

An efficient system was developed to generate megakaryocytes (MKs) from human induced pluripotent stem cells (hiPSCs) under a feeder-free and xeno-free condition, in which all animal-derived products were eliminated. Additional maturation and investigation of hiPSC-derived MKs should provide insights into MK biology and lead to the generation of large numbers of platelets ex vivo.
Keywords: Megakaryocytes, Human induced pluripotent stem cells, Romiplostim, Thrombopoietin, Plasbumin
Abstract
Megakaryocytes (MKs) are rare hematopoietic cells in the adult bone marrow and produce platelets that are critical to vascular hemostasis and wound healing. Ex vivo generation of MKs from human induced pluripotent stem cells (hiPSCs) provides a renewable cell source of platelets for treating thrombocytopenic patients and allows a better understanding of MK/platelet biology. The key requirements in this approach include developing a robust and consistent method to produce functional progeny cells, such as MKs from hiPSCs, and minimizing the risk and variation from the animal-derived products in cell cultures. In this study, we developed an efficient system to generate MKs from hiPSCs under a feeder-free and xeno-free condition, in which all animal-derived products were eliminated. Several crucial reagents were evaluated and replaced with Food and Drug Administration-approved pharmacological reagents, including romiplostim (Nplate, a thrombopoietin analog), oprelvekin (recombinant interleukin-11), and Plasbumin (human albumin). We used this method to induce MK generation from hiPSCs derived from 23 individuals in two steps: generation of CD34+CD45+ hematopoietic progenitor cells (HPCs) for 14 days; and generation and expansion of CD41+CD42a+ MKs from HPCs for an additional 5 days. After 19 days, we observed abundant CD41+CD42a+ MKs that also expressed the MK markers CD42b and CD61 and displayed polyploidy (≥16% of derived cells with DNA contents >4N). Transcriptome analysis by RNA sequencing revealed that megakaryocytic-related genes were highly expressed. Additional maturation and investigation of hiPSC-derived MKs should provide insights into MK biology and lead to the generation of large numbers of platelets ex vivo.
Introduction
The lifespan of enucleated platelets in the blood stream is about 10 days; therefore, continual production of platelets from mature megakaryocytes (MKs) is required for normal hemostasis in a healthy human adult [1, 2]. Platelet transfusion is an effective approach for preventing or treating bleeding and is essential for cancer treatment by radiation and chemotherapy and for treatments involving hematopoietic stem cell (HSC) and bone marrow (BM) transplantation [3]. During embryonic development, immature MKs are produced in the yolk sac [4]. The adult MKs are derived from HSCs in BM, where MKs are the largest (50–100 μm) and one of the rarest (∼0.01%) cells. A unique characteristic of MKs is endomitosis, a unique process of DNA/chromosome replication without cellular mitosis, that leads to the formation of large cells with polyploid DNA (ranging from 4N to 128N, compared with 2N–4N in diploid cells). Platelets are then generated from the cytoplasm of MKs by the demarcation membrane system, in which platelet-specific granules and organelle contents in cytoplasm are packaged and remodeled as proplatelets and then preplatelets before releasing into the circulation as platelets [5]. Enucleated platelets retain surface molecules on the cell membrane such as human leukocyte antigen (HLA) and MK-specific markers, including CD41/CD61 and CD42a/CD42b complexes. Thrombopoietin (TPO), which activates its receptor c-Mpl to stimulate downstream signaling pathways [6–9], is the primary hematopoietic factor to induce HSC and hematopoietic progenitor cell (HPC) differentiation to MKs and platelets [10]. In addition to its role in MK generation and maturation, TPO-Mpl signaling regulates HSC and HPC expansion [11]. Other regulators, in addition to TPO-Mpl signaling, however, participate in platelet generation, because mice lacking either c-Mpl or TPO still have the potential to produce platelets at a lower level [10]. Some cytokines, including interleukin (IL)-3, IL-6, IL-11, stromal cell-derived factor 1 (SDF-1), and fibroblast growth factor 4 (FGF-4), have been identified to be involved in MK generation [12, 13].
The massive production of MKs and platelets ex vivo would be extremely valuable for treating thrombocytopenia; however, currently, two challenges exist. First, MKs, which are present in BM but absent in the peripheral blood (PB), must be differentiated from HSCs or HPCs. The current inability to expand HSCs ex vivo, especially in a clinically compliant setting, has hindered the capacity to massively expand MKs, which have limited proliferation potential. Second, platelet production ex vivo from MKs remains inefficient, and the lack of large numbers of MKs as starting materials has made this approach more difficult. In the past decade, human pluripotent stem cells, including embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs), which are capable of self-renewal and differentiation into all somatic cell types became an attractive cell source. The generation of MKs and platelets from human pluripotent stem cells [14–19], especially from HLA-matched hiPSCs, should provide a renewable cell source to treat patients with thrombocytopenia. Although with tremendous promise, platelet generation in vitro from hESCs or other reprogrammed cell sources is currently inefficient, even in the presence of stromal cells such as 10T1/2 and OP9 mouse cell lines, which were commonly used in early studies [15, 16, 19]. Although mature MKs have limited expansion capabilities, the lineage-committed MK precursors, megakaryoblasts, are proliferative. The generation of robust expandable megakaryoblasts would provide an important cell source of platelets for research and clinical application. A recent study demonstrated that coordinative ectopic expression of c-MYC, BMI1, and BCL-XL genes in hiPSCs produces expandable immature MKs in a procedure that includes coculture with the mouse 10T1/2 stromal cells. Turning off the overexpression of these genes in the immature MKs results in the production of platelets [20]. However, it would be highly desirable if we could generate large numbers of MKs from hiPSCs in the absence of mouse stromal cells and without the need for manipulation of oncogene expression.
A recent study demonstrated that the generation of MKs from HPCs is achievable under feeder-free conditions [18]. However, that study only examined hESCs and used animal-derived products such as bovine serum albumin (BSA) [18, 21]. These xenogeneic and undefined reagents often cause low reproducibility and conflict with the strict requirements of clinical or preclinical applications [22, 23]. To search for a robust and efficient culture condition to generate expandable MK progenitors from hiPSCs, we developed a serum-free and feeder-free system of hiPSC differentiation to MKs with a high level of reproducibility. In this two-step differentiation system [21, 24–26] which first generates CD45+CD34+ definitive HPCs followed by MK differentiation, we were able to effectively generate a cell population enriched for CD41+CD42a+ megakaryoblasts. Moreover, we also used Food and Drug Administration (FDA)-approved pharmacological agents to replace TPO and BSA in the culture medium, an important factor for future clinical applications. Forty-five hiPSC lines from 23 individuals were evaluated in our system, resulting in highly reproducible outcomes.
Materials and Methods
Maintenance and Expansion of hiPSC Lines
Human iPSC lines, BC1 and E2, derived from human adult BM hematopoietic cells and mesenchymal stem cells (MSCs), respectively [27–29], were adapted to feeder-free conditions using the E8 medium (Essential 8 medium commercialized by Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) [30]. The cells were maintained in an undifferentiated state and routinely passaged as small clumps using the EDTA method or as single cells after enzymatic digestion by Accutase (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com). To enhance single cell survival, 10 μM ROCK inhibitor Y27632 (Stemgent, Cambridge, MA, http://www.stemgent.com) was added in the medium for the first 24 hours after seeding.
Other hiPSC lines were derived from peripheral blood mononuclear cells (MNCs) using nonintegrating episomal vectors, as previously described [26–28, 31–33]. After establishment, they were all expanded in the Essential 8 medium on either Matrigel (1:30; BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) or vitronectin (5 μg/cm2, Life Technologies).
Generation of MKs and Platelets From hiPSCs
Human iPSCs were differentiated into definitive CD34+CD45+ HPCs, using the “spin-embryoid body” (spin-EB) method in feeder- and serum-free conditions modified from previously described protocols [24, 26, 34]. Single hiPSCs were suspended in serum-free medium (SFM) (supplemental online Table 1). In brief, the SFM contained components that included insulin-transferrin-selenium or BSA. We added 10 μM Y27632, 10 ng/ml recombinant human FGF-2 (R&D Systems, Minneapolis, MN, http://www.rndsystems.com) and 10 ng/ml bone morphogenetic protein 4 (BMP4; R&D Systems). Cells (3,000) were seeded into nontreated round-bottom 96-well plates (Corning Costar, Acton, MA, http://www.corning.com/lifesciences) in 50 μl per well and centrifuged at 300g for 5 minutes. From day 2 to day 14, the cells were cultured in SFM containing FGF-2 (10 ng/ml), BMP4 (10 ng/ml), stem cell factor (SCF) (50 ng/ml; R&D Systems), and vascular endothelial growth factor A (VEGF-A) (10 ng/ml; PeproTech, Rocky Hill, NJ, http://www.peprotech.com). TPO (20 ng/ml; PeproTech) or romiplostim (Nplate, 20 ng/ml; Amgen, Thousand Oaks, CA, http://www.amgen.com) was added to the medium at day 11. Nplate is a U.S. FDA-approved TPO analog [35]. On day 14, the suspended cells were harvested and then filtered through 100-μm cell strainers (BD Biosciences) to remove the EBs. The harvested single cells were seeded into 6-well plates (1 × 106 cells per well) for MK culture in SFM containing SCF (20 ng/ml), oprelvekin (IL-11, 10 ng/ml; Pfizer, New York, NY, http://www.pfizer.com), and TPO or Nplate (50 ng/ml) for 5 days. We also substituted BSA in the original SFM with FDA-approved human albumin (HuA) made from pooled human venous plasma. One source is Plasbumin (NDC13533-684-20; Grifols, Barcelona, Spain, http://www.grifols.com).
To generate platelets from MKs, we continued MK culture for an additional 18 days in SFM containing oprelvekin and Nplate. The platelet-like particles (PLPs) were labeled with calcein AM (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) for live cell staining and analyzed using flow cytometry for CD41+CD42a+ platelets compared with human peripheral blood (PB) platelets. The activation of hiPSC-PLP was determined by adenosine diphosphate (ADP) (20 μM; Sigma-Aldrich) stimulation, and then CD62P (P-selectin) expression was analyzed using flow cytometry [36, 37].
Flow Cytometry and Fluorescence-Activated Cell Sorting
On days 14 and 19, single cells from EB culture or MK culture were harvested and labeled with anti-human CD34-PE (Miltenyi Biotec, San Diego, CA, http://www.miltenyibiotec.com), CD45-Alexa Fluor 700 (eBioscience, San Diego, CA, http://www.ebioscience.com), CD41-APC (BD Biosciences), CD42-efluor 450 (eBioscience), CD42b-fluorescein isothiocyanate (FITC) (eBioscience), CD62P-PE (BD Biosciences), and CD61-FITC (eBioscience). All samples were analyzed using the FACSCalibur or LSRII flow cytometer (BD Biosciences). Ig isotype controls were used as the control for flow cytometry in each experiment. A representative view of the side scatter/forward scatter dot plot and the Ig control for flow cytometry of the day-14 cells is shown in supplemental online Figure 1A.
RNA-Sequencing Analysis of Transcriptomes
Total RNA was extracted from iPSCs or derived MKs, after which cDNA was prepared from polyadenylated mRNA primed by oligo(dT). The cDNAs are end-polished, A-tailed, and ligated to Illumina adapters (Illumina, San Diego, CA, http://www.illumina.com) and checked for quality at the Genetic Resources Core Facility (Johns Hopkins Medicine, Baltimore, MD, http://www.hopkinsmedicine.com). Sequencing was then performed using an Illumina HS2500 instrument, achieving about 100 million independent reads per sample (paired-end reads of 100 nucleotides). The short read output files were aligned to the reference genome using the Bowtie/TopHat software pipeline (John Hopkins University, Baltimore, MD, http://www.ccb.jhu.edu/software/tophat/). Full-length transcript reads were assembled and quantified using Cufflinks (Johns Hopkins University). Twelve hiPSC lines derived from 6 individuals (n = 12) yielded a median of 103.8 million reads (range 50.9–140.3), and the iPSC-derived MK samples (n = 12) yielded a median of 71 million reads (range 50.6–98.2). For most of the samples, >99% reads passed the quality control examination. A median of 81% reads (range 59%–89%) mapped to the reference genome. The expression values were calculated for each sample according to the number of fragments per kilobase of exon per million fragments mapped (FPKM). The FPKM for the highest expressed gene could reach 4,000–5,000.
Differential gene expression was determined using Cuffmerge, a “meta-assembler,” which merges transfrags together parsimoniously. To calculate the differential gene expression and test the statistical significance of each observed change in expression between the iPSCs and derived MKs, we used the program Cuffdiff.
DNA Content Analysis
For polyploidy analysis, the cells at day 19 were fixed in 70% ethanol for at least 2 hours at room temperature or overnight at 4°C. The cells were then washed once in phosphate-buffered saline, treated with 100 μg/ml RNase A (Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com) for 30 minutes, and stained with 20 μg/ml propidium iodide (PI; Sigma-Aldrich). For the double staining of CD41-FITC and 4′,6-diamidino-2-phenylindole (DAPI), the day-19 cells were labeled with CD41-FITC conjugate (BD Biosciences). After washing, the cells were resuspended in paraformaldehyde (2%) and incubated at room temperature for 10 minutes before the addition of DAPI (1 μg/ml) for another 10 minutes. After washing, the cellular DNA content was analyzed using an LSRII flow cytometer.
Colony-Forming Unit Assay for MKs
The collagen-based MegaCult-C Kit (STEMCELL Technologies, Vancouver, BC, Canada, http://www.stemcell.com) was used for the colony-forming unit (CFU)-MK colony-forming assays. Suspended cells (15,000 cells) at days 14 and 19 were seeded onto 35-mm dishes. After 10 days of semiliquid culture, the collagen gels were dehydrated, fixed, and stained with anti-CD41. The CFU-MK colonies were scored according to the standards provided in the MegaCult-C protocol. Colonies with at least 50 cells were counted.
MK Morphological Analysis After Cytospin and Histological Staining
After 19 days of differentiation, single cells (4 × 104 cells per slide) were used for the cytospin (Thermo Fisher Scientific). The Hema 3 kit (Thermo Fisher Scientific) was used as a Wright-Giemsa-like stain for cell morphology analysis.
Statistical Analysis
The results are presented as the mean ± SD, if not otherwise specified. A two-tailed Student’s t test or two-way analysis of variance was performed using GraphPad Prism, version 5, software (GraphPad Software, Inc., La Jolla, CA, http://www.graphpad.com) for comparisons between two groups or multiple groups, respectively. The significance level was set as p < .05, p < .01, or p < .001.
Results
Two-Step hiPSC Differentiation to Megakaryocytic Precursors
Forced aggregation of hiPSCs by spin-EB formation has been used to induce mesodermal differentiation and generate hematopoietic cells, including megakaryocytic progenitors, in the absence of any input stromal cell lines [18, 21, 25, 26]. We modified the spin-EB method to effectively generate HPCs during 14 days of differentiation and reduce the number of required bioactive molecules (Fig. 1A). Single-cell suspension of hiPSCs (3 × 103 cells per well) was dispensed in 96-well plates in the presence of a ROCK inhibitor, Y27632, to improve single-cell survival. BMP4 and FGF-2 were added at the beginning to induce mesoderm, and VEGF and SCF were used starting at day 2 to induce hematopoietic differentiation. We added TPO at day 11 to enhance HPC formation, megakaryocytic lineage commitment, maintenance, and expansion [11]. At day 14, a significant number of hematopoietic cells were released to suspension from EBs (∼2.5 × 104 cells per well). The hematopoietic progenitor and MK markers CD34, CD45, CD41, and CD42a changed during the differentiation process and were closely monitored (Fig. 1A). On day 14, most of the suspension cells were CD45+ hematopoietic cells (∼80%) (Fig. 1B). Approximately 40% of hematopoietic cells were CD34+CD45+ cells, characteristic of definitive HPCs, and about 30% of the hematopoietic cells at day 14 were positive for CD41 (glycoprotein [GP]IIb, encoded by the ITGA2B gene) and CD42a (GPIX, encoded by the GP9 gene), which are the characteristic megakaryocytic markers. To further induce the differentiation to MKs, we harvested the suspended cells on day 14 and subcultured them (1 × 106 cells per well) in 6-well plates in the presence of SCF, IL-11, and TPO for 5 days. At day 19, approximately 55% of the cells were CD41+CD42a+ MKs, and the proportion of CD34+CD45+ cells had decreased to less than 30% (Fig. 1B). The CD41+CD42a+ MKs on day 19 also expressed other MK markers, such as CD61 and CD42b, on the cell surface (supplemental online Fig. 1B). During the 5 days of MK differentiation, the number of cells tripled, and large size cells emerged (Fig. 1C, 1D). We performed a colony-forming unit (CFU)-MK assay for the suspension cells harvested on days 14 and 19 (Fig. 2A). Compared with the cells on day 19, the HPC population in the cells harvested on day 14 contained significantly higher frequencies of CFU-MK (Fig. 2B), suggesting that after the 5-day subculture in the presence of TPO, IL-11, and SCF, the cells lost their progenitor properties. Furthermore, we performed CFU assays to test the progenitor potential for other hematopoietic lineages using total cells or isolated CD34+ cells from the day-14 and day-19 cells. We found that the CFU counts of the other blood cell types, including myeloid and erythroid colonies, had decreased significantly in the day-19 cells compared with the day-14 cells (supplemental online Fig. 2B). The MKs on day 19 possessed a high rate of polyploidy (Fig. 2C). The percentage of DNA ploidy cells with more than 4N was 16% analyzed using PI staining. The >4N population had slightly increased to 19% when the CD41+ cells were analyzed after double staining with CD41 and DAPI (Fig. 2C). Large multinucleated cells were also observed on day 19 (Fig. 2D).
Figure 1.

Generation of MKs derived from human induced pluripotent stem cells (hiPSCs). (A): Schematic diagram for MK generation and expression of CD34, CD45, CD41, and CD42a during hiPSC differentiation. (B): Representative flow cytometry dot plots of suspension cells on day 14 (left) and day 19 (right). The suspension cells expressed MK markers CD41 and CD42a and hematopoietic cell markers CD34 and CD45. Ig isotype controls for CD41, CD42a, CD34, and CD45 were used to determine the background. (C): Phase contrast images of suspension cells on days 14 and 19. The yellow arrows indicate some of the morphologically larger cells on day 19. (D): The cell number increment of suspension cells from day 14 to day 19 in one well of a 6-well plate. Mean ± SD; n = 3. Scale bars = 100 μm. Abbreviations: BMP4, bone morphogenetic protein 4; D, day; EB, erythroid body; EGF2, epidermal growth factor 2; FGF-2, fibroblast growth factor 2; HPCs, hematopoietic progenitor cells; IL, interleukin; MK, megakaryocyte; SCF, stem cell factor; TPO, thrombopoietin.
Figure 2.

Characterization of human induced pluripotent stem cell-derived MKs. (A): Representative images of CFU-MK colonies taken from D14 (upper) and D19 (lower) suspension cells. All the colonies containing at least 50 CD41+ cells were considered CFU-MKs. (B): The number of CFU-MK colonies from 1.5 × 105 isolated CD34+ cells on days 14 and 19. The colonies were counted after 12 days of culture from one 35-mm dish. Mean ± SD; n = 3; ∗∗, p < .01. (C): DNA content analysis by flow cytometry on day 19. Left: The whole population stained by propidium iodide. Right: Double staining using CD41-APC and DAPI, gated on CD41+ population. (D): Wright-Giemsa staining of the suspension cells on day 19. Scale bars = 100 μm. Abbreviations: CFU, colony-forming unit; D, day; MKs, megakaryocytes.
To determine the presence of other hematopoietic cells in MK culture, we analyzed the mixed cell population on day 19 using flow cytometry (supplemental online Fig. 3) and found that approximately 10% of the cells on day 19 were CD235a+ cells. Additional analysis indicated that CD41+CD235a+ cells (MEPs) were more than 8% and that CD41−CD235a+ erythroid cells were less than 2%, suggesting that MEPs were present after 5 days of MK culture (supplemental online Fig. 3A). To determine myelomonocytic cells, we analyzed CD41−CD11b+CD14+ cells and found that within the CD41− subpopulation, fewer than 1% of the cells were CD11b+CD14+ cells, and the proportion of CD11b+CD14− cells and CD11b−CD14+ cells was 9.5% and 2.8%, respectively (supplemental online Fig. 3B). The lymphoid marker, CD45RA, was not expressed in the CD41+ cells or the CD34+ cells (supplemental online Fig. 3B, 3C). To test whether hiPSC-derived MKs can produce platelets, after 5 days of MK culture, we continued culturing the cells for an additional 18 days in a chemically defined media without feeder cells. We were able to obtain CD41+CD42a+ PLPs, based on size gating and the metabolic activity staining using calcein AM to distinguish the live cells from debris. The immunophenotypes of hiPSC-derived PLPs were similar to human platelets from peripheral blood (supplemental online Fig. 4A). To test the function of hiPSC-derived PLPs, we used the platelet agonist, ADP, to stimulate the platelet activation and then measured the expression of CD62P (P-selectin) [36, 37]. Flow cytometric analysis indicated that approximately 5%–6% of the CD41+CD42a+ PLPs expressed CD62P after ADP stimulation, but no CD62P expression on platelet surface was detected without stimulation (supplemental online Fig. 4B).
Enhancement of HPC and MK Generation by an FDA-Approved TPO Analog
To adapt our procedure for clinical applications, we examined whether an FDA-approved TPO analog, romiplostim (Nplate), is sufficient to replace TPO in one or both steps in our MK production protocol. From day 14 to day 19, we tested the dose responses with Nplate versus TPO. The suspension cells harvested on day 14 were subcultured for 5 days in SFM with different concentrations of Nplate or TPO (0–100 ng/ml) in the presence of SCF and IL-11. Our data have demonstrated that Nplate was more potent than TPO at a low protein concentration (20 ng/ml) (Fig. 3A, 3B). The frequency of CD41+CD42a+ MKs was increased in the presence of TPO or Nplate, even at a low concentration (Fig. 3A). To achieve an efficient production of CD41+CD42a+ MKs, we routinely used Nplate at 50 ng/ml. In the presence of Nplate (50 ng/ml) and SCF (20 ng/ml), the effect of IL-11 was modest in maintaining or increasing the CD41+CD42a+ cell numbers and percentages. We also used a clinical-grade form of IL-11 (oprelvekin) during MK differentiation to establish cell culture conditions as clinically compliant as possible (data not shown).
Figure 3.

Replacement of TPO by Nplate during MK differentiation from hiPSCs. (A): CD41+CD42a+ cells on day 19 suspension cells in different culture conditions with various concentrations of TPO or Nplate were identified by flow cytometry. (B): The yield of CD41+CD42a+ cells on day 19 from 30,000 input cells on day 14. Mean ± SD; n = 3; ∗, p < .05 for Nplate and TPO at 25 ng/ml. (C): Representative flow cytometry plots of hematopoietic markers and MK markers suspension cells with TPO or Nplate at concentration of 20 ng/ml from day 11 to day 14. Flow cytometry was performed on day 14. (D): The total CD34+CD45+ and CD41+CD42a+ cells in suspension on day 14 with TPO or Nplate at concentration of 20 ng/ml from day 11 to day 14. Control groups were in differentiation medium in the absence of TPO or Nplate. Mean ± SD; n = 3; ∗, p < .05. Abbreviations: MK, megakaryocyte; Nplate, romiplostim; TPO, thrombopoietin.
We further evaluated the effect of TPO or Nplate on the differentiation in the first step from day 11 to day 14. As shown by flow cytometry at day 14, not only the percentages of CD34+CD45+ HPCs and CD41+CD42a+ MKs, but also the total cell numbers, were increased in the presence of Nplate or TPO compared with the control (Fig. 3C, 3D). These data indicated that both Nplate and TPO promoted the differentiation of hiPSCs to hematopoietic and megakaryocytic lineages and that Nplate was as effective as TPO. Nplate was sufficient to replace TPO in our differentiation condition.
A Complete Xeno-Free Condition Supplemented With Human Albumin
In our original medium formulation for hematopoietic differentiation, which was adapted from BSA polyvinyl alcohol essential lipids medium reported first by Ng et al. [21], BSA is a critical component in a serum-free medium to nourish the cells and induce efficient differentiation (supplemental online Table 1). However, commercial sources of BSA, which are known as Cohn fraction V and enriched in bovine albumin proteins, also contain other proteins and bioactive substances, including undefined amounts of fatty acids, hormones, and metal ions. We have tested several BSA preparations from different sources and selected one product that gave the best results regarding CD45+CD34+ HPC production. However, different lots of the BSA product gave inconsistent results, even with the same hiPSC line, such as BC1 (Fig. 4). “Good” lots (e.g., BSA lots 1 and 5 in Fig. 4B) led to efficient differentiation (Fig. 4A, left), and “bad” lots (e.g., BSA lots 2–4 in Fig. 4B) caused low viability on day 2, which led to failure of EB formation or large cystic EBs with no release of single HPCs (Fig. 4A, right). Thus, we had difficulties in establishing a standard culture condition for the differentiation of multiple hiPSC lines. To achieve consistent hiPSC differentiation to HPCs, we replaced BSA in serum-free medium with Plasbumin, which is a clinic-grade agent enriched for HuA made from serum plasma. We found that Plasbumin from several lots consistently gave significantly higher efficiency and smaller variation during HPC differentiation to generate CD45+CD34+ cells (1.38 ± 0.17 × 106 cells per plate) compared with the BSA fraction preparation (0.44 ± 0.54 × 106 cells per plate; Fig. 4B; mean ± SD, n = 15). Furthermore, Plasbumin can be used to fully replace BSA in support of MK differentiation under a serum-free culture condition, using either recombinant human proteins or human-sourced proteins.
Figure 4.

Replacing BSA with HuA in serum-free medium (SFM) increased the efficiency and consistency of hematopoietic progenitor cell (HPC) differentiation. (A): Representative images of day 2 and day 14 erythroid bodies from failed (left) and successful (right) differentiations using various lots of BSA and HuA preparations in SFM. Scale bars = 100 μm. (B): CD34+CD45+ HPCs generated from one 96-well plate of BC1 using SFM containing either BSA product or HuA of 5 different lot numbers each. Solid dots represent BSA group, and hollow dots represent HuA group. Mean ± SD; n = 15. Abbreviations: BSA, bovine serum albumin; D, day; HuA, human albumin.
To evaluate the robustness and consistency of this feeder- and xeno-free system for MK differentiation from multiple hiPSC lines derived from different individuals, we used our system to induce MK generation from 44 additional hiPSC lines derived from 22 individuals. These hiPSC cell lines were established and expanded either as clonal lines or as two mixed hiPSC pools. Typically, two hiPSC lines (A and B) from one individual were used for the tests of MK differentiation procedure, as described in Figure 1A. After 19 days of differentiation, all iPSC lines showed successful MK generation, and the percentage of CD41+CD42a+ MKs was ranged from 30% (sample P013) to 94% (sample P026; Fig. 5A). To determine whether this variation was caused by the differences between the cell lines derived from different patients or by the inconsistency of the differentiation conditions, we performed linear regression on the 44 hiPSC lines. It showed good linear correlation between the two iPSC lines derived from the same individual (y = [0.98 ± 0.03]x, with the 95% confidence interval shown as dashed lines, Fig. 5B). These data indicated that the variations in the efficiency of MK generation were much smaller between the hiPSCs derived from the same individual than from different individuals. To further examine the generality of the differentiation system for MK production, we also tested it on an iPSC line (E2) that was derived from human MSCs established from adult BM MNCs (supplemental online Fig. 5). Under the improved hematopoietic differentiation condition, comparable hematopoietic cell yields on day 14 after EB differentiation (1.53 ± 0.40 × 106 cells per 96-well plate, n = 2) and a similar percentage of CD34+CD45+ cells were achieved (supplemental online Fig. 5A). These hematopoietic cells harvested from suspension can also commit to the MK lineage, which has an increased CD41+CD42+ expression (supplemental online Fig. 5B) and polyploidy morphology (supplemental online Fig. 5C). Therefore, our differentiation method also allowed efficient MK differentiation from MSC-derived iPSCs comparable to that of the hematopoietic cell-derived iPSCs as shown above.
Figure 5.

Megakaryocyte generation from 44 hiPSC lines from 22 individuals. (A): The two hiPSC lines derived from one individual were labeled as iPSC-A and iPSC-B. The frequency of CD41+CD42a+ cells from each iPSC line were analyzed by flow cytometry on day 19. (B): Statistical analysis with linear regression of iPSC-A and iPSC-B lines. P013 and P026 marked the iPSC lines that gave the lowest and highest frequency of CD41+CD42a+ cells, respectively. Solid line is the linear regression curve of all data points with constraint of the origin point, and the dashed lines indicate the 95% confidence intervals; y = (0.98 ± 0.03)x. Abbreviation: hiPSC, human induced pluripotent stem cell.
Transcriptome Analysis of hiPSC-Derived MKs
To evaluate mRNA transcript profiles of the MKs derived from hiPSCs, 12 hiPSC lines (from 6 subjects, including BC1) were differentiated into MKs under the described culture condition (Fig. 1A). We analyzed mRNA transcript profiles using whole transcriptome RNA sequencing of both hiPSCs and hiPSC-derived MKs (iPSC-MKs). We found that 14,414 genes were expressed in undifferentiated hiPSCs and 14,300 genes were expressed in the iPSC-MKs. The clustering analysis clearly showed that all the iPSCs displayed a common mRNA expression profile, different from the one shared by iPSC-MKs (Fig. 6). The hiPSC lines derived from the same individuals tended to cluster closer with each other. The iPSC-MK samples also showed this trend (e.g., P002A and P002B), although this was not always the case.
Figure 6.

Dendrogram illustrating the clustering of hiPSCs and iPSC-derived MKs. Twelve hiPSC lines from 7 individuals (each subject has 2 hiPSC lines, except for BC1, with 1 line, and P003, with 3 lines) were differentiated to MKs (iPSC-MKs). The profiles of mRNA transcripts by whole transcriptome RNA sequencing of both hiPSCs and hiPSC-derived MKs (iPSC-MKs) are presented by the dendrogram to demonstrate clustering of gene expression of iPSCs and iPSC-MKs. Abbreviations: hiPSCs, human induced pluripotent stem cells; MKs, megakaryocytes.
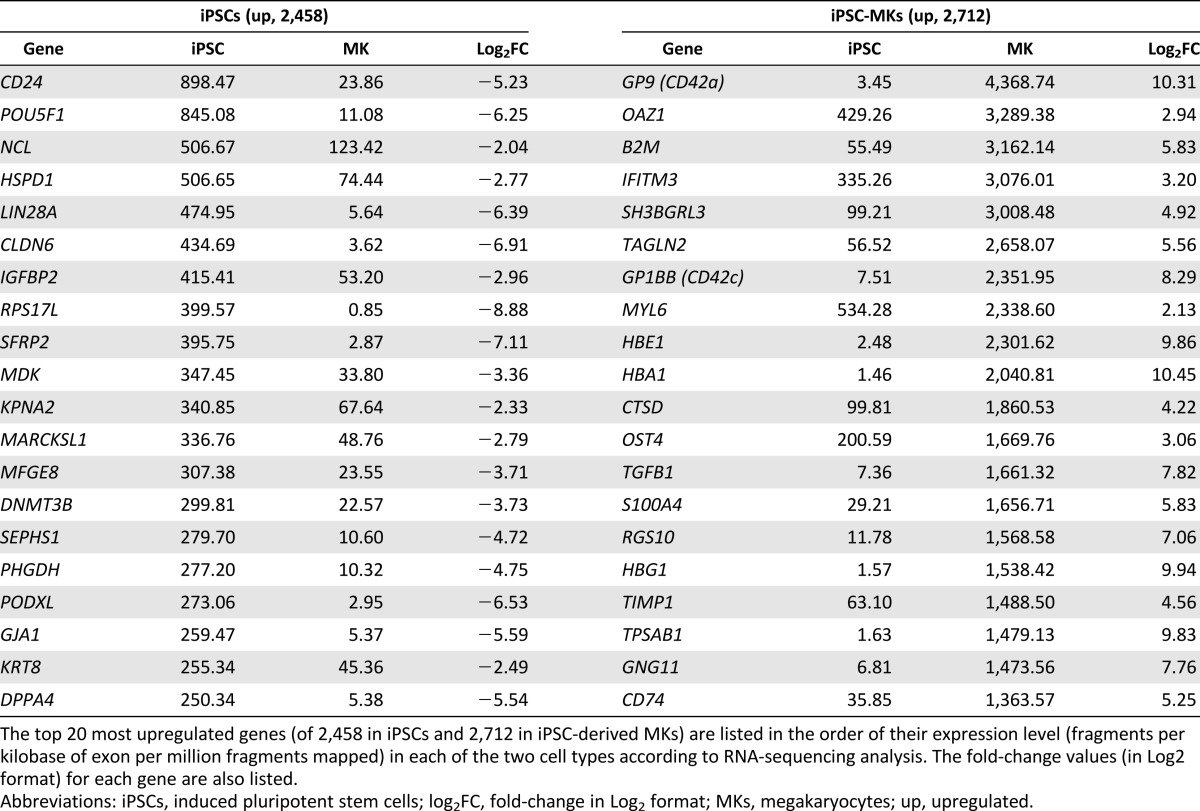
We next analyzed mRNAs that were differentially expressed in the 12 hiPSC-MK culture compared with those in the parental hiPSCs before differentiation. A slightly larger number of genes were upregulated (2,712) than downregulated (2,458) in the hiPSC-MKs. The most differentially expressed 20 genes in hiPSCs and hiPSC-MKs are listed in Table 1. POU5F1 (OCT4), LIN28A, DNMT3B, PODXL, and DDPA4 genes were highly expressed in hiPSCs, as expected. MK-specific genes, GP9 (CD42a) and GP1BB (CD42c) were found in the top 20 most differentially expressed genes in the hiPSC-MKs, followed by ITGB3 (CD61), ITGA2B (CD41), and PF4 in the next 20 genes (data not shown). This global gene expression analysis demonstrated a fundamental biological shift in the gene expression patterns accompanying the differentiation of hiPSCs into MKs.
Table 1.
Top 20 most differentially expressed genes in iPSCs and iPSC-MKs
Discussion
In our study, we developed a two-step differentiation system for efficient MK production under a feeder-free and serum-free condition: HPC generation from hiPSCs and MK generation from HPCs. In the first step of HPC generation, we used a spin-EB method to induce hiPSC differentiation, based on a pioneering study from Ng et al. [21] in 2005. We, and others, have since improved this method [24–26]. For example, Yahata et al. [24] added polyvinyl alcohol (PVA) in the serum-free medium to enhance the aggregation of hESCs and formation of EBs by centrifugation. We found that 3,000–4,000 cells in each 96-well plate were sufficient to form an EB for most hiPSC lines without PVA. By day 14, approximately 2.5 × 104 hematopoietic cells were released into medium surrounding EBs in each well. We observed that the addition of PVA into hiPSCs generated larger EBs but somehow reduced the release of hematopoietic cells, resulting in a low yield of HPCs at day 14 (supplemental online Fig. 6). Therefore, we omitted PVA in the first stage of hiPSC differentiation toward HPC formation.
Although our study was focused on the generation of MKs from hiPSCs, we demonstrated that hiPSC-MKs were capable of generating platelets or PLPs, although the numbers of PLPs were still low (supplemental online Fig. 4). The low efficiency of the generation of hiPSC-PLP in our study likely resulted from the use of a culture system without feeder cells, such as in previous studies of hESCs and hiPSCs [15, 20] and primary CD34+ cells [38]. This basic and defined differentiation system provide a platform for our future effort in the investigation of the regulatory factors and protocol optimization toward generating large numbers of platelets ex vivo.
TPO has been identified as an indispensible regulator of HSCs and HPCs, in addition to its role in megakaryopoiesis [35]. We examined the effect of TPO on the production of CD45+CD34+ HPCs and MK progenitor cells within the HPC population and tested the possibility of replacing TPO with Nplate during hiPSC differentiation. We found that adding TPO on day 11 of hematopoietic differentiation significantly increased the yield of CD45+CD34+ HPCs released from EBs on day 14. As we expected, the number of CD41+CD42a+ cells on day 14 was also higher with TPO stimulation than without TPO stimulation. A second generation of TPO receptor agonists, romiplostim (Nplate) and eltrombopag (an orally active drug), has been developed and approved by the FDA for clinical applications [35]. The major reason to choose romiplostim over TPO is for a clinical applicable strategy and cost-effectiveness, because romiplostim obtained from the clinical research pharmacy is less expensive than recombinant TPO. We found that romiplostim is sufficient to substitute for the same dose of TPO and promote hiPSC differentiation to HPCs and MKs (Fig. 2).
The potential of the cells to form CFU-MK colonies (containing >50 cells) had decreased significantly after 5 days in the MK differentiation process. The cell viability stayed high after culture (>90% by Trypan blue staining, data not shown), suggesting that the low numbers of CFU-MKs cannot be explained by cell death/apoptosis. Actually, the CD34+ population on day 19 not only showed a much lower mean intensity by flow cytometry (supplemental online Fig. 2A), but also generated much fewer CFU colonies of other hematopoietic lineages, especially the multilineage progenitors (CFU-granulocyte macrophage or CFU-granulocyte, erythrocyte, monocyte, megakaryocyte), compared with the CD34+ population on day 14 (supplemental online Fig. 2B). That the day-19 cells could only give rise to small CFU-MK colonies (Fig. 2A) also suggested that the MKs on day 19 had almost lost their proliferative potential. In addition, we analyzed the expression of CD123 (an IL-3 receptor) using flow cytometry and found that CD123 was not expressed in CD34+ cells and CD41+ cells on day 19 (supplemental online Fig. 3B, 3C). A lack of an IL-3 receptor in the cells on day 19 could be the reason for the proliferative potential. We will investigate this possibility in the future.
To develop a clinically applicable procedure, we evaluated critical reagents in terms of reliability and cost effectiveness. More importantly, we introduced reagents already approved by the FDA and available in pharmacy. For example, we used oprelvekin as the source of recombinant IL-11. We replaced unreliable sources of BSA with FDA-approved Plasbumin (Fig. 4). These improvements gave much more consistent results in the generation of CD41+CD42a+ MKs. In terms of the efficiency of MK generation, we observed a high level of consistency from two iPSC lines originated from the same person, based on our results from 44 hiPSC lines derived from 22 individuals (Fig. 5). The differences observed among the hiPSC lines from the different individuals might have been largely due to the genetic variations in the human population rather than the inconsistency of the differentiation conditions (Fig. 5B). This finding and our experimental system should provide a platform to study the genetic influences on megakaryopoiesis in the future.
Under the feeder-free and xeno-free condition, hiPSC-derived MKs expressed not only CD41 and CD42a, but also other MK markers such as CD42b and CD61 (supplemental online Fig. 1B), suggesting a close similarity of hiPSC-derived MKs with BM-derived MKs. However, only a small fraction of these cells at day 19 of culture had polyploid DNA contents (∼16%). Although MK-associated genes such as CD41, CD42a, CD42b, CD42c, CD61, and PF4 were highly upregulated after hiPSC differentiation, we also observed that HBE1, HBG1, and HBA1 were expressed in the cells on day 19. The HBE and HBG genes encode embryonic and fetal globin, respectively, the β-subunit that forms hemoglobin with the α-subunit encoded by the HBA gene. This result was not totally unexpected and is likely unique to hiPSC-initiated hematopoiesis in culture that only lasted for ∼20 days. It is well established that MKs and erythroid cells originate from common bipotential precursors (megakaryocyte-erythroid progenitors [MEPs]), which in turn are generated from HSCs. Therefore, erythroid-associated genes could be either expressed in committed erythroid cells in the current MK culture stimulated by TPO, SCF, and IL-11 or expressed in the MEPs present in culture. Additional analysis of flow cytometry showed that approximately 9% cells were CD41+CD235a+ MEPs on day 19 and that CD41−CD235a+ cells were less than 2%, suggesting that MEPs were present after 5 days of MK culture (supplemental online Fig. 3A). Our data are consistent with previous observations that erythroid cells differentiated from hESCs and hiPSCs predominantly express HBE and HBG genes but not the HBB gene encoding the adult form of β-globin.
These data, together with the observation that platelets were rarely found in our iPSC-MK cultures at day 19, suggest that additional maturation of MKs and/or improving platelet formation is required for clinical applications. For more than two decades, scientists have been working on the development of an efficient culture condition that will allow the formation of platelets (or pro- and preplatelets) from culture-expanded MKs, which could be generated in moderate quantities from primary CD34+CD45+ HPCs in BM and cord blood. This is particularly challenging if stromal cells are absent. During the preparation of this report, a group of interdisciplinary investigators reported that they have developed a biomimetic microfluidic platelet bioreactor, which recapitulates the BM and blood vessel microenvironments to facilitate platelet production from mouse fetal-liver culture-derived MKs [39]. The system appears also applicable to hiPSC-derived MKs, and the details of hiPSC-MKs used in the study are expected to be published soon [39]. Collaboratively, that unpublished study, together with the hiPSC-MK system we have described in the present report, should provide large numbers of cells for us to search for mechanisms that promote platelet formation.
Because of the inability to produce large numbers of platelets from MKs in culture, an alternative approach of transfusing culture-expanded MKs into patients with thrombocytopenia has been proposed in an animal study [40] and even tested in limited clinical studies [41]. This was based on the premise that MKs transfused into a patient with thrombocytopenia will develop into platelets in vivo, perhaps even with a higher efficiency than ex vivo under current culture conditions. Because it is difficult to obtain enough MK progenitors from patients with thrombocytopenia, an allogeneic HSC/HPC source with the best-match HLA genotype is required. MKs generated from hiPSCs that derive from the same thrombocytopenic patient would have a perfect match of HLA and other platelet-specific antigens. Nakamura et al. [20], using hiPSCs, and previous studies [15, 42], using hESCs, showed that hiPSC- or hESC-derived MKs generate functional platelets in mouse models.
Conclusion
Our study on the efficient generation of hiPSC-derived MKs under a feeder-free and xeno-free culture condition using FDA-approved pharmacological reagents will provide a reliable and unlimited source of MKs for future investigations involving either direct transfusion of MKs with subsequent platelet formation in vivo or the generation of platelets ex vivo before transfusion. Our results provide a solid foundation for such important investigations in the future.
Supplementary Material
Acknowledgments
We thank Siddharth Shah for excellent technical assistance at the initial stage of this project. This work was supported in part by the NIH (Grants U01 HL107446 and 2R01 HL-073781) and the Maryland State Stem Research Cell Fund (Grant 2012-MSCRFII-0124). Additional technical support was provided by Xiaoling Zhang in the flow cytometry core and Jin-Shui Fang and David Mohr in the Genetic Resources Core Facility at Johns Hopkins Medicine. L.C. is also supported by the Edythe Harris Lucas and Clara Lucas Lynn Chair in Hematology of Johns Hopkins University.
Author Contributions
Y.L. and Y.W.: collection and assembly of data, manuscript writing; Y.G. and J.A.F.: data collection; R.Q. and L.B.: data analysis and interpretation, contribution of vital new reagents; L.C. and Z.Z.W.: conception and design, data analysis and interpretation, and manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
References
- 1.George JN. Platelets. Lancet. 2000;355:1531–1539. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- 2.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115:3339–3347. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stroncek DF, Rebulla P. Platelet transfusions. Lancet. 2007;370:427–438. doi: 10.1016/S0140-6736(07)61198-2. [DOI] [PubMed] [Google Scholar]
- 4.Tober J, Koniski A, McGrath KE, et al. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood. 2007;109:1433–1441. doi: 10.1182/blood-2006-06-031898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 6.Ito T, Ishida Y, Kashiwagi R, et al. Recombinant human c-Mpl ligand is not a direct stimulator of proplatelet formation in mature human megakaryocytes. Br J Haematol. 1996;94:387–390. doi: 10.1046/j.1365-2141.1996.d01-1813.x. [DOI] [PubMed] [Google Scholar]
- 7.Choi ES, Nichol JL, Hokom MM, et al. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood. 1995;85:402–413. [PubMed] [Google Scholar]
- 8.de Sauvage FJ, Hass PE, Spencer SD, et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature. 1994;369:533–538. doi: 10.1038/369533a0. [see comments] [DOI] [PubMed] [Google Scholar]
- 9.Drachman JG, Griffin JD, Kaushansky K. The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J Biol Chem. 1995;270:4979–4982. doi: 10.1074/jbc.270.10.4979. [DOI] [PubMed] [Google Scholar]
- 10.Bunting S, Widmer R, Lipari T, et al. Normal platelets and megakaryocytes are produced in vivo in the absence of thrombopoietin. Blood. 1997;90:3423–3429. [PubMed] [Google Scholar]
- 11.Fox N, Priestley G, Papayannopoulou T, et al. Thrombopoietin expands hematopoietic stem cells after transplantation. J Clin Invest. 2002;110:389–394. doi: 10.1172/JCI15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avecilla ST, Hattori K, Heissig B, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10:64–71. doi: 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- 13.Tian X, Hexum MK, Penchev VR, et al. Bioluminescent imaging demonstrates that transplanted human embryonic stem cell-derived CD34(+) cells preferentially develop into endothelial cells. Stem Cells. 2009;27:2675–2685. doi: 10.1002/stem.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takayama N, Eto K. In vitro generation of megakaryocytes and platelets from human embryonic stem cells and induced pluripotent stem cells. Methods Mol Biol. 2012;788:205–217. doi: 10.1007/978-1-61779-307-3_15. [DOI] [PubMed] [Google Scholar]
- 15.Lu SJ, Li F, Yin H, et al. Platelets generated from human embryonic stem cells are functional in vitro and in the microcirculation of living mice. Cell Res. 2011;21:530–545. doi: 10.1038/cr.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takayama N, Nishimura S, Nakamura S, et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med. 2010;207:2817–2830. doi: 10.1084/jem.20100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaur M, Kamata T, Wang S, et al. Megakaryocytes derived from human embryonic stem cells: A genetically tractable system to study megakaryocytopoiesis and integrin function. J Thromb Haemost. 2006;4:436–442. doi: 10.1111/j.1538-7836.2006.01744.x. [DOI] [PubMed] [Google Scholar]
- 18.Pick M, Azzola L, Osborne E, et al. Generation of megakaryocytic progenitors from human embryonic stem cells in a feeder- and serum-free medium. PLoS One. 2013;8:e55530. doi: 10.1371/journal.pone.0055530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono Y, Wang Y, Suzuki H, et al. Induction of functional platelets from mouse and human fibroblasts by p45NF-E2/Maf. Blood. 2012;120:3812–3821. doi: 10.1182/blood-2012-02-413617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura S, Takayama N, Hirata S, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14:535–548. doi: 10.1016/j.stem.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Ng ES, Davis RP, Azzola L, et al. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Gulbranson DR, Hou Z, et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hulse WL, Gray J, Forbes RT. Evaluating the inter and intra batch variability of protein aggregation behaviour using Taylor dispersion analysis and dynamic light scattering. Int J Pharm. 2013;453:351–357. doi: 10.1016/j.ijpharm.2013.05.062. [DOI] [PubMed] [Google Scholar]
- 24.Yahata T, Ando K, Nakamura Y, et al. Functional human T lymphocyte development from cord blood CD34+ cells in nonobese diabetic/Shi-SCID, IL-2 receptor gamma null mice. J Immunol. 2002;169:204–209. doi: 10.4049/jimmunol.169.1.204. [DOI] [PubMed] [Google Scholar]
- 25.Hiramatsu H, Nishikomori R, Heike T, et al. Complete reconstitution of human lymphocytes from cord blood CD34+ cells using the NOD/SCID/gammacnull mice model. Blood. 2003;102:873–880. doi: 10.1182/blood-2002-09-2755. [DOI] [PubMed] [Google Scholar]
- 26.Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou BK, Mali P, Huang X, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng L, Hansen NF, Zhao L, et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai J, Miao X, Li Y, et al. Whole-genome sequencing identifies genetic variances in culture-expanded human mesenchymal stem cells. Stem Cell Repots. 2014;3:227–233. doi: 10.1016/j.stemcr.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Chou BK, Dowey S, et al. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem Cell Res (Amst) 2013;11:1103–1116. doi: 10.1016/j.scr.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mali P, Chou BK, Yen J, et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010;28:713–720. doi: 10.1002/stem.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mali P, Ye Z, Hommond HH, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- 33.Dowey SN, Huang X, Chou BK, et al. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc. 2012;7:2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Civin CI, Almeida-Porada G, Lee MJ, et al. Sustained, retransplantable, multilineage engraftment of highly purified adult human bone marrow stem cells in vivo. Blood. 1996;88:4102–4109. [PubMed] [Google Scholar]
- 35.Himburg HA, Harris JR, Ito T, et al. Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Reports. 2012;2:964–975. doi: 10.1016/j.celrep.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michelson AD. Flow cytometry: A clinical test of platelet function. Blood. 1996;87:4925–4936. [PubMed] [Google Scholar]
- 37.Michelson AD, Furman MI. Laboratory markers of platelet activation and their clinical significance. Curr Opin Hematol. 1999;6:342–348. doi: 10.1097/00062752-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 38.Cheng L, Qasba P, Vanguri P, et al. Human mesenchymal stem cells support megakaryocyte and pro-platelet formation from CD34(+) hematopoietic progenitor cells. J Cell Physiol. 2000;184:58–69. doi: 10.1002/(SICI)1097-4652(200007)184:1<58::AID-JCP6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 39.Thon JN, Mazutis L, Wu S, et al. Platelet bioreactor-on-a-chip. Blood. 2014;214:1857–1867. doi: 10.1182/blood-2014-05-574913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 41.Xi J, Zhu H, Liu D, et al. Infusion of megakaryocytic progenitor products generated from cord blood hematopoietic stem/progenitor cells: Results of the phase 1 study. PLoS One. 2013;8:e54941. doi: 10.1371/journal.pone.0054941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takayama N, Nishikii H, Usui J, et al. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood. 2008;111:5298–5306. doi: 10.1182/blood-2007-10-117622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.