishable” by PFGE were nonclonal by WGS. This fraction ranged from a low of 14.1% for VRE to 75.0% for MRSA (P < 0.0001 [2-sample Z-test]), suggesting organism-specific bias.
None of the pairwise strain comparisons deemed “different” by PFGE were defined as clonal by WGS for any of the three organisms. For MRSA and VRE, WGS did not identify clonal relationships among the pairs characterized as “closely related,” “possibly related,” or “different” by PFGE. For A. baumannii, in contrast, 27.3% of the strains identified as “closely related” by PFGE and 14.8% of the “possibly related” strains were clonal by WGS.
DISCUSSION
Here we compared the performance of WGS to that of PFGE for bacterial strain typing, using a scalable and species-independent protocol for sequencing and analysis. The study revealed a number of important differences between these two methods with respect to their suitability for application in clinical molecular epidemiology investigations.
Clinical surveillance and investigation of nosocomial outbreaks are primarily dependent on determining whether biogeographically related pairs of strains are clonal and thus on whether they could have originated from the same source or are genomically distinct and could therefore reflect independent transmission events (1). To enable derivation of a functional definition of clonality (genomic identity) from whole-genome sequence data, we set a maximum of 3 distinguishing variants in order to rule out spurious differences resulting from technical artifacts. Although this quantitative definition should be applicable across many different bacterial species, in theory, the probability of sequencing errors should be proportional to the genome size and, to some extent, to the genomic architecture and composition (43); thus, it may need to be altered for evolutionarily divergent or unusual species. The error rates observed in this study were sufficiently low (Table 3) that the results from most (82%) of the technical replicates were 100% identical. The error rates were nonlimiting in our study, and yet further improvements may be achieved by incorporating methods such as PCR-free library preparation (43) or limitation of variant calls to particular portions of microbial genomes (29), though the latter strategy could theoretically negatively impact sensitivity.
Regardless, the issue of what truly constitutes a “bacterial clone” (47) or a “clonal outbreak” (48) is surprisingly complex. Strains may undergo diversification over the course of infection within an individual patient (31); thus, isolates which are genetically distinct but very closely related may reflect a common origin. Presently, there is no established threshold defining the number of genome-wide polymorphisms which identify isolates as belonging to an outbreak (48). Various studies have estimated that the genomes of most bacterial strains accumulate single nucleotide polymorphisms at a rate of roughly 2 to 10 per year, depending on the organism (31, 32, 48, 49). However, organism-specific guidelines for interpreting WGS data for molecular epidemiology investigations will require dedicated, large scale, and long-term observational studies of defined outbreaks among patients, involving multiple isolates from the same patient, in order to determine the rates at which particular species accrue variants during carriage in human hosts (28, 31). For the time being, and in keeping with the existing classification framework developed for PFGE, we propose that strain comparisons based on WGS should be interpreted generally with respect to the following three actionable outcomes: strains are to be considered “genomically indistinguishable” if they are separated by 3 or fewer variants, “closely related” if they are separated by up to 12 variants (based on existing studies of inter- and intrapatient strain variability [31]), and “unrelated” if they are distinguishable by 13 or more variants. As with interpreting the results of any strain typing, molecular epidemiological investigations will require integration of these classifications with additional information and context, notably spatiotemporal information about isolate collection, in order to determine the plausibility of potential transmission events (5). Detailed phylogenomic reconstructions of strain relationships (see Fig. S1 in the supplemental material) are achievable with whole-genome sequence data and provide insight into the likely order of transmission events (26), providing additional information that is useful in outbreak investigations when coupled with biogeographic and temporal data.
It is worth noting differences between WGS and PFGE with respect to DNA acquisition events. Horizontal gene transfer among strains, mediated by plasmids and other mobile genetic elements, and recombination may significantly alter the genomic and phenotypic properties of a strain through single, discrete events. In the context of molecular strain typing, such events may result in shifts to PFGE patterns that are not proportional to the time or degree of strain divergence. In contrast, point mutations detectable by WGS accumulate at a far more predictable rate (47, 50) and consequently serve as a more reliable molecular clock for molecular epidemiology reconstructions (51). Although more-sophisticated whole-genome sequence analysis methods could potentially identify and catalog novel DNA elements acquired among strains (52), on principle alone, assessing the continuum of strain relatedness may consequently be best achieved by WGS and comparison of shared genomic regions among strains.
As a case in point, A. baumannii is known to undergo significant amounts of horizontal gene transfer, even over brief timescales that are compatible with disease outbreaks (53). In comparison to the other organisms studied here, we observed an elevated number of discrepancies between WGS and PFGE results for A. baumannii (Table 4). Of particular note, a substantial fraction of the pairwise comparisons deemed “closely related” by PFGE (12 of 44) were clonal by next-generation sequencing (Table 4). Horizontal DNA transfer among isolates or from external sources is a plausible explanation for these inconsistencies: identification of single nucleotide variants and indels by WGS would ignore changes resulting from gene acquisition or loss, effectively circumventing the confounding effects of horizontal gene transfer, whereas, in contrast, PFGE would be more susceptible to them (47), with the unintended consequence of exaggerating the magnitude of divergence among strains.
Our data indicate that WGS has significantly improved resolving power compared to PFGE (Tables 2 and 3). On average, 467 mutations had accumulated between pairs of MRSA strains, and 822 mutations between pairs of VRE isolates, before a difference was registered by PFGE. A minimum of 152 and 767 genomic polymorphisms were observed among pairs of MRSA and VRE isolates resolved as nonidentical by PFGE, respectively, while up to 47 variants distinguished MRSA isolate pairs categorized as “indistinguishable” by PFGE. Restriction patterns reflect variations in only a relatively small fraction of an organism's genome (1), and our findings suggest that relatively large numbers of sequence variants can accumulate in bacterial genomes before the underlying differences are registered by PFGE.
Importantly, in light of the numbers of genomic polymorphisms detectable by WGS that were able to distinguish strains deemed identical by PFGE (Tables 3 and 4), we conclude that PFGE is prone to false-positive results (i.e., to declaring genomically distinct strains to be clonally related). In the analysis of A. baumannii isolates, PFGE also demonstrated results that are consistent with false negatives (strains which are essentially genomically identical but which are classified as different from one another). In the context of infection control, either kind of erroneous result would carry adverse consequences. False-negative results would prevent the identification of true outbreaks, an obvious threat to patient well-being. False-positive results may conversely indicate an outbreak or nosocomial transmission when there is actually none, resulting in unnecessary patient isolation and other interventions (54). Such risks are not merely hypothetical: recent work suggests that most MRSA infections observed in hospitals are not the result of nosocomial transmission but rather originate from independent infections by distinct strains (29, 33), a conclusion which is anecdotally consistent with the investigation of a suspected MRSA outbreak in this study (Table 4). Similar patterns have been reported for Clostridium difficile infections (55).
Given these results, we argue that WGS should replace PFGE as the gold standard method for bacterial strain typing in molecular epidemiology applications. Analytically, WGS appears to offer superior resolution of strain types and is less prone to false-positive and false-negative findings. Further, genomic differences distinguishing strains can be precisely measured and are highly reproducible, allowing high-resolution inference of phylogenomic relationships (see Fig. S1 in the supplemental material) and of the sequence of transmission events. There are additional practical benefits and considerations. The digital nature of sequencing data allows sharing of sequence information among laboratories, potentially enabling a system capable of identifying large-scale outbreaks analogous to existing PFGE databases such as PulseNet (6). Genomic-sequencing libraries are prepared with protocols similar to current PCR-based strain typing protocols (52), requiring less technical skill than PFGE. WGS can enable exploration of isolates' virulence genes, antibiotic resistance mechanisms, and other medically relevant factors (56), concordantly with molecular epidemiology investigation. The per-sample cost of next-generation sequencing drops rapidly when amortized over even a modest number of specimens: here, up to 30 isolates were multiplexed for a total per-sample reagent cost of ∼$35. WGS and analysis can be completed rapidly and are likely to become more rapid (57), potentially enabling real-time epidemiological investigations: the methods implemented here are compatible with a turnaround time of 3 working days, and the computational analysis methods implemented here are scalable to encompass even large numbers of specimens. Although present barriers to the universal adoption of WGS by clinical laboratories include relatively high costs of instrumentation and a lack of bioinformatic expertise (52, 58), these beneficial capabilities of WGS will potentially enable epidemiological investigation of bacterial outbreaks in real time and with an unprecedented ability to define strain relationships, significantly impacting the practice of infection control and patient outcomes and justifying the additional efforts required for its implementation.
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by grant UL1TR000423 from the National Center For Advancing Translational Sciences of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.03385-14.
REFERENCES
- 1.Singh A, Goering RV, Simjee S, Foley SL, Zervos MJ. 2006. Application of molecular techniques to the study of hospital infection. Clin Microbiol Rev 19:512–530. doi: 10.1128/CMR.00025-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orsi GB, Ciorba V. 2013. Vancomycin resistant enterococci healthcare associated infections. Ann Ig 25:485–492. doi: 10.7416/ai.2013.1948. [DOI] [PubMed] [Google Scholar]
- 3.Zimlichman E, Henderson D, Tamir O, Franz C, Song P, Yamin CK, Keohane C, Denham CR, Bates DW. 2013. Health care-associated infections: a meta-analysis of costs and financial impact on the US health care system. JAMA Intern Med 173:2039–2046. doi: 10.1001/jamainternmed.2013.9763. [DOI] [PubMed] [Google Scholar]
- 4.Field N, Cohen T, Struelens MJ, Palm D, Cookson B, Glynn JR, Gallo V, Ramsay M, Sonnenberg P, Maccannell D, Charlett A, Egger M, Green J, Vineis P, Abubakar I. 2014. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): an extension of the STROBE statement. Lancet Infect Dis 14:341–352. doi: 10.1016/S1473-3099(13)70324-4. [DOI] [PubMed] [Google Scholar]
- 5.Blanc DS. 2004. The use of molecular typing for epidemiological surveillance and investigation of endemic nosocomial infections. Infect Genet Evol 4:193–197. doi: 10.1016/j.meegid.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Swaminathan B, Barrett TJ, Hunter SB, Tauxe RV, CDC PulseNet Task Force . 2001. PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerg Infect Dis 7:382–389. doi: 10.3201/eid0703.010303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33:2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goering RV. 2010. Pulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious disease. Infect Genet Evol 10:866–875. doi: 10.1016/j.meegid.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 9.Ranjbar R, Karami A, Farshad S, Giammanco GM, Mammina C. 2014. Typing methods used in the molecular epidemiology of microbial pathogens: a how-to guide. New Microbiol 37:1–15. [PubMed] [Google Scholar]
- 10.Foley SL, Lynne AM, Nayak R. 2009. Molecular typing methodologies for microbial source tracking and epidemiological investigations of Gram-negative bacterial foodborne pathogens. Infect Genet Evol 9:430–440. doi: 10.1016/j.meegid.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Harmsen D, Claus H, Witte W, Rothganger J, Claus H, Turnwald D, Vogel U. 2003. Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J Clin Microbiol 41:5442–5448. doi: 10.1128/JCM.41.12.5442-5448.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strandén A, Frei R, Widmer AF. 2003. Molecular typing of methicillin-resistant Staphylococcus aureus: can PCR replace pulsed-field gel electrophoresis? J Clin Microbiol 41:3181–3186. doi: 10.1128/JCM.41.7.3181-3186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.te Witt R, van Belkum A, MacKay WG, Wallace PS, van Leeuwen WB. 2010. External quality assessment of the molecular diagnostics and genotyping of meticillin-resistant Staphylococcus aureus. Eur J Clin Microbiol Infect Dis 29:295–300. doi: 10.1007/s10096-009-0856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung M, de Lencastre H, Matthews P, Tomasz A, Adamsson I, Aires de Sousa M, Camou T, Cocuzza C, Corso A, Couto I, Dominguez A, Gniadkowski M, Goering R, Gomes A, Kikuchi K, Marchese A, Mato R, Melter O, Oliveira D, Palacio R, Sá-Leão R, Santos Sanches I, Song JH, Tassios PT, Villari P, Multilaboratory Project Collaborators . 2000. Molecular typing of methicillin-resistant Staphylococcus aureus by pulsed-field gel electrophoresis: comparison of results obtained in a multilaboratory effort using identical protocols and MRSA strains. Microb Drug Resist 6:189–198. doi: 10.1089/mdr.2000.6.189. [DOI] [PubMed] [Google Scholar]
- 15.Cookson BD, Aparicio P, Deplano A, Struelens M, Goering R, Marples R. 1996. Inter-centre comparison of pulsed-field gel electrophoresis for the typing of methicillin-resistant Staphylococcus aureus. J Med Microbiol 44:179–184. doi: 10.1099/00222615-44-3-179. [DOI] [PubMed] [Google Scholar]
- 16.Davis MA, Hancock DD, Besser TE, Call DR. 2003. Evaluation of pulsed-field gel electrophoresis as a tool for determining the degree of genetic relatedness between strains of Escherichia coli O157:H7. J Clin Microbiol 41:1843–1849. doi: 10.1128/JCM.41.5.1843-1849.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tenover FC, Goering RV. 2009. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J Antimicrob Chemother 64:441–446. doi: 10.1093/jac/dkp241. [DOI] [PubMed] [Google Scholar]
- 18.Thong KL, Goh YL, Radu S, Noorzaleha S, Yasin R, Koh YT, Lim VK, Rusul G, Puthucheary SD. 2002. Genetic diversity of clinical and environmental strains of Salmonella enterica serotype Weltevreden isolated in Malaysia. J Clin Microbiol 40:2498–2503. doi: 10.1128/JCM.40.7.2498-2503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson JK, Arduino SM, Stine OC, Johnson JA, Harris AD. 2007. Multilocus sequence typing compared to pulsed-field gel electrophoresis for molecular typing of Pseudomonas aeruginosa. J Clin Microbiol 45:3707–3712. doi: 10.1128/JCM.00560-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trujillo S, Keys CE, Brown EW. 2011. Evaluation of the taxonomic utility of six-enzyme pulsed-field gel electrophoresis in reconstructing Salmonella subspecies phylogeny. Infect Genet Evol 11:92–102. doi: 10.1016/j.meegid.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Blanc DS, Francioli P, Hauser PM. 2002. Poor value of pulsed-field gel electrophoresis to investigate long-term scale epidemiology of methicillin-resistant Staphylococcus aureus. Infect Genet Evol 2:145–148. doi: 10.1016/S1567-1348(02)00093-X. [DOI] [PubMed] [Google Scholar]
- 22.Nemoy LL, Kotetishvili M, Tigno J, Keefer-Norris A, Harris AD, Perencevich EN, Johnson JA, Torpey D, Sulakvelidze A, Morris JG Jr, Stine OC. 2005. Multilocus sequence typing versus pulsed-field gel electrophoresis for characterization of extended-spectrum beta-lactamase-producing Escherichia coli isolates. J Clin Microbiol 43:1776–1781. doi: 10.1128/JCM.43.4.1776-1781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francois P, Huyghe A, Charbonnier Y, Bento M, Herzig S, Topolski I, Fleury B, Lew D, Vaudaux P, Harbarth S, van Leeuwen W, van Belkum A, Blanc DS, Pittet D, Schrenzel J. 2005. Use of an automated multiple-locus, variable-number tandem repeat-based method for rapid and high-throughput genotyping of Staphylococcus aureus isolates. J Clin Microbiol 43:3346–3355. doi: 10.1128/JCM.43.7.3346-3355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbier N, Saulnier P, Chachaty E, Dumontier S, Andremont A. 1996. Random amplified polymorphic DNA typing versus pulsed-field gel electrophoresis for epidemiological typing of vancomycin-resistant enterococci. J Clin Microbiol 34:1096–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris SR, Cartwright EJ, Torok ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13:130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, Bowden R, Walker AS, Peto TE, Paul J, Llewelyn MJ. 2014. Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin Infect Dis 58:609–618. doi: 10.1093/cid/cit807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Moller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 365:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. 2013. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis 13:137–146. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holden MT, Hsu LY, Kurt K, Weinert LA, Mather AE, Harris SR, Strommenger B, Layer F, Witte W, de Lencastre H, Skov R, Westh H, Zemlickova H, Coombs G, Kearns AM, Hill RL, Edgeworth J, Gould I, Gant V, Cooke J, Edwards GF, McAdam PR, Templeton KE, McCann A, Zhou Z, Castillo-Ramirez S, Feil EJ, Hudson LO, Enright MC, Balloux F, Aanensen DM, Spratt BG, Fitzgerald JR, Parkhill J, Achtman M, Bentley SD, Nubel U. 2013. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res 23:653–664. doi: 10.1101/gr.147710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long SW, Beres SB, Olsen RJ, Musser JM. 2014. Absence of patient-to-patient intrahospital transmission of Staphylococcus aureus as determined by whole-genome sequencing. mBio 5:e01692-14. doi: 10.1128/mBio.01692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salipante SJ, Roach DJ, Kitzman JO, Snyder MW, Stackhouse B, Butler-Wu SM, Lee C, Cookson BT, Shendure J. 4 November 2014, posting date. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res doi: 10.1101/gr.180190.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pendleton S, Hanning I, Biswas D, Ricke SC. 2013. Evaluation of whole-genome sequencing as a genotyping tool for Campylobacter jejuni in comparison with pulsed-field gel electrophoresis and flaA typing. Poult Sci 92:573–580. doi: 10.3382/ps.2012-02695. [DOI] [PubMed] [Google Scholar]
- 36.Leekitcharoenphon P, Nielsen EM, Kaas RS, Lund O, Aarestrup FM. 2014. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One 9:e87991. doi: 10.1371/journal.pone.0087991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goering RV, Winters MA. 1992. Rapid method for epidemiological evaluation of gram-positive cocci by field inversion gel electrophoresis. J Clin Microbiol 30:577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 43.Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods 6:291–295. doi: 10.1038/nmeth.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiatt JB, Pritchard CC, Salipante SJ, O'Roak BJ, Shendure J. 2013. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res 23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Homer N, Nelson SF. 2010. Improved variant discovery through local re-alignment of short-read next-generation sequencing data using SRMA. Genome Biol 11:R99. doi: 10.1186/gb-2010-11-10-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam HY, Clark MJ, Chen R, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, Butte AJ, Ji HP, Snyder M. 2012. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol 30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spratt BG. 2004. Exploring the concept of clonality in bacteria. Methods Mol Biol 266:323–352. [DOI] [PubMed] [Google Scholar]
- 48.Lindsay JA. 7 May 2013, posting date. Evolution of Staphylococcus aureus and MRSA during outbreaks. Infect Genet Evol doi: 10.1016/j.meegid.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 49.Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 50.Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar S. 2005. Molecular clocks: four decades of evolution. Nat Rev Genet 6:654–662. doi: 10.1038/nrg1659. [DOI] [PubMed] [Google Scholar]
- 52.SenGupta DJ, Cummings LA, Hoogestraat DR, Butler-Wu SM, Shendure J, Cookson BT, Salipante SJ. 21 May 2014. Whole-genome sequencing for high-resolution investigation of methicillin-resistant Staphylococcus aureus epidemiology and genome plasticity. J Clin Microbiol doi: 10.1128/JCM.00759-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valenzuela JK, Thomas L, Partridge SR, van der Reijden T, Dijkshoorn L, Iredell J. 2007. Horizontal gene transfer in a polyclonal outbreak of carbapenem-resistant Acinetobacter baumannii. J Clin Microbiol 45:453–460. doi: 10.1128/JCM.01971-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Page K, Graves N, Halton K, Barnett AG. 2013. Humans, ‘things’ and space: costing hospital infection control interventions. J Hosp Infect 84:200–205. doi: 10.1016/j.jhin.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 55.Walker AS, Eyre DW, Wyllie DH, Dingle KE, Harding RM, O'Connor L, Griffiths D, Vaughan A, Finney J, Wilcox MH, Crook DW, Peto TE. 2012. Characterisation of Clostridium difficile hospital ward-based transmission using extensive epidemiological data and molecular typing. PLoS Med 9:e1001172. doi: 10.1371/journal.pmed.1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Chiodini R, Badr A, Zhang G. 2011. The impact of next-generation sequencing on genomics. J Genet Genomics 38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fricke WF, Rasko DA. 26 November 2013, posting date. Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat Rev Genet doi: 10.1038/nrg3624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.