

Background: Protein kinase compartmentalization through anchoring proteins provides spatiotemporal specificity.
Results: Competitive elution combined with cyclic nucleotide affinity enrichment identifies HAP1 as a putative novel PKG anchoring protein (GKAP).
Conclusion: Secondary structure predictions, in vitro binding studies, and site-directed mutagenesis define the binding domain and classify HAP1 as a GKAP specifically anchoring PKG Iβ.
Significance: The repertoire of PKG anchoring proteins is expanded, enforcing that also PKG signaling is tightly spatiotemporally regulated.
Keywords: Cyclic GMP (cGMP), Mass Spectrometry (MS), Protein Kinase G (PKG), Proteomics, Signal Transduction, Chemical Proteomics, HAP1
Abstract
Protein-protein interactions are important in providing compartmentalization and specificity in cellular signal transduction. Many studies have hallmarked the well designed compartmentalization of the cAMP-dependent protein kinase (PKA) through its anchoring proteins. Much less data are available on the compartmentalization of its closest homolog, cGMP-dependent protein kinase (PKG), via its own PKG anchoring proteins (GKAPs). For the enrichment, screening, and discovery of (novel) PKA anchoring proteins, a plethora of methodologies is available, including our previously described chemical proteomics approach based on immobilized cAMP or cGMP. Although this method was demonstrated to be effective, each immobilized cyclic nucleotide did not discriminate in the enrichment for either PKA or PKG and their secondary interactors. Hence, with PKG signaling components being less abundant in most tissues, it turned out to be challenging to enrich and identify GKAPs. Here we extend this cAMP-based chemical proteomics approach using competitive concentrations of free cyclic nucleotides to isolate each kinase and its secondary interactors. Using this approach, we identified Huntingtin-associated protein 1 (HAP1) as a putative novel GKAP. Through sequence alignment with known GKAPs and secondary structure prediction analysis, we defined a small sequence domain mediating the interaction with PKG Iβ but not PKG Iα. In vitro binding studies and site-directed mutagenesis further confirmed the specificity and affinity of HAP1 binding to the PKG Iβ N terminus. These data fully support that HAP1 is a GKAP, anchoring specifically to the cGMP-dependent protein kinase isoform Iβ, and provide further evidence that also PKG spatiotemporal signaling is largely controlled by anchoring proteins.
Introduction
Signaling pathways are largely organized into complex and versatile transduction units, each tailored to respond optimally to a particular signal. These dynamic units consist of compartmentalized anchoring hubs harboring a combination of signaling proteins to ensure spatiotemporal control of signaling. One of the best characterized models for compartmentalization through protein-protein interactions is the large family of protein kinase A-protein kinase A anchoring protein (PKA-AKAP)3 complexes. At present more than 40 mammalian genes encoding AKAPs are known with each AKAP harboring specific isoforms of PKA in addition to other signaling proteins such as phosphodiesterases and phosphatases (1, 2). Unique targeting domains on each AKAP direct PKA signaling modules toward specific subcellular compartments, thereby providing a mechanism that positions PKA in proximity of its anchoring proteins (3, 4). PKA exists as a heterotetramer consisting of a regulatory subunit dimer (PKA-R) of which each PKA-R binds a catalytic subunit (PKA-C). The molecular basis of PKA-AKAP complexes is a hydrophobic groove formed at the dimerization interface of PKA-R. This constitutes a docking site for the hydrophobic edge of a small three- to four-turn amphipathic α-helix present in each AKAP (5–7).
In mammalian cells, the three isoforms of cGMP-dependent protein kinase, also known as protein kinase G (PKG and cGK), are the closest homologs of PKA (8). The domain architecture of PKG is largely identical to PKA except for the fact that PKG exists as a homodimer in which regulatory and catalytic domains reside on a single subunit and, more importantly, its molecular mechanism of dimerization is very different. At the N terminus of PKG, the monomers are held together by a heptad repeat leucine zipper, whereas PKA-R dimerizes with a very different fold called the X-type four-helix bundle. This suggests that, although PKA and PKG are close homologs, they differ entirely in the domains mediating their intracellular localization through binding to anchoring proteins. This difference in localization mechanism may well be the basis for the molecular segregation of cAMP and cGMP signaling in vivo.
Mammalian cells express three PKG isoforms: type I PKG (PKG I), which exists as two different splice variants of the same gene (PKG Iα and PKG Iβ) (9, 10), and type II PKG (PKG II) (11). All the PKG isoforms share the same domain organization with the earlier mentioned N-terminal dimerization domain followed by a regulatory domain with an autoinhibitory sequence, two cooperative cyclic nucleotide binding domains, and a catalytic domain. Although being similar folds, the leucine zipper of the three PKG isoforms share only a little sequence similarity, suggesting different localization behavior of the three isoforms. PKG localization via binding to protein kinase G anchoring proteins (GKAPs) is mediated by its N terminus, and the handful of GKAPs found thus far indeed show specificity for different PKG isoforms (12–16). This is again similar to how AKAPs interact with respect to different isoforms of PKA regulatory subunits (5, 17, 18).
In particular, MYPT1 and GKAP42 bind to PKG Iα (19–21), IRAG and TFII-I are specific for PKG Iβ (12, 22), whereas Rab11b forms a complex with PKG II (23). Given the low number of GKAPs known, there is still no convincing definition of a common PKG I/GKAP binding domain. Therefore, it is essential to identify additional GKAPs, elucidate their binding mechanism(s), and study their role in vivo.
Over the years, overlay assays and yeast two-hybrid screens have been used for the discovery of many new AKAPs (24, 25) and some GKAPs (21) with varying success. More recently, chemical proteomics-based mass spectrometry approaches have provided an alternative to investigate kinase signaling (26, 27). Chemical proteomics enabled the discovery of new anchoring proteins while preserving the native structure of the proteins and their interactions with small molecules and scaffold proteins. Particularly, immobilized cAMP on agarose beads played a pivotal role in the identification and characterization of several new AKAPs. However, all these studies focused on PKA and AKAPs (18, 28, 29) and not on PKG and new GKAPs.
Due to the high similarity between both the second messenger molecules cAMP and cGMP and their main target sites on PKA and PKG, the binding constants of cAMP and cGMP for PKG only differ by ∼100-fold in favor of cAMP (30). The same holds true for PKA and cGMP. Therefore, there has always been speculation about the occurrence of (in vivo) cross-reactivity (31). An argument against this hypothesis is the tight localization of the kinases via anchoring proteins close to their designated pools of cAMP or cGMP, which should largely minimize this event from occurring in vivo. In chemical proteomics experiments, in vitro enrichment by immobilized cAMP or cGMP leads to the simultaneous pulldown of both PKA and PKG possibly due to the high local concentration of the cyclic nucleotide on the resin.
Here we describe an extension of our previously described cAMP-based chemical proteomics method using in-solution competition with a low dose of free cAMP or cGMP, which allows us to dissect PKA- and PKG-driven signaling complexes. Affinity purification followed by mass spectrometry analyses of these competed pulldowns in rat lung tissue led to the selective enrichment of known GKAPs along with a putative novel GKAP candidate, Huntingtin-associated protein 1 (HAP1). Follow-up experiments presented here establish HAP1 as a novel GKAP and highlight the potential of our novel chemical proteomics methodology for discovery of GKAPs in other cells and tissue.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
HEK293 and COS-7 cells were grown at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. HEK293 cells were grown to 80% confluence and then harvested with trypsin. Cells were washed in PBS, snap frozen in liquid nitrogen, and kept at −80 °C until use. COS-7 cells were grown to 70% confluence and transfected using PolyFect according to the manufacturer's protocol for the specific cell type (Qiagen).
Sample Preparation and Pulldowns
HEK293 and COS-7 cells were lysed with a Dounce homogenizer in ice-cold PBS supplemented with 0.1% Tween 20 and protease and phosphatase inhibitors. Lysates were centrifuged at 20,000 × g for 10 min at 4 °C. Prior to the pulldown assay, ADP and GDP were added to the lysate to a final concentration of 10 mm. For the HEK293 cells, the lysate was divided into three equal fractions of 10 mg. One was supplemented with cAMP (10 μm final concentration), one was supplemented with cGMP (10 μm final concentration), and the third sample was used as a control. The three samples were then incubated for 30 min at 4 °C under agitation. 8-AHA-cAMP-agarose beads (Biolog, Germany) were added to the lysates at a ratio of 1:100 (μl of dried beads:μg of protein) and incubated at 4 °C for 2 h. The 8-AHA-linked cAMP-agarose beads have been described before and were chosen because they enable the pulldown of the PKA regulatory subunits and exhibit somewhat lower affinity for PKA than other commercially available cAMP-coupled beads (28). After the incubation, the beads were washed three times with 1 ml of ice-cold lysis buffer containing the above mentioned concentrations of either free cAMP or free cGMP or normal lysis buffer for the control. Beads were subsequently washed three times with 1 ml of ice-cold PBS before protein elution using SDS sample buffer at 95 °C. The eluted proteins were separated by 4–12% SDS-PAGE (Bio-Rad) and subjected to in-gel digestion. Briefly, the proteins were reduced with dithiothreitol (Sigma-Aldrich), alkylated with iodoacetamide (Sigma-Aldrich), and digested with sequencing grade modified trypsin (Promega) prior to analysis by LC-MS/MS (32).
Rat Lung Tissue
Lungs originating from 6-month-old male Wistar rats were excorporated, frozen in liquid nitrogen, and stored at −80 °C until use. Protein isolation was achieved by cooling the tissues in liquid nitrogen followed by pulverization in a precooled custom-made steel mortar. The powdered tissues were then taken up in ice-cold lysis buffer, and 15 mg of protein lysate (1.5 times more than for the HEK293 lysate) was subjected to the same procedure as described above for the HEK293 cell lysates.
LC-MS/MS Analysis
LC-MS/MS analysis was performed using an Agilent 1100 series liquid chromatography system equipped with a 20-mm Aqua C18 (Phenomenex, Torrance, CA) trapping column (packed in house; inner diameter; 100 μm; resin; 5 μm) and a 400-mm ReproSil-Pur C18-AQ analytical column (packed in house; inner diameter; 50 μm; resin; 3 μm). Trapping was performed with 5 μl min−1 solution A (0.6% acetic acid) for 10 min. Peptide elution was achieved with a linear gradient from 13 to 32% solution B (80% acetonitrile, 0.6% acetic acid) in 60 min at a flow rate of 100 nl min−1 obtained by passively splitting the flow from 0.6 ml min−1. Nanospray was achieved using a distally coated fused silica emitter (New Objective, Cambridge, MA; outer diameter, 360 μm; inner diameter, 20 μm, tip inner diameter, 10 μm) biased to 1.8 kV. The LC system was coupled to an LTQ-Orbitrap Discovery or LTQ-Orbitrap Velos mass spectrometer (Thermo Electron, Bremen, Germany). Briefly, the mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS acquisition. Survey full-scan MS spectra were acquired from m/z 350 to m/z 1500 in the Orbitrap. The five most intense ions were fragmented in the linear ion trap using collision-induced dissociation at a target value of 10,000. Peak lists were generated using Proteome Discoverer (version 1.3, Thermo Scientific, Bremen, Germany) using a standardized work flow. Peak lists generated in Proteome Discoverer were searched against the Swiss-Prot database (taxonomy, human; 20,407 protein entries) or the International Protein Index (rat 3.36; 42,689 protein entries) supplemented with frequently observed contaminants using Mascot (version 2.3.02, Matrix Science, London, UK). The database search parameters were as follow: a mass tolerance of 50 ppm for the precursor ions and ±0.6 Da for the fragment ions. Enzyme specificity was set to trypsin with two missed cleavages allowed. Carbamidomethylation of cysteines was set as a fixed modification, and oxidation of methionine was used as a variable modification. Search results were filtered by using a 5% false discovery rate at the peptide-spectrum match (PSM) level (33). In addition, the results were filtered using the following criteria: (i) Mascot ion score of at least 20, (ii) minimum of six amino acid residues per peptide, and (iii) maximum search engine rank of 1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (34) via the PRoteomics IDEntifications (PRIDE) partner repository with the data set identifier PXD001434. The protein interactions from this study have been submitted to the International Molecular Exchange (IMEx) Consortium through IntAct (35) and assigned the identifier IM-23267.
DNA Constructs and Site-directed Mutagenesis
PKG Iα and PKG Iβ plasmids tagged with GFP were gifts from Robert Feil (Universität Tübingen, Tübingen, Germany) (36). HAP1 and a shorter sequence of HAP1, amino acids 157–386, tagged with DsRed were gifts from Edward Schmidt (Montana State University, Bozeman, MT) (37). The double site mutation K567A/R568A was introduced in the HAP1 plasmid using the QuikChange mutagenesis kit using the protocol provided by the manufacturer (Agilent Technologies).
In Vivo Interaction Studies
Transfections were performed in COS-7 cells. HAP1 was co-transfected with PKG Iα or PKG Iβ. The shorter version of HAP1 (amino acids 157–386) and the double point mutant of HAP1 were co-transfected with PKG Iβ. The cells were harvested 24–48 h post-transfection, and pulldowns were performed on 4 mg of protein lysate as described above using 8-AHA-cAMP-agarose beads. After separation by SDS-PAGE, the bound proteins were transferred onto PVDF membrane. Blots were incubated with rabbit anti-PKGI (Abcam; 1:1000) and mouse anti-HAP1 (Novus Biologicals; 1:5000). After several washes, the blots were incubated with appropriate Cy3- and Cy5-labeled secondary antibodies (GE Healthcare), and detection was performed on a Typhoon 9400 imager (Amersham Biosciences).
GKAP Binding Domain Secondary Structure prediction and Docking Study
Secondary structure prediction of IRAG, TFII-I, and HAP1 domains that bind to PKG Iβ were obtained with the NetSurfP program (38). The PKG Iβ crystal structure of the N terminus was retrieved from the Protein Data Bank (code 3NMD) and used with the predicted GKAP secondary structures for docking prediction (39).
Fluorescence Anisotropy
The dimer of the PKG Iβ leucine zipper (LZPKG Iβ; amino acids 4–55), a kind gift from Dr. C. Kim (Baylor College of Medicine, Houston, TX), was purified as described previously (40). The GKAP interaction domains of IRAG (EAKLVSERFLTRRGRKSRSSPGESS; amino acids 164–188) and HAP1 (QQLSNWQDAHSKRQQKQKVVPKDSP; amino acids 556–580) were tagged N-terminally with 5-carboxytetramethylrhodamine. A mutated version of the HAP1 peptide (QQLSNWQDAHSAAQQKQKVVPKDSP; amino acids 556–580) together with a scrambled peptide (EAQEELAWKIAKMIVSDIMQQAQY) were also tagged and used as controls. All peptides were synthesized at the peptide facility of the Netherland Cancer Institute (Amsterdam, The Netherlands). Binding affinity assays between these peptides and the LZPKG Iβ dimeric domains were performed as described by us previously (29).
RESULTS
Competitive Chemical Proteomics Enables the Selective Identification of GKAPs
Previous chemical proteomics experiments based on various immobilized cAMP or cGMP analogs revealed not much specificity for either PKA or PKG (41–43). Often, in cell lines, PKG is expressed at a lower level when compared with PKA. Therefore, the search for novel GKAPs using a chemical proteomics methodology is severely hampered by co-purification of the more abundant PKA pathway components.
Here we investigated the use of in-solution competition with free cAMP to compete for PKA binding to the resin while retaining binding of PKG and its putative binding partners. Because the affinity of cAMP for PKG is ∼100-fold weaker than for PKA and vice versa for cGMP and PKA (30), we sought to selectively occupy the cyclic nucleotide binding motifs of either PKA or PKG prior to the pulldown performed by using the 8-AHA-linked cAMP-agarose beads. Based on these data, we first aimed to identify the appropriate concentration of cAMP. In addition, we also used different competing concentrations of cGMP to investigate the effect on isolating PKA pathway components. A concentration range of cAMP and cGMP was spiked into a HEK293 cell lysate, and subsequently an 8-AHA-cAMP pulldown was performed. A control experiment without added competing cyclic nucleotides was performed in parallel (Fig. 1A). It could be deduced that the most effective concentration of cAMP/cGMP to compete off either PKA or PKG was 10 μm (Fig. 1B). Although in the control experiment both PKA and PKG were captured, supplementing 10 μm cAMP to the lysate resulted in the complete abolishment of PKA binding while maintaining PKG association to the resin. In the reverse experiment, 10 μm cGMP resulted in the opposite effect: full abolishment of PKG binding without affecting PKA interactions (Fig. 1B). To investigate this with more sensitivity, we performed in-gel digestion and mass spectrometry analysis of the control and the two 10 μm cyclic nucleotide (cAMP and cGMP) gel lanes. This resulted in the cumulative identification of 246 proteins under these three different conditions (i.e. no competition, supplemented with cAMP, or supplemented with cGMP) (supplemental Table 1A). To quantify the effectiveness of the competition, we quantitatively compared the number of PSMs between the cAMP-supplemented pulldown and the cGMP-supplemented pulldown (Table 1). This quantitation revealed that PKA and all AKAPs were completely competed off the beads by 10 μm cAMP, whereas PKG binding was entirely retained (Table 1). When comparing with the control experiment, competing for binding of either PKA or PKG also increased our spectral and peptide coverage on the enriched kinase and its binding partners and thus our depth of analysis on both sides of the experiment.
FIGURE 1.
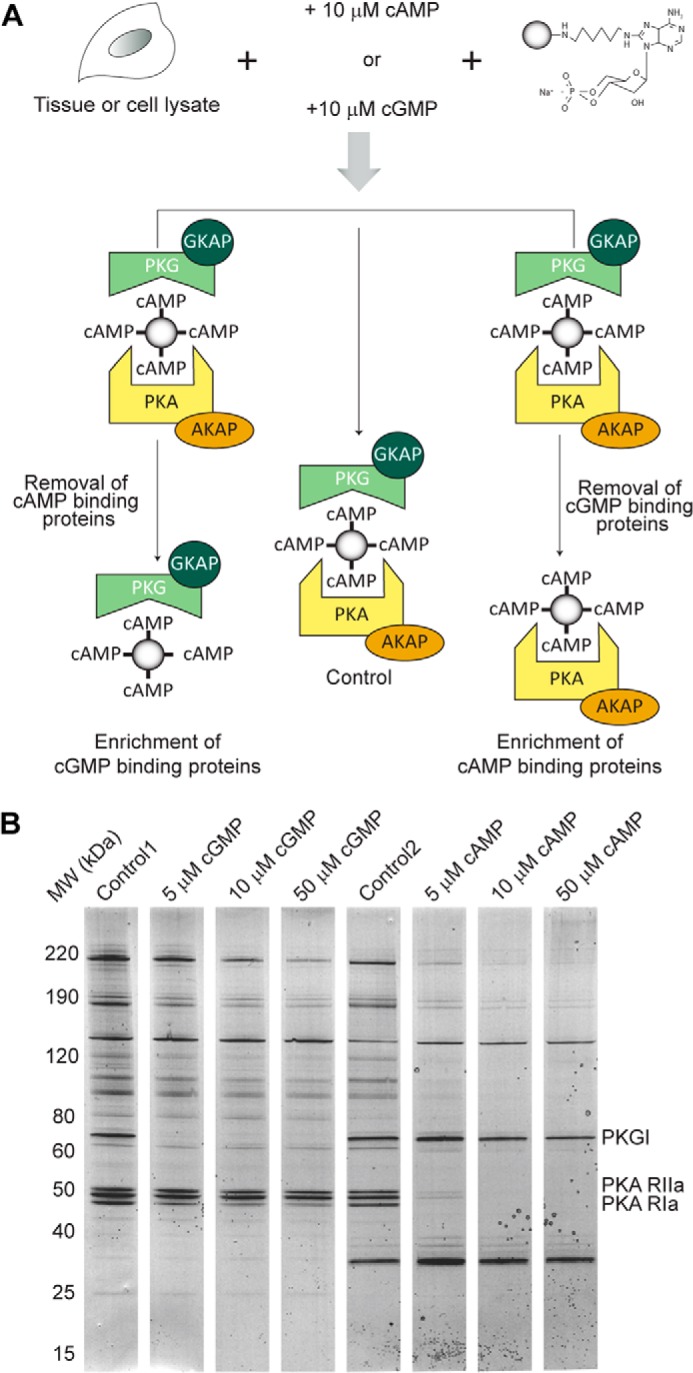
A, competitive chemical proteomics workflow used for the identification of new GKAPs. In the control, no cyclic nucleotide is added to the lysate, whereas cAMP and cGMP are added to the lysate to selectively enrich for PKG and its interactors and PKA and its interactors, respectively. B, Coomassie staining of the proteins pulled down by using 8-AHA-linked cAMP-agarose beads in HEK293 cells with increasing amounts of cyclic nucleotide added to the lysate. Adding 10 μm cGMP resulted in a complete removal of PKG. A similar scenario was observed for the pull downs performed in the presence of 10 μm cAMP, indicating that the PKA regulatory subunits and their interactors were competed off the beads.
TABLE 1.
Specificity of primary and secondary cAMP- and cGMP-interacting proteins enriched by using 8-AHA-cAMP beads in HEK293 lysates
The three columns represent the detection of proteins bound onto the beads and in the control lysates and those supplemented with either 10 μm cAMP (+cAMP) or 10 μm cGMP (+cGMP). Listed are the numbers of unique peptides and PSMs found for each protein in each of the experiments. Preferential cGMP targets are reported in green, whereas cAMP-preferred targets are shown in orange. -indicates undetected proteins.
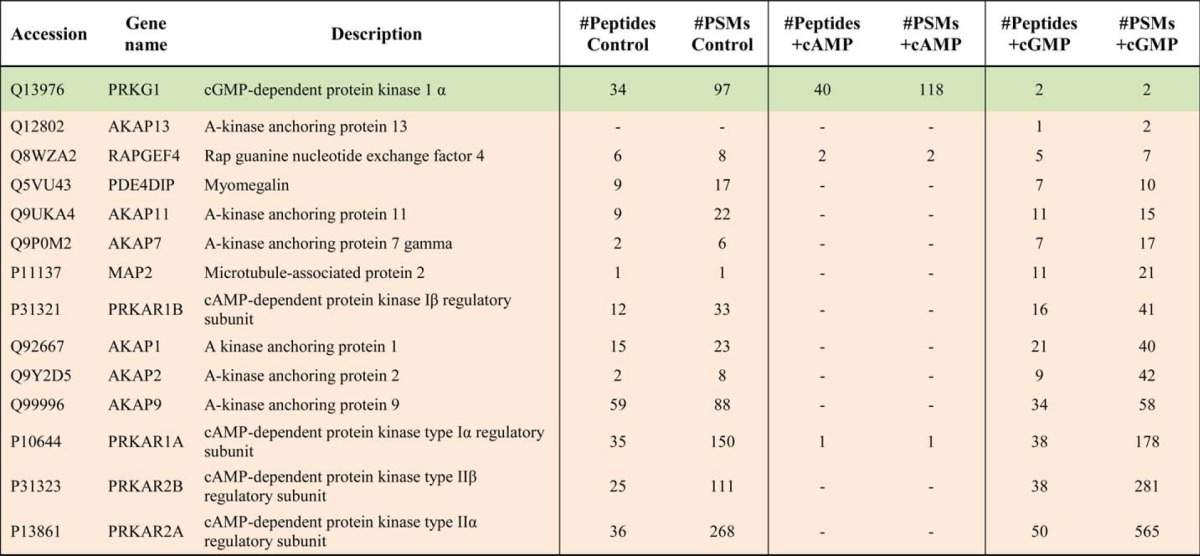
These experiments in HEK293 cells revealed that selectivity could be achieved in our pulldowns as numerous AKAPs could be specifically pulled down; albeit we did not identify any (putative) GKAPs in these experiments. Nevertheless, these data indicate that enriching for PKG while competing for PKA/AKAP binding is a promising tool to screen cells and tissues for novel putative GKAPs.
Following the optimization of the protocol in HEK293 cells, we extended our analysis to (rat) lung tissue, which attractively also has PKG present at a relatively high abundance. The setup was identical to that used for the HEK293 experiments (Fig. 1A) such that again the pulldowns supplemented with cAMP and cGMP could be quantitatively compared using spectral counts (Table 2). Fortuitously, we were now able to selectively enrich not only PKG but also a known GKAP, IRAG. Among the other proteins competed by 10 μm cGMP and thus co-enriched with PKG (supplemental Table 1B), we identified HAP1. HAP1 is involved in a protein complex with Huntingtin and the type 1 inositol 1,4,5-trisphosphate receptor in an altered neuronal Ca2+ signaling pathway (44). Interestingly, IRAG and thus also PKG are located nearby the type 1 inositol 1,4,5-trisphosphate receptor, which upon phosphorylation by PKG reduces the intracellular calcium release from intracellular storage sites (12).
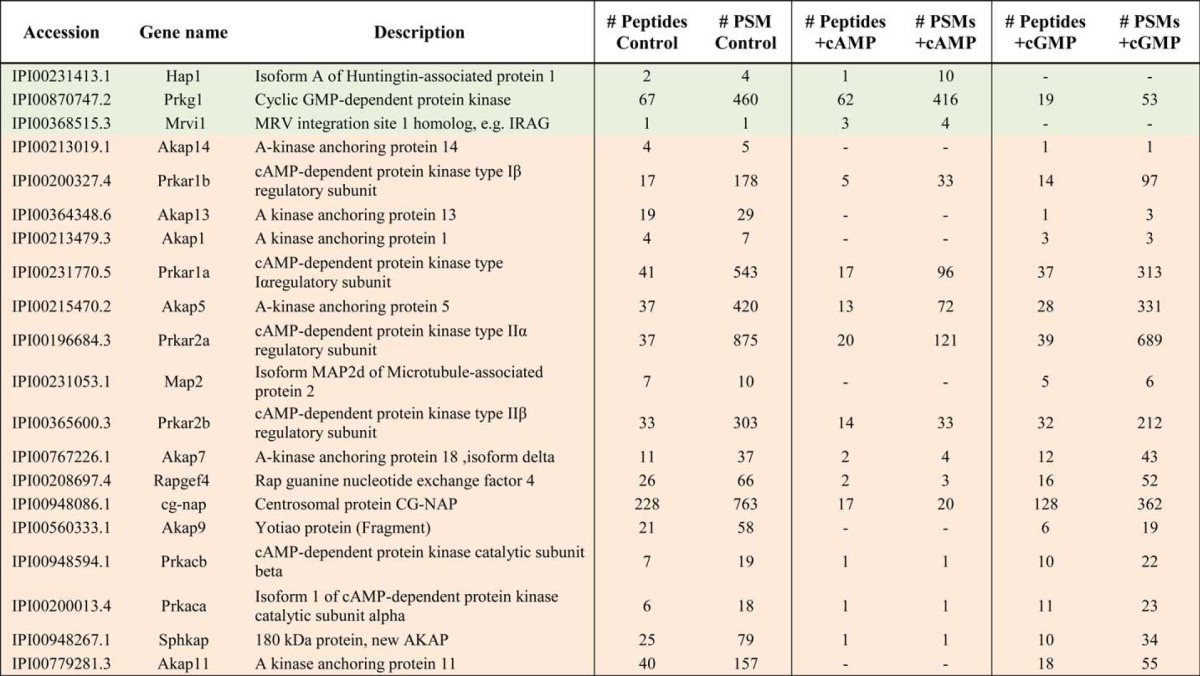
TABLE 2.
Specificity of primary and secondary cAMP- and cGMP-interacting proteins enriched by using 8-AHA-cAMP beads in rat lung tissue lysates
The three columns represent the detection of proteins bound onto the beads and in the control lysates and those supplemented with either 10 μm cAMP (+cAMP) or 10 μm cGMP (+cGMP). Listed are the numbers of unique peptides and PSMs found for each protein in each of the experiments. Preferential cGMP targets are reported in green, whereas cAMP-preferred targets are shown in orange. Within the preferred cGMP targets, we identified Mrvi1 (IRAG), the known interactor of PKG Iβ and HAP1, the here presented putative novel GKAP. -indicates undetected proteins.
To further validate the interaction between HAP1 and PKG, HAP1 was co-expressed with either PKG Iα or PKG Iβ in COS-7 cells. Subsequently, a pulldown using 8-AHA-cAMP beads was performed. The proteins eluted from the beads were analyzed by SDS-PAGE and immunoblotting with antibodies against HAP1 and PKG I. These assays clearly revealed that HAP1 was not enriched by the cAMP-resin when co-expressed with PKG Iα, but it was selectively enriched in the presence of PKG Iβ (Fig. 2). This suggests that HAP1 and PKG Iβ are interacting in the context of the cell and that the interaction found in the proteomics studies is specific.
FIGURE 2.
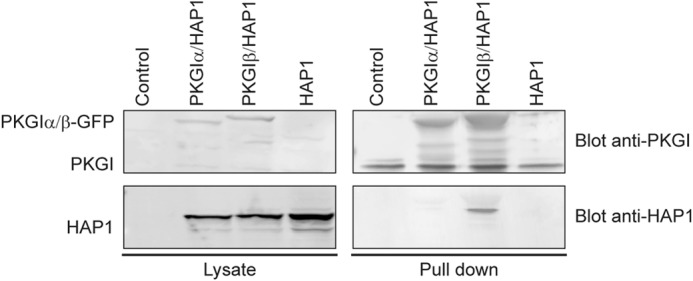
HAP1 interacts specifically with the PKG Iβ isoform. Western blot analysis of pulldowns performed in COS-7 cells co-expressing HAP1 alone or with either PKG Iα or PKG Iβ is shown. The blots from the lysate show the expression of the proteins in the cells, whereas the pulldown with the 8-AHA-linked cAMP-agarose beads shows that HAP1 co-precipitates selectively with PKG Iβ.
HAP1 Harbors a Domain with Sequence Similarity to Known PKG Iβ-selective GKAPs
As only the N terminus is spliced differently between PKG Iα and PKG Iβ, it may harbor the basis for the observed GKAP isoform specificity. Ammendola et al. mapped the interaction between PKG Iβ and IRAG in detail (45), whereas Casteel et al. identified the region in TFII-I interacting with PKG Iβ (22). Mutating basic residues (into alanine) in the IRAG anchoring domain caused disruption in PKG binding, suggesting that electrostatic interactions play an important role in the PKG Iβ-GKAP interaction. In addition, the basic residues in the TFII-I binding domain interact with the acidic residues in the N terminus of PKG Iβ. Although the anchoring domains of IRAG and TFII-I are known, with only two such domains, it is not yet possible to clearly define a PKG binding motif using bioinformatics tools such as the hidden Markov model, which successfully defined the PKA binding motif for several AKAPs (28, 46, 47). Considering the fact that HAP1 binds only to the β isoform of PKG and knowing the anchoring sequences of IRAG and TFII-I, we aligned HAP1 with the binding domains of IRAG and TFII-I to screen for a region that showed at least a similar charge distribution. The alignment revealed the presence of a region rich in basic residues close to the C terminus of HAP1 (amino acids 565–580) with resemblance to the IRAG sequence and to the TFII-I binding domain, albeit for the latter in a reverse configuration (Fig. 3A), providing us the hypothesis that this could be the HAP1 anchoring motif to PKG Iβ. The alignment of the three protein sequences displays a clear pattern of basic residues that are involved in the interaction with PKG Iβ. BLAST analysis of this HAP1 domain revealed strong sequence conservation across several mammals, suggesting the importance of these basic amino acid residues (Fig. 3B).
FIGURE 3.
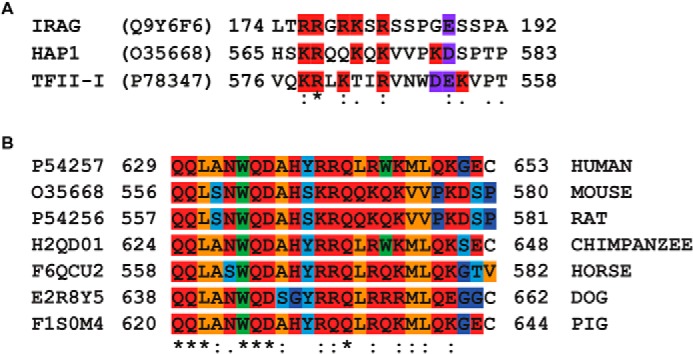
A, alignment of the established IRAG and the reverse TFII-I binding domains to PKG Iβ with the HAP1 region predicted to bind to PKG Iβ. These two sequences show a high degree of similarity regarding the position of the basic and acidic residues. The basic residues are highlighted in red, and the acidic residues are highlighted in purple. B, sequence alignment of the human HAP1 region binding to PKG Iβ with various orthologs in other mammalian species. : and . indicate similarity, and * indicates identity. The residues are color-coded as follows: orange, aliphatic; red, polar; light blue, Ser/Thr/Tyr; green, bulky; and blue, Gly/Pro.
Docking of IRAG, TFII-I, and HAP1 Anchoring Domains onto PKG Iβ
Recently the crystal structure of LZPKG Iβ has been reported (40). We used this structure to first model the binding of the two known Iβ-specific GKAPs, IRAG and TFII-I, onto the N terminus of PKG Iβ using molecular dynamic calculations by HADDOCK (39). We then compared these results with the docking of the proposed interaction site of HAP1. The modeling data suggested that TFII-I exhibits the strongest modeled affinity (HADDOCK score, −102 ± −9) for PKG Iβ followed by HAP1 (−92 ± 5) and IRAG (−86 ± 1) (Fig. 4A). As the anchoring domain of each GKAP is positioned a bit diagonally across the leucine zipper interface, they do not make similar contacts on either monomer (Fig. 4A). Nonetheless, the interaction sites of PKG Iβ with each GKAP studied here were identical: Glu-27 and Glu-31 on one monomer and Glu-29, Asp-33, and Asp-36 (Fig. 4, B–D) on the other monomer of LZPKG Iβ. Moreover, TFII-I shows an additional interaction with Asp-26 as also reported by Casteel et al. (22). Our prediction shows not only that TFII-I has the strongest docking score but that it is also bound backward in sequence with respect to HAP1 and IRAG in agreement with our (reverse) sequence alignment.
FIGURE 4.

Known and predicted GKAP anchoring domains modeled onto the surface of PKG Iβ. A, the overlay of HAP1(red), TFII-I (purple), and IRAG (yellow) shows a binding mode similar to the leucine zipper domain of PKG Iβ (blue). None of the GKAP anchoring domains show a full helical structure. Close-up views of HAP1 (B), TFII-I (C), and IRAG (D) interacting with the dimerization domain of PKG Iβ are shown. The interactions take place at Glu-29 and Asp-36 of PKG with the positive charged residues of the GKAPs. E, helicity prediction of the PKG Iβ binding domains of HAP1, TFII-I, and IRAG. The plot shows how the helical potential decreases after the KR/RR residues in all the protein sequences (residues 572/571, 567/568, and 175/176, respectively). Mutation of the basic residues can influence the helical potential of the proteins and be part of the cause of the disruption of the binding of the GKAPs to PKG Iβ.
Interaction of HAP1 with the Leucine Zipper of PKG Iβ Takes Place at the C Terminus of HAP1
To firmly establish that the HAP1 region at the C terminus interacts with the N terminus of PKG Iβ as suggested by HADDOCK, we performed in vitro binding affinity assays. We probed the binding between LZPKG Iβ and the 25-residue-long 5-carboxytetramethylrhodamine-tagged GKAP anchoring sequences of HAP1 (QQLSNWQDAHSKRQQKQKVVPKDSP), IRAG (EAKLVSERFLTRRGRKSRSSPGESS), a scrambled peptide (EAQEELAWKIAKMIVSDIMQQAQY), and the anchoring sequence of HAP1 with the Lys-12 and Arg-13 mutated into alanine (QQLSNWQDAHSAAQQKQKVVPKDSP) as control peptides using fluorescence anisotropy. The in vitro assay, carried out in triplicate, clearly demonstrated the high affinity binding of HAP1 peptide fragment to the N-terminal domain of PKG Iβ (Fig. 5A) with a Kd of around 15 nm, i.e. in the range of that of the peptide used for IRAG (positive control; 25 nm), whereas the scrambled peptide (negative control) and the mutated HAP1 peptide did not bind to PKG Iβ (Fig. 5B) in line with the modeling data.
FIGURE 5.
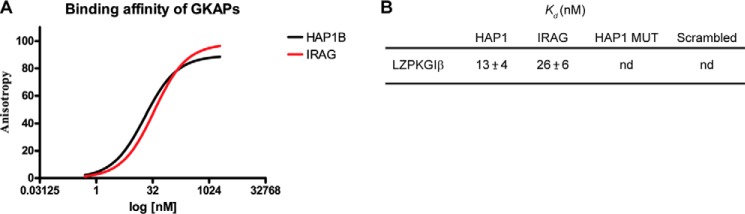
A, binding affinity of HAP1 peptide (black) mimicking its proposed anchoring domain to the leucine zipper of PKG Iβ by fluorescence anisotropy. IRAG peptide binding to PKG Iβ was used as a positive control. The peptides were tagged with 5-carboxytetramethylrhodamine with excitation at 535 nm and emission at 580 nm. B, Kd values and relative standard deviations (n = 3). MUT, mutant.
After establishment of the putative anchoring domain in vitro, we sought to verify that the HAP1 region from residues 556 to 580 is essential for the binding to PKG Iβ also in a cellular context. Following an approach similar to that of Casteel et al. (22) determining the residues involved in the binding of IRAG and TFII-I to PKG Iβ, we decided to create a double mutant of the HAP1 construct to show that the interaction of HAP1 with the N terminus of PKG Iβ relies on particular electrostatic interactions. Therefore, we mutated two consecutive basic residues, K567A and R568A, in the HAP1 anchoring domain. Also a truncated version of HAP1 lacking the C terminus (amino acids 157–386) was used. PKG Iβ was co-expressed either with HAP1 (control), mutated HAP1 (K567A and R568A), or truncated HAP1 in COS-7 cells. Subsequently, a cAMP pulldown was performed (Fig. 6). The double mutated HAP1 and the truncated version of HAP1 did not bind to PKG Iβ, further establishing that the basic amino acid stretch located at the C terminus of HAP1 is crucial for binding to PKG (Fig. 6). All these data combined led us to the conclusion that HAP1 is a novel genuine GKAP with the same specificity and docking sites as the related GKAPs IRAG and TFII-I.
FIGURE 6.

PKG Iβ binds exclusively to HAP1 harboring the predicted anchoring domain. Western blot analysis of lysates and proteins pulled down with the 8-AHA-linked cAMP-agarose beads in COS-7 cells co-expressing PKG Iβ-GFP plasmid with (i) the HAP1-DSRed construct (Control), (ii) the HAP1-DSRed bearing the double mutation K567A/R568A, and (iii) the C-terminal truncated HAP1-DSRed construct (HAP1 amino acids 157–386) is shown. Pulldowns also were performed in non-transfected cells and cells transfected with only PKG Iβ or HAP1 as negative controls.
DISCUSSION
Although there is clear proof that they do exist, evidence for protein scaffolds involved in spatiotemporal cGMP/PKG I signaling is still scarce. Here, by using an efficient competitive chemical proteomics approach, we identified a new GKAP expressed in mammalian (rat) lung tissue. The affinity capture method used is based on cAMP- and cGMP-coupled resins that were shown previously to enrich both PKA and PKG and their respective interactors. This is caused by the high concentration of the immobilized cyclic nucleotide (6 mm) on the agarose beads needed to achieve efficient pulldown results (28). Such concentrations exceed the physiological cyclic nucleotide concentration by at least 3 orders of magnitude (48) as well as the Ka for both PKA and PKG (30). Therefore, here we set out to use a specific elution protocol with free cyclic nucleotides to selectively isolate the interactome of PKG. After optimization of the protocol in HEK293 cells, we were able to extend it to rat tissue and enrich for and identify HAP1 as a putative GKAP along with the known GKAP IRAG.
The few described GKAPs are often found to interact selectively with one of the two isoforms of PKG I (12, 19, 21, 49), and the specificity depends on the charge distribution at the PKG I N terminus. Indeed, Casteel et al. (22) investigated in detail how PKG Iβ interacts with its GKAPs TFII-I and IRAG, showing that the presence of the negatively charged residues (Asp-26, Glu-27, Glu-29, and Glu-31) on the PKG Iβ N terminus is crucial for the interaction with the positively charged residues on the GKAPs. The N-terminal domain of PKG Iα contains a highly charged sequence with basic residues with the consequent different overall charge distribution and topology when compared with the PKG Iβ isoform. Still, the positively charged residues on PKG Iα (Lys-37 and Lys-39) in the LZ domain also are essential for the formation of the complex with the PKG Iα-specific GKAP MYPT1 (13).
These differences between the two isoforms with the opposite charge state distribution may be the origin of the specificity of GKAPs for a single PKG I isoform. An additional proof of the relative importance of the charge distribution on the LZ domain of PKG determining the interaction with GKAPs has been published recently (23). PKG II shows a completely different amino acid sequence and charge distribution when compared with the PKG I isoforms, and the crystal structure of the PKG II leucine zipper-Rab11b complex shows that PKG II binds to Rab11b mainly through van der Waals forces instead of electrostatic interactions.
Here we demonstrated that HAP1, similarly to the previously discovered GKAPs, is selective for a single isoform, PKG Iβ. Using molecular modeling, we hypothesized how the negatively charged residues on PKG Iβ may interact with a positively charged sequence in the known GKAPs (IRAG and TFII-I) to accommodate the binding. With these data as a starting point and the crystal structure of the PKG Iβ N-terminal leucine zipper (40), we used HADDOCK to perform docking studies with the HAP1 binding sequence and compared it with the two known PKG Iβ-specific GKAPs. After overlaying the three docking studies, we propose that TFII-I binds backward in comparison with IRAG and HAP1. This is further supported by the alignment of TFII-I with IRAG and HAP1 where there is much more consensus for the reverse TFII-I sequence (Fig. 3A). Our modeling results reveal that the interactions of each GKAP with PKG Iβ take place in a similar manner. Indeed, the Glu-29, Asp-33, and Asp-36 on one PKG I monomer interact with Gln-572 and Lys-573 on HAP1 (Fig. 4B); with Arg-566, Lys-569, and Arg-571 on TFII-I (Fig. 4C); and with Arg-174 and Ser-176 on IRAG (Fig. 4D).
The presence and order of these basic residues in this region of HAP1 structurally align with those present in the PKG Iβ binding domains of IRAG and TFII-I (Figs. 3A and 4). A quite surprising result was that neither of the GKAP anchoring sequences displayed a full helix as each of them contains a small knick. Indeed, the helical potential of each GKAP obtained from the secondary structure prediction (38) decreases radically after the initial KR/RR sequence (Fig. 4E). We hypothesize that, besides the change in charge-bearing amino acids, the mutations we made onto the HAP1 sequence may increase the helical potential of the sequence stretch, which would then further disturb the binding of the GKAPs to PKG. Through the double mutation of Lys-567 and Arg-568 to alanine or the deletion of the HAP1 C terminus, we showed that this region is essential for binding to PKG Iβ.
In summary, the here presented chemical proteomics-mass spectrometry-based approach is a valid method for the identification of novel PKG anchoring proteins directly in any cell or tissue lysate. Defining HAP1 as a novel GKAP, anchoring specifically to the cGMP-dependent protein kinase isoform Iβ, provides further proof for the fact that PKG spatiotemporal signaling is, in analogy to that for its sister kinase PKA, principally controlled by a wide family of known and yet to be discovered protein kinase G anchoring proteins.
Supplementary Material
Acknowledgments
We thank Prof. Dr. Robert Feil (Universität Tübingen, Tübingen, Germany) for providing the PKG Iα and PKG Iβ constructs. We also thank Dr. Edward Schmidt (Montana State University, Bozeman, MT) for supplying the HAP1 constructs and Dr. Choel Kim (Baylor College of Medicine, Houston, TX) for donating the LZPKG Iβ.
Part of this work was performed within the framework of PRIME-XS Project Grant 262067 funded by the European Union 7th Framework Program and the Netherlands Organization for Scientific Research (NWO)-supported large scale proteomics facility Proteins@Work (Project 184.032.201) embedded in the Netherlands Proteomics Centre.

This article contains supplemental Table 1.
- AKAP
- protein kinase A anchoring protein
- GKAP
- PKG anchoring protein
- HAP1
- Huntingtin-associated protein 1
- PKA-R
- PKA regulatory subunit dimer
- IRAG
- inositol 1,4,5-trisphosphate receptor-associated PKG substrate
- TF
- transcription factor
- 8-AHA
- 8-(6-aminohexyl)aminoadenosine
- PSM
- peptide-spectrum match
- LZ
- leucine zipper.
REFERENCES
- 1. Welch E. J., Jones B. W., Scott J. D. (2010) Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interv. 10, 86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stangherlin A., Zaccolo M. (2011) Local termination of 3′–5′-cyclic adenosine monophosphate signals: the role of A kinase anchoring protein-tethered phosphodiesterases. J. Cardiovasc. Pharmacol. 58, 345–353 [DOI] [PubMed] [Google Scholar]
- 3. Wong W., Scott J. D. (2004) AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5, 959–970 [DOI] [PubMed] [Google Scholar]
- 4. Skroblin P., Grossmann S., Schäfer G., Rosenthal W., Klussmann E. (2010) Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 283, 235–330 [DOI] [PubMed] [Google Scholar]
- 5. Carr D. W., Stofko-Hahn R. E., Fraser I. D., Bishop S. M., Acott T. S., Brennan R. G., Scott J. D. (1991) Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 266, 14188–14192 [PubMed] [Google Scholar]
- 6. Kim C., Xuong N. H., Taylor S. S. (2005) Crystal structure of a complex between the catalytic and regulatory (RIα) subunits of PKA. Science 307, 690–696 [DOI] [PubMed] [Google Scholar]
- 7. Kinderman F. S., Kim C., von Daake S., Ma Y., Pham B. Q., Spraggon G., Xuong N. H., Jennings P. A., Taylor S. S. (2006) A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell 24, 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Francis S. H., Corbin J. D. (1999) Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 36, 275–328 [DOI] [PubMed] [Google Scholar]
- 9. Wernet W., Flockerzi V., Hofmann F. (1989) The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 251, 191–196 [DOI] [PubMed] [Google Scholar]
- 10. Wolfe L., Francis S. H., Corbin J. D. (1989) Properties of a cGMP-dependent monomeric protein kinase from bovine aorta. J. Biol. Chem. 264, 4157–4162 [PubMed] [Google Scholar]
- 11. de Jonge H. R. (1981) Cyclic GMP-dependent protein kinase in intestinal brushborders. Adv. Cyclic Nucleotide Res. 14, 315–333 [PubMed] [Google Scholar]
- 12. Schlossmann J., Ammendola A., Ashman K., Zong X., Huber A., Neubauer G., Wang G. X., Allescher H. D., Korth M., Wilm M., Hofmann F., Ruth P. (2000) Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 404, 197–201 [DOI] [PubMed] [Google Scholar]
- 13. Sharma A. K., Zhou G. P., Kupferman J., Surks H. K., Christensen E. N., Chou J. J., Mendelsohn M. E., Rigby A. C. (2008) Probing the interaction between the coiled coil leucine zipper of cGMP-dependent protein kinase Iα and the C terminus of the myosin binding subunit of the myosin light chain phosphatase. J. Biol. Chem. 283, 32860–32869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richie-Jannetta R., Francis S. H., Corbin J. D. (2003) Dimerization of cGMP-dependent protein kinase Iβ is mediated by an extensive amino-terminal leucine zipper motif, and dimerization modulates enzyme function. J. Biol. Chem. 278, 50070–50079 [DOI] [PubMed] [Google Scholar]
- 15. French P. J., Bijman J., Edixhoven M., Vaandrager A. B., Scholte B. J., Lohmann S. M., Nairn A. C., de Jonge H. R. (1995) Isotype-specific activation of cystic fibrosis transmembrane conductance regulator-chloride channels by cGMP-dependent protein kinase II. J. Biol. Chem. 270, 26626–26631 [DOI] [PubMed] [Google Scholar]
- 16. Yuasa K., Yamagami S., Nagahama M., Tsuji A. (2008) Trafficking of cGMP-dependent protein kinase II via interaction with Rab11. Biochem. Biophys. Res. Commun. 374, 522–526 [DOI] [PubMed] [Google Scholar]
- 17. Gold M. G., Lygren B., Dokurno P., Hoshi N., McConnachie G., Taskén K., Carlson C. R., Scott J. D., Barford D. (2006) Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell 24, 383–395 [DOI] [PubMed] [Google Scholar]
- 18. Kovanich D., van der Heyden M. A., Aye T. T., van Veen T. A., Heck A. J., Scholten A. (2010) Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. Chembiochem 11, 963–971 [DOI] [PubMed] [Google Scholar]
- 19. Vo N. K., Gettemy J. M., Coghlan V. M. (1998) Identification of cGMP-dependent protein kinase anchoring proteins (GKAPs). Biochem. Biophys. Res. Commun. 246, 831–835 [DOI] [PubMed] [Google Scholar]
- 20. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Iα. Science 286, 1583–1587 [DOI] [PubMed] [Google Scholar]
- 21. Yuasa K., Omori K., Yanaka N. (2000) Binding and phosphorylation of a novel male germ cell-specific cGMP-dependent protein kinase-anchoring protein by cGMP-dependent protein kinase Iα. J. Biol. Chem. 275, 4897–4905 [DOI] [PubMed] [Google Scholar]
- 22. Casteel D. E., Boss G. R., Pilz R. B. (2005) Identification of the interface between cGMP-dependent protein kinase Iβ and its interaction partners TFII-I and IRAG reveals a common interaction motif. J. Biol. Chem. 280, 38211–38218 [DOI] [PubMed] [Google Scholar]
- 23. Reger A. S., Yang M. P., Koide-Yoshida S., Guo E., Mehta S., Yuasa K., Liu A., Casteel D. E., Kim C. (2014) Crystal structure of the cGMP-dependent protein kinase II leucine zipper and Rab11b protein complex reveals molecular details of G-kinase-specific interactions. J. Biol. Chem. 289, 25393–25403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lohmann S. M., Walter U., Miller P. E., Greengard P., De Camilli P. (1981) Immunohistochemical localization of cyclic GMP-dependent protein kinase in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 78, 653–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alto N. M., Soderling J., Scott J. D. (2002) Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 158, 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bantscheff M., Scholten A., Heck A. J. (2009) Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov. Today 14, 1021–1029 [DOI] [PubMed] [Google Scholar]
- 27. Rix U., Superti-Furga G. (2009) Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 5, 616–624 [DOI] [PubMed] [Google Scholar]
- 28. Scholten A., Poh M. K., van Veen T. A., van Breukelen B., Vos M. A., Heck A. J. (2006) Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J. Proteome Res. 5, 1435–1447 [DOI] [PubMed] [Google Scholar]
- 29. Burgers P. P., Ma Y., Margarucci L., Mackey M., van der Heyden M. A., Ellisman M., Scholten A., Taylor S. S., Heck A. J. (2012) A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J. Biol. Chem. 287, 43789–43797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X. B., Christensen A. E., Schwede F., Genieser H. G., Bos J. L., Doskeland S. O., Beavo J. A., Butt E. (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278 [DOI] [PubMed] [Google Scholar]
- 31. Pelligrino D. A., Wang Q. (1998) Cyclic nucleotide crosstalk and the regulation of cerebral vasodilation. Prog. Neurobiol. 56, 1–18 [DOI] [PubMed] [Google Scholar]
- 32. Shevchenko A., Jensen O. N., Podtelejnikov A. V., Sagliocco F., Wilm M., Vorm O., Mortensen P., Shevchenko A., Boucherie H., Mann M. (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. U.S.A. 93, 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 34. Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Ríos D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N. H., Chavali G., Chen C., del-Toro N., Duesbury M., Dumousseau M., Galeota E., Hinz U., Iannuccelli M., Jagannathan S., Jimenez R., Khadake J., Lagreid A., Licata L., Lovering R. C., Meldal B., Melidoni A. N., Milagros M., Peluso D., Perfetto L., Porras P., Raghunath A., Ricard-Blum S., Roechert B., Stutz A., Tognolli M., van Roey K., Cesareni G., Hermjakob H. (2014) The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feil R. (2002) Functional reconstitution of vascular smooth muscle cells with cGMP-dependent protein kinase I isoforms. Circ. Res. 90, 1080–1086 [DOI] [PubMed] [Google Scholar]
- 37. Prigge J. R., Schmidt E. E. (2007) HAP1 can sequester a subset of TBP in cytoplasmic inclusions via specific interaction with the conserved TBP(CORE). BMC Mol. Biol. 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petersen B., Petersen T. N., Andersen P., Nielsen M., Lundegaard C. (2009) A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 40. Casteel D. E., Smith-Nguyen E. V., Sankaran B., Roh S. H., Pilz R. B., Kim C. (2010) A crystal structure of the cyclic GMP-dependent protein kinase Iβ dimerization/docking domain reveals molecular details of isoform-specific anchoring. J. Biol. Chem. 285, 32684–32688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aye T. T., Soni S., van Veen T. A., van der Heyden M. A., Cappadona S., Varro A., de Weger R. A., de Jonge N., Vos M. A., Heck A. J., Scholten A. (2012) Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 52, 511–518 [DOI] [PubMed] [Google Scholar]
- 42. Scholten A., van Veen T. A., Vos M. A., Heck A. J. (2007) Diversity of cAMP-dependent protein kinase isoforms and their anchoring proteins in mouse ventricular tissue. J. Proteome Res. 6, 1705–1717 [DOI] [PubMed] [Google Scholar]
- 43. Wong J. W., McRedmond J. P., Cagney G. (2009) Activity profiling of platelets by chemical proteomics. Proteomics 9, 40–50 [DOI] [PubMed] [Google Scholar]
- 44. Tang T. S., Tu H., Chan E. Y., Maximov A., Wang Z., Wellington C. L., Hayden M. R., Bezprozvanny I. (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5)triphosphate receptor type 1. Neuron 39, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ammendola A., Geiselhöringer A., Hofmann F., Schlossmann J. (2001) Molecular determinants of the interaction between the inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Iβ. J. Biol. Chem. 276, 24153–24159 [DOI] [PubMed] [Google Scholar]
- 46. Hundsrucker C., Skroblin P., Christian F., Zenn H. M., Popara V., Joshi M., Eichhorst J., Wiesner B., Herberg F. W., Reif B., Rosenthal W., Klussmann E. (2010) Glycogen synthase kinase 3β interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 285, 5507–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Burgers P. P., van der Heyden M. A., Kok B., Heck A. J., Scholten A. (2015) A systematic evaluation of protein kinase A-A-kinase anchoring protein interaction motifs. Biochemistry 54, 11–21 [DOI] [PubMed] [Google Scholar]
- 48. Eigenthaler M., Nolte C., Halbrügge M., Walter U. (1992) Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur. J. Biochem. 205, 471–481 [DOI] [PubMed] [Google Scholar]
- 49. Casteel D. E., Zhuang S., Gudi T., Tang J., Vuica M., Desiderio S., Pilz R. B. (2002) cGMP-dependent protein kinase Iβ physically and functionally interacts with the transcriptional regulator TFII-I. J. Biol. Chem. 277, 32003–32014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.