

Abstract
Nucleosome is a histone–DNA complex known as the fundamental repeating unit of chromatin. Up to 90% of eukaryotic DNA is wrapped around consecutive octamers made of the core histones H2A, H2B, H3 and H4. Nucleosome positioning affects numerous cellular processes that require robust and timely access to genomic DNA, which is packaged into the tight confines of the cell nucleus. In living cells, nucleosome positions are determined by intrinsic histone–DNA sequence preferences, competition between histones and other DNA-binding proteins for genomic sequence, and ATP-dependent chromatin remodelers. We discuss the major energetic contributions to nucleosome formation and remodeling, focusing especially on partial DNA unwrapping off the histone octamer surface. DNA unwrapping enables efficient access to nucleosome-buried binding sites and mediates rapid nucleosome removal through concerted action of two or more DNA-binding factors. High-resolution, genome-scale maps of distances between neighboring nucleosomes have shown that DNA unwrapping and nucleosome crowding (mutual invasion of nucleosome territories) are much more common than previously thought. Ultimately, constraints imposed by nucleosome energetics on the rates of ATP-dependent and spontaneous chromatin remodeling determine nucleosome occupancy genome-wide, and shape pathways of cellular response to environmental stresses.
Keywords: chromatin, nucleosome, DNA, transcription factor, gene regulation, stress response
INTRODUCTION
Genetic information encoded in DNA sequence needs to be manipulated efficiently and robustly to execute built-in cellular programs and respond to challenges that a cell faces throughout its life cycle. This requirement, which implies DNA accessibility, is especially stringent in eukaryotes, where all chromosomes have to be packaged inside a cell nucleus. The simplest polymer model of DNA represents each chromosome by a freely jointed chain (Gaussian coil) of stiff links with the Kuhn length l of 100 nm, or ∼300 bp for DNA at physiological temperatures and salt concentrations [1, 2]. Gaussian coils are compact, with the average radius given by , where N is the number of Kuhn segments in the DNA chain. In diploid human cells, there are 23 pairs of chromosomes, ranging in length from 48 to 249 million bp, or 16–85 mm when fully stretched. Left to itself, the radius of even the smallest chromosome would be around 40 μm according to the Gaussian coil model, eight times larger than the 5 μm radius of a cell nucleus in a typical human cell. Furthermore, all 46 chromosomes with the total DNA length of ∼6 billion bp (2 m) need to share the same nuclear volume.
Fortunately, Gaussian coils have rather low densities: a full diploid set of human chromosomes would occupy only ∼25 μm3 in a densely packed state, a fraction of the available volume. Thus it is possible to package all genomic DNA into the nucleus before incurring insurmountable energy costs. To achieve the required levels of compactification, cells have developed elaborate mechanisms for controlling DNA accessibility and manipulating DNA packaging. Thus, eukaryotic genomes are found in a condensed chromatin state composed primarily of DNA and histone proteins. Under physiological conditions, histones form an octamer made of two copies of the four core histones: H2A, H2B, H3 and H4 [3]. A 147 bp long DNA segment wraps around each histone octamer in ∼1.75 turns of a left-handed superhelix, following a positively charged groove on the histone octamer surface [4]. The resulting histone–DNA complex is called a nucleosome core particle, or simply a nucleosome. Nucleosomes have a 2-fold axis of symmetry, called the dyad axis. The 71 bp DNA centered on the dyad is in contact with the H3-H4 tetramer, while the flanking sequences bend around H2A-H2B dimers on each side [3]. The N-termini of all core histones and the C-terminus of histone H2A are flexible hydrophylic tails, which serve as substrates for numerous posttranslational modifications (methylation, acetylation, phosphorylation and ubiquitination, among others), providing recognition motifs for chromatin-binding proteins [5, 6].
Depending on the organism and cell type, 75–90% of genomic DNA is packaged into nucleosomes—the fundamental units of chromatin [3, 7]. At low ionic strengths, the resulting chromatin fiber is extended and resembles beads on a string, with adjacent nucleosomes separated by relatively short stretches of linker DNA (Figure 1A, left panel; this picture neglects effects of nucleosome crowding discussed in detail below). At physiological salt concentrations, the fiber folds back onto itself, forming higher-order structures [9]. The functional role of these structures, their dynamics and their effect on DNA accessibility remain poorly understood. In contrast, much more is known about the nucleosome core particle. In accordance with earlier predictions [4], nucleosome crystal structures show that the histone octamer forms a molecular spool with ∼8.5 nm diameter and 5.5 nm height [10–12]. The 147 bp long DNA wrapped around it (whose length, incidentally, is very close to the DNA persistence length of 150 bp) is severely twisted, bent and sheared compared with B-DNA [10], due to mechanical stresses imposed by favorable interactions between the positively charged histone octamer surface and the negatively charged DNA.
Figure 1:
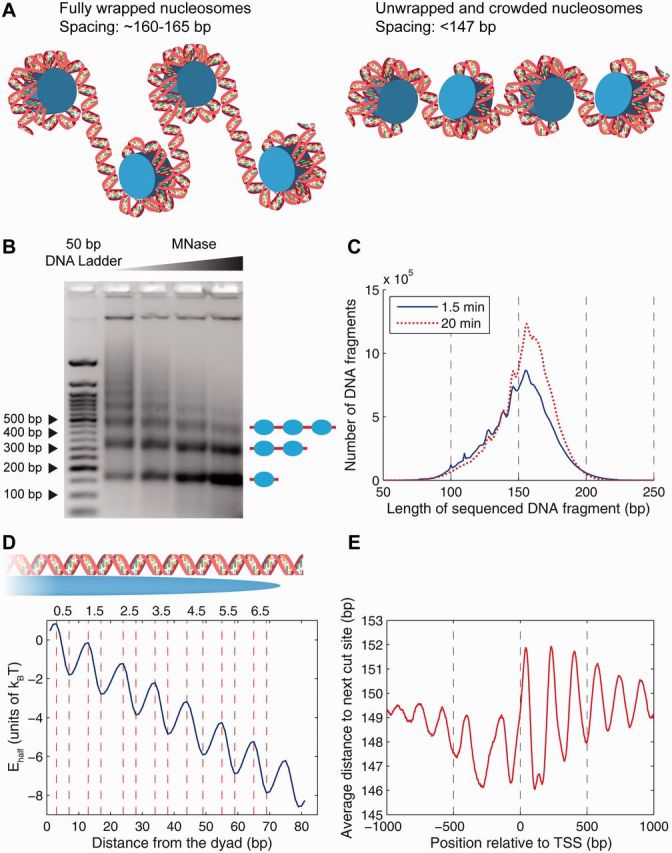
Energetics of DNA unwrapping and nucleosome spacing in the yeast genome. (A) Left panel: fully wrapped nucleosomes with ∼160–165 bp spacing between neighboring dyads. Right panel: partially unwrapped and crowded nucleosomes with <147 bp spacing between neighboring dyads. (B) Nucleosome ladder on an agarose gel. Wild-type yeast was grown in synthetic complete medium. DNA purified from nuclei digested with increasing amounts of MNase was analyzed in a 2% agarose gel stained with ethidium bromide. Leftmost lane: 50 bp DNA ladder (New England Biolabs). Image courtesy of Drs Josefina Ocampo and David J. Clark (NICHD, NIH). (C) Histogram of DNA fragment lengths from the high-resolution chemical map, 1.5 min (blue, solid) and 20 min (red, dotted) after the addition of hydrogen peroxide [8]. (D) Histone–DNA unwrapping and higher-order structure energy profile. The total energy of a partially wrapped nucleosome with x bp of DNA to the left of the dyad and y bp of DNA to the right of the dyad is given by . The minima and maxima of the energy landscape are based on crystal structures of nucleosome core particles [9, 10]. Histone–DNA contact patches, corresponding to the regions where the minor groove of the DNA double helix faces inward, are labeled 0.5, 1.5, 2.5, 3.5, 4.5, 5.5 and 6.5. (E) Average distance between hydroxyl cut sites marking neighboring dyads in the vicinity of the TSS. The genome-wide average distance is 150.9 bp. (A colour version of this figure is available online at: http://bfg.oxfordjournals.org)
Because a large fraction of genomic DNA is occluded and conformationally distorted due to interactions with histones, nucleosome formation creates a barrier for any function mediated by DNA-binding factors, including gene regulation and transcription. Therefore, nucleosome positioning and dynamics have profound biological consequences. Important aspects of nucleosome dynamics include spontaneous or assisted DNA unwrapping at the ends of the nucleosome core particle, kinetics of histone–DNA association and disassociation (i.e. nucleosome formation and removal) and nucleosome translocation along DNA.
NUCLEOSOME ENERGETICS
Nucleosome formation energy
Nucleosomal DNA interacts with the histone octamer through 14 distinct contact patches; 12 of these patches are in contact with the inner 121 bp of nucleosomal DNA [9, 13]. At each binding site, histone–DNA contacts occur through favorable electrostatic interactions and hydrogen bonds between amino acid side chains and the phosphate backbone of the DNA minor groove. The number of hydrogen bonds and their locations differ from site to site, and the total free energy of binding at each site is unknown. However, a rough estimate based on an assay that measured accessibility of nucleosomal DNA to restriction enzymes yields 1.5–2.0 kBT per patch, where kB is the Boltzmann constant and T is room temperature (1 kBT = 0.59 kcal/mol) [14].
This energy is smaller than the free energy gain of forming a single hydrogen bond [15]. However, this is the net balance between the elastic energy cost of bending DNA into the nucleosomal superhelix, loss of DNA conformational entropy and formation of favorable histone–DNA interactions. A rough estimate of DNA elastic energy based on a worm-like chain model of DNA yields ∼60 kBT to form a nucleosomal superhelix [14]. Together, this estimate and the total free energy of binding inferred from the restriction enzyme assay suggest that each contact patch provides around 6 kBT of favorable energy. This calculation neglects effects of DNA sequence: flexible DNA is expected to be easier to bend into the distorted shape found in nucleosomal crystal structures. This effect is counteracted by the concomitant loss of DNA conformational entropy, which is correlated with DNA flexibility [16]. Thus more flexible sequences are easier to bend but at the same time lose more entropy upon nucleosome formation. Moreover, intrinsic DNA curvature and bending anisotropy may make it easier for some DNA sequences to adopt the nucleosomal shape. Taken together, these contributions determine which DNA sequences are best and worst nucleosome formers.
Based on the net free energy gained at each contact patch, the total energy of nucleosome formation is roughly 21−28 , in good agreement with the 23.8 estimate obtained from single-nucleosome unzipping experiments [17]. The nucleosome used in those experiments was assembled on the so-called 601 sequence selected in vitro for high affinity [18]. Because the range of free energies of nucleosome formation is 7.0 between DNA sequences with the strongest and weakest propensity for histone octamer binding [19], we expect absolute formation energies to be between 17 and 24 kBT. The difference between the 601 high-affinity sequence and DNA sequences from chicken genome is smaller yet at [19], yielding nucleosome formation energies of ∼18 for bulk genomic sequences. The relatively low sequence specificity is consistent with the fact that nucleosomes can form on any DNA segment of sufficient length, fulfilling their role as a universal compaction factor of eukaryotic DNA.
Nucleosome unwrapping and translocation
Because nucleosome formation energies are much greater than , the characteristic scale of thermal fluctuations, spontaneous nucleosome unfolding occurs too slowly to matter on cellular time scales. The same is true about nucleosome repositioning or ‘sliding’ along the DNA: although it has been observed in vitro, it takes minutes to hours, even at elevated temperatures [20, 21]. Furthermore, in vitro nucleosome mobility can be suppressed by adding linker histones H1 or H5 to the system [22]. Linker histones make a separate histone class; they likely bind both nucleosomal and linker DNA, stabilizing the entire histone–DNA complex [23, 24]. Although the height of the free energy barrier that must be surmounted to reposition a nucleosome is not exactly known, two possible mechanisms have been proposed: (i) Diffusion of loops or DNA bulges [25], whereby a ‘loop defect’ is created by unwrapping some DNA on one end of the nucleosome core particle, pulling in extra bp of DNA, and forming new contacts with the histone octamer surface. Propagation of this loop defect to the other end of the particle is equivalent to shifting the nucleosome by bp (optimal should be a multiple of DNA helical twist to preserve DNA orientation with respect to the histone core). (ii) Diffusion of ‘twist defects’ [26], whereby DNA becomes undertwisted or overtwisted through thermal fluctuations, creating a 1 bp defect. Propagation of this defect to the other end of the nucleosome will result in a net corkscrew motion with ∼36° magnitude. Although only approximate estimates of free energies of loop and twist defects are available [14, 25, 26], the energetic cost appears to be ≫, in agreement with slow nucleosome repositioning observed in experiments [20, 21]. Note, however, that this cost is less than that entailed by complete nucleosome unfolding and assembly at a new position.
Because spontaneous nucleosome repositioning activated by thermal fluctuations is too slow and random to be of much biological significance, cells depend on ATP-dependent chromatin remodelers to catalyze the repositioning reaction. Chromatin remodelers are multi-subunit proteins capable of translocating nucleosomes along the DNA, evicting nucleosomes or changing the nucleosome histone composition (i.e. exchanging canonical core histones for histone variants) [27, 28]. All remodelers have ATPase subunits, which hydrolyze ATP to gain 12.3 kBT per ATPADP reaction. In principle, the energy of ATP hydrolysis is sufficient to keep the entire population of genomic nucleosomes out of equilibrium. Indeed, the total concentration of ATP in Saccharomyces cerevisiae is ∼2.5 mM [29], and the rate of ATP synthesis is ∼1 mM/s [30]. Assuming the volume of the yeast cell to be ∼60 μm3, we see that the number of new ATP molecules synthesized per cell each second is around 106. On the other hand, the rate of ATP consumption was estimated to be 75 per minute for the nucleosome remodeler ISWI, and the number of remodelers per yeast cell is around 6000–7000, with one remodeler per ∼10 nucleosomes [31]. Thus remodelers are capable of hydrolyzing ∼7500–9000 ATP molecules per second, a tiny fraction of the total output. Interestingly, even with perfect efficiency a remodeler will take several seconds to unfold or reposition a single nucleosome, consuming several ATPs in the process; waiting times, which depend on the remodeler-to-nucleosome ratio, may be an order-of-magnitude greater still.
Unlike nucleosome repositioning, DNA unwrapping off the ends of the nucleosome core particle requires activation energies on the order of several , as only one or a few histone–DNA contact patches have to be involved. Transient DNA unwrapping induced by thermal fluctuations has been observed in vitro, through competitive protein binding to nucleosomal DNA and fluorescence resonance energy transfer measurements [32–34]. In these and other studies, nucleosome unwrapping (as opposed to relatively costly nucleosome repositioning) has been thought to assist binding of nucleosomal DNA by transcription factors (TFs). However, until recently its significance in genome-wide chromatin organization has been unclear.
Extracting nucleosome energetics from high-throughput maps
Large-scale mapping of nucleosomes assembled in vitro on genomic or synthetic DNA, or extracted from living cells has become a standard technique for studying chromatin structure. Typically, chromatin is treated with micrococcal nuclease (MNase)—an endo-exonuclease that preferentially digests non-nucleosomal DNA (optionally, nucleosomes are folmaldehyde-crosslinked on the DNA before the MNase treatment). After the MNase digestion step, the DNA is purified and resolved on an agarose gel. The mononucleosomal band is excised and sequenced using one of the next-generation sequencing technologies. High-throughput sequencing results in a collection of relatively short, 35–95 bp reads (either single- or paired-end), which are mapped to the reference genome (for recent reviews, see [35, 36]). In the end, this procedure yields a nucleosome density profile—the number of nucleosomes starting at each genomic bp.
With single-end nucleosome maps, the actual length of mapped DNA fragments is unknown and commonly assumed to be equal to the canonical nucleosome length of 147 bp [37]. This approach neglects nucleosome unwrapping as well as under- or overdigestion of nucleosomal DNA by MNase. When paired-end nucleosome maps (in which both ends of each DNA fragment are sequenced simultaneously) became available, the resulting distribution of ‘mononucleosome’ fragment lengths turned out to be rather broad and markedly dependent on the extent of chromatin exposure to MNase (see e.g. [38]). Unfortunately, in such experiments, MNase-related artifacts such as partial digestion of nucleosomal DNA cannot be decoupled from DNA unwrapping. Similarly, both incomplete digestion of linker DNA by MNase and linker histone binding would yield longer DNA fragments. In addition, MNase–DNA interactions are sequence-specific, which biases the nucleosome maps [39].
These difficulties were overcome in a recent experiment in which both nucleosome dyad positions and distances between dyads of neighboring nucleosomes were mapped with high precision in S. cerevisiae [8, 40]. In this so-called chemical method, mutant H4 histones (S47C) were modified by covalent attachment of a sulfhydryl-reactive copper-chelating label to the cysteines. With the addition of copper and hydrogen peroxide, a localized cloud of hydroxyl radicals was produced, which cleaved the DNA backbone at specific sites flanking nucleosome dyads. The cleavage products that corresponded to DNA fragments linking neighboring nucleosomes were then size-selected on an agarose gel, purified, sequenced using paired-end reads and mapped to the S. cerevisiae genome. As a result, each read marks a dyad position, and each mate pair yields a measurement of the distance between dyads of neighboring nucleosomes positioned on the same chromosome and in the same cell. Although more precise than methods using MNase digestion, chemical mapping is subject to unknown hydroxyl radical cutting preferences for two alternate sites on each DNA strand, at −1 and +6 bp with respect to the dyad [8, 41]. If DNA cuts at both sites are equally likely, averaging over hydroxyl cleavage preferences shows that the average interdyad distances are 5 bp longer than the average distances between adjacent hydroxyl cut sites [42].
Large-scale nucleosome positioning data obtained by MNase or chemical mapping can be used to predict nucleosome energetics. Sequence reads mapped to genomic coordinates produce one-dimensional nucleosome density profiles, in which nucleosome-enriched regions and nucleosome-depleted regions (NDRs) are marked with more and fewer reads, respectively. These density profiles can be used to infer sequence-dependent nucleosome formation energies, in a rigorous procedure that uses exact results from physics of one-dimensional liquids, and is capable of disentangling steric exclusion between neighboring nucleosomes from intrinsic histone–DNA sequence preferences [42, 43]. The earlier version of this work, which used single-end data sets, had to assume 147 bp canonical nucleosome length. These predictions, based on maps of nucleosomes assembled in vitro on genomic DNA, supplemented direct measurements of histone–DNA interaction energies available only for a handful of sequences [19]. More recently, with the advent of paired-end nucleosome maps and high-resolution chemical mapping, it has become possible to model energetics of nucleosome unwrapping using genome-scale data [42].
Nucleosome crowding and unwrapping
An important feature of the chemical mapping approach is that in addition to single-nucleosome positions it provides information about distances between neighboring nucleosomes. Before this work, inter-nucleosome distances were commonly estimated using a nucleosome ladder on an agarose gel (Figure 1B). This method yields an average nucleosome repeat length of 160–165 bp [3]; as MNase concentration increases, the distances become shorter owing to more extensive digestion at DNA fragment ends. Strikingly, the high-resolution picture obtained by chemical mapping reveals that for many nucleosomes, distances between neighboring dyads are <147 bp (Figure 1C). These nucleosomes must be partially unwrapped and crowded, as opposed to separate ‘beads-on-a-string’ (compare left and right panels in Figure 1A).
Moreover, the histogram of distances between adjacent hydroxyl cut sites exhibits prominent oscillations consistent with the 10–11 bp periodic model of DNA unwrapping based on nucleosome crystal structures (Figure 1D) [42]. The model, which yields an energy profile averaged over sequence-dependent effects, is obtained by a parametric fit to the 1.5 min distribution in Figure 1C. Interestingly, to reproduce this distribution, the energy profile had to be extended beyond the nucleosome edge, thus accounting for both DNA unwrapping and higher-order chromatin structure/linker histone deposition, which lead to discretization of linker DNA lengths [44]. The total free energy of nucleosome unwrapping is according to this model, reasonably close to the estimates described above.
When distances between neighboring dyads are averaged over all yeast genes aligned by their transcription start sites (TSS), a prominent oscillatory pattern emerges (Figure 1E). Even with the +5 bp correction discussed above, the interdyad distances appear so close to 147 bp in the minima of the curve that some degree of nucleosome unwrapping is inevitable, especially if one recalls that the shortest linker capable of alleviating 3D steric clashes between neighboring nucleosome core particles is around 5 bp in length [45, 46]. Thus, many yeast nucleosomes are crowded and partially unwrapped, as illustrated in the right panel of Figure 1A. This conclusion cannot be reached using single-nucleosome data alone: a characteristic oscillatory profile of dyad counts averaged over all yeast genes yields ∼160–165 bp between neighboring peaks (Figure 2; the genes are aligned by the +1 nucleosome immediately downstream of the nucleosome-depleted promoter region).
Figure 2:
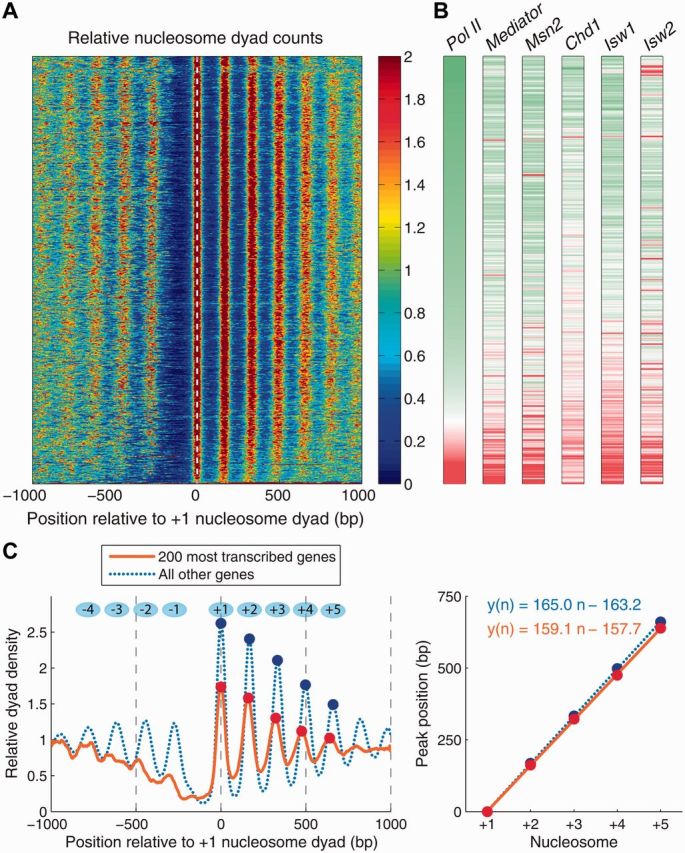
Chromatin organization, gene transcription and remodeler activity in S. cerevisiae. (A) Heatmap of nucleosome dyad counts in S. cerevisiae in the vicinity of coding regions; genes are sorted by Pol II occupancy averaged in the [TSS,TTS] range (TTS: transcription termination site; nucleosome and Pol II data: wild-type cells in glucose [47]). (B) DNA occupancy levels for Pol II, Mediator, TF Msn2 [47] and chromatin remodelers Chd1, Isw1, Isw2 [48]. Genes are sorted as in (A). Pol II and Mediator: wild-type yeast in glucose; Msn2: wild-type yeast 20 min after a glucose-to-glycerol switch; Chd1, Isw1, Isw2: wild-type yeast grown in yeast extract peptone dextrose (YPD) medium [48]. For each factor, the occupancy was averaged in the [TSS-300, TSS] range (Mediator, Msn2), or in the [TSS, TTS] range (Pol II, Chd1, Isw1, Isw2). Distributions of average occupancies over all yeast genes were converted into z-scores, and the color scheme in each vertical bar was set so that genes in the bottom 5 percentile (negative z-scores) are green, genes in the top 5 percentile (positive z-scores) are red and genes with zero z-scores are white. (C) Left panel: average nucleosome dyad density in the 200 most transcribed genes (orange, solid) and in the rest of the yeast genes (blue, dotted). Dots mark peaks of nucleosome dyad density in the coding region. Right panel: linear fit to the positions of the nucleosome density peaks shown in the left panel. The slope of the fit is an estimate of the inter-nucleosome spacing based on single-nucleosome data. (A colour version of this figure is available online at: http://bfg.oxfordjournals.org)
Nucleosome positioning and gene regulation
Nucleosomes occluding genomic DNA in promoters and coding regions may present a significant barrier to gene regulation, which requires sequence-specific TF binding and assembly/disassembly of transcriptional machinery. In S. cerevisiae, nutrient limitations elicit a common pattern of expression changes in ∼900 genes, many of which also change expression in response to a wide variety of environmental stresses [49]. The TF Msn2 mediates a significant proportion of this stress response [49, 50]. As can be expected from considerable energetic costs associated with nucleosome repositioning, large-scale gene activation and repression following stress is, with some exceptions, not accompanied by widespread nucleosome removal and translocation [47, 51, 52]. Rather, for yeast cells growing at steady-state in glucose a canonical oscillatory pattern of nucleosome occupancy [35] is observed, with ordered nucleosome arrays upstream and downstream of the NDR (Figure 2A).
However, in the most transcribed genes (as measured by Pol II occupancy, see Figure 2B), promoter nucleosomes are further depleted, making NDRs wider, and nucleosomes covering coding regions are more closely spaced (Figure 2C) [53]. The occupancy of Mediator (a multi-protein complex that functions as a transcriptional co-activator in eukaryotes) is also higher than average in the promoters of these genes. Interestingly, 20 min after cells are stressed by being transferred from glucose to glycerol, Msn2 binding is also predominantly, but not exclusively, concentrated in promoters of genes with wider NDRs that were actively transcribed in glucose, consistent with its dual role of gene activator and repressor [47]. Note that Msn2 occupancy in the nucleus is negligible before stress [54]; after stress, it appears to take advantage of better promoter accessibility of actively transcribed genes to exert its repressive function. Its activating function on the other hand requires binding to promoters of initially inactive genes and is accompanied by limited nucleosome rearrangements. Finally, occupancy of ATP-dependent chromatin remodelers Chd1, Isw1 and Isw2 [48] is also higher in the coding regions of actively transcribed genes (Figure 2B). This suggests that a distinct pattern of nucleosome occupancy observed in highly transcribed genes may be due to the action of ATP-dependent chromatin remodeling enzymes. RSC, an essential chromatin-remodeling complex, also plays a critical role in organizing yeast chromatin. RSC depletion results in narrower and partially filled NDRs and is accompanied by global repositioning of nucleosomal arrays [55].
Accessibility of nucleosomal DNA and TF binding
Partial DNA unwrapping off the histone octamer surface leads to differential accessibility of protein-binding sites: sites closer to nucleosomal edges are easier to bind (Figure 3A) [32, 56]. Indeed, DNA unwrapping makes such sites more readily available compared with sites buried inside the nucleosome. Once bound by a TF or a restriction enzyme, nucleosomal DNA cannot rewrap, completing the process of nucleosome invasion. DNA unwrapping may be spontaneous (without additional energy input) or assisted by ATP-dependent chromatin remodelers. In either case, as partial unwrapping requires breaking of only a few histone–DNA contact patches, it is likely preferable to repositioning of the entire nucleosome. Indeed, Msn2 occupancy is highest in linker DNA (where nucleosomes do not interfere at all) and decreases gradually toward the dyad [47]; low nucleosome occupancy over TF binding sites was observed for other factors as well [37].
Figure 3:

DNA accessibility and TF binding to nucleosomal DNA. (A) Restriction enzyme sites inserted into the 601 nucleosome sequence at locations indicated by black arrows [56]. Each group of three bars represents independent measurements in which the 601 sequence was flanked by different DNA sequences. The height of each bar is the equilibrium constant for site exposure averaged over multiple experiments; error bars show standard deviations. (B) Nucleosome-induced cooperativity between two DNA-binding factors. Left panel: two TFs bound simultaneously to nucleosomal DNA; the nucleosome is partially unwrapped. Right panel: TF binding probability at a site located at bp 31–40 from the edge of the fully wrapped nucleosome, in the absence (blue, solid) or presence (red, dotted) of a second site for the same TF located on the same side of the dyad, at bp 11–20. The free energy of a fully wrapped nucleosome is ; histone chemical potential is ; TF binding energy is for its cognate sites and for all other sites [57]. The model of nucleosome unwrapping is as described in [42]. (A colour version of this figure is available online at: http://bfg.oxfordjournals.org)
When two or more binding sites are covered by a single nucleosome, binding of one of the factors makes it easier for the other site(s) to become bound, even if the sites are too widely spaced on DNA to allow for direct protein–protein contacts. This phenomenon, known as nucleosome-induced cooperativity, has been observed both in vitro and in vivo [58, 59, 60]. Nucleosome-induced cooperativity mediated by DNA unwrapping requires that the binding sites be on the same side of the nucleosome dyad [60]; a representative example is shown in Figure 3B. This synergistic competition of multiple TF molecules against a nucleosome may play an important role in gaining access to nucleosome-covered binding sites in eukaryotic regulatory regions.
CONCLUSION
In eukaryotic cells, all DNA-mediated biological processes occur on the chromatin template. Since nucleosomes cover most of the genomic DNA, it is important to understand how their energetics constrains vital cellular functions such as gene regulation and DNA repair. In this review, we have discussed free energy costs of nucleosome formation, removal, repositioning and partial DNA unwrapping off the histone octamer surface. Although there is more than enough ATP in the cell to keep nucleosomes permanently out of equilibrium via ATP-dependent chromatin remodeler activity, cells that have evolved to minimize the extent of chromatin remodeling and use most efficient remodeling pathways will have an evolutionary upper hand. Thus nucleosome energetics has likely affected evolutionary past of eukaryotic cells and shaped their present state.
As nucleosomal DNA easily unwraps at the ends owing to thermal fluctuations or the action of chromatin remodeling enzymes, nucleosomes are dynamic entities rather than static ‘beads-on-a-string’. With many nucleosomes forming simultaneously, DNA unwrapping may lead to crowding, with neighboring nucleosomes located too closely to each other to be fully wrapped. This may occur as a result of initial nucleosome formation in partially wrapped states, or subsequent translocation and unwrapping. The crowding phenomenon has been recently observed in S. cerevisiae using high-resolution mapping of interdyad distances, and is probably common to all eukaryotes. Nucleosome unwrapping also provides an efficient way of gaining access to nucleosome-covered DNA sites, which can get bound by proteins while they are transiently accessible. Furthermore, two or more proteins binding simultaneously to nucleosomal DNA aid each other in nucleosomal destabilization. Many complex factors, including competition of nucleosomes with TFs and other DNA-binding proteins (which may be bound specifically or non-specifically to genomic DNA [61, 62]), ATP-dependent chromatin remodeling and intrinsic histone–DNA sequence specificity, combine to produce patterns of nucleosome occupancy observed in living cells. Computational modeling of dynamic chromatin states presents a conceptual challenge that is only beginning to be addressed.
Key points.
Nucleosome energetics shape and constrain chromatin remodeling pathways.
Many S. cerevisiae nucleosomes are crowded and partially unwrapped.
Actively transcribed yeast genes are characterized by a distinct nucleosome signature.
Chromatin remodeling enzymes act in concert with RNA polymerase.
Partial DNA unwrapping enables access of TFs to their nucleosome-occluded sites.
Acknowledgments
We thank Dr David J. Clark and Dr Josefina Ocampo for making the image from Figure 1B available before publication, and for helpful discussions. We also thank Dr Yuri Moshkin for his help with estimates of chromatin remodeling energetics.
Biographies
Răzvan V. Chereji holds a Ph.D. degree in Physics from Rutgers University and is a Postdoctoral Fellow at the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH. He studies the action of chromatin remodeling factors and the role of chromatin organization in transcriptional regulation.
Alexandre V. Morozov is an Associate Professor at the Department of Physics and Astronomy, Rutgers University. His research interests include chromatin structure and function, eukaryotic gene regulation, molecular evolution and population genetics, and the theory of olfaction.
FUNDING
A.V.M. was supported by National Institutes of Health (R01 HG004708) and an Alfred P. Sloan Research Fellowship.
References
- 1.Grosberg AY, Khokhlov AR. Statistical Physics of Macromolecules. Woodbury, NY: AIP Press; 1994. [Google Scholar]
- 2.Frank-Kamenetskii MD. Biophysics of the DNA molecule. Phys Rep. 1997;288:13–60. [Google Scholar]
- 3.van Holde KE. Chromatin. New York: Springer; 1989. [Google Scholar]
- 4.Arents G, Moudrianakis EN. Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc Natl Acad Sci USA. 1993;90:10489–93. doi: 10.1073/pnas.90.22.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McBryant SJ, Adams VH, Hansen JC. Chromatin architectural proteins. Chromosome Res. 2006;14:39–51. doi: 10.1007/s10577-006-1025-x. [DOI] [PubMed] [Google Scholar]
- 6.Rippe K, Mazurkiewicz J, Kepper N. Interactions of histones with DNA: nucleosome assembly, stability, dynamics, and higher order structure. In: Dias R, Lindman B, editors. DNA Interactions with Polymers and Surfactants. John Wiley & Sons, Isnc.; 2008. pp. 135–72. [Google Scholar]
- 7.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–71. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 8.Brogaard K, Xi L, Wang JP, et al. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–53. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 10.Davey C, Sargent D, Luger K, et al. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. 2002;319:1097–13. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 11.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–50. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 12.Vasudevan D, Chua EYD, Davey C. Crystal structures of nucleosome core particles containing the 601 strong positioning sequence. J Mol Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 13.Luger K, Mäder AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 14.Schiessel H. The physics of chromatin. J Phys Cond Matter. 2003;15:R699–774. doi: 10.1088/0953-8984/27/6/060301. [DOI] [PubMed] [Google Scholar]
- 15.Morozov AV, Kortemme T, Tsemekhman K, et al. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc Natl Acad Sci USA. 2004;101:6946–51. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Travers AA, Vaillant C, Arneodo A, et al. DNA structure, nucleosome placement and chromatin remodelling: a perspective. Biochem Soc Trans. 2012;40:335–40. doi: 10.1042/BST20110757. [DOI] [PubMed] [Google Scholar]
- 17.Forties R, North J, Javaid S, et al. A quantitative model of nucleosome dynamics. Nucleic Acids Res. 2011;39:8306–13. doi: 10.1093/nar/gkr422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 19.Thåström A, Lowary P, Widom J. Measurement of histone–DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 20.Pennings S, Meersseman G, Bradbury EM. Mobility of positioned nucleosomes on 5 S rDNA. J Mol Biol. 1991;220:101–10. doi: 10.1016/0022-2836(91)90384-i. [DOI] [PubMed] [Google Scholar]
- 21.Flaus A, Richmond TJ. Positioning and stability of nucleosomes on MMTV 3’LTR sequences. J Mol Biol. 1998;275:427–41. doi: 10.1006/jmbi.1997.1464. [DOI] [PubMed] [Google Scholar]
- 22.Pennings S, Meersseman G, Bradbury EM. Linker histones H1 and H5 prevent the mobility of positioned nucleosomes. Proc Natl Acad Sci USA. 1994;91:10275–9. doi: 10.1073/pnas.91.22.10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zlatanova J, Seebart C, Tomschik M. The linker-protein network: control of nucleosomal DNA accessibility. Trends Biochem Sci. 2008;33:247–53. doi: 10.1016/j.tibs.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Syed SH, Goutte-Gattat D, Becker N, et al. Single-base resolution mapping of H1–nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci USA. 2010;107:9620–5. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiessel H, Widom J, Bruinsma R, et al. Polymer reptation and nucleosome repositioning. Phys Rev Lett. 2001;86:4414–7. doi: 10.1103/PhysRevLett.86.4414. [DOI] [PubMed] [Google Scholar]
- 26.Kulic I, Schiessel H. Chromatin dynamics: nucleosomes go mobile through twist defects. Phys Rev Lett. 2003;91:148103. doi: 10.1103/PhysRevLett.91.148103. [DOI] [PubMed] [Google Scholar]
- 27.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006;7:437–47. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 28.Erdel F, Krug J, Längst G, et al. Targeting chromatin remodelers: signals and search mechanisms. Biochim Biophys Acta. 2011;1809:497–508. doi: 10.1016/j.bbagrm.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Ozalp VC, Pedersen TR, Nielsen LJ, et al. Time-resolved measurements of intracellular ATP in the yeast Saccharomyces cerevisiae using a new type of nanobiosensor. J Biol Chem. 2010;285:37579–88. doi: 10.1074/jbc.M110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheldon JG, Williams SP, Fulton AM, et al. 31P NMR magnetization transfer study of the control of ATP turnover in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1996;93:6399–404. doi: 10.1073/pnas.93.13.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rippe K, Schrader A, Riede P, et al. DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc Natl Acad Sci USA. 2007;104:15635–40. doi: 10.1073/pnas.0702430104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–49. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 33.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–9. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 34.Li G, Levitus M, Bustamante C, et al. Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol. 2004;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 35.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–72. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolkunov D, Morozov AV. Genomic studies and computational predictions of nucleosome positions and formation energies. Adv Prot Chem Struc Biol. 2010;79:1–57. doi: 10.1016/S1876-1623(10)79001-5. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan N, Moore IK, Fondufe-Mittendorf Y, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 458:362–6. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole HA, Howard BH, Clark DJ. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res. 2011;39:9521–35. doi: 10.1093/nar/gkr643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung HR, Dunkel I, Heise F, et al. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS One. 2010;5:e15754. doi: 10.1371/journal.pone.0015754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brogaard K, Xi L, Wang JP, et al. A chemical approach to mapping nucleosomes at base pair resolution. Meth Enzymol. 2012;513:315–34. doi: 10.1016/B978-0-12-391938-0.00014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flaus A, Luger K, Tan S, et al. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci USA. 1996;93:1370–5. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chereji RV, Morozov AV. Ubiquitous nucleosome crowding in the yeast genome. Proc Natl Acad Sci USA. 2014;111:5236–41. doi: 10.1073/pnas.1321001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Locke G, Tolkunov D, Moqtaderi Z, et al. High-throughput sequencing reveals a simple model of nucleosome energetics. Proc Natl Acad Sci USA. 2010;107:20998–1003. doi: 10.1073/pnas.1003838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Widom J. A relationship between the helical twist of DNA and the ordered positioning of nucleosomes in all eukaryotic cells. Proc Natl Acad Sci USA. 1992;89:1095–9. doi: 10.1073/pnas.89.3.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang JP, Fondufe-Mittendorf Y, Xi L, et al. Preferentially quantized linker DNA lengths in Saccharomyces cerevisiae. PLoS Comput Biol. 2008;4:e1000175. doi: 10.1371/journal.pcbi.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chereji RV, Tolkunov D, Locke G, et al. Statistical mechanics of nucleosome ordering by chromatin-structure-induced two-body interactions. Phys Rev E. 2011;83:050903. doi: 10.1103/PhysRevE.83.050903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elfving N, Chereji RV, Bharatula V, et al. A dynamic interplay of nucleosome and Msn2 binding regulates kinetics of gene activation and repression following stress. Nucleic Acids Res. 2014;42:5468–82. doi: 10.1093/nar/gku176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zentner GE, Tsukiyama T, Henikoff S. ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet. 2013;9:e1003317. doi: 10.1371/journal.pgen.1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gasch AP, Spellman PT, Kao CM, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–57. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez-Pastor M, Marchler G, Schuller C, et al. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–35. [PMC free article] [PubMed] [Google Scholar]
- 51.Zawadzki KA, Morozov AV, Broach JR. Chromatin-dependent transcription factor accessibility rather than nucleosome remodeling predominates during global transcriptional restructuring in Saccharomyces cerevisiae. Mol Biol Cell. 2009;20:3503–13. doi: 10.1091/mbc.E09-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolkunov D, Zawadzki KA, Singer C, et al. Chromatin remodelers clear nucleosomes from intrinsically unfavorable sites to establish nucleosome-depleted regions at promoters. Mol Biol Cell. 2011;22:2106–18. doi: 10.1091/mbc.E10-10-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiner A, Hughes A, Yassour M, et al. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20:90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrenko N, Chereji RV, McClean MN, et al. Noise and interlocking signaling pathways promote distinct transcription factor dynamics in response to different stresses. Mol Biol Cell. 2013;24:2045–57. doi: 10.1091/mbc.E12-12-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ganguli D, Chereji RV, Iben JR, et al. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res. 2014 doi: 10.1101/gr.177014.114. gr.177014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson JD, Thåström A, Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol Cell Biol. 2002;22:7147–57. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teif VB, Ettig R, Rippe K. A lattice model for transcription factor access to nucleosomal DNA. Biophys J. 2010;99:2597–607. doi: 10.1016/j.bpj.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15:1405–21. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller JA, Widom J. Collaborative competition mechanism for gene activation in vivo. Mol Cell Biol. 2003;23:1623–32. doi: 10.1128/MCB.23.5.1623-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moyle-Heyrman G, Tims HS, Widom J. Structural constraints in collaborative competition of transcription factors against the nucleosome. J Mol Biol. 2011;412:634–46. doi: 10.1016/j.jmb.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Afek A, Sela I, Musa-Lempel N, et al. Nonspecific transcription-factor-DNA binding influences nucleosome occupancy in yeast. Biophys J. 2011;101:2465–75. doi: 10.1016/j.bpj.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Afek A, Lukatsky DB. Genome-wide organization of eukaryotic preinitiation complex is influenced by nonconsensus protein-DNA binding. Biophys J. 2013;104:1107–15. doi: 10.1016/j.bpj.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]