

Background: G-quadruplexes (G4s) play a variety of roles in DNA transactions.
Results: Pif1-catalyzed duplex DNA unwinding was greatly stimulated by G4s in several aspects, including the unwinding rate and amplitude and the chemical-mechanical coupling efficiency.
Conclusion: G4s significantly activate helicase Pif1-catalyzed duplex DNA unwinding through a mechanism of G4-induced dimerization.
Significance: The G4-activating effect may be implicated in the rescue of stalled replication forks, activating of replication origins, and lagging strand maturation.
Keywords: DNA Helicase, DNA Replication, G-quadruplex, Genomic Instability, Kinetics, Replication Origin
Abstract
The evolutionarily conserved G-quadruplexes (G4s) are faithfully inherited and serve a variety of cellular functions such as telomere maintenance, gene regulation, DNA replication initiation, and epigenetic regulation. Different from the Watson-Crick base-pairing found in duplex DNA, G4s are formed via Hoogsteen base pairing and are very stable and compact DNA structures. Failure of untangling them in the cell impedes DNA-based transactions and leads to genome instability. Cells have evolved highly specific helicases to resolve G4 structures. We used a recombinant nuclear form of Saccharomyces cerevisiae Pif1 to characterize Pif1-mediated DNA unwinding with a substrate mimicking an ongoing lagging strand synthesis stalled by G4s, which resembles a replication origin and a G4-structured flap in Okazaki fragment maturation. We find that the presence of G4 may greatly stimulate the Pif1 helicase to unwind duplex DNA. Further studies reveal that this stimulation results from G4-enhanced Pif1 dimerization, which is required for duplex DNA unwinding. This finding provides new insights into the properties and functions of G4s. We discuss the observed activation phenomenon in relation to the possible regulatory role of G4s in the rapid rescue of the stalled lagging strand synthesis by helping the replicator recognize and activate the replication origin as well as by quickly removing the G4-structured flap during Okazaki fragment maturation.
Introduction
G-quadruplexes (G4s)4 are four-stranded secondary structures held together by noncanonical G-G base pairs (1, 2). Once formed, G4s are highly stable with a melting temperature of higher than 95 °C under physiological conditions (3). Conceivably, such stable G4s will lead to deleterious consequences on genomic stability. Indeed, recent genetic studies have provided strong evidence that G4 structures can form within cells from single endogenous G4 motifs, obstruct replication fork movement, and increase frequencies of spontaneous chromosomal rearrangements (4–9). The G4-induced replication fork arrest also disturbs histone extrusion and the assembly of chromatin, impeding faithful transmission of epigenetic information (10–14). Previous studies have shown that cells have evolved mechanisms for regulating G4 structures to counteract their intrinsic recombinogenic effects. It has been shown that several helicases such as those in the RecQ (15), DEAH-box (16, 17), and Pif families (18, 19) possess G4 resolving activity.
If G4s have merely detrimental effects in cells, why are the G4-containing sequences well conserved in evolution? In recent years, the notion that G4s may play critical and beneficial functions in cells has emerged (20). G4s formed from the G-rich telomeric overhang will prevent DNA damage surveillance mechanisms from recognizing telomeres as a DNA damage signal (21–24). Furthermore, it has long been recognized that G4 motifs are very abundant in promoter regions and at the 5′ end of the first intron (25–27). The observations that differential expression characterized mRNAs transcribed from genes enriched in G4s support the view that G4s can play essential roles in gene expression in vivo (28–31).
An interesting concept put forward in recent years is that G4s may determine the precise position and efficiency of the replication start sites. Several mappings of genome-wide locations of replication origins in Drosophila (32), mouse (32), and human cells (33–35) have identified a strong correlation between the consensus G4-forming DNA motifs and the replication origins. A further study has shown that it is a G4 structure, but not the poly(G) sequence, that contributes to the directionality of replication forks (36). These observations indicated that G4s are structural elements determining or activating replication origins. However, how G4s act to influence origin usage remains completely unknown.
Here, we report that G4s can significantly stimulate helicase-catalyzed duplex DNA unwinding when they are present at a stalled replication fork, upstream of a replication origin, and in a flap in the Okazaki fragment. This finding may broaden our understanding of the properties and functions of G4s. The phenomenon that G4 DNA activates Pif1 helicase and enhances its duplex unwinding may thus be biologically meaningful. The activation mechanism by G4 could be used to rapidly rescue the stalled leading strand, helping the replicator recognize and activate the replication origin, as well as to quickly remove a G4-structured flap in the Okazaki fragment that is immune to cleavage by nucleases FEN1/Dna2.
EXPERIMENTAL PROCEDURES
Reagents, Buffers, and Oligonucleotide Substrates
All chemicals were reagent grade, and all buffers were prepared in high quality deionized water from a Milli-Q ultrapure water purification system (Millipore) having resistivity greater than 18.2 megohms/cm. All chemicals were from Sigma, except for TMPyP4 which was purchased from Merck. The DNA substrates used in the unwinding, binding, and dynamic light scattering were purchased from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China). Their structures and sequences are shown in Tables 1 and 2. The protein trap used for single-turnover kinetic experiments was 56-nt poly(dT) (dT56). All synthetic oligonucleotides were purified by denaturing PAGE before storage in 10 mm Tris-HCl (pH 8.0), 1 mm EDTA at −20 °C. Concentrations of single-stranded oligodeoxynucleotides were determined spectrophotometrically based on extinction coefficients calculated by the nearest neighbor method.
TABLE 1.
DNA substrates used in DNA binding assay
| DNA name | Structure and sequence (5′ to 3′) |
|---|---|
| G4 | Fa-TGGGTTAGGGTTAGGGTTAGG |
| G4-control (21-nucleotide ssDNA) | F-TTTTTTTTTTTTTTTTCATGGG |
a F is fluorescein.
TABLE 2.
DNA substrates used in DNA unwinding assay


a F indicates fluorescein.
b HF indicates hexachlorofluorescein.
* G4 motifs are shown in boldface.
Preparation of G4s-containing DNA Substrates
A 2 μm working stock solution of G4-containing DNA or G4/duplex DNA was prepared by mixing equal concentrations of complementary single-stranded oligonucleotides in a 20 mm Tris-HCl buffer (pH 7.5 at 25 °C) containing 100 mm KCl, followed by heating to 95 °C. After equilibrating for 5 min, annealing was allowed by slow cooling to room temperature. The quality of the resulting DNA substrates was inspected by native-PAGE and circular dichroism (CD) spectroscopy. DNA concentration was further determined by absorbance of 260 nm light using a Smart Spec 3000 UV spectrophotometer (Bio-Rad). The DNA substrates were stored at −20 °C.
Protein Expression and Purification
The nuclear form of yeast Pif1 (amino acids 40–859) expression vector was a generous gift from V. Zakian and was expressed according to Boule and Zakian (37) with some minor modifications. Briefly, a pET28 vector (Novagen) was transformed into the BL21 Rosetta Escherichia coli strain, and cultures were grown at 37 °C until an A600 of ∼0.6 and then incubated overnight with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C. After centrifugation, the cell pellets were resuspended in lysis buffer (20 mm Tris-HCl at pH 8.0, 500 mm NaCl, 10 mm imidazole, and 10% glycerol). The cells were sonicated and then centrifuged at 14,000 rpm for 30 min. Prior to loading on the Ni2+-charged IMAC column (GE Healthcare), the samples were filtered through a 0.45-μm filter. The protein was eluted from the Ni2+ affinity column by running a gradient from 20 to 500 mm imidazole in a buffer containing 20 mm Tris-HCl at pH 8.0, 500 mm NaCl, and 10% glycerol. The eluted protein was dialyzed against SP buffer (20 mm Tris (pH 7.4), 200 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol (DTT), 5% glycerol) and was loaded on a Source SP column (GE Healthcare), The protein was eluted with a 5-column volume gradient (SP buffer containing 1 m NaCl) and was further purified by gel filtration with Superdex 200 10/300 GL column. The final purified protein is 95% pure as determined by SDS-PAGE (Fig. 1A) and was stored at −80 °C. The purified Pif1 exhibits equivalent unwinding rate and amplitude as the studies reported previously (37, 38).
FIGURE 1.

Purified Pif1 helicase and biologically relevant G4 DNA constructs. A, SDS-PAGE analyses of the purified nuclear form of S. cerevisiae Pif1 protein (amino acids 40–859) and its ATPase-dead protein Pif1E303A. 20 μg of protein was loaded onto a 10% polyacrylamide gel. Lanes 1 and 2 are native and mutated Pif1, respectively. B, schematic presentations of G4s formed at the stalled replication fork and upstream of replication origin (top panel) and the constructions of the corresponding biologically relevant G4/duplex DNA substrates (bottom panel). G4 motif was connected with ssDNA or dsDNA at its 5′ end and a duplex labeled with fluorescent donor (fluorescein, F) and acceptor (hexachlorofluorescein, HF) at its 3′ end (bottom panel). The sequences are shown in the Table 2.
To produce the ATPase activity-deficient mutant, the conserved glutamic acid at position 303 in the ATP hydrolysis motif II (DEIS) was mutated to alanine and resulting mutant of Pif1E303A. The point mutation was constructed using recombinant PCR, with the desired mutations introduced in the internal mutagenic primers (forward E303A, 5′-ttggttgtcgatgcaatatcaatgt, and reverse E303A, 5′-acattgatattgcatcgacaaccaa). To ensure that only the desired mutation was introduced, the PCR portions were sequenced with the dideoxy-DNA sequencing method. The procedure of the expression and purification of the mutant Pif1E303A are essentially the same as wild-type protein.
Stopped-flow Fluorescence Measurements
The stopped-flow assays were carried out using a Bio-Logic SFM-400 mixer with a 1.5 × 1.5-mm cell (FC-15, Bio-Logic), and the Bio-Logic MOS450/AF-CD optical system was equipped with a 150-watt mercury-xenon lamp (39). Fluorescein was excited at 492 nm (2-nm slit width), and its emission was monitored at 525 nm using a high pass filter with a 20-nm bandwidth (D525/20; Chroma Technology Co.). Unwinding kinetics were measured in a two-syringe mode, where Pif1 helicase and DNA substrates were preincubated at 25 °C in syringe 1 for 5 min, and ATP with or without protein trap were in syringe 4. Each syringe contained unwinding reaction buffer, and the unwinding reaction was initiated by rapid mixing. All concentrations listed are after mixing unless noted otherwise. For converting the output data from volts to percentage unwinding, a calibration experiment was performed in a four-syringe mode, where helicase in syringe 1, hexachlorofluorescein-labeled single-stranded oligonucleotides in syringe 2, and fluorescein-labeled single-stranded oligonucleotides in syringe 3 were incubated in unwinding reaction buffer, and the solution in syringe 4 was the same as in the above unwinding experiment. The fluorescent signal of the mixed solution from the four syringes corresponded to 100% unwinding. All of the solutions were filtered and extensively degassed immediately before they were used. The stopped-flow temperature was controlled by means of an external thermostated water bath (Ministat 125; Huber) and a high flux pump to circulate the water between the bath and the stopped-flow apparatus. The standard reaction was usually performed with 2 nm DNA substrates and 80 nm Pif1 in buffer A (25 mm Tris-HCl (pH 7.5), 50 mm NaCl, 2 mm MgCl2, 2 mm DTT) at 25 °C.
Kinetic Data Analysis
All stopped-flow kinetic traces were an average of over 10 individual traces. The kinetic traces were analyzed using Bio-Kine (version 4.26; Bio-Logic).
To determine the step size with the n-step sequential model (40, 41), the single-turnover unwinding data curves are fit with Equation 1,
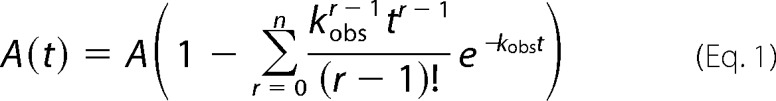 |
where A(t) represents the unwinding amplitude as a function of time; A is the total unwinding amplitude, and kobs is the unwinding rate constant.
Determination of Specific Constants (kcat/Km)
Pif1-mediated single-turnover DNA unwinding with excess of enzyme over DNA substrate was analyzed according to Reaction 1,
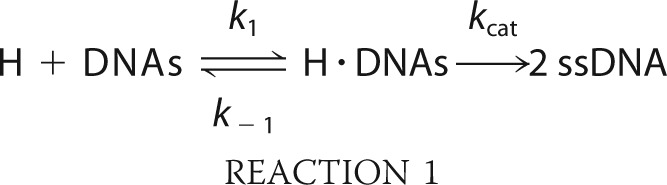 |
where H and ssDNA designate helicase and unwound single-stranded DNA, respectively. The conservation relationship for DNA substrate can be written as shown in Equation 2,
where [DNA]tot represents the total concentration of DNA.
Equilibrium DNA Binding Assay
We use a steady-state fluorescence anisotropy binding assay to determine Pif1 binding activity (60). The binding processes were analyzed according to Reaction 2,
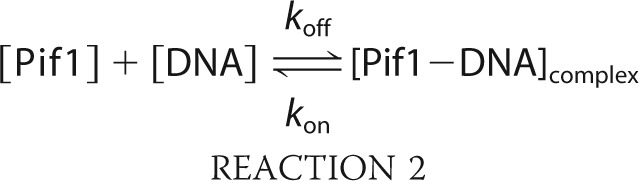 |
when the binding reaction has achieved equilibrium, the equilibrium dissociation constant is shown in Equation 3,
 |
The determined isothermal binding curves were fit by a fractional saturation function shown in Equation 4,
 |
where γ represents measured binding signal (γ is anisotropy signal in this study); Δγ = γcomplex − γDNA, free designs the binding capacity at a different concentration of protein; Δγmax = γcomplex, saturation − γDNA, free designs the maximum binding capacity. Then the binding fraction function is shown in Equation 5,
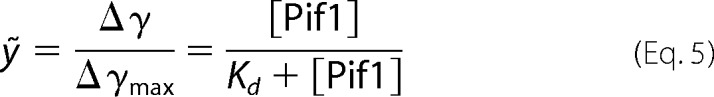 |
If the binding curve is in sigmoidal shape, the curve can be fit by Hill Equation 6:
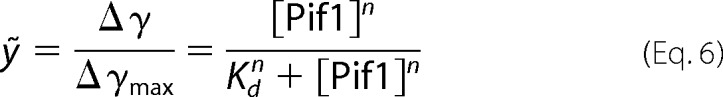 |
where n is Hill coefficient, describing cooperativity. If n > 1, it indicates that more than one DNA can bind to the enzyme.
The isothermal binding curves were determined by fluorescence polarization assay using Infinite F200 (TECAN). Fluorescein-labeled DNA substrates were used in this study. Varying amounts of protein were added to a 150-μl aliquot of binding buffer A (25 mm Tris-HCl (pH 7.5), 50 mm NaCl, 2 mm MgCl2, and 2 mm DTT) containing 5 nm labeled DNA. Each sample was allowed to equilibrate in solution for 5 min, after which fluorescence polarization was measured. A second reading was taken after 10 min, to ensure that the mixture was well equilibrated. Less than 5% change was observed between the 5- and 10-min measurements, indicating that equilibrium was reached in 5 min. The equilibrium dissociation constant was determined by fitting the data to the saturation fraction Equation 5 or the Hill Equation 6 using KaleidaGraph (Synergy Software).
Kinetics of Pif1 Binding to ssDNA and G4 Motif
Experiments were performed according to Kozlov and Lohman (42) with some modifications by the same stopped-flow equipment as mentioned above. The fluorescence emission from FAM was monitored using a 525 nm interference filter (Oriel). All slit widths were 2 mm. The binding solutions (buffer A) were put on ice and then incubated in the reservoir syringes of the stopped-flow instrument at 25 °C for at least 5 min prior to mixing. Longer equilibration times had no effect on the results.
For the reversible protein-DNA binding kinetic, we analyze the kinetics time course according to Reaction 3,
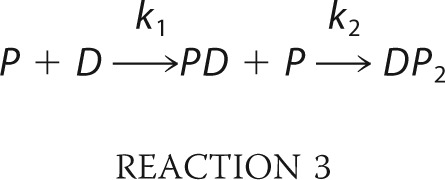 |
All kinetic traces used in the fluorescence represent an average of 10 individual traces. Where applicable, the fluorescence time courses were fit to either single or two exponentials using Origin8.0 software according to Equation 7,
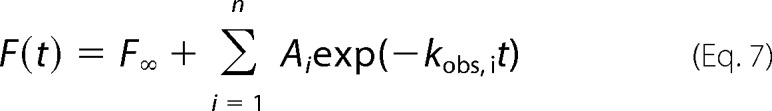 |
where F(t) is the fluorescence intensity at time t; F∞ is the fluorescence intensity at infinite time; Ai is the amplitude of the ith binding process; kobs, i is the observed rate characterizing the ith binding process, and n = 1 or 2.
Stopped-flow ATPase Experiments
Single cysteine mutant (A197C) phosphate-binding protein (PBP) was expressed in E. coli and purified as described previously (43). It was then conjugated to the fluorophore N-[2-(1-maleimidyl)-ethyl]-7-diethylamino) coumarin-3-carboxamide (MDCC) in the presence of a coupled enzymatic phosphate mop composed of 0.2 unit/ml purine nucleoside phosphorylase with 200 μm 7-methylguanosine and purified as described previously (43). The labeled protein (PBP-MDCC) after purification had a 280:430 nm absorbance ratio of 1.6, indicating the majority of the PBP was labeled. PBP-MDCC displayed a 6-fold increase in fluorescence emission at 465 nm upon binding inorganic phosphate (Pi). Stopped-flow kinetic measurements of ATP hydrolysis were performed according to Fischer et al. (44) with minor modifications.
Dynamic Light Scattering
Dynamic light scattering measurements were performed using a DynaPro NanoStar instrument (Wyatt Technology Corp.) equipped with a thermostated cell holder (45). All solutions were filtered using disposable filters (0.1-μm filters) (UVette, Eppendorf). The protein concentration was at 1.5–2.0 μm in buffer DS (50 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm DTT) (total volume, 30 μl). The scattered light was collected at an angle of 90°. Recording times were typically between 3 and 5 min (20–30 cycles in average of 10 s each). The analysis was performed with the Dynamics 7.0 software using regularization methods (Wyatt Technology Corp.). The diffusion coefficient (D) of the molecules was calculated by fitting the experimental data, and the hydrodynamic radius (Rh) of the molecules can be calculated from D = kT/6πη0Rh, where k is Boltzmann constant; T is temperature, and η0 is solvent viscosity. The molecular weight was calculated from the hydrodynamic radius using empirical Equation 8,
where M and Rh represent the molecular mass (in kDa) and the hydrodynamic radius (nm), respectively.
RESULTS
Experimental Design for Kinetic Characterization of Pif1-mediated G-quadruplex DNA Unwinding
Pif1 was reported previously as a low or nonprocessive helicase for unwinding duplex DNA (19, 38, 46), but it is highly active for unwinding G4s (19, 47). In fact, the G4s do not exist alone as an isolated motif, but instead they are usually connected to double-stranded DNA at one or both sides, such as the G4s implicated in stalled replication forks and in replication origins (Fig. 1B). To better mimic G4 unfolding events in the cell and facilitate rapid kinetic and mechanistic studies of helicase-mediated G4 unfolding, we designed two types of DNA structures. The first mimics the stalled replication fork by G4, and the other represents a G4 upstream of a DNA replication start site (Fig. 1B). To construct these substrates, a G4 was linked with ssDNA or dsDNA at its 5′ end, and a fluorescently labeled duplex oligodeoxynucleotide at its 3′ end, respectively. By using the nuclear form of yeast Pif1 (amino acids 40–859), the helicase-catalyzed DNA unwinding was monitored continuously and in real time by measuring the change in fluorescence signal. Circular dichroism analyses confirmed that the G4 structure was well formed. Electrophoretic mobility shift assays (EMSA) further showed that the well folded compact unimolecular G4s migrated faster than tetramolecular G4s (data not shown), as expected.
To confirm that the above designed unimolecular G4 substrates are reliable for studying helicase-mediated G4 unfolding, we performed additional control experiments. First, the unwinding activity was measured with an increasing concentration of TMPyP4, a potent and specific G4-DNA ligand. We observed that the Pif1-catalyzed G4 unfolding activity was significantly decreased with increasing concentrations of TMPyP4 (Fig. 2, A and C). The effect of TMPyP4 on the unwinding of partial duplex DNA alone was negligible (Fig. 2B). In addition, we have confirmed that the binding activity of Pif1 was not inhibited in the presence of 5 μm TMPyP4 (data not shown). These experiments indicate that this assay can be used for characterizing Pif1-mediated G4 unwinding. Second, we measured the G4 unwinding amplitude and unwinding rate with increasing concentrations of an engineered, structure-specific antibody (BG4) that binds with high selectivity to G4 structures (48). Consistent with the above experiments, both the rate and amplitude of unwinding of G4/duplex DNA were significantly decreased as the concentration of the antibody increased (Fig. 2D). Note that the effect of BG4 on the unwinding of a partial duplex DNA alone was negligible (Fig. 2B). These results demonstrate that the observed unwinding activity truly reflects Pif1-catalyzed G4 unfolding. Therefore, the designed DNA structures and the established fluorescence method are reliable for studying Pif1-mediated G4 and duplex DNA unwinding.
FIGURE 2.

Established fluorescence methods are reliable for measuring G4/duplex DNA unwinding. A, Pif1-mediated G4-DNA/duplex DNA (substrate S26G4D17) unwinding in the absence and in the presence of 5 μm TMPyP4. B, no inhibitory effect of TMPyP4 and BG4 on the duplex DNA (S26D17) unwinding by Pif1. C, influence of the concentration of TMPyP4 on Pif1-mediated G4/duplex DNA unwinding. D, influence of the concentration of the G4 antibody (BG4) on Pif1-mediated G4/duplex DNA unwinding. The above kinetics were performed with 80 nm Pif1 and 2 nm fluorescence-labeled DNA substrates under experimental conditions as indicated under “Experimental Procedures.”
Pif1-catalyzed G-quadruplex DNA Unwinding Is ATP-dependent
Previous studies have suggested that Bloom syndrome helicase unfolds G4 DNA in an ATP-independent manner (49). To study whether Pif1 needs the energy derived from ATP hydrolysis to unfold G4 structures, we first measured the unwinding reaction in the presence of the nonhydrolyzable ATP analogues ATPγS and AMP-PNP. The results show that G4 DNA was only unfolded in the presence of 1 mm ATP but not in the presence of 1 mm ATPγS or AMP-PNP (Fig. 3A). To further confirm this observation, we prepared an ATPase-dead Pif1 (Pif1E303A), in which the residue glutamic acid 303 in the conserved ATP hydrolysis motif II (DEIS) was mutated into alanine. Consistent with the above results, G4 cannot be unfolded in the presence of 80 nm Pif1E303A and 1 mm ATP (Fig. 3A). Altogether, these results indicate that both protein binding and ATP hydrolysis are required for Pif1-catalyzed G4 unfolding, but not merely helicase or ATP binding.
FIGURE 3.

G4 position relative to duplex DNA determines the G4 activation effect. A, omission of ATP or substitutions of ATP by 1 mm nonhydrolyzable ATP analogues AMP-PNP and ATPγS abolishes G4 unfolding. The ATPase-dead Pif1E303A cannot catalyze G4 unfolding in the presence of 1 mm ATP. B, substrate containing G4 at ss/dsDNA junction (substrate S26G4D17) significantly activates Pif-catalyzed duplex DNA unwinding (S26D17). C, substrate harboring a G-triplex at ss/dsDNA junction (substrate S26G3D17) does not activate duplex DNA unwinding. D, G4 upstream (substrate S26G4D17), but not downstream of duplex DNA (substrate S26D17G4), and G4 motif separated by 5 nucleotides of ssDNA from duplex DNA (substrate S26G4–5nt-D17) activate duplex DNA unwinding.
G-quadruplex DNA Strongly Stimulates Pif1 Helicase-mediated Duplex DNA Unwinding
Because previous studies have established that G4 DNA is very stable and Pif1 unwinds duplex DNA with low efficiency (38), it was expected that the unwinding activity of Pif1 with the above designed G4/duplex DNA substrate should be negligibly low. Very surprisingly, we found that the presence of G4 at the ss/dsDNA junction did not reduce but instead increased significantly the unwinding amplitude and the unwinding rate as follows: from 25 to 87% and from 3.37 to 15.82 s−1, respectively (Fig. 3B). This indicates that Pif1-mediated duplex DNA unwinding is strongly activated by G4s. More interestingly, the G4-stimulating phenomenon can be observed even at low concentrations of Pif1 (from 5 to 20 nm). To avoid overestimating the G4-stimulating effects, we performed the following experiments under conditions in which the duplex DNA alone was unwound at a maximal extent with the appropriate Pif1 concentration (80 nm).
To further confirm the activation phenomenon and to determine which structural property of G4 is necessary, we replaced the G4 motif with a G-triplex (G3) (TTAGGG)3. The G-triplex structure, like the G-quadruplex structure, is also stabilized by Hoogsteen-like hydrogen bonds that contain different layers of G-triads (50). This structure was first characterized as an intermediate form of the G-quadruplex but with a stable conformation. Its existence in solution has been directly visualized (50), and its three-dimensional structure in solution has been determined. The correct formation of G3 used in this study was confirmed by CD and EMSA (data not shown). By measuring the unwinding activity of G3/duplex DNA (Fig. 3C), we found that in sharp contrast to the reaction with the G4 motif, Pif1-mediated duplex DNA unwinding is not influenced by the presence of the G3 motif, both in terms of unwinding rate and amplitude (Fig. 3C). This experiment indicated that Pif1-catalyzed duplex DNA unwinding cannot be activated by G3, suggesting that the integral G-columns are very important for G4 activation.
A previous study has suggested that G4 localized near a duplex DNA may influence the stability of duplex DNA (51). To exclude the possibility that the observed stimulation effect is due to G4-induced instability of duplex DNA, we studied the unwinding activity with two G4/duplex DNA substrates as follows: one in which the G4 and duplex were spaced by five nucleotides to mitigate the possible G4 destabilizing effect, and the other in which the G4 motif was placed downstream of duplex DNA rather than at the ss/dsDNA junction. We found that although strong activation of Pif1-mediated duplex unwinding still exists with the first substrate, there was no detectable activation effect with the second substrate (Fig. 3D), indicating that Pif1 must be first activated by G4 to enhance duplex DNA unwinding. Altogether, these experiments established that G4 greatly activated Pif1-mediated duplex DNA unwinding, and the integral G-quadruplex structure is necessary for this activation effect.
We next tested the possibility that G4 may stimulate duplex DNA unwinding in trans. To this end, we first co-incubated Pif1 with isolated G4 DNA and then with a partial duplex DNA. The partial duplex DNA unwinding was monitored after addition of ATP. We found that there was no detectable stimulatory effect when G4 was not covalently linked with the duplex DNA on the same molecule (Fig. 4A). Other experiments performed with different protocols, such as varying the ratio between dsDNA and G4 DNA and simultaneous preincubation of Pif1 with G4 and dsDNA, gave the same or similar results.
FIGURE 4.

G4 fails to stimulate duplex DNA unwinding in trans, but activates origin replication and flap maturation. A, isolated G4 cannot stimulate Pif1-catalyzed duplex DNA unwinding. 80 nm Pif1 was first preincubated with 80 nm nonlabeled G4 DNA at 25 °C for 5 min, and 4 nm labeled partial duplex DNA was added and further incubated for 5 min; the unwinding reaction was initiated by adding 1 mm ATP. B, G4 upstream of a model replication origin (med 14) in chicken stimulates duplex DNA unwinding. Experiments were performed with substrates G4-Origin and D52-Origin. C, schematic presentation of the back-fold and G4-structured flaps that can be formed during DNA replication. D, experiment shows that Pif1 removes more rapidly a G4-structured flap (substrate G4-structured flap) than a fold-back flap (substrate which has been reported to be removed by Pif1 with radiometric assay). All experiments were performed under the conditions indicated under “Experimental Procedures.”
We next probed the activation effect on a duplex DNA with the G4 motif located at its middle position, which resembles a DNA sequence with G4 upstream of a replication origin (Fig. 4B). To assess G4's function to stimulate the unwinding of origin replication duplex DNA, we used a chicken model origin (med14) as the unwinding substrate in which the original G4 sequence was present at the middle region of duplex DNA. Consistent with the above observations, although such a G4/duplex DNA substrate was unwound with unwinding amplitude as high as 75%, no significant unwinding was observed with a duplex DNA in the same nucleotide length (Fig. 4B). This experiment further confirms that G4 stimulates Pif1 to unwind duplex DNA in the context of G4 upstream of the replication origin.
It was reported that Pif1 helicase enhances Okazaki fragment maturation by removing the displaced flaps that may possess secondary structures, such as a fold-back flap (52). We noticed that if the template of the leading strand forms a G4 structure, its complementary sequences in the lagging strand template must contain a poly(C) sequence, and therefore the replicated Okazaki fragment from such poly(C)-rich lagging strand templates may result in a G4-structured displaced flap in the Okazaki fragment (Fig. 4C). Structured flaps, including the fold-back and G4s, are inhibitory for nuclease cleavage by FEN1 and Dna2 (51) and must be removed for Okazaki fragment maturation. To study whether Pif1 could remove such a G4-structured flap as it does for a fold-back flap (52), we designed an internal G4-structured flap substrate that is reminiscent of a fold-back flap flanked by the adjacent Okazaki fragment (Fig. 4, C and D). By appropriately labeling the above two types of flap substrates (fold-back and G4-structured flaps) and using a stopped-flow method to follow the flap displacement rate and amplitude, we found that the fold-back flap was poorly removed from the Okazaki fragment within 4 s, which is comparable with the previous observations (52). However, the G4-structured flap was removed with a removing rate and amplitude as high as 1.83 s−1 and 40% under the same experimental conditions (Fig. 4D). These results indicated that Pif1 can quickly remove the G4-structured flap through a G4-activated duplex unwinding mechanism.
G4s Activate Duplex DNA Unwinding by Increasing the Unwinding Rate, Step-size, and Processivity
To further assess the G4-dependent stimulating effect on duplex DNA unwinding and to determine at which step and in what manner this takes place, we determined the kinetic unwinding parameters with a set of DNA substrates in which G4 is located at the ss/dsDNA junction, although the duplex length is varied. As a control, parallel analyses were also performed with the same set of duplex DNA but without a G4 structure. The unwinding kinetics was measured under single-turnover conditions. The time courses were highly reproducible and have been repeated at least four times with the above substrates. The resulting kinetic parameters were fit to the n-step sequential mechanism (40). Although the step-size determined with G4/duplex DNA is 2.04 ± 0.03 bp, the one with duplex DNA alone is 1.0 ± 0.05 bp (Fig. 5, A–C). Furthermore, the deduced processivity with the former (≈0.97 ± 0.02) is higher than that with the latter (≈0.92 ± 0.01) (see Table 3). These results not only further emphasize the stimulating effect of the G4 structure for duplex DNA unwinding, they also demonstrate that the activated Pif1 couples more efficiently the energy of ATP hydrolysis to DNA unwinding (as can be seen from the 2-bp step-size in the presence of G4).
FIGURE 5.

G4s increase unwinding rate, amplitude, step-size, and processivity. A and B, single-turnover unwinding kinetics with DNA of short duplex lengths. The assay was performed with 4 nm DNA, 80 nm Pif1, 5 μm dT56, and 1 mm ATP. The solid lines in A are best fits of the time courses with Equation 1, with n = 8, 12, and 16 for the three data curves, respectively. The values of A are 21.6, 18.1, and 10.5% with kobs of 79.5 ± 0.1, 80.9 ± 0.1, and 76.5 ± 0.2 s−1, respectively, for the 12-, 16-, and 20-bp duplex substrates. The substrates used are S26D12, S26D16, and S26D20 for A, and S26G4D10, S26G4D16, and S26G4D20 for B, respectively. C, number of steps obtained above as a function of duplex length. The slopes of the straight lines are 1.00 bp−1 (−G4) and 0.49 bp−1 (+G4). D, unwinding efficiencies with partial duplex and G4/duplex DNA substrates over a large duplex length range (12 to 239 bp). The length of the 5′-ssDNA tails is 26 nucleotides. The assay was performed under multiple-turnover conditions with 4 nm DNA, 80 nm Pif1, and 1 mm ATP (DNA sequences used are shown in Table 2).
TABLE 3.
Unwinding and ATPase activities in the presence and absence of G4 motif in duplex DNA
The values are averages of results from 3 to 4 experiments.
| Unwinding rate | Step size | Processivity | ATPase activity (kcat) | |
|---|---|---|---|---|
| s−1 | bp | s−1 | ||
| S26G4D17 | 15.82 ± 0.28 | 2.04 ± 0.03 | 0.97 ± 0.02 | 3.72 ± 0.08 |
| S26D17 | 3.37 ± 0.08 | 1.0 ± 0.05 | 0.92 ± 0.01 | 3.89 ± 0.09 |
| S47D17 | 4.13 ± 0.14 | 3.97 ± 0.06 |
A previous study has shown that the kinetic step-size for Pif1 is 1 bp with 6 bp of the substrates melting spontaneously (38). In this study, although the determined step-size is about 1, the spontaneous base pair melting is about 4, as determined from the x intercept of the plot draw from unwinding step versus substrate length (Fig. 5C). The differences in spontaneous base pair melting may be due to different experimental conditions such as different concentrations of Mg2+ and unwinding activity determination methods (the rapid chemical quench-flow versus the stopped-flow). Especially, the forked substrates used in the previous study may be more apt to melt spontaneously than the partial duplexes used in this study.
In the above single-turnover experiments, we cannot use substrates with a long duplex because of the limited processivity of Pif1. Thus, we next performed multiple-turnover experiments while using DNA substrates with a large range of duplex lengths (12–239 bp). We observed that there was a significant difference in the unwinding amplitudes between the two kinds of substrates (Fig. 5D). In the case of G4/duplex DNA, when the duplex length increased from 12 to 239 bp, the unwinding amplitude decreased from ∼95 to ∼5%. In the case of partial duplex DNA, however, the unwinding amplitude decreased rapidly to zero when the duplex length increased to only 38 bp. These results clearly show that G4-stimulated Pif1 displays significantly enhanced unwinding efficiency and processivity.
Mechanistic Studies of G4 Activation of Pif1 Helicase
Having established that G4s can greatly stimulate Pif1-catalyzed duplex unwinding activity, we wondered what could be the underlying mechanism. It was previously shown that T4 DNA helicase activity may be stimulated by the action of several individual molecular motors on the same strand of DNA (53). Therefore, it is possible that our observed stimulation effect is simply due to the fact that Pif1 unfolds G4 and releases a long strand of ssDNA, allowing more Pif1 molecules to bind to the 5′-ssDNA tail and thus enhancing the unwinding activity. To examine this possibility, we designed a DNA substrate in which the G4 sequence was replaced by another one with the same nucleotide length (21 nucleotides) that is incapable of forming G4 structure. We found that in this case, the duplex unwinding efficiency was much lower than that for the G4/duplex DNA (data not shown). This confirms that it is not the increased ssDNA length but the presence of a G4 structure that is essential for stimulating the helicase activity of Pif1.
It remains possible that G4 binding and/or unfolding causes Pif1 to assume an active conformation, by which Pif1 hydrolyzes ATP and unwinds duplex DNA more efficiently. Therefore, we measured the ATPase activity of Pif1 with both a partial duplex and a G4/duplex DNA, but we found no significant difference in terms of catalytic efficiency between the two cases (Table 3). We then measured the specific constant of unwinding with increasing protein concentrations under single-turnover conditions, although the catalyst (Pif1) is in excess (Fig. 6, A–D). The determined specific constant (kcat/Km) is about five times higher for G4/duplex DNA (1.14 nm−1 s−1) than for a partial duplex DNA (0.23 nm−1 s−1). It is noteworthy that the higher specific constant of Pif1 for G4/duplex DNA is mainly due to a lower Km value.
FIGURE 6.

G4 induces Pif1 dimerization to activate duplex DNA unwinding. A and B, determination of specificity constants of the partial duplex (A) and G4/duplex DNA (B) substrates with S26D17 and S26G4D17, respectively. Single-turnover kinetics of Pif1-catalyzed DNA unwinding with increasing concentrations of enzyme. The solid lines are double-exponential fits. C, kobs as a function of Pif1 concentration as determined from A and B. Fitting of the data using kobs = kcat/(Km/[Pif1] + 1) yields kcat = 15.7 ± 1.0 s−1 and Km = 69.3 ± 7.6 nm for partial duplex DNA (black) and kcat = 10.6 ± 0.3 s−1 and Km = 9.6 ± 1.1 nm for G4/duplex (red) DNA. D, plots of 1/kcat as a function of 1/[Pif1]. Linear fits yield kcat/Km = 0.20 ± 0.01 nm−1 s−1 for partial duplex DNA (black) and 1.09 ± 0.07 nm−1 s−1 for G4/duplex (red) DNA, respectively. E, DNA binding curves were determined with polarization anisotropy assay using 5 nm FAM-labeled DNA and increasing Pif1 concentration under equilibrium conditions. The titration curve for ssDNA was fit by Equation 5 with a Kd of 10.1 ± 1.1 nm, whereas that for G4 was fit by Hill Equation 6 with a Kd of 25.9 ± 0.6 nm and a Hill coefficient of 2.16 ± 0.09. F, stopped-flow kinetic time courses for Pif1 binding to ssDNA and G4 DNA. Fluorescence signal (λemi = 525 nm) of FAM-labeled DNA was recorded upon addition of 100 nm Pif1 and 500 μm ADP-AlF3. The time course for ssDNA was fit to a single exponential, yielding k1 = 1.72 ± 0.03 s−1. The time course for G4 was fit to a double exponential, yielding k1 = 38.50 ± 0.01 s−1 and k2 = 0.51 ± 0.16 s−1.
We therefore further investigated the DNA binding properties of Pif1 under equilibrium conditions. Although the binding isotherm with ssDNA was fit well by Equation 5 with a Kd of 10.1 nm, the binding isotherm with G4 exhibited a sigmoid curve pattern that was fit with a Hill coefficient of 2.16 and a Kd of 25.9 nm (Fig. 6E). These results suggest that G4 binding may induce a dimerization of Pif1. Because protein-DNA binding kinetics may provide additional information about the different structural transition during binding (54, 55), we then studied the DNA binding kinetics of Pif1 with ssDNA and G4s DNA. By following extrinsic fluorescence on DNA that allows us to monitor Pif1 binding kinetics, we found that the formation of the ssDNA-Pif1 complex could be described by a single-step reaction occurring with a rate constant of ∼1.72 s−1 (Fig. 6F). The kinetic binding curve for the G4-Pif1 complex, however, was best fit by a two-step process. The first step is very fast, with a rate of 38.5 s−1, and is followed by a second step with a lower rate (0.51 s−1) (Fig. 6F). The two-step kinetics suggest that Pif1 indeed goes through a dimerization process after binding to G4. Taken together, the above equilibrium and kinetic binding measurements indicate that the G4-stimulated DNA unwinding activity of Pif1 should result from G4-induced dimerization of the helicase.
We then used dynamic light scattering analysis to determine the conformational states of Pif1 in complex with ssDNA or G4s. In the absence of cofactors (a nonhydrolyzable ATP analogue, ATPγS, and DNA), the determined hydrodynamic radius of apo-Pif1 is about 4.6 ± 0.5 nm (Table 4). This corresponds to a protein with a molecular mass of 117 kDa, which is higher than the expected molecular mass calculated from its amino acid sequence. Because the hydrodynamic size depends on both mass and shape (conformation), this deviation may suggest that Pif1 does not assume a perfect globular conformation in solution. In the presence of ATPγS and/or 21 nucleotides of ssDNA, the observed hydrodynamic radii varied between 4.9 ± 0.2 and 5.4 ± 0.3 nm (Table 4). Considering the fact that binding of the above factors will certainly increase the hydrated radius of the complex, the complexes still remain in their monomeric state as judged from the above determined hydrodynamic radius. In the presence of G4, however, the observed hydrodynamic radius becomes as large as 6.4–6.6 nm and corresponds to Pif1 with a molecular mass of 262–276 kDa, suggesting the formation of a dimer of Pif1 (Table 4). This agrees well with the previous observations and strongly suggests that G4 DNA induces the dimerization of Pif1 and thus enhances the duplex DNA unwinding.
TABLE 4.
Determined Rh and molecular weight of apoprotein and proteins in complex with ATPγS and/or DNA using dynamic light scattering measurements
The values are averages and standard deviations from 2 to 4 independent determinations. The sequence of the ssDNA is 21 nucleotides (5′-CGCTGATGTCGCCTGGTGCAT).
| Substrate | Concentration | Mass | Rh | Calculated mass |
|---|---|---|---|---|
| % | nm | kDa | ||
| Apo | 1 μm | 93.5 ± 1.9 | 4.6 ± 0.5 | 117.8 ± 27.8 |
| +ATPγS | 1 mm | 92.6 ± 1.3 | 4.9 ± 0.2 | 137.8 ± 13.2 |
| +ssDNA | 1 μm | 100.0 | 5.4 ± 0.3 | 177.2 ± 21.0 |
| +ssDNA/ATPγS | 1 μm/1 mm | 99.8 ± 0.2 | 5.1 ± 0.8 | 156.5 ± 50.0 |
| +G4 | 1 μm | 100.0 | 6.6 ± 0.7 | 276.5 ± 66.5 |
| +G4/ATPγS | 1 μm/1 mm | 98.9 ± 2.1 | 6.4 ± 0.3 | 262.0 ± 24.0 |
DISCUSSION
Recently, two smFRET studies have shown that Pif1 monomer unwinds intramolecular G4 DNA substrate by a periodic patrolling mechanism that keeps the G4 unfolded but cannot unwind duplex DNA (56, 57). In this study, we reported a previously unobserved phenomenon that G4, which is generally considered as a roadblock for motor proteins translocating along DNA, greatly stimulates Pif1-catalyzed duplex DNA unwinding. The activation needs the integral structure of G4 DNA, because deletion of one G column (G3) suppresses the G4 activation effects. It is noteworthy that a G4 upstream of the duplex DNA is necessary for the observed activation phenomenon, further supporting that Pif1 is first activated by G4 to enhance duplex DNA unwinding.
Careful characterization and comparison of the kinetic parameters obtained with a simple duplex DNA and one harboring a G4 motif revealed that the G4-activated Pif1 displays greater unwinding rate and processivity than nonactivated Pif1. More interestingly, although Pif1 unwinds duplex DNA with a step-size of 1 bp, the activated Pif1 unwinds duplex DNA with a step-size of 2 bp, indicating that G4 activation leads to Pif1 assuming a conformation that couples more efficiently the chemical energy derived from ATP hydrolysis to mechanical force in duplex unwinding. These data indicate that the G4 activation phenomenon is different from the substrate preference displayed by a helicase. Upon activation, Pif1-mediated duplex DNA unwinding was enhanced in several aspects, including unwinding rate, unwinding amplitude, and chemical-mechanical coupling efficiency.
By studying the possible mechanism of activation, several lines of evidence suggest that the activation of duplex DNA unwinding occurs through a mechanism in which the G4 upstream of a duplex DNA enhances Pif1 dimerization as follows: (i) study of DNA binding under equilibrium conditions have shown that although Pif1 binds ssDNA as a monomer, it binds to G4 as a dimer; (ii) the DNA binding kinetics studies indicated that the kinetic binding curves for ssDNA-Pif1 and G4-Pif1 were best fit by single- and two-step models, respectively, indicating that Pif1 indeed goes through a dimerization process after binding to G4; and (iii) direct characterization of Pif1 oligomeric states by dynamic light scattering shows that the hydrodynamic radius of the ssDNA-Pif1 complex corresponds to a monomer and that the one of G4-Pif1 corresponds to a dimer. The structural basis for G4 enhancing Pif1 dimerization is evident. G4 has a four-stranded structure possessing the structural characteristics and spatial configuration to allow more than one Pif1 monomer to bind G4. This structural feature enhances Pif1 dimerization, thereby stimulating its unwinding activity for duplex DNA downstream of G4. Barranco-Medina and Galletto (58) have studied the binding properties of Pif1 with various DNA substrates, including ssDNA, dsDNA, and 3′- and 5′-tailed duplex DNA. They found that only the forked DNA induced a more stable dimer, which can be detected by ultracentrifugation and fluorescence titration methods (58). However, whether G4 could induce dimerization of Pif1 was not addressed in the above study. In this study, we found that Pif1 is a monomer in solution and that G4 DNA induced Pif1 dimerization (Table 4), which greatly enhanced duplex DNA unwinding. Furthermore, Ramanagoudr-Bhojappa et al. (38) showed that Pif1 unwound more efficiently the forked duplex DNA than the partial duplex DNA. Taken together, these results underscore the link between the dimerization of Pif1 and efficient duplex DNA unwinding.
What might be the biological advantages of G4 activation of Pif1 in duplex DNA unwinding? First, G4s can be formed along the lagging strand and/or leading strand templates due to transit uncoupling of the minichromosome maintenance helicase from the DNA polymerase, which could stall or block high fidelity polymerases, thus inhibiting DNA replication (8–10). If a G4 structure is formed in the leading strand template, the polymerases can become uncoupled, and continued template unwinding allows nascent lagging-strand synthesis to proceed past the blocked nascent leading strand, resulting in a stalled fork. In the case in which a G4 is located in the lagging-strand template, synthesis of the Okazaki fragment is blocked, but the two polymerases remain physically associated, allowing leading-strand synthesis to continue. The nonresumed Okazaki fragment will then leave a daughter-strand gap in the nascent DNA. For rapid rescue of the stalled leading/lagging-strand replication, in addition to a timely unfolding of G4, efficient unwinding of certain base pairs of the duplex DNA downstream of the G4 should be necessary. The unwound duplex DNA will leave 3′-ssDNA, which is accessible to nucleases and will be degraded. Once enough ssDNA regions are available, the replisome is recruited to assembly for completion of chromosomal replication. Moreover, the higher duplex DNA unwinding rate may be necessary to compensate for the stall time to rapidly rescue the synthesis of leading or lagging strand in a synchronous manner.
Second, if G4s are merely roadblocks for replication fork progression and constitute a lethal threat to the cell, why isn't the over-representations of G4s in mammalian genome eliminated in evolution? Recent studies show that over 90% of replication origins are associated with G4 motifs, a phenomenon observed from large scale genome-wide analyses with Drosophila, mouse, and human cells (33). Further analysis has shown that initiation of DNA synthesis takes place precisely at 160–280 bp from the G4 motif (33). More interestingly, the distribution of G4 motifs is asymmetric on each of the two DNA strands within the origin (36). The fact that the density of the G4 motif is higher in early rather than in late replication domains and the G4 motif orientation determines the replication start site position strongly suggests that G4s are one of the main determinants of replication initiation (34). How G4 determines the replication position remains largely unknown, but a logical possibility is that a G4-resolving enzyme could recognize the G4 motif, rapidly unfold a G4-activated duplex DNA as shown in the above study, and thus provide a platform for loading of replication initiation factors. In this context, our observation that G4s greatly stimulate Pif1-catalyzed duplex DNA unwinding may suggest that Pif1, or an unidentified G4-resolving enzyme that may function as Pif1 family helicases, rapidly recognizes the G4 motif and accelerates origin duplex DNA unwinding. It remains to be shown whether mammalian Pif1 helicase participates directly in the initiation of DNA replication by using a similar mechanism to enhance the loading of a pre-replication complex.
Third, computational analyses have revealed that there are >375,000 G4 motifs in the human genome. The synthesis of genes from lagging strand in a G4 motif area will probably produce G4-structured flap, which is immune to cleavage by FEN1/Dna2 nucleases (51). Impediment of Okazaki fragment maturation poses a threat to genomic stability. Genetic studies have shown that the Pif1 helicase, implicated in Okazaki fragment maturation, improved flap displacement (59). Consistent with this idea, we have shown in this study that a G4-structured flap significantly stimulates Pif1 to remove a G4-structured Okazaki fragment, which allows polymerase δ to extend the upstream Okazaki fragment. The upstream and the downstream Okazaki fragments will then finally be ligated by ligase as revealed in previous studies. Alternatively, even though the G4-structured flap is not removed entirely, it can still enhance Pif1 to rapidly unwind the duplex DNA downstream of the flap, leaving enough length of ssDNA and allowing FEN1 and Dna2 nucleases to cleave the G4-structured Okazaki fragment. It is interesting to note that the Pif1-mediated removing rate and amplitude of a G4-structured flap were significantly higher than the rate previously reported for removing a fold-back flap by Pif1 (52). Two recent smFRET studies have shown that the unfolded G4 by Pif1 refolds immediately, although Pif1 translocates in the 5′ to 3′ direction (56, 57). Pif1 unfolds G4 repetitively and patrols DNA to keep its unwound form. In our study, Pif1 is unlikely to unfold repetitively the G4-structured flap due to its 5′ to 3′ polarity. However, the immediate refolding of G4 will not be problematic for continuing removal of the G4-strucutred flap, because the refolded G4 on the flap will continue activating Pif1 to unwind the downstream duplex DNA until the entire flap is removed.
Finally, it was previously shown that Pif1 helicase displaces telomerase from telomeres or the 3′-tail on DNA double strand break resection to regulate telomere length and/or inhibit de novo telomere addition (47). The core telomerase holoenzyme is an RNA-dependent DNA polymerase (TERT) associated with an RNA molecule (TER) that pairs with telomeres and serves as a template for the addition of a telomeric sequence. In addition, the region of TERT comprising the N-terminal domain has been implicated in the formation of the telomerase “anchor site,” which is thought to bind telomeric DNA upstream of the telomere 3′ end. Therefore, the telomerase forms a very stable complex with the telomere through RNA-DNA and DNA-protein interactions. To dislodge the telomerase from the telomere, Pif1 should function as a strong molecular motor to disrupt efficiently the RNA-DNA-protein interactions. However, we and others have found that Pif1 is not processive and displays a low unwinding rate with 5′-ssDNA-tailed duplex DNA. Because the telomere region contains G4 motifs, it is reasonable to suppose that the activated Pif1 by G4 may be necessary to more efficiently couple the chemical energy and produce a higher mechanical force to disrupt the telomerase from the DNA end.
Acknowledgments
We thank Drs. V. Zakian and S. Balasubramanian for kindly providing the plasmids encoding S. cerevisiae Pif1 and G4 antibody (BG4) expression, respectively. We thank Dr. E. Deprez for insightful discussion and Dr. B. Scalvi for carefully reading the manuscript and commenting on it.
This work was supported by the National Natural Science Foundation of China Grants 31370798, 11304252, and 31301632 and Projects 985 and 211 from the Ministry of Education of China. The research was partially financed by a grant from CNRS and conducted within the context of the “LIA helicase-mediated G-quadruplex DNA unwinding and genome stability.”
- G4
- G-quadruplex
- PBP
- phosphate-binding protein
- MDCC
- N-[2-(1-maleimidyl)-ethyl]-7-diethylamino) coumarin-3-carboxamide
- ssDNA
- single-stranded DNA
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- TMPyP4
- meso-tetra(N-methyl-4-pyridyl)porphine.
REFERENCES
- 1. Neidle S., Parkinson G. N. (2008) Quadruplex DNA crystal structures and drug design. Biochimie 90, 1184–1196 [DOI] [PubMed] [Google Scholar]
- 2. Balasubramanian S., Hurley L. H., Neidle S. (2011) Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discov. 10, 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burge S., Parkinson G. N., Hazel P., Todd A. K., Neidle S. (2006) Quadruplex DNA: sequence, topology, and structure. Nucleic Acids Res., 34, 5402–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kruisselbrink E., Guryev V., Brouwer K., Pontier D. B., Cuppen E., Tijsterman M. (2008) Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr. Biol. 18, 900–905 [DOI] [PubMed] [Google Scholar]
- 5. Youds J. L., Barber L. J., Ward J. D., Collis S. J., O'Neil N. J., Boulton S. J., Rose A. M. (2008) DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol. Cell. Biol. 28, 1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ribeyre C., Lopes J., Boulé J. B., Piazza A., Guédin A., Zakian V. A., Mergny J. L., Nicolas A. (2009) The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 5, e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piazza A., Boulé J. B., Lopes J., Mingo K., Largy E., Teulade-Fichou M. P., Nicolas A. (2010) Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 38, 4337–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M. P., Foiani M., Nicolas A. (2011) G-quadruplex-induced instability during leading-strand replication. EMBO J. 30, 4033–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsang E., Carr A. M. (2008) Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair 7, 1613–1623 [DOI] [PubMed] [Google Scholar]
- 10. Verma A., Yadav V. K., Basundra R., Kumar A., Chowdhury S. (2009) Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 37, 4194–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarkies P., Reams C., Simpson L. J., Sale J. E. (2010) Epigenetic instability due to defective replication of structured DNA. Mol. Cell 40, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez R., Miller K. M., Forment J. V., Bradshaw C. R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S. P. (2012) Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 8, 301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whitehouse I., Smith D. J. (2013) Chromatin dynamics at the replication fork: there's more to life than histones. Curr. Opin. Genet. Dev. 23, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwab R. A., Nieminuszczy J., Shin-ya K., Niedzwiedz W. (2013) FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J. Cell Biol. 201, 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larsen N. B., Hickson I. D. (2013) RecQ Helicases: conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 767, 161–184 [DOI] [PubMed] [Google Scholar]
- 16. Lattmann S., Stadler M. B., Vaughn J. P., Akman S. A., Nagamine Y. (2011) The DEAH-box RNA helicase RHAU binds an intramolecular RNA G-quadruplex in TERC and associates with telomerase holoenzyme. Nucleic Acids Res. 39, 9390–9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giri B., Smaldino P. J., Thys R. G., Creacy S. D., Routh E. D., Hantgan R. R., Lattmann S., Nagamine Y., Akman S. A., Vaughn J. P. (2011) G4 resolvase 1 tightly binds and unwinds unimolecular G4-DNA. Nucleic Acids Res. 39, 7161–7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bochman M. L., Sabouri N., Zakian V. A. (2010) Unwinding the functions of the Pif1 family helicases. DNA Repair 9, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanders C. M. (2010) Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 430, 119–128 [DOI] [PubMed] [Google Scholar]
- 20. Tarsounas M., Tijsterman M. (2013) Genomes and G-quadruplexes: for better or for worse. J. Mol. Biol. 425, 4782–4789 [DOI] [PubMed] [Google Scholar]
- 21. Salvati E., Scarsella M., Porru M., Rizzo A., Iachettini S., Tentori L., Graziani G., D'Incalci M., Stevens M. F., Orlandi A., Passeri D., Gilson E., Zupi G., Leonetti C., Biroccio A. (2010) PARP1 is activated at telomeres upon G4 stabilization: possible target for telomere-based therapy. Oncogene 29, 6280–6293 [DOI] [PubMed] [Google Scholar]
- 22. Gomez D., Wenner T., Brassart B., Douarre C., O'Donohue M. F., El Khoury V., Shin-Ya K., Morjani H., Trentesaux C., Riou J. F. (2006) Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J. Biol. Chem. 281, 38721–38729 [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez R., Müller S., Yeoman J. A., Trentesaux C., Riou J. F., Balasubramanian S. (2008) A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J. Am. Chem. Soc. 130, 15758–15759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Izbicka E., Wheelhouse R. T., Raymond E., Davidson K. K., Lawrence R. A., Sun D., Windle B. E., Hurley L. H., Von Hoff D. D. (1999) Effects of cationic porphyrins as G-quadruplex interactive agents in human tumor cells. Cancer Res. 59, 639–644 [PubMed] [Google Scholar]
- 25. Oei S. L., Babich V. S., Kazakov V. I., Usmanova N. M., Kropotov A. V., Tomilin N. V. (2004) Clusters of regulatory signals for RNA polymerase II transcription associated with Alu family repeats and CpG islands in human promoters. Genomics 83, 873–882 [DOI] [PubMed] [Google Scholar]
- 26. Krumm A., Meulia T., Brunvand M., Groudine M. (1992) The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 6, 2201–2213 [DOI] [PubMed] [Google Scholar]
- 27. Eddy J., Maizels N. (2008) Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 36, 1321–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verma A., Halder K., Halder R., Yadav V. K., Rawal P., Thakur R. K., Mohd F., Sharma A., Chowdhury S. (2008) Genome-wide computational and expression analyses reveal G-quadruplex DNA motifs as conserved cis-regulatory elements in human and related species. J. Med. Chem. 51, 5641–5649 [DOI] [PubMed] [Google Scholar]
- 29. Du Z., Zhao Y., Li N. (2009) Genome-wide colonization of gene regulatory elements by G4 DNA motifs. Nucleic Acids Res. 37, 6784–6798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nguyen G. H., Tang W., Robles A. I., Beyer R. P., Gray L. T., Welsh J. A., Schetter A. J., Kumamoto K., Wang X. W., Hickson I. D., Maizels N., Monnat R. J., Jr., Harris C. C. (2014) Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc. Natl. Acad. Sci. U.S.A. 111, 9905–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siddiqui-Jain A., Grand C. L., Bearss D. J., Hurley L. H. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 99, 11593–11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cayrou C., Coulombe P., Vigneron A., Stanojcic S., Ganier O., Peiffer I., Rivals E., Puy A., Laurent-Chabalier S., Desprat R., Méchali M. (2011) Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 21, 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cayrou C., Coulombe P., Puy A., Rialle S., Kaplan N., Segal E., Méchali M. (2012) New insights into replication origin characteristics in metazoans. Cell Cycle 11, 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Besnard E., Babled A., Lapasset L., Milhavet O., Parrinello H., Dantec C., Marin J. M., Lemaitre J. M. (2012) Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 19, 837–844 [DOI] [PubMed] [Google Scholar]
- 35. Cadoret J. C., Meisch F., Hassan-Zadeh V., Luyten I., Guillet C., Duret L., Quesneville H., Prioleau M. N. (2008) Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 15837–15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valton A. L., Hassan-Zadeh V., Lema I., Boggetto N., Alberti P., Saintomé C., Riou J. F., Prioleau M. N. (2014) G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 33, 732–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boulé J. B., Zakian V. A. (2007) The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 35, 5809–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramanagoudr-Bhojappa R., Chib S., Byrd A. K., Aarattuthodiyil S., Pandey M., Patel S. S., Raney K. D. (2013) Yeast Pif1 helicase exhibits a one base-pair stepping mechanism for unwinding duplex DNA. J. Biol. Chem. 288, 16185–16195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X. D., Dou S. X., Xie P., Hu J. S., Wang P. Y., Xi X. G. (2006) Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 281, 12655–12663 [DOI] [PubMed] [Google Scholar]
- 40. Lucius A. L., Maluf N. K., Fischer C. J., Lohman T. M. (2003) General methods for analysis of sequential “n-step” kinetic mechanisms: application to single turnover kinetics of helicase-catalyzed DNA unwinding. Biophys. J. 85, 2224–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou Y., Zhuang X. (2007) Kinetic analysis of sequential multistep reactions. J. Phys. Chem. B 111, 13600–13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kozlov A. G., Lohman T. M. (2002) Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry 41, 6032–6044 [DOI] [PubMed] [Google Scholar]
- 43. Okoh M. P., Hunter J. L., Corrie J. E., Webb M. R. (2006) A biosensor for inorganic phosphate using a rhodamine-labeled phosphate binding protein. Biochemistry 45, 14764–14771 [DOI] [PubMed] [Google Scholar]
- 44. Fischer C. J., Saha A., Cairns B. R. (2007) Kinetic model for the ATP-dependent translocation of Saccharomyces cerevisiae RSC along double-stranded DNA. Biochemistry 46, 12416–12426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu Y. N., Bazeille N., Ding X. Y., Lu X. M., Wang P. Y., Bugnard E., Grondin V., Dou S. X., Xi X. G. (2012) Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 40, 9802–9814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. George T., Wen Q., Griffiths R., Ganesh A., Meuth M., Sanders C. M. (2009) Human Pif1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 37, 6491–6502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paeschke K., Bochman M. L., Garcia P. D., Cejka P., Friedman K. L., Kowalczykowski S. C., Zakian V. A. (2013) Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 497, 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Biffi G., Tannahill D., McCafferty J., Balasubramanian S. (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 5, 182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Budhathoki J. B., Ray S., Urban V., Janscak P., Yodh J. G., Balci H. (2014) RecQ-core of BLM unfolds telomeric G-quadruplex in the absence of ATP. Nucleic Acids Res. 42, 11528–11545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rajendran A., Endo M., Hidaka K., Sugiyama H. (2014) Direct and single-molecule visualization of the solution-state structures of G-hairpin and G-triplex intermediates. Angew. Chem. Int. Ed. Engl. 53, 4107–4112 [DOI] [PubMed] [Google Scholar]
- 51. König S. L., Huppert J. L., Sigel R. K., Evans A. C. (2013) Distance-dependent duplex DNA destabilization proximal to G-quadruplex/i-motif sequences. Nucleic Acids Res. 41, 7453–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pike J. E., Henry R. A., Burgers P. M., Campbell J. L., Bambara R. A. (2010) An alternative pathway for Okazaki fragment processing: resolution of fold-back flaps by Pif1 helicase. J. Biol. Chem. 285, 41712–41723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Byrd A. K., Raney K. D. (2004) Protein displacement by an assembly of helicase molecules aligned along single-stranded DNA. Nat. Struct. Mol. Biol. 11, 531–538 [DOI] [PubMed] [Google Scholar]
- 54. Bjornson K. P., Moore K. J., Lohman T. M. (1996) Kinetic mechanism of DNA binding and DNA-induced dimerization of the Escherichia coli Rep helicase. Biochemistry 35, 2268–2282 [DOI] [PubMed] [Google Scholar]
- 55. Basu A., Schoeffler A. J., Berger J. M., Bryant Z. (2012) ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA. Nat. Struct. Mol. Biol. 19, 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou R., Zhang J., Bochman M. L., Zakian V. A., Ha T. (2014) Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife 3, e02190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hou X. M., Wu W. Q., Duan X. L., Liu N. N., Li H. H., Fu J., Dou S. X., Li M., Xi X. G. (2014) Molecular mechanism of G-quadruplex unwinding helicase: sequential and repetitive unfolding of G-quadruplex by Pif1 helicase [J]. Biochem. J. 10.1042/BJ20140997 [DOI] [PubMed] [Google Scholar]
- 58. Barranco-Medina S., Galletto R. (2010) DNA binding induces dimerization of Saccharomyces cerevisiae Pif1. Biochemistry 49, 8445–8454 [DOI] [PubMed] [Google Scholar]
- 59. Budd M. E., Reis C. C., Smith S., Myung K., Campbell J. L. (2006) Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase δ. Mol. Cell. Biol. 26, 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dou S. X., Wang P. Y., Xu H. Q., Xi X. G. (2004) The DNA binding properties of the Escherichia coli RecQ helicase. J. Biol. Chem. 279, 6354–6363 [DOI] [PubMed] [Google Scholar]