

Abstract
In bacteria, disulfide bonds confer stability on many proteins exported to the cell envelope or beyond. These proteins include numerous bacterial virulence factors. Thus, bacterial enzymes that promote disulfide bond formation represent targets for compounds inhibiting bacterial virulence. Here, we describe a novel target- and cell-based screening methodology for identifying compounds that inhibit the disulfide bond-forming enzymes E. coli DsbB (EcDsbB) or M. tuberculosis VKOR (MtbVKOR). MtbVKOR can replace EcDsbB although the two are not homologues. Initial screening of 51,487 compounds yielded six specifically inhibiting EcDsbB. These compounds share a structural motif and do not inhibit MtbVKOR. A medicinal chemistry approach led us to select related compounds some of which are much more effective DsbB inhibitors than those found in the screen. These compounds inhibit purified DsbB and prevent anaerobic E. coli growth. Furthermore, these compounds inhibit all but one of the DsbBs of nine other gram-negative pathogenic bacteria tested.
Introduction
Disulfide bonds between pairs of cysteines contribute importantly to the folding and stability of many proteins. In bacteria, this advantageous modification is generally limited to proteins that are exported to the cell envelope or beyond. Many proteins involved in bacterial virulence (such as toxins, adhesins, flagella, fimbriae, pili, and type II and III secretion systems) require disulfide bonds for their stability and activity1. Thus, inactivation of enzymes that make protein disulfide bonds interferes with the activity of multiple bacterial virulence factors. Inhibitors of these enzymes could have profound effects on pathogen virulence.
In Gram-negative bacteria, disulfide bonds are introduced into substrate proteins as they cross through the cytoplasmic membrane into the cell envelope2,3. The periplasmic enzyme DsbA, a member of the thioredoxin family, oxidizes pairs of cysteines in substrate proteins through its Cys-X-X-Cys active site4. The resulting reduced DsbA is re-oxidized by the membrane protein DsbB, regenerating DsbA’s activity. DsbB itself is reoxidized by membrane-imbedded quinones, from which electrons are transferred to the electron transport chain (Figure 1).
Figure 1.
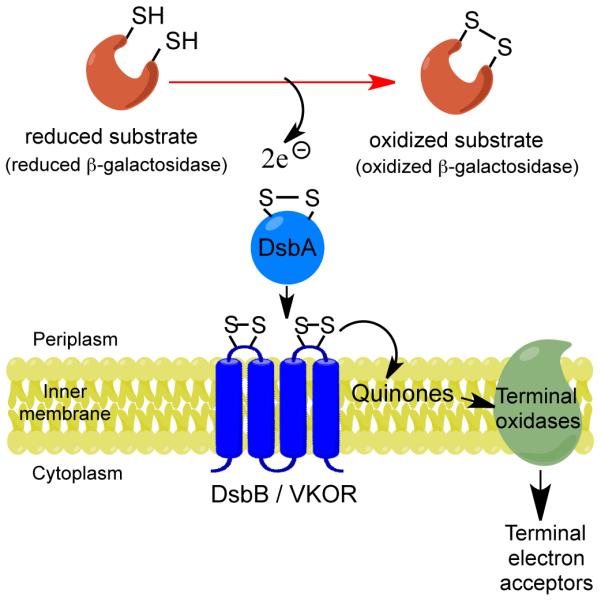
DsbB pathway and screening basis. E. coli disulfide bond formation pathway with endogenous EcDsbB or exogenous MtbVKOR enzyme. Black arrows indicate the flow of electrons. One DsbA substrate in the screening strain is β-Galdbs (see text for details). Contrary to the natural DsbA-substrates, β-Galdbs is inactive when oxidized; hence colonies are white when growing on X-Gal agar growth media.
While the DsbB/DsbA system is widespread in bacteria, some bacteria, e.g. the Actinobacteria and Cyanobacteria, use a somewhat different pathway5. This alternate pathway retains a DsbA-like protein, but uses the membrane protein VKOR instead of DsbB to oxidize DsbA6-8. Bacterial VKOR is a homologue of the vertebrate protein, vitamin K epoxide reductase, an endoplasmic reticular enzyme involved in blood coagulation and the target of the anticoagulant warfarin (Coumadin©). While bacterial VKORs show no homology to DsbB, like DsbB they encode two extra-cytoplasmic soluble domains containing essential pairs of cysteines, one of which is a Cys-X-X-Cys motif. The interactions between the redox-active cysteines of DsbA and VKOR proceed by the same steps seen between DsbA and DsbB7. Therefore, a vkor gene from Mycobacterium tuberculosis (MtbVKOR) expressed in E. coli complements a dsbB null mutant; while E. coli DsbB (EcDsbB) complements a vkor null mutant of Mycobacterium smegmatis9.
Although some bacterial VKORs, like their eukaryotic homologues, are sensitive to coumarin-based anticoagulants, EcDsbB is not6,9. Furthermore, VKOR is essential for Mycobacterial growth9,10, while neither DsbB nor DsbA is essential for aerobic E. coli growth.
The fact that EcDsbB and MtbVKOR are functionally homologous but do not share amino acid homology suggested a methodology for identifying specific inhibitors of either enzyme. This methodology is greatly facilitated by the ability to detect inhibitory effects on disulfide bond formation in growing E. coli cells. The methodology is further enhanced by a sensitive assay for disulfide bond formation in E. coli, provided by a version of the enzyme β-galactosidase that is exported to the E. coli periplasm where it is inactivated by the introduction of non-native disulfide bonds11. This disulfide-sensitive β-galactosidase (β-Galdbs ) is the product of a hybrid gene encoding a β-galactosidase fused to a periplasmic domain of the membrane protein MalF4,12. In E. coli cells with an intact disulfide bond pathway, the activity of β-Galdbs is two to three orders of magnitude lower than when disulfide bond-forming enzymes are absent. Thus, wild-type cells expressing the β-Galdbs form white colonies on agar media that contain the chromogenic β-galactosidase indicator, X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) while dsb mutant colonies expressing the same β-Galdbs are blue. Importantly for limiting the array of targets in this screen, our previous genetic studies revealed that only null mutations in dsbA or dsbB restore high levels of β-galactosidase activity. Much weaker restoration of β-galactosidase activity results from certain non-null mutations of the dsbA or dsbB genes or in genes encoding proteins required for cytoplasmic membrane protein assembly12,13. These latter mutations restore only ~1% of the β-galactosidase activity, presumably because strong mutations in these genes are lethal.
Employing this highly sensitive assay of disulfide bond activity, we carried out a High Throughput Screen (HTS) to identify compounds potentially useful in the development of antibiotics. The rationale follows: 1)Disulfide-bonded proteins are important for bacterial virulence; 2)Detecting inhibition of MtbVKOR or EcDsbB can be achieved in growing E. coli cells; 3)Detecting high levels of β-Galdbs activity requires strong inhibition of either MtbVKOR or EcDsbB; 4) Strong inhibitors of other pathways that restore β-galactosidase activity are not likely to be detected; 5)Screening compounds in parallel for inhibition of the non-homologous MtbVKOR and EcDsbB enzymes provides reciprocal controls, allowing us to tentatively eliminate inhibitors that are influencing β-galactosidase activity by affecting membrane protein assembly or acting directly on DsbA. Such inhibitors would show up as hits in the screen for both strains while specific inhibitors would only register as hits against one strain or the other.
Here, we report the results of a screen of 51,487 compounds that yielded six specific inhibitors of EcDsbB, but none of MtbVKOR. Chemical modifications led to more potent inhibitors that directly inhibit purified EcDsbB activity and inhibit anaerobic growth of E. coli. Furthermore, these small molecules inhibit DsbBs of other gram-negative pathogenic bacteria.
Results
Target- and cell-based screen using agar-multiwell plates
We screened inhibitors of disulfide bond formation in two E. coli strains in parallel. One expresses chromosomally encoded EcDsbB and the other is deleted for the dsbB gene but complemented by the Mtbvkor gene expressed from an IPTG-inducible promoter. Both strains express the β-Galdbs from the chromosome. In these strains, strong inhibition of disulfide bond formation should lead to a substantial increase in β-galactosidase activity4,12. Since the enzyme directly responsible for disulfide bond formation in both strains is DsbA, inhibitors of DsbA or of other processes enhancing the activity of β-galactosidase would raise the activity in both MtbVKOR- and EcDsbB-based strains. In contrast, compounds that specifically inhibit EcDsbB would increase β-galactosidase activity in the EcDsbB-dependent strain but not in the MtbVKOR-dependent strain and vice versa. Assaying the effects of compounds on the two strains in parallel allowed us to pick candidate inhibitors that were specific to either EcDsbB or MtbVKOR. Therefore, we eliminate from further consideration compounds giving positive signals with both strains, even though these might include compounds that inhibit both enzymes directly.
Initially, we compared screens with 384-well plates in which wells contained liquid or agar-based growth media. It appeared that aeration interfered with growth in the liquid media leading us to choose the agar media where growth occurred on the media surface. When these mini-agar wells included X-Gal, high levels of β-galactosidase activity in the presence of an inhibitor were readily seen in the blue color resulting from X-Gal hydrolysis (Supplementary Results, Supplementary Figure 1). Using such agar-filled wells required special procedures that quickly became routine (Online Methods).
We observed that while the EcDsbB strain exhibited no visible trace of blue in the wells, the MtbVKOR-complemented strain did show a pale blue color, indicating that the MtbVKOR did not completely restore levels of disulfide bond formation9. This difference did not interfere with the principles of the screen, but did result in a greater number of false positives in the MtbVKOR screen.
The HTS was carried out with 50,374 compounds from the collection of Harvard University’s Institute for Chemistry and Chemical Biology (ICCB) and 1,113 compounds from the National Institute of Allergy and Infectious Diseases (NIAID) collection of inhibitors of M. tuberculosis H37Rv growth14. Each compound was assayed in duplicate for each strain. Only a small number of EcDsbB hits were observed, seen easily by the clear blue/white difference. Because of the weaker expression of MtbVKOR, real potential hits with the MtbVKOR-based strain were harder to distinguish from the light blue background color. We initially set a very low threshold for assigning hits (i.e. anything bluer than the background) and identified 11 (hit rate of 0.02%) and 150 (hit rate of 0.29%) potential inhibitors of EcDsbB and MtbVKOR, respectively. The much higher rate in the case of MtbVKOR is explained by the lower effectiveness in its oxidation of DsbA mentioned above and in the Discussion.
To verify as potential inhibitors the compounds identified in the screen, we retested them in the same assay. Failure to retest positively reduced the numbers of inhibitors of EcDsbB and MtbVKOR to 8 and 62, respectively. Other candidate compounds were discarded after careful retesting showed inhibition of both EcDsbB and MtbVKOR. Evaluation of the possible chemical reactivity and stability of the hits helped us further reduce our sets of candidates, leaving us with four potential inhibitors of MtbVKOR (#1-4) and seven of EcDsbB (#5-11).
Testing for candidate inhibitors of MtbVKOR
We examined the potential MtbVKOR inhibitors to determine whether any might be lead compounds for antibiotic development against tuberculosis since MtbVKOR is essential for M. tuberculosis growth. We tested these compounds (#1-4) and the potential EcDsbB inhibitors (#5-11) for their effects on M. tuberculosis growth in three different media (Supplementary Results, Supplementary Table 1). Compound 4, obtained from the NIAID collection, strongly inhibited M. tuberculosis growth, but was also a weak inhibitor of EcDsbB, eliminating it from further study. Compounds 2 and 3 were eliminated since they did not affect M. tuberculosis growth. The EcDsbB inhibitors (#5-10) showed little to no inhibition of M.tuberculosis growth as expected.
For a potential MtbVKOR inhibitor to be a useful antibiotic, it should not have anticoagulant activity (as does warfarin). To test for anticoagulant activity, we assessed the activity of compound 1 in inhibiting the VKOR activity present in mouse liver (Supplementary Results, Supplementary Figure 2a). We also assessed the effect of compound 1 on insulin reduction by human Protein Disulfide Isomerase (PDI) which is a thiol-redox active enzyme (Supplementary Results, Supplementary Figure 2b). While compound 1 inhibits both enzymes, the strong inhibitory effect of compound 1 on the off-target PDI eliminates it as a specific MtbVKOR inhibitor.
These tests eliminated all potential inhibitors of MtbVKOR from further study. However, in a screen with bioactive compounds, one specific hit against MtbVKOR was brominedione, a known anticoagulant inhibitor of human VKORc1. This positive result indicates that the screen should identify new classes of anticoagulants as well as specific M. tuberculosis inhibitors.
Sorting out potential EcDsbB inhibitors
Of the seven inhibitors in the EcDsbB screen, compound 11 was eliminated because it showed inhibitory activity in the MtbVKOR re-test and caused growth defects with M. tuberculosis (Supplementary Results, Supplementary Table 1). Hence, the false discovery rate is 45% (5/11) for EcDsbB screen. The remaining 6 potential EcDsbB inhibitors (#5-10) did not inhibit the MtbVKOR-dependent strain, making it likely that they directly inhibit EcDsbB. These compounds shared a pyridazinone ring structure (Supplementary Results, Supplementary Table 1).
Inhibition of purified EcDsbB
We tested compounds 5-10 for their inhibitory effect on EcDsbB-mediated ubiquinone reduction using purified enzymes. In this assay, reduced DsbA provides the source of electrons that are used by EcDsbB to reduce ubiquinone-5. The compounds show dose-dependent inhibition of EcDsbB with Inhibitory Concentrations 50 (IC50s) in the low μM range (Supplementary Results, Supplementary Table 2). The lowest IC50, 1.7 μM, was exhibited by compound 9 (Figure 2a). Enzyme kinetics analysis of compound 9 revealed a KI of 46 ± 20 nM (The Km for ubiquinone-5 is 1.03 ± 0.12 μM, Supplementary Results, Supplementary Figure 3).
Figure 2.

In vivo and in vitro inhibition of EcDsbB by compound 9 and 12. (a) In vitro inhibition of purified EcDsbB enzyme by compounds 9 (left) and 12 (right). Results are average of at least two independent experiments ± SD. (b) In vivo accumulation of reduced DsbA (EcDsbB substrate) caused by compounds 9 (top) and 12 (bottom). Cells were grown aerobically with different concentrations of drug and precipitated proteins were treated with 4-acetamido-4’-maleimidylstilbene-2,2’-disulphonic acid (AMS, 0.5kDa). Samples were run by reducing SDS-PAGE and immunoblotted against anti-DsbA. Dithiothreitol (DTT) was used for reducing disulfide bonds. “ox” refers to the position of the oxidized protein which is the same as that of the reduced protein with no alkylating agent present. “red” refers to bands where the positions of the protein with reduced cysteines are detected because of the alkylation which adds to the molecular weight. Pictures are representative immunoblots of at least two independent experiments. See complete pictures in Supplementary Information. (c) Inhibition of E. coli anaerobic growth with compound 12. Growth curve of wild-type E. coli (black) and dsbB mutant (red) under anaerobic conditions in the absence (solid lines) or presence (dotted lines) of 10 μM compound 12. Results are the average of three independent experiments ± SD.
Inhibition of disulfide bond formation in vivo
We recognized that the agar-based 384-well plates did not allow calculation of real concentrations of the compounds in the wells since aliquots of the compounds are dropped onto solidified agar and might concentrate on top of the agar and/or diffuse slowly into the agar. Therefore, we attempted to determine in aerated liquid media the minimal inhibitory concentration (MIC) of the compounds necessary to restore detectable β-galactosidase activity. However, we did not observe any increase in β-galactosidase activity during growth in liquid media with the strongest of the inhibitors, compound 9, at concentrations up to between 50-100 μM where the compound begins to precipitate. In the HTS agar assay, the “calculated” (see above) but likely underestimated MIC for compound 9 required to show any blue color in the agar assay was 5.7 μM. Thus, the HTS agar assay is a more sensitive but still specific detector of inhibitors than an HTS liquid assay. If we had used a liquid assay in the HTS, we would never have detected these compounds as EcDsbB inhibitors.
To directly determine compound concentrations inhibitory of EcDsbB in liquid media, we assayed the oxidation of its substrate (DsbA). A standard alkylation assay that distinguishes oxidized (disulfide-bonded) from reduced (free cysteines) DsbA, showed that the IC50 of compound 9, at which oxidation of DsbA was lowered by 50%, is 8.5 ± 0.6 μM (Figure 2b). This disparity between observable effects on β-galactosidase activity and effects on DsbA oxidation in liquid media is probably due to the high sensitivity of β-galactosidase to disulfide bond formation resulting from the presence of 16 cysteines in the protein; oxidation of any pair of these to disulfide bonds is likely to cause misfolding and lack of activity of the enzyme15. Furthermore, the aeration of the liquid cultures could increase the air oxidation of DsbA, increasing the likelihood that β-galactosidase would be inactivated.
Nevertheless, while the agar assay itself cannot be used to obtain an accurate measure of MICs or IC50s for individual compounds, it allows us both to detect inhibitors that would not be found in a liquid assay and to rank a collection of compounds for their inhibitory potency. For instance, from dilutions of compounds and assaying effects in the agar assay, we estimate that compound 10, the weakest found in the HTS, is 6-fold less inhibitory than compound 9. Since compound 9 fully inhibits DsbB at 20-50 μM, our HTS is detecting compounds (e.g. 10) that fully inhibit DsbB only at high micromolar concentrations. This result means that the nature of the HTS is such that we would probably not detect compounds that inhibit fully only in the high micromolar or millimolar range.
In experiments that follow which estimate MICs from the agar-based assay for calculating inhibitor concentrations, we will refer to these presumably overestimated concentrations as “agar-based calculated” MICs.
Approach to obtain more effective EcDsbB inhibitors
The common structural features of the six EcDsbB inhibitors obtained led us to seek available compounds with similar structures amongst which we might find stronger inhibitors. We first asked whether any of the 51,487 compounds screened that were not inhibitors of EcDsbB had pyridazinone core structures. We found 46 such compounds, all but four of which failed to inhibit DsbB in the agar assay. These four were weaker inhibitors than any of those detected in the HTS and their inhibitory activity could only be detected by assays in a strain expressing lower levels of EcDsbB than wild-type (Online Methods). We also examined the list of the remaining 549,283 ICCB-compounds from which we found only five with related structures, three of which inhibited EcDsbB in our assay.
From a survey of compounds that were not inhibitors and from the commonalities found among the effective inhibitors, we selected 24 additional compounds that were commercially available and more similar to the active members of the pyridazinone family. Amongst the criteria we used to choose compounds to order were 1) variation in the groups linked to the pyridazinone; 2) introducing halogen or other groups in the benzyl ring; 3) substitution of the benzyl ring of the pyridazinone by phenyl. Amongst these, 6 compounds exhibited EcDsbB inhibitory activity as well as or better than compound 9 (Table 1).
Table 1.
Inhibition of EcDsbB by pyridazinone compounds.
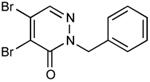
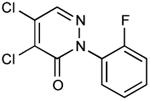
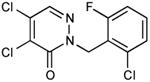
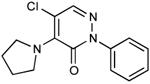
| ID Number | Structure |
RIC50 Ratio
(RIC50 compound #9 / RIC 50) |
|---|---|---|
| 12 |

|
23.03 |
| 13 |

|
10.77 |
| 14 |

|
3.80 |
| 15 |
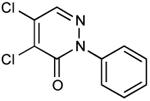
|
2.49 |
| 16 |

|
1.65 |
| 17 |
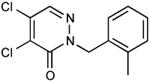
|
1.43 |
| 9 |

|
1.00 |
| 18 |
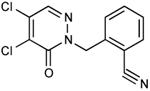
|
0.39 |
| 19 |
|
0.38 |
| 20 |
|
0.37 |
| 21 |

|
0.37 |
| 22 |
|
0.28 |
| 7 |

|
0.20 |
| 8 |
|
0.18 |
| 6 |
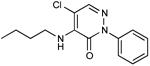
|
0.17 |
| 23 |

|
0.13 |
| 5 |
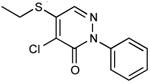
|
0.08 |
| 10 |

|
0.07 |
| 24 |
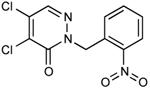
|
0.05 |
| 25 |

|
0.02 |
| 26 |

|
0.01 |
| 27 |
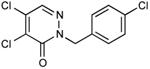
|
<<0.01 |
| 28 |
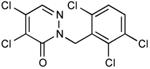
|
<<0.01 |
| 29 |
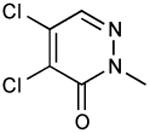
|
<<0.01 |
| 30 |
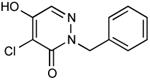
|
<<0.01 |
| 31 |

|
<<0.01 |
| 32 |

|
<<0.01 |
| 33 |
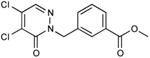
|
<<0.01 |
| 34 |

|
<<<0.01 |
| 35 |
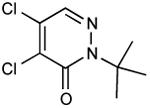
|
<<<0.01 |
In vivo inhibition of EcDsbB was measured by effects on β-galactosidase activity in liquid cultures of a strain expressing β-Galdbs and EcdsbB under a weak IPTG-inducible promoter. Relative Inhibitory Concentration 50 (RIC50) was calculated as the concentration of compound required to reach 50% of βgalactosidase activity compared to the full β-galactosidase activity of the dsbB mutant. RIC50 ratio was calculated to rank the compound potency relative to compound 9. The RIC50 average of at least three independent experiments was used to calculate the RIC50 ratios.
Mechanism of EcDsbB inhibition by compound 12
Among the compounds discussed above, we found two EcDsbB inhibitors (13 and 12), that showed 10- and 23-fold more inhibitory activity than #9, respectively (Table 1). Compound 12 has a Ki of 0.8 ± 0.1 nM (IC50 of 18.85 nM) in the in vitro assay (Figure 2a and Supplementary Results, Supplementary Figure 3) and an IC50 of 0.9 ± 0.5 μM in inhibiting DsbA oxidation in aerobically growing cells (Figure 2b). Additionally, when the in vivo redox state of the cysteines of EcDsbB is probed with the cysteine alkylating agent MaleimidePEG-2k (ME2k) after treatment of cells with compound 12, EcDsbB shows only one ME2k modification (Supplementary Results, Supplementary Figure 4a). This indicates that of the four essential EcDsbB cysteines (the two non-essential cysteines are mutated to alanine and valine, respectively), two are in the disulfide state, one in the reduced state (labeled by ME2k) and the forth is unavailable to react with ME2k.
Further, we noticed that the slightly pinkish color of purified DsbB turns yellow when treated with compound 12. The pink color represents a small population of DsbB that is in the charge-transfer complex state with ubiquinone, which absorbs strongly at 500 nm15-17. When DsbAC33A is used as a substrate for DsbB, it forms a stable mixed disulfide complex in which Cys44 is trapped in the charge-transfer complex state. Likewise, when the compound is added to the DsbB-DsbAC33A dimer, its characteristic color turns yellow (Supplementary Results, Supplementary Figure 4b). This is not due to the dissociation of the dimer since non-reducing SDS-PAGE shows the complex intact. Moreover, when compound 12 is added to DsbB before the addition of DsbAC33A, the pink color quickly develops but does not persist. These results suggested that the compound influences the interaction between Cys44 of DsbB and ubiquinone.
To determine whether the interaction between compound 12 and the DsbB-DsbAC33A is due to a covalent bond, we performed ion-trap mass spectrometry which revealed an adduct of 253.6 Da with the DsbB-DsbAC33A dimer (Supplementary Results, Supplementary Figure 4c). Since the theoretical Mw of compound 12 is 289.54 Da, the 35.9 Da mass loss is possibly representing a leaving chloride ion. This mass adduct was not observed when treating either DsbB (oxidized), DsbA (reduced) or DsbAC33A alone with the compound (Supplementary Results, Supplementary Figure 4c). These data indicate a covalent modification of DsbB by compound 12 which is occurring after the formation of the charge-transfer complex during ubiquinone reduction. Consistent with this expectation, high resolution tandem mass spectrometry of chymotrypsin digested DsbB-DsbAC33A complex treated with the compound shows that Cys44 of DsbB has an adduct of 252.995 Da (Supplementary Results, Supplementary Figure 4d).
To test for off-target reactivity of compound 12 towards other thiol-containing redox enzymes, we assessed its inhibitory effect on the reduction of insulin by PDI. Compound12 did not show significant inhibitory effect on PDI at concentrations up to 100 µM (Supplementary Results, Supplementary Figure 2b) in contrast to compound 1. The possibility that compound 12 might form a covalent adduct with DsbA sufficient to inhibit disulfide bond formation is ruled out by the finding that this compound does not inhibit disulfide bond formation in the MtbVKOR-dependent E. coli strain in which DsbA is functional in disulfide bond formation. Moreover, electrospray-ionization mass spectrometry of reduced DsbA shows no difference in mass upon addition of the compound (Supplementary Results, Supplementary Figure 4c).
Other properties of DsbB inhibitors
Since E. coli mutants lacking DsbA or DsbB do not grow anaerobically, we tested the effect of compound 12 on anaerobic E. coli growth in minimal medium and found that it fully inhibits anaerobic growth of a wild-type strain at a concentration of 10 μM (Figure 2c).
We tested all pyridazinone inhibitors for their effects on MtbVKOR expressed in the E. coli dsbB null strain using the agar-filled well plates. We observed no inhibition of MtbVKOR at “agar-based calculated” MICs up to at least 100 μM. An additional method for demonstrating inhibitor specificity for EcDsbB is made possible by our finding that a vkor-deletion strain of M. smegmatis grows very poorly but that growth is restored when the strain is complemented by a plasmid expressing either MtbVKOR or EcDsbB9. Therefore, we added compound 12 to test the growth of EcDsbB-dependent and MtbVKOR-dependent M. smegmatis. Compound 12 completely inhibited growth of the EcDsbB-dependent M. smegmatis strain at 400 nM, while the VKOR-dependent strain grew normally at concentrations up to and including 100 μM. Although neither the relative expression levels of EcDsbB and MtbVKOR in these two strains nor their relative efficiencies in oxidizing the MsmegDsbA can be assessed, these vast differences in sensitivity to compound 12 are consistent with the specificity of the compound found in the E. coli studies.
Inhibition of DsbBs from other gram-negative pathogens
We further tested the EcDsbB inhibitors for their ability to inhibit DsbBs from other gram-negative pathogens when expressed in E. coli. We cloned the dsbB genes from Acinetobacter baumanni, Klebsiella pneumonia, Vibrio cholerae, Hemophilus influenza, Francisella tularensis, two dsbB homologues from Pseudomonas aeruginosa (dsbB and dsbH), and two dsbB homologues from Salmonella typhimurium (dsbB and dsbI) under an IPTG-inducible promoter. All DsbB homologues complemented the dsbB null strain in maintaining the β-Galdbs in the disulfide-bonded state as indicated by the absence of blue color in agar growth assay (Online Methods). We then tested the complemented strains for their sensitivity to the collection of EcDsbB inhibitors and to the related non-inhibitory compounds. Several dilutions of these inhibitors were dropped onto the agar media in 384-well plates with the complemented strains, thus allowing us to rank the different inhibitors in terms of their ability to inhibit each DsbB. For the most part, the seven compounds that did not inhibit EcDsbB also did not inhibit the DsbBs of the other gram-negative bacteria, while at least one of those that inhibited EcDsbB also inhibited the other DsbBs. Interestingly, while the most effective inhibitor of several of the organisms was #12, other DsbBs were more effectively inhibited by other compounds of this group in the agar assay (Figure 3). The only DsbB homologue that was not inhibited by any of these compounds, StDsbI, is an unusual homologue that appears to be involved in a specialized pathway of disulfide bond formation18,19. Although we can rank the inhibitors in terms of strong vs weak, it is not possible to compare effectiveness of their action on different DsbBs without knowing for each DsbB the expression level and effectiveness of oxidizing EcDsbA.
Figure 3.

In vivo inhibition of DsbB enzymes from gram-negative bacteria expressed in E. coli. E. coli dsbB mutant strains expressing β-Galdbs and dsbB genes from Salmonella typhimurium (St), Klebsiella pneumoniae (Kp), Vibrio cholerae (Vc), Haemophilus influenzae (Hi), Pseudomonas aeruginosa (Pa), Acinetobacter baumannii (Ab), Francisella tularensis (Ft) as well as two DsbB-homologs of P. aeruginosa (dsbH) and S. typhimurium (dsbI) and a non-homolog vkor from Mycobacterium tuberculosis (Mtb) were tested against pyridazinone-like compounds. Inhibition range from strong to weak is relative to each DsbB-expressing strain and was obtained by dividing the MIC of each compound between the lowest MIC observed for each particular strain. Results are the average of three independent experiments. Compounds that did not inhibit at the highest concentration tested are shown as black. Identity (%) compared to EcDsbB and protein length (amino acid) are indicated.
In order to begin determining whether those compounds that inhibited other gram-negative DsbBs would act to inhibit disulfide bond formation in the organism from which they were cloned, we tested the effect of compound 12 on the property of twitching motility in P. aeruginosa. Twitching motility allows certain pathogens to attach to the cell surface and colonize the host. P. aeruginosa motility depends on Type IV pili and on the presence of a disulfide loop at the C-terminus of PilA20. A dsbA mutant of P. aeruginosa shows decreased virulence and is defective in twitching motility21,22. Similarly, the double mutant dsbB,dsbH of P. aeruginosa is defective in twitching motility23. Therefore, we tested P. aeruginosa motility in the presence of inhibitor and observed that the wild-type strain in the presence of 50 μM of compound 12 is completely defective in motility (Supplementary Results, Supplementary Figure 5). Growth itself was not affected as indicated by colony size on agar media. While the phenotype is consistent with an inhibition of disulfide bond formation, we cannot rule out the possibility that the compound inhibits a different process affecting motility.
Discussion
The HTS procedure described here allows detection of compounds that inhibit the DsbB protein of the gram-negative bacterium E. coli and presumably in further screenings can do the same for the VKOR of M. tuberculosis. Since inhibitors of these enzymes cause reduced activity of virulence factors, defects in anaerobic growth and in the integrity of the bacterial outer membrane, they may be models for a new class of antibiotics or antivirulents.
The novel aspects of our approach may be useful in other types of screens. First, employing agar media-filled wells in which compounds are dropped in the well may concentrate the compounds at the already solidified agar surface, making the assay much more sensitive than a liquid assay would have been. Inhibitors were readily detected in the strains carrying β-Galdbs as causing blue color in the wells. For a cellular process for which β-galactosidase can be used as a reporter for effects on components of the process, the agar well assay could provide a more sensitive approach for detecting inhibitors.
Second, assessing potential inhibition of two proteins in parallel (EcDsbB and MtbVKOR) that perform the same function, but are not homologues, allows detection of inhibitors that are specific to one or the other protein, making it a target-based in vivo screen. This approach may be extended to pathways with similar advantages or may be made possible by assaying a native and an alternative laboratory-generated pathway in parallel screens. For example, distinct pathways for disulfide bond formation in either the E. coli cytoplasm or cell envelope have been evolved in the laboratory, presenting possible screens like ours that focus on components of those pathways24,25.
Third, the screening assay in E. coli which depends on the target protein being an enzyme that can regenerate active DsbA, has allowed screens in which the native DsbB is replaced by DsbBs from numerous gram-negative bacteria or with the enzyme VKOR from M. tuberculosis. With such complemented strains one can test existing and developed inhibitors of EcDsbB for their effects on these DsbBs and VKORs or proceed to HTS in E. coli with them. Positive results with inhibitors in E. coli could then be followed by tests for their effects on the organisms from which the DsbB or VKOR was cloned. Moreover, if human VKOR can be expressed in E. coli as a functional replacement for EcDsbB, such a strain could be used to screen for new classes of potential blood anticoagulants.
Since the completion of this first screening, we have constructed a more effective MtbVKOR-complemented strain in which DsbA is efficiently oxidized. We expect that planned further screens for MtbVKOR inhibitors in this improved strain will allow more ready detection of strong inhibitors of MtbVKOR and not cause the detection of so many false positives.
From the EcDsbB screen and subsequent medicinal chemistry analysis, we have obtained a collection of pyridazinone-like compounds that inhibit EcDsbB-based but not MtbVKOR-based disulfide bond formation in E. coli. For instance, comparing sensitivity to compound 12 in the agar-filled wells, we found no inhibition of the VKOR-based strain at concentrations of at least two orders of magnitude higher than the minimal amount of #12 needed to give blue color with the DsbB-based strain. Since our MtbVKOR strain is already weaker in terms of inactivating β-Galdbs, and thus would be more sensitive to inhibitors, the difference is certainly greater than seen from this last result. Further, compound 12 inhibits M. smegmatis growth in a strain which we have made dependent on EcDsbB for disulfide bond formation, but it does not inhibit M. smegmatis growth when disulfide bond formation is dependent on MtbVKOR. These findings together indicate that the EcDsbB inhibitors obtained are specific to EcDsbB in that they do not inhibit MtbVKOR despite the similar mechanisms of action of the two thiol-redox proteins.
Since disulfide bond-formation in many bacteria is important for their virulence, it might seem that DsbA would also be a useful target for antivirulents. However, DsbA belongs to the widespread thioredoxin family, while DsbB has no known homologues in eukaryotes, raising questions about DsbA as a target for potential antivirulents. Inhibitors of DsbA may also inhibit members of the thioredoxin family that in humans contribute to the reducing environment of the cytoplasm and to protein folding in the endoplasmic reticulum. Admittedly, VKOR does have two homologues in humans, one (VKORc1) involved in blood coagulation and the other (VKORc1L1) suggested to be involved in redox-maintenance26,27. Thus any inhibitors of MtbVKOR detected in further HTS must be assayed for their effects on the two VKORs found in most vertebrates.
A recent report using a target immobilized NMR screening with DsbB solubilized in micelles and nanodiscs describes inhibitors of DsbB appearing from a collection of 1071 drug fragments28. This study allowed the identification of 93 fragments that bind to DsbB, 8 of which inhibit electron transfer to quinone either by competing with quinone binding or disturbing the affinity of DsbB and quinone. We note that the pyridazinone-like compounds found in our in vivo screening appear to interfere with the mode of interaction of quinone with DsbB.
Our primary intention in this screen was to develop DsbB inhibitors as anti-virulents, since inactivation of the disulfide bond formation pathway interferes with the activity of multiple virulence factors. Others have suggested that targeting the overall virulence rather than bacterial viability could provide a successful strategy for developing new antibiotics. Such compounds could result in weaker selective pressure to develop resistance by disarming the pathogen, which may then be cleared by the host immune response1,29,30. However, since compound 12 inhibits anaerobic growth of E. coli, the development of these compounds to effectively inhibit disulfide bond formation in vivo may directly prevent infections, since many Gram-negative pathogens infect tissues in low oxygen environments31,32. In such pathogens, the inhibitors may function as antibiotics while simultaneously reducing virulence. In addition, since reducing disulfide bond formation has multiple effects on the bacterial cell envelope, these compounds may exhibit synergistic effects with other known antibiotics.
We show that compound 12 binds covalently to the DsbB-DsbA complex in vitro via DsbB’s Cys44, apparently losing a single chlorine atom in the process. This finding is consistent with our results indicating that the compound acts by interfering with the quinone-Cys44 charge-transfer complex. Compounds that bind covalently to periplasmic substrates, such as β-lactams, have proved quite successful as antibiotics. Recent review articles have promoted the search for compounds that covalently bind to their targets as an important approach to drug development, in general33,34.
Finally, we point out that the HTS for potential new antibiotics or anti-virulents described here would not have been possible without the many years of basic research on disulfide bond formation in bacteria.
Online Methods
Bacterial strains and growth conditions
The strains and plasmids used in this study are listed in Supplementary Table 3. The malF-lacZ102 fusion (referred to as β-Galdbs) with Kanamycin resistance derived from pNG10211 was integrated into the chromosome of HK295 and HK320 strains by the λInCh method35 to generate HK314 and HK325 strains, respectively. MER672 and DHB7658 were constructed inserting pTrc99a at the recombined λatt site by λInCh into the chromosome of HK314 and HK325 strains, respectively. In order to generate the DHB7657 strain, instead pTrc99aMtbVKOR was moved to HK325 by λInCh. In order to stabilize both insertions at the λ attachment site, the recA− mutation (BW10724, Keio collection) was moved by P1 transduction into the three strains. Strains DHB7935 and DHB7936 were constructed by introducing into the chromosome plasmids pDSW206dsbB and pDSW206 at the ϕ80 attachment site of HK325 as described previously36. Pseudomonas aeruginosa dsbB and Klebsiella pneumoniae dsbB genes were placed into the chromosome of E. coli due to toxicity problems when growing cells in glucose-maltose M63 minimal media. All dsbB homologs from other gram-negative bacteria were expressed from plasmids. In order to generate CL315 and CL320, a PCR product that extends from the lacI gene to the ampicillin-resistance cassette (primers Cl65 and Cl66, Supplementary Table 4) of pCL25 (PadsbB) and pCL24 (KpdsbB) plasmids were introduced into the ΔdsbB loci of HK320 strain using λ−Red proteins expressed from pCL58 plasmid. Then, each insertion was moved to HK325 strain by P1 transduction. The other strains expressing different dsbB genes were obtained by transformation of the respective plasmid into HK325. All of the dsbB-complemented strains were verified by their motility in 0.3% agar minimal media and adjusted in X-Gal minimal media plates to levels of IPTG that resulted in white colonies, i.e. complementing the dsbB mutant phenotype. Thus, the IPTG concentrations used for agar assays were: 50 μM for the E. coli strain expressing PadsbH and AbdsbB, 75 μM for the strain expressing StdsbI and 2 mM for the strain expressing FtdsbB. For strains expressing KpdsbB, PadsbB, StdsbB, VcdsbB and HidsbB genes the basal levels of expression were enough to complement so, no IPTG was required to add.
All strains were grown in NZ or in M63 broth and agar media at 30ºC when indicated. The antibiotic concentrations used were: ampicillin 25 μg/mL (chromosomal) or 100 μg/mL, kanamycin 40 μg/mL and chloramphenicol 10 μg/mL.
Agar Screening Plate Preparation
A Matrix Wellmate (Thermo Scientific) fitted with a small-bore tubing cartridge was used to dispense 50 μL of hot agar M63 medium containing 0.2% glucose and 0.9% agar, supplemented with kanamycin (40 μg/mL), ampicillin (50 μg/mL), IPTG (1 mM), and X-Gal (120 μg/mL) to 384-well tissue culture-treated plates (BD Falcon #353961). In order to prevent agar solidification in the Wellmate tubing or inactivation of the antibiotics and X-Gal, the medium was maintained at 57 ºC in a water bath throughout the pouring process. In addition, the Wellmate tubing was pre-warmed by washing with sterile hot water immediately prior to loading the agar medium, and the plates were poured as quickly as possible. We were able to prepare up to 80 uniform 384-well screening plates at a time. After the agar solidified, the plates were stored overnight at 4 °C in which condition the blue color of the wells is enhanced.
High-Throughput Chemical Screen
Most of the compound collections were supplied by the Institute of Chemistry and Cell Biology (ICCB) at Harvard Medical School. The initial screen included 50,374 compounds from several commercial small molecule libraries (Asinex 1, ChemBridge 3, ChemDiv 4, Life Chemicals 1, and Enamine 2) and small libraries of known bioactive molecules and natural products. In addition, a 1113-compound library of M. tuberculosis H37Rv growth inhibitors provided by the National Institute of Allergy and Infectious Diseases14 (http://urlm.co/www.taacf.org) was assayed. Aliquots (100 nL) of library compounds typically 5 mg/mL in dimethyl sulfoxide (DMSO, Sigma), were transferred to the agar surface in each well of the screening plates by pin transfer (EPSON compound transfer robot). It was important to place the compounds on top of the medium rather than injecting them deep within the agar, where contact with bacteria would be extremely limited. The agar concentration was optimized (0.9%) to balance this requirement with the need for the medium to remain in the liquid state while pouring the plates.
Overnight E. coli cultures grown in minimal media were diluted to an OD600 of 0.05 with M63 minimal medium containing 0.2% glucose, 40 μg/mL kanamycin, 50 μg/mL ampicillin, and 1 mM IPTG. The diluted bacteria were added to each well in 10 μL aliquots with a Matrix Wellmate dispenser. Positive (a strain lacking DsbB) and negative (DMSO no inhibitor) controls were included on each plate. The plates were sealed with breathable sealing film (Axygen BF-400) and incubated for three days at 30 °C in a humidified box. To enhance the blue color, the plates were incubated for 12 to 24 h at 4°C (Suplementary Table 5).
The compounds identified as inhibitors in the first round of screening were retested in a cherry-pick assay. The experiment was identical to the initial screen, except that the compounds were added to aliquots of bacteria with PocketTips (Thermo Scientific) rather than directly to the plates, and then the bacteria-compound mixtures were transferred to 384-well agar plates.
Compound Resupply for Retesting
Compounds 1, 2, 4, 5, 6, 8, 9, 10, 14, 21, 28 and 30 were purchased from ChemBridge (San Diego, CA); 11 from Asinex Ltd. (Moscow, Russia); 3 from Sequoia Research Products Ltd. (Pangbourne, UK); 7 from Key Organics Ltd. (Camelford, UK); 15 and 29 from AK Scientific (Union City, CA); 12, 16, 23, 27, and 31 from Ambinter (Orleans, France); 20 from Ryan Scientific (Mt. Pleasant, SC); and compounds 13, 17, 18, 19, 22, 24, 25, 26, 32, 33, 34 and 35 from Enamine (Ukraine). Larger quantities of compound 12 were obtained by custom synthesis from Enamine (Ukraine, purity over 95%, Supplementary Information). All purchased compounds were analyzed by mass spectrometry (LCMS) to verify their molecular weights and to confirm their purity (over 90%).
M. tuberculosis H37Rv growth inhibition
The bacteria were grown to stationary phase (OD600 of 2.0) and diluted to OD600 of 0.003. Chemical compounds were dissolved in growth medium and subsequent serial two-fold dilutions, 0.12 μg/mL being the lowest concentration, were performed in 96-well plates. A bacterial inoculum of 50 μL was added to each well, yielding a final volume of 100 μL/well. This was done in triplicate and repeated with bacteria grown in Middlebrook 7H9 medium (Difco), 7H9 medium supplemented with 0.2% glycerol, 5% Tween 80 (Sigma) and OADC (Becton Dickinson), and Sauton’s medium (Modified from 37 pH 7.4 and filter sterilized instead of autoclaving). The plates were sealed with breathable film and incubated in a shaker at 37°C. After five days, 10 μL of Alamar Blue reagent (Biosource) was added to each well and the plates were incubated for 24 h at 37°C. The MIC was defined as the lowest drug concentration that prevented a color change from blue to pink.
Preparation of mouse liver microsomes
Mouse hepatic microsomes were obtained from mouse liver by homogenization in PBS/20% glycerol/protease inhibitor cocktail (Calbiochem, 1x final concentration) using a Potter tissue grinder with an attached power unit (Con-Torque/Eberbach). Mouse liver (10 g) was homogenized with ten strokes of the tissue grinder 4 times with cooling on ice after each set of strokes. The sample was centrifuged at 10,780 × g for 10 min at 4ºC. The supernatant was collected and the remaining pellet was subjected to another cycle of homogenization as before. After two more cycles the four supernatants were pooled and subjected to centrifugation at 38,000 × g for 1 h at 4º C. The pellet was resuspended in PBS/20% glycerol/PIC/0.2% phosphatidycholine/0.5% CHAPS and sonicated twice with a Microson XL sonicator (Misonix) at power level 4 with cooling on ice after each sonication. The sample was centrifuged at 38,000 × g for 1 h at 4ºC. The supernatant from this centrifugation containing the solubilized liver microsomes was stored at −80 C.
Assay for vertebrate VKOR enzymatic activity
Solubilized mouse liver microsomes (20 μL) were added to 180 μL of buffer (25 mM N-[Tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid, pH 8.6 in 150 mM NaCl/30% glycerol). When inhibitors were used they were added at the indicated concentrations to the solubilized microsomes in buffer and the mixture incubated for 10 min at 4°C. The substrate, 4 μL of 12 mM vitamin K epoxide (VKO) in isopropanol, was added to the microsomes and 5 μL of 200 mM DTT was added to start the reaction. The reaction was incubated for 24 h at room temperature protected from light and it was stopped by adding 500 μL of a mixture of 0.05 M AgNO3 in isopropanol (5:9 v/v). The mixture was vortexed for 1 min and centrifuged to separate the phases. The upper organic phase (400 μL) was transferred to a brown vial and dried with a gentle stream of nitrogen. The dried sample was dissolved in acetonitrile:isopropanol:water (100:7:2 v/v) which also served as the mobile phase for HPLC. The concentration of VKO was determined by HPLC analysis on a C18 column (Vydac) and the amount of VKO converted to vitamin K calculated using a known concentration of VKO as a standard.
Insulin reductase activity assay
The activity of human Protein Disulfide Isomerase was tested using the insulin reduction assay (ProteoStat™, Enzo life Sciences) with minor modifications. The catalyzed reduction of insulin was measured in the presence of tris(2-carboxyethyl)phosphine (TCEP) instead of DTT in order to avoid possible reaction of DTT with compounds. The reaction mixture contained 200 µM TCEP and 330 µM insulin in phosphate/EDTA buffer (pH 7.4). PDI was used at a final concentration of 1 μM. Inhibitor compounds at the indicated concentrations were included in the reaction mixture up to 100 µM. The reaction was initiated by the addition of TCEP in 96-well plates (100 µl final volume) for 30 min at 25º C in triplicates. The uncatalyzed reduction of insulin was subtracted from the reaction containing PDI.
In vitro assay of DsbB inhibition
Potential E. coli DsbB inhibitors were evaluated in vitro by a ubiquinone-5 (UbQ-5) reduction assay at 275 nm. DsbB was produced in E.coli using fermentation15 and purified as described (Purity over 90% as determined by SDS-PAGE, Supplementary Information). Purified DsbA or DsbAC33A were reduced by 10 mM DTT for 30 min on ice. DTT was subsequently removed by gel filtration using a PD10 column and reduced DsbA was used as a source of electrons for DsbB-mediated reduction of UbQ-5. For determination of inhibition constants (IC50 and KI), various amounts of inhibitors were mixed with DsbB (2 nM) in phosphate buffer (pH 6.5) containing 0.1% DDM (Affymetrix Inc.; Santa Clara, CA, USA), 100 mM NaCl and UbQ-5 (1-50 μM). Reactions were started at room temperature by the addition of small amounts of highly concentrated DsbA solution to give a final concentration of 20 μM.
Mass spectrometry analysis
DsbB, DsbAC33A and DsbB-DsbAC33A dimer (each at 100 μM) were mixed with compound 12 on ice at 1:2 ratio in 50 mM Tris buffer (pH:8) containing 300 mM NaCl and 0.05% DDM. Compound 12 was added by two increments of 100 μM. For DsbB-DsbAC33A dimer, when absorbance at 500 nm reached a minimum upon addition of compound (around 4 minutes). Samples were directly diluted 5-fold with aqueous solution of 0.1% trifluoroacetic acid and sent for electrospray ionization mass spectrometry analysis using LTQ Velos Pro ion-trap mass spectrometer (ThermoFisher, San Jose, CA). In order to determine the site of modification, DsbB-DsbAC33A dimer treated with compound 12 was reduced on ice with 50 mM DTT for 30 min, loaded on a HPLC column and eluted fractions corresponding to DsbB were digested with chymotrypsin. Chymotryptic peptides were subsequently pressure loaded onto a HPLC column. Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions and analyzed using HCD LTQ-Orbitrap at 15,000 resolution (ThermoFisher, San Jose, CA).
In vivo chemical alkylation of E. coli DsbA and DsbB
The in vivo chemical alkylation procedure was done as described previously38. Precipitated proteins (reduced or not) were solubilized in 50 μL of 100 mM Tris.HCl, pH 6.8 containing 1% SDS and 5 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS, Life technologies) or 12.5 mM maleimide-PEG2000 (ME2k, NOF Corporation, Japan). Reducing 5X-SDS sample buffer was then added and 2 μL (for DsbA detection) or 5 μL (for DsbB detection) of sample was applied to SDS-PAGE directly. Tris-HCl polyacrylamide (12%) gels were used. The proteins were transferred onto PVDF membranes and immunoblotted with α-DsbA antibody4 or α-DsbB antibody39. The IC50s of compound 9 and 12 were obtained by measuring the relative amount of the reduced and oxidized DsbA-bands by Image J (NIH). The amount of reduced-DsbA and concentration of inhibitor were used to extrapolate the 50% reduced-DsbA with trend function in Excel from at least two independent experiments.
Medicinal chemistry approach of pyridazinone inhibitors
A substructure search of compounds with a pyridazinone core was performed to detect molecules similar to compound 9 (DsbB inhibitor) among the ICCB-libraries of compounds tested and not tested in the agar screening. The obtained list of similar molecules indicated that compounds with substitutions at position 6 of the pyridazinone were discarded since it was detrimental to the inhibitory activity of the compound. Compounds that did have a single change either at position 2, 4 or 5 were selected as candidates. To determine if those candidates were commercially available, a substructure search for the pyridazinone was done using SciFinder software (American Chemical Society). 24 compounds out of the 57 commercially available candidates were purchased (“Compound resupply” section) and tested in liquid media against E. coli DHB7935 strain (Table 1), which expresses dsbB gene from a weaken Trc promoter40, making it more sensitive to weak inhibitors. This strain allowed us to easily rank the compounds. The Relative Inhibitory Concentration 50 (RIC50) for each compound was determined by quantifying the β-galactosidase activity of DHB7935 strain in the presence of different concentrations of compound. The RIC50 was defined as the concentration of compound in which the strain reaches 50% β-galactosidase activity compared to the 100% obtained in ΔdsbB mutant strain (DHB7936). To measure β-galactosidase activity, the velocity of hydrolysis of o-nitrophenyl- β-galactoside (ONPG, Sigma) was determined. The assay was done in a flat bottom 96-well plate (Thermo Scientific) as described previously41. Briefly, DHB7935 cells were inoculated to an OD600 of 0.01 in 200 μL of M63 with 0.2% glucose as a carbon source, 0.2% maltose to induce the expression of β-Galdbs and with serial dilutions of inhibitor. The cells were incubated for 12 hours at 30ºC, 80% humidity and 900 rpms in an orbital shaker (Multitron, ATR). 100 μL of cells were lysed using 10 μL of PopCulture reagent (Novagen) with 400 U/mL lyzosyme and incubated with 90 μL of 4 mg/mL ONPG at 28ºC in a microplate reader (VERSAmax). The OD420 was measured every minute during 1 hour to follow the kinetics of ONPG hydrolysis and the velocity of the reaction was calculated by SoftMax®Pro software (Molecular Devices, LLC). Miller Units were determined using 1.81 (CF1), 2.45 (CF2) and 3.05 (CF3) as constants and the relative β-galactosidase activity was calculated normalizing to the full activity obtained for the dsbB mutant (100%). Finally, the RIC50 was calculated by GraphPad Prism Software (La Jolla California, USA) with non-linear log dose-response normalized curve using 4 parameters. The RIC50 values and 95% confidence intervals were obtained using data of at least three independent experiments. In order to observe the difference in activity and have more meaningful idea of the inhibitor potency, all the RIC50 values were compared to the RIC50 of compound 9, the stronger compound before this analysis. So, the inhibition ratio was calculated for each dividing the RIC50 mean of compound 9 (5.10 μM) between the RIC50 mean of the compound (Table 1).
E. coli growth inhibition under anaerobic conditions
To test the sensitivity of E. coli to inhibitors under anaerobic conditions, strains were first grown aerobically overnight in M63 glucose medium. These cultures were diluted 1:100 into fresh medium and grown aerobically to mid-log phase. All cultures were adjusted to an OD600 of 0.1 and transferred into a Coy anaerobic chamber. Cultures were then diluted 1:1,000 into anaerobically-prepared M63 glucose medium containing 40 mM fumarate (± compound) and grown without shaking at 37°C. Samples were taken over time, serially diluted, and plated aerobically on NZ medium for enumeration.
M. smegmatis growth inhibition
To determine the MIC for compound 12 against M. smegmatis expressing E. coli DsbB or MtbVKOR, strains RD263 and RD265 were grown for two days in 7H9 medium containing 0.05% Tween 80. These dense cultures were diluted 1:200 into fresh medium, grown to mid-log phase, and then diluted to a starting OD600 of 0.05. Cultures were aliquoted into 96-well plates containing 2-fold dilutions of compound 12 and then placed in an ATR Multitron low-orbital shaker. Cells were grown at 37°C in 90% humidity and shaken at 900 rpm for 24 h. Growth was considered inhibited if the OD600 <0.1 at this time point.
Testing DsbB inhibitors against other gram-negative DsbB
In order to test E. coli strains expressing the dsbB genes from other organisms, the 384-well plates were prepared in the same way as in the HTS only differing in that 0.2% maltose was included in the media to induce the expression of the β-Galdbs and different concentrations of IPTG indicated in “Bacterial strain and growth conditions” section.
Two compound plates (Corning 384-well storage plates, polypropylene round bottom) were prepared with the entire collection of compounds purchased as a result of the medicinal chemistry approach (Compound resupply section). Dilutions of the compounds were dispensed in the 384-well plate ranging from 30 mM to 0.6 μM. 100 nL aliquot of the compounds were transferred to solidified-agar plates by pin transfer (EPSON compound transfer robot) in order to have a final concentration ranging from 50 μM to 0.001 μM of compound, except compounds 27 and 23 which highest concentration started at 28.9 μM and 26.4 μM, respectively. Then, 10 μL of the bacterial cultures at 0.05 of OD600 were added to the agar plates with a Matrix Wellmate (Thermo Scientific). Plates were sealed with breathable sealing film (Axygen BF-400) and incubated at 30ºC for one day in humidified boxes and one day at 4ºC to analyze the results. MER672 was used as EcDsbB expressing strain and CL379 was included as a positive control in each plate. The minimal inhibitory concentration (MIC) of each compound that caused the bacteria to turn light blue was registered for all of the strains expressing dsbB genes from pathogenic bacteria. Since the MIC is related to the expression of each DsbB in E. coli, we ranked the compounds from strong to weak in each DsbB-expressing strain normalizing the data for each strain. Thus, we divided the MIC observed for each compound between the lowest MIC observed for that particular strain expressing DsbB, so the most potent compounds have a ratio of 1 (Figure 3). We obtained the ranking ratio of three independent experiments and the ratio average was calculated and plotted in a 3-rule color-coded table (beige as 1, olive green as 10 and black as 1500) using Excel. For the compounds that did not inhibit at the maximum concentration tested, an arbitrary ratio was assigned (1500) to have the highest value and darkest color in the table (black, non-inhibitor).
Twitching motility assays in Pseudomonas aeruginosa
Twitching motility of P. aeruginosa was assayed on a thin-layer solid surface42. Briefly, 100 × 15 mm plates were filled with 10 mL of M63 minimal media with 1% agarose (instead of 1.5% agar, since agarose is a more purified version). The compound was dissolved in the media before pouring and immediately after solidification one fresh colony was stabbed in the plates without and with 50 μM of compound 12. The plates were incubated for 2 days at 37ºC in humidified boxes.
Statistical Analysis
All results presented are reported as average ± S.D. and represent data from a minimum of three independent experiments, unless otherwise stated in figure legends. RIC50 values were calculated from a minimum of three independent experiments and their 95% confidence intervals were estimated by non-linear log dose-response normalized curve using 4 parameters (GraphPad Prism, La Jolla). Then, inhibition ratios presented in Table 1 were calculated for each compound dividing the RIC50 mean of compound 16 between the RIC50 mean of each compound. KI values were estimated from Dixon plots by taking the average values of intersection points.
Supplementary Material
Acknowledgments
We thank the ICCB-Longwood screening facility for access to their compound libraries, equipment, screening supplies and to the staff including Dr. Su Chiang, Dr. Kyungae Lee, Stewart Rudnicki and Doug Flood for helpful advice and data handling. We thank Dr. Robert Goldman of the National Institute of Allergy and Infectious disease for the collection of M. tuberculosis-growth inhibitors. We thank Dr. Ross Tomaino of Taplin Mass Spectrometry Core Facility at Harvard Medical School for his assistance in sample analysis. This work was supported by National Institute of General Medical Sciences grants GMO41883 (to J.B. and D.B.), Harvard Catalyst Pilot Grant Harvard #149734 (to J.B), NIH Grant #5-UL1RR02568-01 (to J.B), Blavatnik Biomedical Accelerator at Harvard University (to J.B), NIH Grant 3P01-A1AI074805-04S1 Subaward R01638 (to E.J.R), NIH Grant PO1-HL087203 (to B.F), NIH Grant RO1-HL092125 (to B.C.F) and NERCE-BEID Grant U54-AI057159. C.L. was partially supported by CONACYT postdoctoral fellowship. J.L.B. was supported by T32-HL07917 Grant (to B.F). B.M.M. was supported by Ruth L. Kirschstein National Research Service Award. M.E. was supported by New England BioLabs Grant. A.M. was supported by HSPH Yerby postdoctoral fellowship. J.B. is an American Cancer Society Professor.
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Heras B, et al. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7:215–25. doi: 10.1038/nrmicro2087. [DOI] [PubMed] [Google Scholar]
- 2.Depuydt M, Messens J, Collet JF. How proteins form disulfide bonds. Antioxid Redox Signal. 2011;15:49–66. doi: 10.1089/ars.2010.3575. [DOI] [PubMed] [Google Scholar]
- 3.Kadokura H, Beckwith J. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid Redox Signal. 2010;13:1231–46. doi: 10.1089/ars.2010.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–9. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 5.Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A. 2008;105:11933–8. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, et al. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–12. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Dutton RJ, Beckwith J, Boyd D. Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid Redox Signal. 2011;14:1413–20. doi: 10.1089/ars.2010.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Premkumar L, et al. Rv2969c, essential for optimal growth in Mycobacterium tuberculosis, is a DsbA-like enzyme that interacts with VKOR-derived peptides and has atypical features of DsbA-like disulfide oxidases. Acta Crystallogr D Biol Crystallogr. 2013;69:1981–94. doi: 10.1107/S0907444913017800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutton RJ, et al. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc Natl Acad Sci U S A. 2010;107:297–301. doi: 10.1073/pnas.0912952107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 11.Froshauer S, Green GN, Boyd D, McGovern K, Beckwith J. Genetic analysis of the membrane insertion and topology of MalF, a cytoplasmic membrane protein of Escherichia coli. J Mol Biol. 1988;200:501–11. doi: 10.1016/0022-2836(88)90539-6. [DOI] [PubMed] [Google Scholar]
- 12.Tian H, Boyd D, Beckwith J. A mutant hunt for defects in membrane protein assembly yields mutations affecting the bacterial signal recognition particle and Sec machinery. Proc Natl Acad Sci U S A. 2000;97:4730–5. doi: 10.1073/pnas.090087297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian H, Beckwith J. Genetic screen yields mutations in genes encoding all known components of the Escherichia coli signal recognition particle pathway. J Bacteriol. 2002;184:111–8. doi: 10.1128/JB.184.1.111-118.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldman RC, Laughon BE. Discovery and validation of new antitubercular compounds as potential drug leads and probes. Tuberculosis (Edinb) 2009;89:331–3. doi: 10.1016/j.tube.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regeimbal J, et al. Disulfide bond formation involves a quinhydrone-type charge-transfer complex. Proc Natl Acad Sci U S A. 2003;100:13779–84. doi: 10.1073/pnas.1935988100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inaba K, et al. DsbB elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J Biol Chem. 2004;279:6761–8. doi: 10.1074/jbc.M310765200. [DOI] [PubMed] [Google Scholar]
- 17.Inaba K, Takahashi YH, Ito K, Hayashi S. Critical role of a thiolate-quinone charge transfer complex and its adduct form in de novo disulfide bond generation by DsbB. Proc Natl Acad Sci U S A. 2006;103:287–92. doi: 10.1073/pnas.0507570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimshaw JP, et al. DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J Mol Biol. 2008;380:667–80. doi: 10.1016/j.jmb.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 19.Lin D, Kim B, Slauch JM. DsbL and DsbI contribute to periplasmic disulfide bond formation in Salmonella enterica serovar Typhimurium. Microbiology. 2009;155:4014–24. doi: 10.1099/mic.0.032904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey H, Habash M, Aidoo F, Burrows LL. Single-residue changes in the C-terminal disulfide-bonded loop of the Pseudomonas aeruginosa type IV pilin influence pilus assembly and twitching motility. J Bacteriol. 2009;191:6513–24. doi: 10.1128/JB.00943-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha UH, Wang Y, Jin S. DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect Immun. 2003;71:1590–5. doi: 10.1128/IAI.71.3.1590-1595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SH, Park SY, Heo YJ, Cho YH. Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor, HudA. Infect Immun. 2008;76:4152–62. doi: 10.1128/IAI.01637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arts IS, et al. Dissecting the machinery that introduces disulfide bonds in Pseudomonas aeruginosa. MBio. 2013;4:e00912–13. doi: 10.1128/mBio.00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatahet F, Boyd D, Beckwith J. Disulfide bond formation in prokaryotes: History, diversity and design. Biochim Biophys Acta. 2014;1844:1402–1414. doi: 10.1016/j.bbapap.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatahet F, Ruddock LW. Topological plasticity of enzymes involved in disulfide bond formation allows catalysis in either the periplasm or the cytoplasm. J Mol Biol. 2013;425:3268–76. doi: 10.1016/j.jmb.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 26.Li T, et al. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427:541–4. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 27.Westhofen P, et al. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286:15085–94. doi: 10.1074/jbc.M110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fruh V, et al. Application of fragment-based drug discovery to membrane proteins: identification of ligands of the integral membrane enzyme DsbB. Chem Biol. 2010;17:881–91. doi: 10.1016/j.chembiol.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–8. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 30.O'Loughlin CT, et al. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A. 2013;110:17981–6. doi: 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassett DJ, et al. Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 2009;17:130–8. doi: 10.1016/j.tim.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Kolpen M, et al. Nitrous Oxide Production in Sputum from Cystic Fibrosis Patients with Chronic Pseudomonas aeruginosa Lung Infection. PLoS One. 2014;9:e84353. doi: 10.1371/journal.pone.0084353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–17. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 34.Mah R, Thomas JR, Shafer CM. Drug discovery considerations in the development of covalent inhibitors. Bioorg Med Chem Lett. 2014;24:33–9. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Boyd D, Weiss DS, Chen JC, Beckwith J. Towards single-copy gene expression systems making gene cloning physiologically relevant: lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J Bacteriol. 2000;182:842–7. doi: 10.1128/jb.182.3.842-847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haldimann A, Wanner BL. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001;183:6384–93. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larsen MH, Biermann K, Jacobs WR., Jr. Laboratory maintenance of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007 doi: 10.1002/9780471729259.mc10a01s6. Chapter 10, Unit 10A 1. [DOI] [PubMed] [Google Scholar]
- 38.Chng SS, et al. Overexpression of the rhodanese PspE, a single cysteine-containing protein, restores disulphide bond formation to an Escherichia coli strain lacking DsbA. Mol Microbiol. 2012;85:996–1006. doi: 10.1111/j.1365-2958.2012.08157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadokura H, Bader M, Tian H, Bardwell JC, Beckwith J. Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:10884–9. doi: 10.1073/pnas.97.20.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol. 1999;181:508–20. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thibodeau SA, Fang R, Joung JK. High-throughput beta-galactosidase assay for bacterial cell-based reporter systems. Biotechniques. 2004;36:410–5. doi: 10.2144/04363BM07. [DOI] [PubMed] [Google Scholar]
- 42.Semmler AB, Whitchurch CB, Mattick JS. A re-examination of twitching motility in Pseudomonas aeruginosa. Microbiology. 1999;145:2863–73. doi: 10.1099/00221287-145-10-2863. Pt 10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.