

Abstract
Rare conditions are sometimes ignored in biomedical research because of difficulties in obtaining specimens and limited interest from fund raisers. However, the study of rare diseases such as unusual cancers has again and again led to breakthroughs in our understanding of more common diseases. It is therefore unsurprising that with the development and accessibility of next-generation sequencing, much has been learnt from studying cancers that are rare and in particular those with uniform biological and clinical behavior. Herein, we describe how shotgun sequencing of cancers such as granulosa cell tumor, endometrial stromal sarcoma, epithelioid hemangioendothelioma, ameloblastoma, small-cell carcinoma of the ovary, clear-cell carcinoma of the ovary, nonepithelial ovarian tumors, chondroblastoma, and giant cell tumor of the bone has led to rapidly translatable discoveries in diagnostics and tumor taxonomies, as well as providing insights into cancer biology.
Keywords: Next-generation sequencing, Formefruste, Rare tumors, Genomics
Impact of study of rare tumors
The history of biomedical research has repeatedly shown that with the advent of new methodologies, the study of rare, but clinically well-defined disease entities has led to the generation of a disproportionate amount of knowledge. For instance in the eighteenth century, the peculiar scrotal skin cancers of adolescent males with a history of occupation as chimney sweeps led Percivall Pott to describe one of the earliest associations of workplace hazards and cancer [1]. His study shed light on the potential role of environmental factors in cancer formation and helped lay the foundations for the science of epidemiology. Another example happened in 1961 when Sir Anthony Epstein came across D.P. Burkitt’s description of an unusual new children’s cancer in Africa with a geographic distribution related to certain rainfall and temperature patterns [2]. Epstein thought of a possible climate-dependent vector and initiated a study, during a time of great advancements in molecular biology and virology, that led to the discovery of the Epstein Barr virus, the first proven human cancer virus [3]. Retinoblastoma was another relatively rare condition that led to the conceptualization of tumor suppressor genes. The comparison of kinetics of unilateral sporadic versus bilateral familial cases led Alfred Knudson Jr. to describe the two-hit hypothesis, which revolutionized cancer biology [4].
It has long been clear that mutations play a critical role in the development of cancer. However, many common cancers are both biologically and clinically complicated and their mutational landscape reflects such complexity [5, 6]. On the other hand, some perhaps more obscure tumors, especially ones affecting younger patients and specific sites, reveal simpler genomic landscapes with characteristic mutations that allow a more focused look at the oncogenic processes. That is not to say that such scenarios are absent in more common cancers, especially when examined as specific subtypes. However, rare cancers should not simply be ignored because of their rarity or the logistical difficulty of working with them. These anomalies in nature, because of the very fact of their unusual patterns, can hold the key to understanding more common tumors. We term such neoplasms forme fruste tumors. Forme fruste refers to an attenuated manifestation of disease. We recognize that these tumors are true neoplasms and in many cases, cancers. However, they do not have the genomic baggage and heterogeneity due to genomic instability as seen in common cancers and it is this attenuated genomic phenotype that makes them such tractable and useful targets for genomic research. In this review, we will go over recent sequencing studies of some forme fruste tumors which led to discoveries of profound importance.
Era of next-generation sequencing
Ever since the discovery of DNA and its association with human cancer, scientists and clinicians have dreamt of the possibility to scrutinize it base by base. The ability to sequence DNA, which quickly developed into a robust method by Sanger sequencing [7], was a solid step toward this goal. With the Human Genome Project establishing a map of the human genetic code and rapid advances in computer technology, everything seemed to be in place other than cost and efficiency. Billions of dollars and years of multi-institutional efforts would not make nucleotide sequencing an accessible tool for scientists to ask questions on a regular basis, and the limited resources were not earmarked for the study of rare specimens. The limitations of Sanger sequencing were in the termination of polymerase reactions as well as in the need to separate the products of these reactions by gel or other electrophoretic systems [8]. Additionally, preparation of sequencing libraries was necessary via transformation in E. coli or by an incredibly large number of separate PCR reactions. However, with massively parallel sequencing platforms, the first shortcoming was overcome by reversible fluorescent nucleotide addition and imaging (used in Illumina platforms) or through monitoring nucleotide addition via ion detection (used in Ion Torrent platforms (Life Technologies)) both achieved by cyclic manipulation of polymerase or ligase enzymes [9]. Moreover, the second shortcoming was resolved by in vitro library preparation via techniques such as emulsion PCR [10] (Ion Torrent) or bridge PCR on solid surfaces [11, 12] (Illumina). With these improvements, the sequencing cost and time requirements have been vastly reduced. Advancements in bioinformatics and the ability to more readily distinguish signal from noise have also increased the feasibility of large- and small-scale genomic studies. Thus, today, sequencing whole genomes and transcriptomes is more accessible and has become a reality for individual laboratories. We argue that we are in an incredibly exciting era of molecular medicine where a new “molecular microscope” in the form of massively parallel sequencing, also commonly referred to as next-generation sequencing (NGS) or second-generation sequencing, is giving rise to a whole new paradigm for the understanding of human diseases.
We will discuss recent attempts to study forme fruste tumors using NGS. The general approach and representative bioinformatic tools employed in such studies are summarized in Fig. 1. Specific forme fruste tumor types will be discussed that exemplify the impact of such studies in three categories: a better definition of an already known disease, establishment of new disease subtypes, and development of novel insights into oncogenic mechanisms. Table 1 includes a more comprehensive summary of discoveries in forme fruste tumors with a focus on recent NGS findings and their study designs. In addition, Fig. 2, accompanied by Table 2, demonstrates the broad range of pathways affected in such tumors. Lastly, we will go over some of the challenges and scenarios where the study of certain forme fruste-like tumors shows that their genomic behavior is not always straightforward. In addition to the broad array of discoveries made, the small number of cases used in each successful discovery process is perhaps noteworthy (Table 1) with the single case of endometrial stromal sarcoma where, as described below, sequencing led to a diagnostic and formal disease reclassification.
Fig. 1.
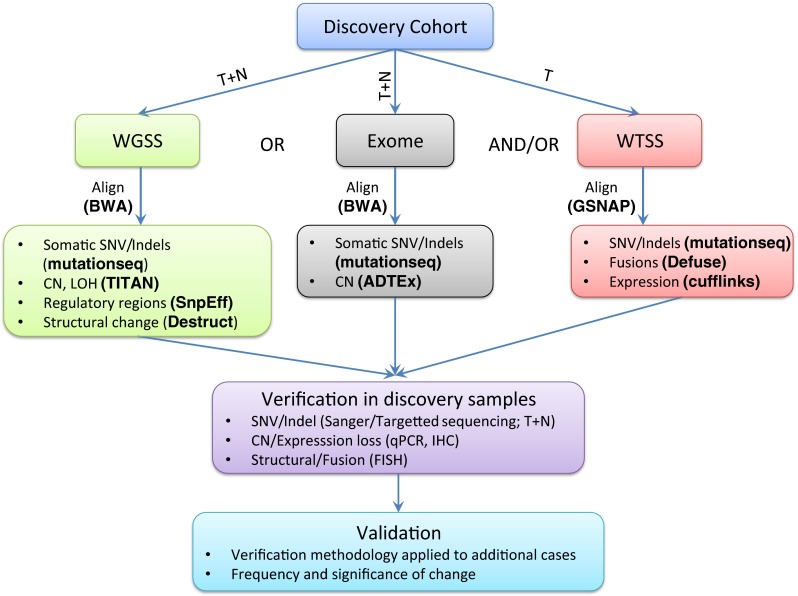
A flowchart of the typical approach to a NGS study to discover novel mutations. Representative bioinformatic programs are in parenthesis and in bold (further details can be found in [75–81]). For somatic mutations, tumor (T) and matched normal (N) samples, obtained from blood or adjacent normal tissue, are used in whole genome (WGSS) or exome sequencing to look for somatic mutations and copy number changes (CN). Transcriptome analysis (WTSS) of tumor samples will enable assessment of expressed mutations and fusions as well as expression patterns. Confirmation of the NGS findings using a different platform such as Sanger sequencing to eliminate false positives would be the next step. Finally, to understand the frequency of the findings in the disease of interest, analysis on a larger validation cohort of tumor samples should be completed. For hotspot mutations, sequencing; for inactivating mutations, sequencing or immunohistochemistry (IHC); and for fusions, fluorescent in situ hybridization (FISH) could be methods of choice for verification and validation
Table 1.
A list of mutations in pathologically specific tumors with a focus on recent NGS discoveries, including discovery methodology, and validation results
| Disease | Mutation | Discovery methodology | Discovery cohort size | Frequency in validation cohort (number of cases) | Reference |
|---|---|---|---|---|---|
| Congenital fibrosarcoma | ETV6-NTRK3 (fusion) | FISH positional cloning, RACE | 4 primary tumors grown in culture | Same as discovery, 100 % (3) | [95] |
| Granulosa cell tumor | FOXL2 (hotspot) | WTSS | 4 tumors | 97 % (89) | [18] |
| CCC | ARID1A (loss) | WTSS | 19 tumors | 46 % (119) | [44] |
| Exome capture, NGS | 8 tumor enriched samples (magnetic beads) | 57 % (42) | [45] | ||
| Epithelioid hemangioendothelioma | WWTR1-CAMTA1 (fusion) | WTSS | 1 case with t(1;3)(p36;q25), snap | 87–89 % (45–47) | [25] |
| FISH positional cloning | 17 FFPE | Same as discovery, 100 % (17) | [28] | ||
| Nonepithelial ovarian tumors | DICER1 (hotspot) | WTSS and exome capture | 14 samples, 5 with matched normal | 20 % (145) | [59] |
| EWSR1-ETS-negative small round cell bone sarcoma | BCOR-CCNB3 (fusion) | WTSS | 4 tumors | 4 % of fusion negative sarcomas (594) | [35] |
| High-grade endometrial stromal tumors | YWHAE-FAM22 (fusion) | WTSS | 1 cell line | 100 % (12) | [32] |
| Chondrosarcoma | COL2A1 (inactivating) | Exome | 49 tumors with matched normal | 44 % (26) | [111] |
| Solitary fibrous tumor | NAB2-STAT6 (fusion) | Exome | 17 tumors with matched normal | 55 % (29) | [112] |
| NF2-negative familial multiple spinal meningioma | SMARCE1 (loss) | Exome | 3 unrelated individuals | 33 % (6) | [54] |
| Chondroblastoma | H3F3B (hotspot) | WGSS | 6 tumor/normal matched | 95 % (77) | [69] |
| Giant cell tumors of bone | H3F3A (hotspot) | Sanger sequencing | 92 % (53) | ||
| DIPG/pediatric GBM | H3F3A (hotspot) | WGSS | 7 with matched normal | 78 % (43) | [64] |
| H3F3A (hotspot) | Exome | 48, 6 with matched normal | 36 % (42) | [65] | |
| ATRX (loss) | Sequencing 33 % (42), IHC 35 % (113) | ||||
| ACVR1 (hotspot) | WGSS, exome, WTSS | 42 HGG tumors with matched normal | 32 % of DIPG(80) | [100] | |
| WGSS, exome, microarray | 36 frozen tumors and matched normal | 20 % (25) | [101] | ||
| WGSS, exome | 26 tumors and matched normal | 21 % (26) | [102] | ||
| Exome, WTSS | 39 tumors with matched normal | 13 % (same as discovery cohort) | [103] | ||
| Glomus tumor | MIR143-NOTCH (fusion) | WTSS | 3 tumors | 64 % (33) | [88] |
| Familial infantile myofibromatosis | PDGFRB (hotspot) | Exome | 11 germline (familial, 9 families) | 89 % (9 families) | [84] |
| Exome, WTSS | 2 germline (exome), 1 tumor (WTSS) | 100 % of familial (8), 0 % of simplex (5) | [85] | ||
| SCCOHT | SMARCA4 (loss) | WGSS, Exome | 13 tumor/normal pairs | 82 % (17) | [49] |
| Exome | 6 tumor/normal pairs (familial, 3 families) | 90 % (20) | [50] | ||
| Exome | 12 tumor/normal pairs | 88 % (43) | [51] | ||
| Sanger sequencing, IHC | 2 FFPE cases | None | [52] | ||
| Chondromyxoid fibroma | GRM1 (fusion) | SNP Arrays, WGSS, WTSS | 8 tumors , 2 with both WGSS and WTSS data | RT-PCR 90 % (20) | [82] |
| Biphenotypicsinonasal sarcoma | PAX3-MAML3 (fusion) | WTSS | 1 tumor | 96 % (25) | [83] |
| Maxillary ameloblastoma | SMO (hotspot) | WTSS | 2 FFPE cases | 82 % (11) | [39] |
| Mandibular ameloblastoma | BRAF (hotspot) | 69 % (13) | [39] | ||
| MPNST | SUZ12, EED (inactivating) | WGSS or exome | 8 tumors, 5 with matched normal | SUZ12 in 26 % (42) | [86] |
| Exome, WTSS | 15 tumors with matched normal | SUZ12 in 49 % and EED in 38 % (37) | [87] | ||
| PLGA | PRKD1 (hotspot) | WTSS, Exome | 6 tumors, 3 with matched normal | 72 % (53) | [89] |
| ALK-negative inflammatory myofibroblastic tumor | ALK, ROS1 and PDGFRB (fusions) | Targeted NGS | 1 FFPE (tumor) | 73 % (11) | [93] |
| Thymoma | GTF2I (hotspot) | Exome | 28 tumors with normal | 82 % type A, 74 % type AB (274) | [96] |
| Cortisol-producing adrenal tumors | PRKACA (hotspot) | Exome | 25 tumors and matched normal | 21 % (63) | [97] |
| Exome | 8 tumors with matched normal | 53 % (57) | [104] | ||
| ERMS | MYOD1 (hotspot) | Exome and WTSS | 20 tumors (8 with both WTSS, exome) | 10 % (104) | [70] |
| Brainstem gliomas | PPM1D Exon 6 truncation, activating | Exome | 26 tumors with matched normal | 37.5 % of H3F3A harboring BSG (24) | [98] |
| Diffuse gastric carcinoma | RHOA (hotspot, GOF) | Exome | 30 tumors with matched normal | 25 % (87) | [99] |
| AITL | RHOA (hotspot, inactivating) | Exome, WTSS, aCGH | 5 tumor/normal pairs, 4 tumor only | 53 % (45) | [105] |
| Exome | 12 PTCL tumor/normal pairs (3 AITL) | Allele-specific PCR mutation assay 67 % (43 AITLs) | [107] | ||
| Exome | 3 tumor/normal pairs | 71 % (72) | [108] | ||
| PMBCL | PTPN1 (inactivating) | WGSS, WTSS | 10 tumors, 2 with matched normal | 22 % (77) | [106] |
| Ewing sarcoma | STAG2 (inactivating) | WGSS | 6 tumors with matched normal | IHC 12 % (154) | [90] |
| Exome | 26 matched tumors, 66 tumor only, 11 lines | 15 % (73), IHC | [91] | ||
| WGSS | 112 tumors with matched normal | Capture sequencing 13 % (199) | [92] | ||
| SNP array | 1 cell line, U138MG | IHC 21 % (53) | [94] | ||
| IGHV4-34+ HCL | MAP2K1 (hotspot) | Exome | 10 tumors, 6 with matched normal | 48 % (21) | [109] |
| Familial schwannomatosis | LZTR1 (inactivating) | Targeted deep sequencing | 16 unrelated cases, germline mutations | 75 % (12) | [110] |
References: [18, 25, 28, 32, 35, 39, 44, 45, 49–52, 54, 64, 65, 69, 70, 82–112]
CCC clear cell carcinoma of the ovary, DIPG diffuse intrinsic pontineglioma, GBM glioblastoma multiforme, SCCOHT small-cell carcinoma of the ovary of the hypercalcemic type, MPNST malignant peripheral nerve sheath tumor, ERMS embryonal rhabdomyosarcoma, PLGA polymorphous low-grade adenocarcinoma of salivary gland, AITL angioimmunoblastic T cell lymphoma, PMBCL primary mediastinal B cell lymphoma, GOF gain of function, FISH fluorescent in situ hybridization, RACE rapid amplification of cDNA ends, WGSS whole genome shotgun sequencing, WTSS whole transcriptome shotgun sequencing, IHC immunohistochemistry, aCGH array comparative genomic hybridization
Fig. 2.
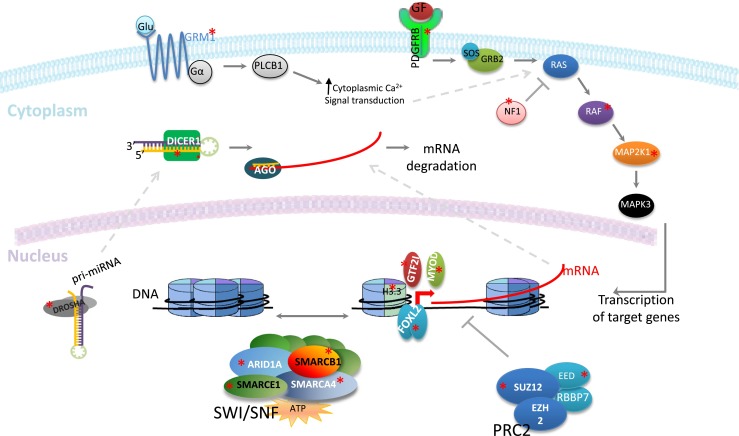
Recent discoveries of mutations in forme fruste tumors revealing a broad range of pathways involved. Driver mutations from membrane receptors, to signal transducers, chromatin modifying, and remodeling complexes, as well as transcription factors and microRNA processing factors have been described in a variety of specific tumor pathologies, which may occur at low frequencies in the population. However, such insight, when followed by an understanding of the tumorigenic mechanisms involved, can vastly improve understanding of more common cancers and tumor biology in general. This figure is accompanied by Table 2 which includes a list of such tumors as well as the indicated mutations (marked by asterisks)
Table 2.
List of mutations indicated in Fig. 2
| Pathway | Target | Mode of dysregulation | Tumor example |
|---|---|---|---|
| SWI/SNF | SMARCA4 | Inactivated | SCCOHT, medulloblastoma, Burkitt’s lymphoma, NSCLC |
| SMARCB1 | Inactivated | Rhabdoid tumors, epithelioid sarcoma, CRINET, SBC, schwannomatosis, renal medullary carcinoma, gastrointestinal neoplasms | |
| SMARCE1 | Inactivated | Familial multiple spinal meningiomas | |
| ARID1A | Inactivated | CCC, EC, GC, neuroblastoma | |
| Histone 3.3 | H3F3A | Hotspot | GCBT, DIIPG |
| H3F3B | Hotspot | Chondroblastoma | |
| Transcription factors | GTF2I | Hotspot | Thymoma |
| MYOD1 | Hotspot | ERMS | |
| FOXL2 | Hotspot | GCT | |
| PRC2 complex | SUZ12 | Inactivated | MPNST |
| EED | Inactivated | MPNST | |
| miRNA processing | DICER | Hotspot | NEOC, e.g., SLCT |
| DROSHA | Hotspot | WT | |
| RTK signal transduction | PDGFRB | Hotspot | IMT |
| Fusion | FIM | ||
| NF1 | Inactivated | MPNST | |
| RAF | Hotspot | M. Ameloblastoma | |
| MAP2K1 | Hotspot | IGHV4-34+ HCL | |
| GPCR | GRM1 | Fusion | Chondromyxoid fibroma |
RTK receptor tyrosine kinase, SCCHOT small-cell carcinoma of the ovary of the hypercalcemic type, NSCLC nonsmall-cell lung cancer, CRINET cribriform neuroepithelial tumor, SBC sinonasal basaloid carcinoma, RMC renal medullary carcinomas, CCC clear-cell carcinoma of the ovary, EC endometriod carcinoma of ovary, GC gastric cancer, GCBT giant cell bone tumor, DIPG diffuse intrinsic pontineglioma, ERMS embryonal rhabdomyosarcoma, GCT granulosa cell tumor, MPNST malignant peripheral nerve sheath tumor, NEOC nonepithelial ovarian cancer, SLCT Sertoli-Leydig cell tumor, WT Wilm’s tumor, IMT inflammatory myofibroblastic tumor, FIM familial infantile myofibromatosis, M. Ameloblastoma mandibular ameloblastoma, HCL hairy cell leukemia
Better definition of an existing pathology
Pathognomic mutations allow a more specific definition of a previously established histological diagnosis. Below, we will discuss the cases of the FOXL2 mutation in adult-type granulosa cell tumors (GCTs) as well as the WTR1-CAMTA1 fusion in epithelioid hemangioendotheliomas (EHEs).
Granulosa cell tumors and FOXL2 mutations
GCTs are rare, constituting only about 5 % of ovarian tumors [13]. Until 2009, the molecular biology of this tumor remained a mystery and hence, there was limited success in development of therapeutics for aggressive cases [14]. There are two subtypes of this tumor: adult-type and juvenile. These have similar biomarker profiles but occur in different age groups and have different histopathological features [13]. Adult-type GCTs fit the concept of a forme fruste cancer: cytogentics had shown a more stable genome compared to other ovarian tumors [15], and the tumor subtypes have a consistent pathological presentation with cells that have maintained some levels of differentiation expressing follicule-stimulating hormone receptors and inhibin [16]. There were no associations between expression of common oncogenes and tumor suppressors, such as MYC, TP53, ERBB2, or RAS family and outcomes in GCTs [17]. Based on these facts, Shah et al. reasoned that with sequencing of very few adult-type GCT cases, rather than the massive sample sets needed for the more common genetically complex tumors, considerable insight into the biology of GCTs could be attained. Thus, only four samples of adult-type GCTs were used for whole transciptome sequencing as a discovery cohort and 11 other ovarian tumors were sequenced as a comparative cohort [18]. After alignment and removal of previously reported germline insertions and deletions, there were between 289 and 495 somatic nonsynonymous variants in the GCTs. Genes with mutations in at least three of the four cases that were not mutated in the comparative cohort were considered for further follow-up [18]. The only potential mutation found in all four cases was a C134W mutation in FOXL2 [18]. The resulting mutant protein was still expressed in GCTs as observed by immunohistochemistry in cases with apparent homozygosity (likely through loss of the normal allele), meaning most likely there was a gain/switch of function. In a validation cohort of an additional 95 sex-cord stromal ovarian tumors, specificity and sensitivity of the C134W mutation in adult-type GCTs was established [18]. This study was significant in three aspects: it was the first time that a consistent genetic event was associated with GCTs, the first time FOXL2 had been indicated to have an oncogenic role in any tumor, and the first example of a novel disease-defining pathognomonic driver mutation being discovered using massively parallel sequencing.
Although FOXL2 is known to be critical for the development of ovaries and is one of the early differentiation markers [19], somatic mutations in this transcription factor had not been linked to pathology before this study. The diagnostic implications of the C134W mutant FOXL2 have already become apparent [20, 21], and some studies have since looked at potential mechanistic pathways. It has been suggested that the hotspot FOXL2 mutation might have very particular effect in a specific context: the mutant FOXL2 reduces the expression of gonadotropin-releasing hormone (GnRH) receptor and limits the GnRH-induced apoptosis seen in normal human granulosa cells [22]. This finding shows that tissue-specific pathways may be the bottlenecks that limit driver mutations that can arise in a specific cell of origin. Furthermore, the mutant FOXL2 has also been suggested to be less stable because of increased phosphorylation via GSK3β and MDM2-mediated ubiquitination and proteasome degradation [23]. Hence, inhibition of GSK3β has already been identified as a therapeutic target that stabilizes mutant FOXL2 and this stabilization may in turn lead to increased apoptosis.
WWTR1-CAMTA1 in EHE
Another example of a disease-defining mutation came with EHE, a rare tumor that can present diagnostic challenges. The tumor is a vascular sarcoma with epithelial-looking cells that show vascular differentiation with positivity for platelet endothelial cell adhesion molecule and CD34 [24]. Through the use of a single index case, Tanas and colleagues were able to identify a fusion of WWTR1 to CAMTA1 and establish it as a specific event in EHE [25]. In a validation cohort of 47 cases, they showed that rearrangements of the involved genes happened 87–89 % of the time whereas none of 118 cases of other vascular tumors showed these rearrangements [25]. Part of the success in identifying this pathognomonic fusion was due to the already known recurrent translocation involving chromosomes 1 and 3 in EHE [26, 27]. This meant that during the bioinformatic analysis, focus was limited to the predicted fusions involving genes on these chromosomes. Simultaneously, another group was also able to use the more traditional method of fluorescent in situ hybridization (FISH) positional cloning in 17 cases of EHE and also discovered the fusion partners WWTR1 and CAMTA1 [28].
WWTR1 (also known as TAZ) encodes a transcriptional coactivator containing the WW domain. This domain, which is named as such because of the two conserved tryptophans, mediates specific protein-protein interactions and has been implicated in Hippo signaling, a critical pathway in regulating organ size and keeping proliferation in check. WWTR1 is phosphorylated by lats tumor suppressor kinases (LATS2) which are key components of Hippo signaling and this in turn leads to binding by 14-3-3 proteins which lead to cytoplasmic localization and hence inactivation of WWTR1 [29]. Interestingly, in the EHE fusion, the 14-3-3 binding domain of WWTR1 is maintained; however, one of the critical LATS2 phosphorylation sites, namely Ser311, is lost. This perhaps could render the fusion protein partly resistant to inhibition by Hippo signaling. CAMTA1 is a transcriptional regulatory protein with the capacity to bind DNA. Because WWTR1 has no known DNA-binding motifs and since the DNA-binding domain of CAMTA1 is maintained, the new fusion protein might giveWWTR1 a new ability to bind DNA [25].
Establishment of new classification/subtype
In addition to identifying pathognomonic mutations by deep sequencing a few samples, sequencing studies of forme fruste tumors have led to new classification and subtype establishments. Three examples are discussed as follows.
YWHAE fusions in high-grade endometrial stromal sarcoma
Endometrial stromal sarcoma (ESS) is a malignancy of the uterus that had been previously linked with recurrent fusions: the fusion of JAZF1, a transcriptional repressor, with members of the polycomb complex including SUZ12, PHF1, and EPC1 [30, 31]. Yet, there remained a subset of ESS tumors, often with a higher histologic grade, that could not be demonstrated to carry fusions involving these genes. Lee and colleagues thus decided to look in depth at the genomics of such cases and ended up discovering recurrent fusions involving YWHAE and the FAM22 family [32]. As in earlier work, Lee and colleagues first drew on results from cytogenetic studies and noted a recurrent t(10;17)(q22;p13). Similar to the case with the WWTR1-CAMTA1 fusion in EHEs, this karyotype information greatly aided the analysis of the next-generation sequencing data such that, by the use of just one sample, they were able to focus on the YWHAE-FAM22A translocation event, later showing that in cases missing this particular fusion, YWHAE was fused to homologs of FAM22A such as FAM22B [32]. This work established a new entity of higher-grade endometrial stromal sarcomas with a molecular defining feature that distinguishes them from other endometrial stromal sarcomas [33]. In fact, soon after the discovery of this novel fusion, the World Health Organization incorporated the presence of YWHAE-FAM22 translocations into the classification of endometrial stromal sarcomas [34].
BCOR-CCNB3 bone sarcoma
An exemplary case of NGS defining a new pathology came through the study of peculiar small round cell bone sarcomas that lacked the EWSR1-ETS fusions of the top candidate in the differential diagnosis, Ewing sarcoma. Four index cases were used for RNA-seq with fusion analyses, and out of these strong evidence for fusion transcripts was seen in two cases: one that had an atypical Ewing fusion of FUS-FEV and another with a completely novel fusion of exon 15 of BCOR to exon 5 of CCNB3 [35]. The authors then carried out a comprehensive RT-PCR screening of 594 sarcomas lacking fusions classically sought in diagnostics laboratories. They were able to identify an additional 24 cases of sarcomas with the BCOR-CCNB3 fusion. Microarray expression profiling of ten such cases showed that these tumors had a different profile than other tumors in the differential diagnosis such as Ewing sarcoma, and hence, a whole new bone sarcoma was established [35]. BCOR is thought to encode a ubiquitously expressed protein with a role in repression of transcription through epigenetic mechanisms and in mesenchymal stem cell function [36]. On the other hand, CCNB3 expression is restricted to testis and the encoded protein is a cyclin expressed during spermatogenesis [37]. The ectopic expression of CCNB3 as a result of the fusion event could be the driver of oncogenesis in this novel sarcoma. Indeed, expression of both the truncated and BCOR fused CCNB3 in fibroblast lines leads to increased proliferative capacity [35].
Maxillary versus mandibular ameloblastomas
Another recent study established that ameloblastomas, rare benign tumors of the jaw thought to originate from ameloblasts [38], have distinct recurrent mutations depending on whether they arise in the maxilla versus the mandible. The maxillary ameloblastomas harbor a SMO hotspot mutation, and the mandibular tumors have BRAF hotspot mutations [39]. Although ameloblastomas are benign and rare tumors, this study emphasizes the mutational heterogeneity of histologically indistinguishable tumors depending on their location and highlights the significance of molecular classification. As mutant BRAF, commonly seen in melanomas, can be targeted with new therapies, this finding also has immediate therapeutic implications. Associations of tumor location and defining mutations have also been identified, for instance, in mengiomas: those that arise in the lateral and posterior regions bear NF2 mutation whereas those in the anterior and medial regions do not [40]. Even in rare tumors with seemingly distinctive histology, there exist subsets defined by specific molecular aberrations. The new disease subclassifications thereby identified may be of great significance for development and application of targeted therapeutics.
Insights into cancer mechanisms
The study of rare tumors has also expanded our knowledge about cancer pathways. We will focus on recent findings of recurrent mutations in chromatin remodelers, microRNA processors, and histones.
SWI/SNF mutations in ovarian epithelial tumors and meningiomas
Clear-cell ovarian carcinomas are the second most common type of ovarian cancer [41] and until 2010 were not very well studied despite evidence of relative genomic stability [42, 43]. With whole transcriptome sequencing/exome sequencing, recurrent mutations in ARID1A, a member of the already established SWI/SNF chromatin remodeling complex were found [44, 45]. The mutations were spread across the ARID1A gene and led to its inactivation, thus suggesting that this gene may function as a tumor suppressor. Although other core members of the SWI/SNF complex had been linked to cancer previously (SMARCB1 and SMARCA4 are known to have lost expression in a variety of tumors), this study showed that noncanonical members of the SWI/SNF complex could also play important roles in tumorigenesis. Furthermore, lack of evidence for mutations in other members of the complex hinted at a context-specific tumor suppressor role for the individual members of the SWI/SNF complex. Additionally, Wiegand and colleagues also showed that the mutation was present in precursor atypical endometriotic lesions of the tumor, and thus was likely an early driver of ovarian clear-cell carcinoma. ARID1A mutations were later found in a variety of other more common types of cancer including gastric adenocarcinomas [46] and colorectal cancers [47].
Small-cell carcinoma of the ovary of the hypercalcemic type (SCCOHT) is another rare but genetically stable tumor [48] that was discovered to have abnormalities in the SWI/SNF complex. In this case, the core enzymatic unit of the protein complex, SMARCA4, was mutated in an inactivating fashion in the majority of cases, and almost all tumors of this specific diagnosis stained negatively for SMARCA4’s protein product BRG1 [49–52]. Although mutations in SMARCA4 have been described in more common cancers such as lung adenocarcinomas [53], they occur in a fraction of cases and are not the obvious drivers of oncogenesis. The studies in SCCOHT with loss of SMARCA4 in almost all cases emphasized the driver role of SMARCA4 loss. Another example of a critical driver role of SWI/SNF mutations came through the NGS study of familial multiple spinal meningiomas [54]. In familial cases, which tested negative for previously described NF2 or SMARCB1 mutations, germline SMARCE1 mutations were identified through exome sequencing. Again, the protein was lost in the tumor samples but not in normal tissue, thus suggesting a classic Kundson biallelic inactivation and a tumor suppressor role of SMARCE1.
It should be noted that the reason for disease specificity of SWI/SNF member mutations and indeed the steps in tumorigenesis associated with their loss are not clear. SWI/SNF is thought to regulate the expression of many genes and interacts with many critical cancer pathways from cell cycle regulation to hedgehog and Wnt signaling. Indeed, it has been suggested that perhaps the remaining complex, which still assembles without the mutated members, might act as an oncoprotein and drive tumorigenesis [55]. Thus, much is still to be clarified in this area; however, since the establishment of the association of the SWI/SNF complex with cancer in rather rare entities, we know today that about 20 % of all cancers have mutations in this complex [56]. However, the impact of these mutations is by and large yet to be established.
MicroRNA processing mutations in nonepithelial ovarian tumors
Given abnormalities in microRNA levels in certain cancer, it was thought that the genes encoding proteins involved in microRNA processing might also be of significance in oncogenesis. Germline mutations in DICER1 were identified in the rare familial pleuropulmonary blastoma–family tumor and dysplasia syndrome [57]. However, the first evidence for somatic oncogenic mutations of DICER1 came from the study of nonepithelial ovarian tumors [58]. Recurrent somatic hotspot mutations in DICER1 were identified across nonepithelial ovarian tumor types and were most predominantly seen in Sertoli-Leydig cell tumors [59]. Although low expression of DICER1 has been previously associated with worse prognosis in breast cancer [60] and ovarian tumors [61], the study of these nonepithelial ovarian tumors changed the paradigm as for the first time it was found that a hotspot genetic aberration in DICER1 can drive cancer through the combination of loss of one allele and a functionally deficient protein, this is an aberration of the classic two-hit hypothesis [59]. In actuality, DICER1 in nonepithelial ovarian tumors does not fit traditional tumor suppressor or oncogene models. Rather, there seems to be a mix of the two models involved in tumorigenesis. There is an inherited inactivation of one copy of the genes, and the remaining allele is not totally inactivated somatically, which would be lethal in most cells rather is hypomorphic via hotspot mutations (Fig. 2). The hotspot mutations are found in the RNaseIIIb metal-binding site, reducing RNaseIIIb activity and leading to a global loss in the processing of mature 5p microRNAs but maintenance of 3p processing [62]. Later studies showed that oncogenic mutations in DROSHA, another microRNA processing gene, and associated global microRNA changes also occur in Wilm’s tumor [63]. Therefore, processors of microRNA represent another family of cancer-associated proteins and forme fruste tumors were significant in this realization.
Histone mutation in bone and central nervous system tumors
Another prime example of insights into cancer biology comes from the identification of mutations in histones in forme fruste tumors. Mutations in H3F3A, which encodes histone 3.3, were identified in pediatric diffuse intrinsic pontinegliomas (DIPGs) [64] and pediatric glioblastomas [65]. Histone 3.3 is a member of the histone 3 family which is associated with active chromatin and is incorporated into chromatin throughout the cell cycle [66–68]. Interestingly, an independently regulated gene named H3F3B also seemingly encodes the same histone 3.3 protein; however, mutations in this gene were not identified in DIPGs or glioblastomas. In a seminal study, Behjati et al. described H3F3A driver mutations in another tumor type: chondroblastomas [69]. Additionally, they also discovered novel H3F3B mutations in giant cell tumors of bone [69]. Chondroblastomas and giant cell tumors of bone have similarities such as clinical presentation in the bone epiphysis and the presence of large numbers of osteoclastic giant cells; however, they tend to affect different age groups and have different clinical outcomes. As mentioned, the two genes encode the same protein, yet Behjati et al. showed a clear predilection toward H3F3A or H3F3B depending on tumor type. Since there is no expression difference between these genes in giant cell bone tumor versus chondroblastomas, temporal expression, for instance at the time of tumor formation, is a possibility suggested by the authors [69]. The above studies were of great value shifting the focus from histone modifying complexes to histones themselves and showing that mutations in histones can be driver mutations.
Concluding remarks
It should be noted that rare tumors with homogenous clinical behavior are not always easy to study, and the examples used above are success stories that have benefitted from the relative ease of interpreting NGS results when the tumors are truly simple genomically. Embryonic rhabdomyosarcomas have clinical and morphologic features of forme fruste tumors but ended up revealing a complex genome with various tumorigenic mechanisms identified in different cases, unlike the more consistent drivers seen in the tumor types described above [70, 71]. Similarly, our own group’s study of epithelioid sarcoma has revealed that despite its unique and consistent pathology and biology, this tumor has a relatively complex genome.
Yet, as a whole, forme fruste tumors have been particularly informative in deep sequencing studies, expanding on our knowledge of cancer biology in a resource-efficient manner. Here, we have cited several successful examples of recent findings that have lead to the discovery of pathognomonic mutations, the establishing new subtypes and classifications, such as the case of high-grade ESS, and providing insight into mechanisms of cancer formation such as findings of SWI/SNF and microRNA processing gene abnormalities. This is not to say that such discoveries are not possible in more common, genetically complex cancers, but in forme fruste tumors, the reduced complexities in the genome allows for identification of driver oncogenic events with the use of very few samples. Part of the reason for success in studying these tumors can also be attributed to the fact that they have tended to be understudied and not so much is known about them. However, their rarity comes with the challenge of a lack of banked samples appropriate for the nucleic acid extractions needed for deep sequencing. Recent advancements in sequencing technologies mean that formalin-fixed paraffin-embedded tissues can now be also used for deep sequencing, and hence, some of the challenges in studying forme fruste tumors are already being overcome [39, 72].
Beyond next-generation sequencing
The focus of this review has been on next-generation sequencing and its role as a molecular microscope helping define tumors in a new way. However, sequencing technologies and associated analytic capacities are advancing at a rapid rate. The ability to study clonal evolution and diversity, which has been successfully utilized in the breast cancer field for instance [73], can be of great value if applied to rare conditions to see the degree of genomic heterogeneity in these seemingly simpler tumors (which should have a more interpretable signal-to-noise ratio for subclone tracking). Additionally, tools such as single-cell RT-PCR and single-cell next-generation sequencing that are in development and reviewed elsewhere [74] can reveal a whole new window on intratumoral heterogeneity, and forme fruste tumors, in particular biphasic cancers like synovial sarcoma, can again provide models that may well prove easier to study. We believe that with all these developing methodologies, rare tumors can be a source of breakthroughs that give clearer answers at lower cost, with fewer samples needed to make discoveries.
Acknowledgments
We would also like to thank Ali Bashashati, Leah Prentice, Niki Boyd, Anthony Karnezis, Michael Anglesio, and Sohrab Shah for their help and input in preparation of this manuscript. TON is supported by the Canadian Cancer Society Research Institute (Grant#701582). FJ is supported by the Vancouver Coastal Health/Canadian Institute of Health Research MD/PhD studentship award. FJ, TON, DGH are supported The Terry Fox Research Institute New Frontiers in Cancer.
Contributor Information
Farzad Jamshidi, Email: jamshidi.f.k@gmail.com.
David G. Huntsman, Email: dhuntsma@bccancer.bc.ca
References
- 1.Pott P (1775) Cancer Scroti. Hawes, Clarke, Collins, London
- 2.Epstein A. Burkitt lymphoma and the discovery of Epstein-Barr virus. Br J Haematol. 2012;156:777–779. doi: 10.1111/j.1365-2141.2011.09008.x. [DOI] [PubMed] [Google Scholar]
- 3.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;1:702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 4.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 8.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 10.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 2006;34:e22. doi: 10.1093/nar/gnj023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adessi C, Matton G, Ayala G, Turcatti G, Mermod JJ, Mayer P, Kawashima E. Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucleic Acids Res. 2000;28:E87. doi: 10.1093/nar/28.20.e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumer ST, Cannistra SA. Granulosa cell tumor of the ovary. J Clin Oncol. 2003;21:1180–1189. doi: 10.1200/JCO.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Colombo N, Parma G, Zanagnolo V, Insinga A. Management of ovarian stromal cell tumors. J Clin Oncol. 2007;25:2944–2951. doi: 10.1200/JCO.2007.11.1005. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher JA, Gibas Z, Donovan K, Perez-Atayde A, Genest D, Morton CC, Lage JM. Ovarian granulosa-stromal cell tumors are characterized by trisomy 12. Am J Pathol. 1991;138:515–520. [PMC free article] [PubMed] [Google Scholar]
- 16.Fuller PJ, Chu S. Signalling pathways in the molecular pathogenesis of ovarian granulosa cell tumours. Trends Endocrinol Metab. 2004;15:122–128. doi: 10.1016/j.tem.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 17.King LA, Okagaki T, Gallup DG, Twiggs LB, Messing MJ, Carson LF. Mitotic count, nuclear atypia, and immunohistochemical determination of Ki-67, c-myc, p21-ras, c-erbB2, and p53 expression in granulosa cell tumors of the ovary: mitotic count and Ki-67 are indicators of poor prognosis. Gynecol Oncol. 1996;61:227–232. doi: 10.1006/gyno.1996.0130. [DOI] [PubMed] [Google Scholar]
- 18.Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, Leung G, Zayed A, Mehl E, Kalloger SE, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360:2719–2729. doi: 10.1056/NEJMoa0902542. [DOI] [PubMed] [Google Scholar]
- 19.Pisarska MD, Bae J, Klein C, Hsueh AJ. Forkhead l2 is expressed in the ovary and represses the promoter activity of the steroidogenic acute regulatory gene. Endocrinology. 2004;145:3424–3433. doi: 10.1210/en.2003-1141. [DOI] [PubMed] [Google Scholar]
- 20.Kommoss S, Anglesio MS, Mackenzie R, Yang W, Senz J, Ho J, Bell L, Lee S, Lorette J, Huntsman DG, et al. FOXL2 molecular testing in ovarian neoplasms: diagnostic approach and procedural guidelines. Mod Pathol. 2013;26:860–867. doi: 10.1038/modpathol.2012.226. [DOI] [PubMed] [Google Scholar]
- 21.Kim MS, Hur SY, Yoo NJ, Lee SH. Mutational analysis of FOXL2 codon 134 in granulosa cell tumour of ovary and other human cancers. J Pathol. 2010;221:147–152. doi: 10.1002/path.2688. [DOI] [PubMed] [Google Scholar]
- 22.Cheng JC, Klausen C, Leung PC. Overexpression of wild-type but not C134W mutant FOXL2 enhances GnRH-induced cell apoptosis by increasing GnRH receptor expression in human granulosa cell tumors. PLoS One. 2013;8:e55099. doi: 10.1371/journal.pone.0055099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JH, Kim YH, Kim HM, Park HO, Ha NC, Kim TH, Park M, Lee K, Bae J. FOXL2 posttranslational modifications mediated by GSK3beta determine the growth of granulosa cell tumours. Nat Commun. 2014;5:2936. doi: 10.1038/ncomms3936. [DOI] [PubMed] [Google Scholar]
- 24.Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer. 1982;50:970–981. doi: 10.1002/1097-0142(19820901)50:5<970::aid-cncr2820500527>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 25.Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, Flanagan J, Luo Y, Fenwick K, Natrajan R et al (2011) Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 3:98ra82 [DOI] [PubMed]
- 26.Boudousquie AC, Lawce HJ, Sherman R, Olson S, Magenis RE, Corless CL. Complex translocation [7;22] identified in an epithelioid hemangioendothelioma. Cancer Genet Cytogenet. 1996;92:116–121. doi: 10.1016/s0165-4608(96)00175-6. [DOI] [PubMed] [Google Scholar]
- 27.Mendlick MR, Nelson M, Pickering D, Johansson SL, Seemayer TA, Neff JR, Vergara G, Rosenthal H, Bridge JA. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol. 2001;25:684–687. doi: 10.1097/00000478-200105000-00019. [DOI] [PubMed] [Google Scholar]
- 28.Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, Healey JH, Antonescu CR. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644–653. doi: 10.1002/gcc.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Dal Cin P, Fletcher JA, Sklar J. Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc Natl Acad Sci U S A. 2001;98:6348–6353. doi: 10.1073/pnas.101132598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micci F, Panagopoulos I, Bjerkehagen B, Heim S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006;66:107–112. doi: 10.1158/0008-5472.CAN-05-2485. [DOI] [PubMed] [Google Scholar]
- 32.Lee CH, Ou WB, Marino-Enriquez A, Zhu M, Mayeda M, Wang Y, Guo X, Brunner AL, Amant F, French CA, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci U S A. 2012;109:929–934. doi: 10.1073/pnas.1115528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CH, Marino-Enriquez A, Ou W, Zhu M, Ali RH, Chiang S, Amant F, Gilks CB, van de Rijn M, Oliva E, et al. The clinicopathologic features of YWHAE-FAM22 endometrial stromal sarcomas: a histologically high-grade and clinically aggressive tumor. Am J Surg Pathol. 2012;36:641–653. doi: 10.1097/PAS.0b013e31824a7b1a. [DOI] [PubMed] [Google Scholar]
- 34.Kurman RJ, Carcangiu ML, Herrington CS, Young RH (2014) WHO classification of tumours of female reproductive organs, WHO Classification of Tumours, vol 6. IARC Press, Lyon
- 35.Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, Perrin V, Coindre JM, Delattre O. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–466. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 36.Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol Cell Biol. 2006;26:6880–6889. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallant P, Nigg EA. Identification of a novel vertebrate cyclin: cyclin B3 shares properties with both A- and B-type cyclins. EMBO J. 1994;13:595–605. doi: 10.1002/j.1460-2075.1994.tb06297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koukourakis GV, Miliadou A, Sotiropoulou-Lontou A. Ameloblastoma, a rare benign odontogenic tumour: an interesting tumour review targeting the role of radiation therapy. Clin Transl Oncol. 2011;13:793–797. doi: 10.1007/s12094-011-0735-5. [DOI] [PubMed] [Google Scholar]
- 39.Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, Qu K, Gong X, Ng T, Jones CD, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 2014;46:722–725. doi: 10.1038/ng.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, Avsar T, Li J, Murray PB, Henegariu O, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobel M, Kalloger SE, Huntsman DG, Santos JL, Swenerton KD, Seidman JD, Gilks CB, Cheryl Brown Ovarian Cancer Outcomes Unit of the British Columbia Cancer Agency VBC Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int J Gynecol Pathol. 2010;29:203–211. doi: 10.1097/PGP.0b013e3181c042b6. [DOI] [PubMed] [Google Scholar]
- 42.Gilks CB. Molecular abnormalities in ovarian cancer subtypes other than high-grade serous carcinoma. J Oncol. 2010;2010:740968. doi: 10.1155/2010/740968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dent J, Hall GD, Wilkinson N, Perren TJ, Richmond I, Markham AF, Murphy H, Bell SM. Cytogenetic alterations in ovarian clear cell carcinoma detected by comparative genomic hybridisation. Br J Cancer. 2003;88:1578–1583. doi: 10.1038/sj.bjc.6600896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 47.Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33:100–103. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young RH, Oliva E, Scully RE. Small cell carcinoma of the hypercalcemic type in the ovary. Gynecol Oncol. 1995;57:7–8. doi: 10.1006/gyno.1995.1092. [DOI] [PubMed] [Google Scholar]
- 49.Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K, Yang Y, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427–429. doi: 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J, Rivera B, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438–443. doi: 10.1038/ng.2931. [DOI] [PubMed] [Google Scholar]
- 51.Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424–426. doi: 10.1038/ng.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, Plisiecka-Halasa J, Szafron L, Podgorska A, Rzepecka IK, Konopka B, Budzilowska A, Rembiszewska A, et al. Ovarian small cell carcinoma of hypercalcemic type—evidence of germline origin and SMARCA4 gene inactivation. a pilot study. Pol J Pathol. 2013;64:238–246. doi: 10.5114/pjp.2013.39331. [DOI] [PubMed] [Google Scholar]
- 53.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, Sharif S, Eccles D, Fitzpatrick D, Rawluk D, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet. 2013;45:295–298. doi: 10.1038/ng.2552. [DOI] [PubMed] [Google Scholar]
- 55.Helming KC, Wang X, Roberts CW. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell. 2014;26:309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Colombo N, Peiretti M, Castiglione M, Group EGW. Non-epithelial ovarian cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):24–26. doi: 10.1093/annonc/mdp118. [DOI] [PubMed] [Google Scholar]
- 59.Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, Fejes AP, Chow C, Tone A, Kalloger SE, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–242. doi: 10.1056/NEJMoa1102903. [DOI] [PubMed] [Google Scholar]
- 60.Grelier G, Voirin N, Ay AS, Cox DG, Chabaud S, Treilleux I, Leon-Goddard S, Rimokh R, Mikaelian I, Venoux C, et al. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br J Cancer. 2009;101:673–683. doi: 10.1038/sj.bjc.6605193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, Urbauer D, Pennacchio LA, Cheng JF, Nick AM, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A, Salamanca C, Maines-Bandiera S, Huntsman DG, Morin GB. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. 2013;229:400–409. doi: 10.1002/path.4135. [DOI] [PubMed] [Google Scholar]
- 63.Torrezan GT, Ferreira EN, Nakahata AM, Barros BD, Castro MT, Correa BR, Krepischi AC, Olivieri EH, Cunha IW, Tabori U, et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun. 2014;5:4039. doi: 10.1038/ncomms5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 66.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 67.Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LE, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 68.Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–1200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 69.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, Lim D, Marchetti A, Viale A, Pirun M, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sweeney RT, Zhang B, Zhu SX, Varma S, Smith KS, Montgomery SB, van de Rijn M, Zehnder J, West RB. Desktop transcriptome sequencing from archival tissue to identify clinically relevant translocations. Am J Surg Pathol. 2013;37:796–803. doi: 10.1097/PAS.0b013e31827ad9b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 74.Lecault V, White AK, Singhal A, Hansen CL. Microfluidic single cell analysis: from promise to practice. Curr Opin Chem Biol. 2012;16:381–390. doi: 10.1016/j.cbpa.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 75.Ding J, Bashashati A, Roth A, Oloumi A, Tse K, Zeng T, Haffari G, Hirst M, Marra MA, Condon A, et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics. 2012;28:167–175. doi: 10.1093/bioinformatics/btr629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22:1995–2007. doi: 10.1101/gr.137570.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N et al (2011) deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol 7:e1001138 [DOI] [PMC free article] [PubMed]
- 78.Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amarasinghe KC, Li J, Hunter SM, Ryland GL, Cowin PA, Campbell IG, Halgamuge SK. Inferring copy number and genotype in tumour exome data. BMC Genomics. 2014;15:732. doi: 10.1186/1471-2164-15-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nord KH, Lilljebjorn H, Vezzi F, Nilsson J, Magnusson L, Tayebwa J, de Jong D, Bovee JV, Hogendoorn PC, Szuhai K. GRM1 is upregulated through gene fusion and promoter swapping in chondromyxoid fibroma. Nat Genet. 2014;46:474–477. doi: 10.1038/ng.2927. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, Lewis JT, Chou MM, Yaszemski MJ, Jen J, et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. 2014;46:666–668. doi: 10.1038/ng.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, Guo Y, Zand DJ, Bernstein AM, Masur SK, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet. 2013;92:1001–1007. doi: 10.1016/j.ajhg.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH, Guo J, Qi M, Guan Y, Albrecht S, et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet. 2013;92:996–1000. doi: 10.1016/j.ajhg.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014 doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014 doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mosquera JM, Sboner A, Zhang L, Chen CL, Sung YS, Chen HW, Agaram NP, Briskin D, Basha BM, Singer S, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer. 2013;52:1075–1087. doi: 10.1002/gcc.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CK, Perez-Ordonez B, Harding NJ, Alfaro J, Chu KC, Viale A, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014 doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10:e1004475. doi: 10.1371/journal.pgen.1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, et al. The genomic landscape of pediatric Ewing Sarcoma. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- 92.Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, et al. Genomic landscape of Ewing Sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, Borinstein SC, Ross JS, Stephens PJ, Miller VA, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–895. doi: 10.1158/2159-8290.CD-14-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–1043. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- 96.Petrini I, Meltzer PS, Kim IK, Lucchi M, Park KS, Fontanini G, Gao J, Zucali PA, Calabrese F, Favaretto A, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet. 2014;46:844–849. doi: 10.1038/ng.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Korah R, Suttorp AC, Dietrich D, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014;46:613–617. doi: 10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, Yang S, Jones S, Wang S, Zhou W, et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet. 2014;46:726–730. doi: 10.1038/ng.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–587. doi: 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 100.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46:451–456. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46:457–461. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46:462–466. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science. 2014;344:917–920. doi: 10.1126/science.1252328. [DOI] [PubMed] [Google Scholar]
- 105.Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee JE, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371–375. doi: 10.1038/ng.2916. [DOI] [PubMed] [Google Scholar]
- 106.Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, Ben-Neriah S, Mottok A, Lim RS, Boyle M, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014;46:329–335. doi: 10.1038/ng.2900. [DOI] [PubMed] [Google Scholar]
- 107.Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–170. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–175. doi: 10.1038/ng.2872. [DOI] [PubMed] [Google Scholar]
- 109.Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, Abaan OD, Davis SR, Kreitman RJ, Meltzer PS. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10. doi: 10.1038/ng.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, Fu C, Crowley MR, Crossman DK, Armstrong L, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46:182–187. doi: 10.1038/ng.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O’Meara S, Davies H, Nik-Zainal S, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923–926. doi: 10.1038/ng.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chmielecki J, Crago AM, Rosenberg M, O’Connor R, Walker SR, Ambrogio L, Auclair D, McKenna A, Heinrich MC, Frank DA, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet. 2013;45:131–132. doi: 10.1038/ng.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]