

Abstract
Background:
Traumatic memories have been resilient to therapeutic approaches targeting their permanent attenuation. One of the potentially promising pharmacological strategies under investigation is the search for safe reconsolidation blockers. However, preclinical studies focusing on this matter have scarcely addressed abnormal aversive memories and related outcomes.
Methods:
By mimicking the enhanced noradrenergic activity reported after traumatic events in humans, here we sought to generate a suitable condition to establish whether some clinically approved drugs able to disrupt the reconsolidation of conditioned fear memories in rodents would still be effective.
Results:
We report that the α2-adrenoceptor antagonist yohimbine was able to induce an inability to restrict behavioral (fear) and cardiovascular (increased systolic blood pressure) responses to the paired context when administered immediately after acquisition, but not 6h later, indicating the formation of a generalized fear memory, which endured for over 29 days and was less susceptible to suppression by extinction. It was also resistant to reconsolidation disruption by the α2-adrenoceptor agonist clonidine or cannabidiol, the major non-psychotomimetic component of Cannabis sativa. Since signaling at N-methyl-D-aspartate (NMDA) receptors is important for memory labilization and because a dysfunctional memory may be less labile than is necessary to trigger reconsolidation on its brief retrieval and reactivation, we then investigated and demonstrated that pre-retrieval administration of the partial NMDA agonist D-cycloserine allowed the disrupting effects of clonidine and cannabidiol on reconsolidation.
Conclusions:
These findings highlight the effectiveness of a dual-step pharmacological intervention to mitigate an aberrant and enduring aversive memory similar to that underlying the post-traumatic stress disorder.
Keywords: fear generalization, memory overconsolidation, norepinephrine, PTSD
Introduction
Patients suffering from post-traumatic stress disorder (PTSD) may present disruption in the inhibitory action mediated through α2-adrenoceptors (Perry et al., 1987) and enhanced noradrenergic activity (Liberzon et al., 1999), particularly shortly after the occurrence of the traumatic event (Pitman, 1989; Yehuda et al., 1998), a period in which the consolidation of the corresponding memory is taking place (Dudai, 2004). Possibly owing to the overconsolidation of this aversive learning, these patients tend to retrieve the memory spontaneously and recurrently (Ehlers et al., 2004) and express generalized fear/anxiety responses (Jovanovic et al., 2009). However, the premise that an abnormal aversive memory formation would lead to the development of features related to PTSD, besides impaired extinction, has frequently been neglected at a preclinical level. Moreover, studies aimed at modeling this psychiatric condition to investigate interventions that could have clinical relevance to its treatment have predominantly dealt with normal fear memories, including those acquired through classical conditioning and which induce behavioral and cardiovascular changes on the animal’s re-exposure to the paired context, for instance.
Based on the above, it would be of potential interest to manipulate the noradrenaline level during the contextual fear memory consolidation in laboratory animals to mimic aspects of this clinical condition, which could improve the translational value of the findings. In this regard, yohimbine can be a useful pharmacological tool because it antagonizes the inhibitory action mediated through somatic and/or dendritic pre-synaptic α2-adrenoceptors, stimulating the locus coeruleus (Ivanov and Aston-Jones, 1995), which is known to increase the noradrenaline release in several brain regions necessary for emotional memory processing (Sara, 2009). Furthermore, as yohimbine acts as an indirect sympathomimetic agent, the endogenous neurotransmitter is the one that ultimately intensifies the noradrenergic transmission, an outcome that could be considered a non-artificial way of inducing PTSD-like memories.
One of the potential strategies under investigation for PTSD management is to uncover/develop a safe method to uncouple, attenuate, or even erase the negative valence associated with aberrant and enduring aversive memories underlying the core of this psychiatric condition (Cain et al., 2012; Debiec, 2012; Parsons and Ressler, 2013). Of potential relevance to this matter is convergent evidence demonstrating that an established contextual fear memory, for instance, can again be rendered labile and susceptible to interference after its retrieval and reactivation (Alberini, 2011; Dudai, 2012). Memory reactivation can be followed by a new phase of stabilization referred to as reconsolidation, which, in turn, is thought to re-establish and maintain it over time (Nader et al., 2000; Sara, 2000; Stern et al., 2013). Various drugs are able to disrupt the reconsolidation of conditioned fear memories in laboratory animals and healthy humans (Przybyslawski et al., 1999; Bustos et al., 2010; Gamache et al., 2012; Schwabe et al., 2012). Taking into account that an abnormal aversive memory may be less prone to intervention (Soeter and Kindt, 2013), it would be important to verify whether clinically-approved drugs that are also reported to be reconsolidation blockers, such as the α2-adrenoceptor agonist clonidine (Gamache et al., 2012; Gazarini et al., 2013) and cannabidiol (Stern et al., 2012), the major non-psychotomimetic component of Cannabis sativa that indirectly potentiates the cannabinoid type-1 receptor-mediated transmission (Campos et al., 2012), would still be sufficiently effective in this case, which could encourage further investigation of their efficacy in PTSD patients.
The present study sought to investigate the applicability of associating contextual fear conditioning with a single and systemic administration of yohimbine, while the aversive learning was being consolidated, to induce concurrent PTSD-related features that would allow a more effective search for potential therapeutic interventions. The working hypothesis was that mimicking the enhanced noradrenergic activity reported in PTSD patients could generate an enduring PTSD-like memory in laboratory animals, leading to the inability to restrict responses to the appropriate context, and that it would have a differential susceptibility to suppression by extinction or reconsolidation disruption by clonidine and cannabidiol. Thereafter, since signaling at N-methyl-D-aspartate (NMDA) glutamate receptors is known to be important for contextual fear memory labilization (Bustos et al., 2010), and because a PTSD-like memory may be less prone to labilization than is necessary to trigger the reconsolidation process on its retrieval (Soeter and Kindt, 2013), we hypothesized that the partial NMDA agonist D-cycloserine would be able to allow the disrupting effects of drugs on reconsolidation when administered before retrieving a contextual fear memory consolidated under the yohimbine influence.
Method
Animals
Experiments were performed in male Wistar rats aged 12–14 weeks, kept grouped on a 12h light/dark cycle, and with food and water ad libitum. All procedures were approved by the Institutional Ethical Committee for the care and use of laboratory animals of our university in compliance with Brazilian legislation.
Drugs
Yohimbine HCl (Tocris) was administered in a dose (1.0mg/kg) able to potentiate fear memory consolidation (Gazarini et al., 2013). The lipophilic β-adrenoceptor antagonist propranolol HCl (10mg/kg) and the hydrophilic β-adrenoceptor antagonist nadolol (10mg/kg) were purchased from Sigma-Aldrich and used to investigate the role of central and peripheral β-adrenoceptors in the enhanced noradrenergic activity induced by yohimbine. In any case, the dose selected was able to prevent the facilitating effects of other β-adrenoceptor agonists on fear memory consolidation (Gazarini et al., 2013). Clonidine HCl (Sigma-Aldrich; 0.3mg/kg) and cannabidiol (THC-Pharma; 10mg/kg) were administered at putative memory reconsolidation-disrupting doses (Stern et al., 2012, Gazarini et al., 2013). D-cycloserine (Sigma-Aldrich) was administered in a dose (15mg/kg) able to potentiate fear memory labilization (Bustos et al., 2010). All drugs were dissolved in 0.9% NaCl, except for cannabidiol, which was dissolved in NaCl 0.9% containing 5% of Tween 80® (Vetec) and administered systemically in a volume of 1.0ml/kg.
Fear Conditioning Apparatus
Fear conditioning was performed in a rectangular chamber (35 x 20 x 30cm), with aluminum sidewalls and a front wall and ceiling-door made of Plexiglas, designated herein as Context A. Its grid floor, made of stainless steel bars, was connected to a circuit board and a shock generator (Insight) to enable the delivery of controlled electrical footshocks as detailed subsequently. A second chamber (30 x 30 x 30cm), designated herein as Context B, was made of glass and had a grid lid and transparent walls and floor, to provide contextual cues as different as possible from those of the footshock-paired Context A.
Behavioral Procedures and Data Collection
Behavioral testing was conducted under 70 lux, from 1:00 to 6:00 PM. In all experiments, the animal was placed in Context A and allowed to explore it for 3min, as an initial familiarization session, and returned to its home cage. For fear conditioning, the animal was again placed in Context A, during which it received, after an initial 30 s delay, the unconditioned stimulus (three electrical footshocks of 0.7 mA, 60 Hz, for 3 s, with a 30 s inter-trial period). The animal remained in this chamber for another 30 s before returning to its home cage. In Test A, the animal was re-exposed to Context A for 3min in the absence of unconditioned stimulus presentation, whereas in Test B it was exposed to Context B, also for 3min.
Overall, the interval among these experimental sessions was of one day. However, in Experiment 2 the interval between conditioning and Test A was of 14 or 28 days. In Experiment 4 there were Tests A and B before and after an extinction session (re-exposure to Context A for 15min in the absence of unconditioned stimulus presentation), and in Experiment 5 there was a session of memory retrieval (re-exposure to Context A for 3min in the absence of unconditioned stimulus presentation) between conditioning and Test A. A scheme of the experimental design used in each experiment is presented either above the graph or in the legend of the table depicting its data.
Freezing behavior was continuously recorded during the experimental sessions by a video camera. The freezing time was subsequently quantified in seconds and expressed as the percentage of total session time. The time was measured by a trained observer (inter- and intra-rater reliabilities ≥ 90%) blind to the experimental groups.
Systolic Blood Pressure Measurement
Two hours after Test B, each animal was immobilized in a restraint chamber, placed on a heater pad, and a tail-cuff was placed proximal to its tail. The tail-cuff occluder was used to stop the blood flow on inflation, so that the integrated sensor could detect its return on each deflation cycle using an acquisition system (AD Instruments). After 5min of stabilization, a typical run involved four repetitions of the inflation-deflation cycle. The average of these measures was calculated and used as the indirect systolic blood pressure value.
Statistical Analysis
The results are expressed as mean ± standard error (SEM). After ensuring the assumptions of normality and homoscedasticity, the freezing times scored in Contexts A and B were subjected to separate one-way, factorial, and/or repeated-measures analysis of variance (ANOVA). For the systolic blood pressure data, a one-way ANOVA was conducted. When ANOVA revealed a significant interaction between independent variables under study, the F values from their main effects were omitted. The Newman-Keuls test was used for post hoc comparisons. When there were two groups and no context re-exposure was performed, an unpaired two-sample student’s t-test was conducted. The statistical significance level was set at p < 0.05.
Results
Experiment 1: Facilitating Effect of Yohimbine on Memory Consolidation Induces Generalized Fear Expression Accompanied by Increased Systolic Blood Pressure
To investigate whether the facilitating effect of yohimbine on memory consolidation could induce generalized fear expression, 36 contextually fear-conditioned rats were randomly allocated to four groups (n = 8–10/group) based on the systemic treatment (vehicle or yohimbine) and the moment it was given (immediately or 6h post-conditioning session).
The two-way ANOVA showed a significant drug treatment versus moment of drug injection interaction (F 1,32 = 46.0; p < 0.00001) for freezing time on exposure to the unpaired Context B (Test B). As shown in Figure 1, animals treated with yohimbine immediately after conditioning expressed significantly more freezing than controls during Test B, indicating fear generalization. All groups presented a comparable (F 1,32 = 0.07; p = 0.78) amount of freezing time when re-exposed to the paired Context A (Test A), probably owing to the achievement of an asymptotic level of this conditioned behavior under our experimental conditions, in which Context A was paired with three shocks.
Figure 1.
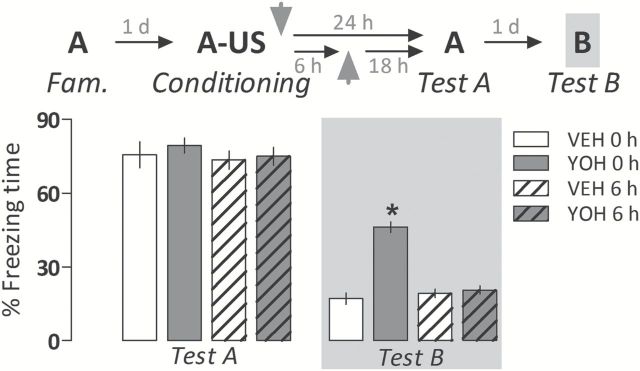
Yohimbine (YOH; 1.0mg/kg i.p.) induces a generalized fear expression when administered immediately after acquisition of a contextual fear memory in rats. This effect was no longer observed when it was given after the completion of the process of memory consolidation (6h post-acquisition). Animals were subjected to Tests A and B undrugged. The scheme above the graph represents the experimental design adopted. Arrowheads indicate the moment of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 8–10/group). *p < 0.05 from the respective control group (VEH).
To support the assumption that pairing Context A with three or more shocks induces a similar level of freezing during Test A, 30 naive rats were randomly allocated to three groups (n = 10/group) based on the number of shocks delivered during conditioning (1, 3, or 5).
The one-way ANOVA showed a significant effect of the number of shocks for freezing time on the re-exposed rats to paired Context A (F 2,27 = 31.8; p < 0.000001). As shown in Table 1, the 1-shock group expressed statistically less freezing than both the 3- and 5-shock groups, while no statistically-significant differences between the two latter groups were observed. This result suggests that this conditioned behavioral response has indeed achieved an asymptotic level. Moreover, there was no significant effect of the number of shocks for freezing time during Test B (F 2,27 = 1.7, p = 0.20).
Table 1.
Effects of the number of shocks (0.7 mA, 60 Hz, 3s) delivered during conditioning on the amount of freezing time expressed during re-exposure to the paired Context A (Test A) and exposure to the unpaired Context B (Test B). Animals in which Context A was paired with 3 or 5 shocks behaved equally in both cases. Data are presented as mean ± S.E.M. (n = 10/group).
| Number of shocks |
1 | 3 | 5 |
|---|---|---|---|
| Test A | 28.3±5.7* | 73.2±4.5 | 75.3±3.7 |
| Test B | 10.2±3.0 | 18.5±2.5 | 19.3±5.4 |
*p < 0.05 from the three-shock group (one-way ANOVA followed by Newman Keuls test).
To investigate whether systolic blood pressure rises when animals treated with yohimbine after conditioning are exposed to the unpaired Context B (Test B), 28 rats were randomly allocated to three groups (n = 9–10/group) based on the treatment and/or the moment it was given (vehicle immediately post-conditioning and yohimbine immediately or 6h post-conditioning).
The one-way ANOVA showed a significant drug treatment effect (F 2,25 = 34.9, p < 0.0000001) for this cardiovascular parameter. As shown in Table 2, it was significantly increased relative to controls only in the group in which yohimbine induced generalized fear expression: i.e., when the drug was administered immediately after conditioning.
Table 2.
Fear generalization profile is accompanied by increased systolic blood Pressure. Yohimbine (1.0 mg/kg i.p.) was administrated immediately after pairing Context A with 3 shocks and animals were re-exposed to Context A (Test A) or to the unpaired Context B (Test B) on subsequent days (the experimental design was similar to that depicted in Figure 1). Two hours after Test B, the systolic blood pressure was measured in restraint animals with a tail-cuff. Animals were undrugged during all test sessions. No statistically significant changes on freezing time were observed during Context A re-exposure (Test A). Data are presented as mean ± S.E.M. (n = 9–10/group).
| Parameter | % freezing time on Test A | % freezing time on Test B | Systolic blood pressure in mmHg |
|---|---|---|---|
| Vehicle 0h post-conditioning | 79.8±2.9 | 18.5±2.8 | 112.6±2.6 |
| Yohimbine 0h post-conditioning | 79.8±2.1 | 51.3±3.7* | 137.6±2.2* |
| Yohimbine 6h post-conditioning | 77.1±1.9 | 18.6±4.8 | 114.7±2.4 |
*p < 0.05 from respective control group (one-way ANOVA followed by Newman-Keuls test).
Experiment 2: Generalized Fear Expression is Long-Lasting
To investigate whether generalized fear expression associated with the facilitating effect of yohimbine on memory consolidation is an enduring feature, 39 animals were randomly allocated to four groups (n = 9–10/group) based on the treatment (vehicle or yohimbine) given post-conditioning and the interval between conditioning and Context B exposure (15 or 29 days).
Two-way ANOVA showed a significant drug treatment effect (F 1,35 = 88.1, p < 0.000001) for freezing time during Context B exposure. As shown in Figure 2, the two yohimbine-treated groups expressed significantly more freezing time than respective controls in Test B. All groups presented a comparable amount of freezing time during Test A (F 1,35 = 0.03, p = 0.86).
Figure 2.
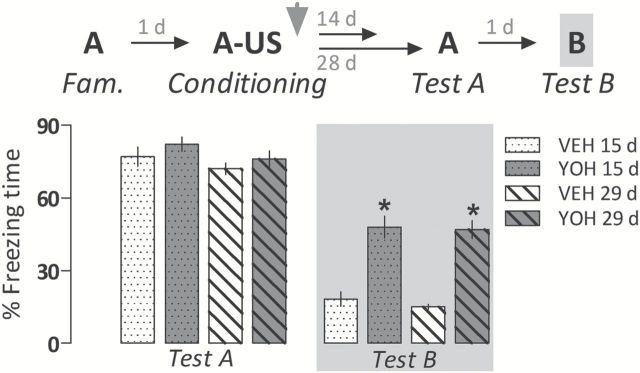
Generalized fear expression induced by yohimbine (YOH; 1.0mg/kg i.p.) persists over 29 days. Animals were subjected to Tests A and B undrugged. The scheme above the graph represents the experimental design adopted. The arrowhead indicates the moment of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 9–10/group). *p < 0.05 from the respective control group (VEH).
Experiment 3: Fear Generalization is Associated with Activation of Brain β-Adrenoceptors
To investigate the relative contribution of central and peripheral β-adrenoceptors to yohimbine-induced enhanced noradrenergic activity and generalized fear expression, 53 animals were randomly allocated to six groups (n = 8–10/group) based on the pretreatment given immediately after conditioning (vehicle, the central β-adrenoceptor antagonist propranolol, or the peripheral β-adrenoceptor antagonist nadolol) and the treatment given 10min later (vehicle or yohimbine). The systemic administration of these drugs was conducted in this interval of time, instead of administrating them concurrently and in the same solution, to minimize the possibility of occurrence of a chemical or pharmacokinetic interaction.
The two-way ANOVA showed a significant drug pretreatment versus treatment interaction (F 2,47 = 12.6; p < 0.0001) for freezing time during Test B. As shown in Figure 3, vehicle- and nadolol-pretreated animals administered with yohimbine expressed significantly more freezing than their respective controls on Context B exposure. The yohimbine-induced fear generalization, however, was no longer observed in propranolol-pretreated animals. All groups presented a comparable amount of freezing time during Test A (F 2,47 = 0.09, p = 0.91).
Figure 3.
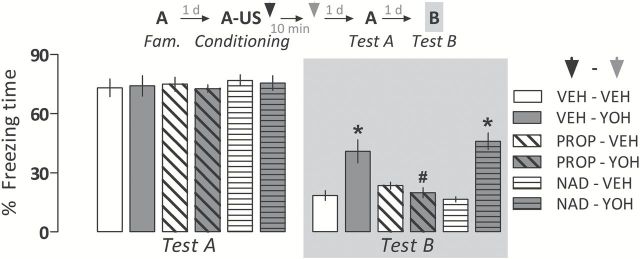
Pretreatment with the central β-adrenoceptor antagonist propranolol (PROP; 10mg/kg), but not the peripheral β-adrenoceptor antagonist nadolol (NAD; 10mg/kg), prevents the generalized fear expression induced by yohimbine (YOH; 1.0mg/kg i.p.) two days later. Animals were subjected to Tests A and B undrugged. The scheme above the graph represents the experimental design adopted. Arrowheads indicate the moment of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 8–10/group). *p < 0.05 from controls (VEH-VEH and NAD-VEH, respectively); # Significant difference from the VEH-YOH group.
Experiment 4: Fear Memory Consolidated Under the Yohimbine Influence is More Resistant to Extinction
To investigate whether overconsolidation of a fear memory induced by yohimbine could affect its later extinction, 22 animals (n = 11/group) were randomly allocated to receive vehicle or yohimbine immediately after conditioning.
As demonstrated earlier, yohimbine-treated animals presented significantly more (t 20 = 26.7; p < 0.0001) freezing than controls during Test B1 (Figure 4A). Both groups expressed comparable (t 20 = 0.0003; p = 0.98) amounts of freezing time during Test A1 (Figure 4A).
Figure 4.
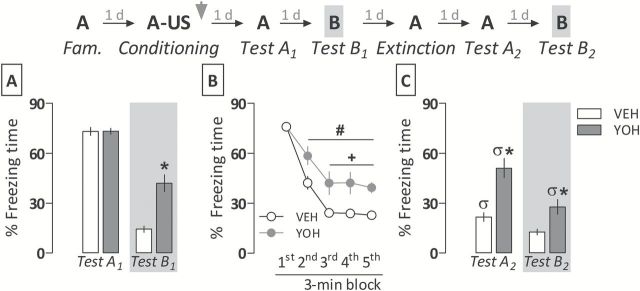
Evidence of a relative resistance to extinction of a contextual fear memory consolidated under the yohimbine (YOH) influence. (A) YOH (1.0mg/kg i.p.) induces generalized fear expression when given immediately after the conditioning session, as demonstrated in Experiment 1. (B) In YOH-treated animals, fear memory extinction was acquired more slowly than in controls (VEH). (C) YOH-treated animals did not retain extinction learning as well as controls. They also displayed higher levels of generalized fear two days following a 15min fear extinction session. Animals were subjected to Tests A1, A2, B1, B2, and the extinction session undrugged. The scheme above the graph represents the experimental design adopted. The arrowhead indicates the moment of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 11/group). *p < 0.05 from the respective control group; #significant difference from the first 3min extinction session block in the same group; +significant difference from controls in the same session block; σsignificant difference from respective Test A1 or Test B1.
The repeated-measures ANOVA showed a significant drug treatment versus time-bin interaction (F 4,80 = 4.2; p < 0.01) for freezing time during a 15min session of extinction. As shown in Figure 4B, vehicle- and yohimbine-treated animals expressed significantly less freezing from the second to the fifth 3min session block relative to the first block, but the extinction acquisition rate in yohimbine-treated animals was significantly slower than controls from the third to the fifth 3min block. Moreover, they expressed significantly more freezing time than controls during Test A2 (t 20 = 22.2; p < 0.001) and Test B2 (t 20 = 10.0; p < 0.01; Figure 4C).
When data depicted in Figure 4A and C are compared, the repeated-measures ANOVA showed a significant interaction between drug treatment and re-exposure to Context A (F 1,20 = 24.2; p < 0.0001) or Context B (F 1,20 = 4.2; p < 0.05). Both vehicle- and yohimbine-treated groups spent significantly less time performing freezing after (Test A2) than before (Test A1) fear memory extinction, but this response was virtually absent in the control group only: i.e., yohimbine-treated animals did not retain extinction learning as well as the control group. There was also a significant attenuation in the amount of freezing time expressed by yohimbine-treated animals on Test B2 when compared with Test B1, although they still presented significantly more freezing time than controls.
Experiment 5: Fear Memory Consolidated Under the Yohimbine Influence Becomes Susceptible to Reconsolidation Disruption After Potentiating its Labilization
To investigate whether the overconsolidation of a fear memory induced by yohimbine could affect the susceptibility of having its reconsolidation disrupted, 58 animals were randomly allocated to six groups (n = 9–10/group) based on the treatment given post-conditioning (vehicle or yohimbine) and the treatment given immediately after a brief retrieval session on the following day (vehicle, clonidine, or cannabidiol).
The repeated-measures ANOVA showed a significant drug pretreatment versus treatment versus Context A exposure interaction for freezing time (F 2,52 = 24.4, p < 0.000001). As shown in Figure 5A, both clonidine and cannabidiol significantly disrupted the reconsolidation process, because reduced freezing time was observed during Test A in vehicle- but not yohimbine-pretreated animals. For freezing time during Test B, the two-way ANOVA showed a significant drug pretreatment effect (F 1,52 = 235.2, p < 0.000001). As shown in Figure 5A, all yohimbine-pretreated groups expressed significantly more freezing than respective controls, indicating fear generalization.
Figure 5.

Comparative effects of clonidine (CLO; 0.3mg/kg) and cannabidiol (CBD; 10mg/kg) given alone or associated with D-cycloserine (DCS; 15mg/kg) on the reconsolidation of a fear memory consolidated under the influence of yohimbine (YOH; 1.0mg/kg). (A) In comparison with the control group (VEH-VEH), both VEH-CLO and VEH-CBD groups expressed less fear when re-exposed to the paired context (Test A). However, the disruptive effect of CLO and CBD on memory reconsolidation was no longer observed in YOH-treated animals, which also expressed fear generalization to an unpaired context (Test B). (B) Administrating DCS 30min prior to memory retrieval allows the memory reconsolidation-disrupting effects of CLO and CBD and prevents the generalized fear expression in YOH-treated animals. Animals were subjected to Tests A and B undrugged. Schemes above graphs represent the experimental design of each one of the experiments. Arrowheads indicate the moments of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 7–10/group). *p < 0.05 from the respective controls; #significant difference from the YOH-DCS-VEH group.
To investigate whether potentiating the labilization of the overconsolidated fear memory with D-cycloserine could allow the disrupting effects of clonidine and cannabidiol on reconsolidation, 51 rats were randomly allocated to six groups (n = 7–9/group) based on the pretreatment given immediately after conditioning (vehicle or yohimbine) and the treatment given post-retrieval (vehicle, clonidine, or cannabidiol). All groups received D-cycloserine 30min before memory retrieval.
The repeated-measures ANOVA showed a significant drug treatment versus Context A exposure interaction for freezing time (F 2,45 = 49.9; p < 0.000001). As shown in Figure 5B, the disrupting effects of clonidine and cannabidiol on reconsolidation were now observed during Test A in both vehicle- and yohimbine-pretreated groups, as they expressed significantly less freezing than respective controls. For freezing time during Test B, the two-way ANOVA showed a significant drug pretreatment versus treatment interaction (F 2,45 = 10.3; p < 0.001). As shown in Figure 5B, both drugs significantly attenuated fear generalization in yohimbine-pretreated animals relative to respective controls.
Importantly, as shown in Figure 6, at the dose tested D-cycloserine did not interfere, by itself, with memory retrieval/expression or extinction, as no statistically-significant differences were observed (retrieval session: t 16 = 0.02, p = 0.89; Test A: t 16 = 0.19, p = 0.67; and Test B: t 16 = 0.38, p = 0.54).
Figure 6.

D-cycloserine (DCS; 15mg/kg) does not change the amount of freezing time expressed during re-exposures to the paired Context A (retrieval and Test A sessions) or exposure to the unpaired Context B (Test B) relative to controls (VEH) when given 30min before briefly retrieving a contextual fear memory. Animals were subjected to Tests A and B undrugged. The scheme above the graph represents the experimental design adopted. The arrowhead indicates the moment of drug administration. Bars represent the percentage of total time spent freezing. Values are expressed as mean ± SEM (n = 9/group).
Discussion
The administration of yohimbine during contextual fear memory formation induced an inability to restrict freezing behavior and increased systolic blood pressure to the paired context. These results suggest that enhanced noradrenergic activity while an aversive memory is being consolidated leads to the occurrence of these maladaptive behavioral and cardiovascular outcomes, as observed 48–50h later. As a significant concentration of yohimbine in the rat brain only persists up to 8h (Hubbard et al., 1988), it is believed these findings are not directly attributable to the anxiogenic and/or the hypertensive property of this α2-adrenoceptor antagonist (Soeter and Kindt, 2012; Gazarini et al., 2013). It is worth mentioning that vehicle- and yohimbine-treated animals had a similar time of freezing on the re-exposure to Context A (Test A). A potential explanation for this finding is that this conditioned behavioral response has achieved an asymptotic (in our case, nearly the ceiling) level in controls. Corroborating this assumption, pairing Context A with 3 or 5 footshocks induced a comparable amount of freezing time during Test A, a result that corresponds to that reported in rats subjected to olfactory fear conditioning (Kroon and Carobrez, 2009).
The potentiating effect of yohimbine on fear memory was observed when it was administered immediately after acquisition, but not 6h later. This result reinforces the premise that drug interference is specific to consolidation, as neither fear generalization nor increased systolic blood pressure could be observed when it was given at a time point when this memory phase had already been completed (Dudai, 2004). The absence of changes in these parameters of animals tested 42–44h after being treated with yohimbine also rules out the possibility that the anxiogenic and/or hypertensive action of this drug could explain the results observed. Moreover, the inability to restrict fear to the appropriate context was an enduring feature, being maintained for at least 29 days. This indirectly suggests that the abnormal (generalized) fear memory induced by enhanced noradrenergic activity persists over time, agreeing with the findings reported for PTSD-related memories (Pitman, 1989; Elzinga and Bremner, 2002; Zoladz and Diamond, 2013).
In this study, the antagonism of central rather than peripheral β-adrenoceptors immediately after conditioning was able to prevent the subsequent fear generalization to a neutral and unpaired context induced by yohimbine. Despite the fact that a possible drug-drug interaction between yohimbine and propranolol cannot be discarded, this result is consistent with that reported in healthy humans (Soeter and Kindt, 2012), and substantiates the premise that these receptors are involved in aversive memory formation (Adamec et al., 2007). As β-adrenoceptors are expressed in brain regions related to contextual fear memory consolidation, such as the hippocampus (Ji et al., 2003), amygdala (LaLumiere et al., 2003) and medial prefrontal cortex (Tronel et al., 2004), they could potentially be the sites where these adrenoceptors are activated after enhancing noradrenergic transmission with yohimbine. Interestingly, these interconnected areas are functionally affected in PTSD patients (Pitman et al., 2012; Parsons and Ressler, 2013; Zoladz and Diamond, 2013), a condition that could contribute to the formation and maintenance of their aberrant and enduring aversive memories (Maren et al., 2013). The relative contributions of the hippocampus, amygdala, and medial prefrontal cortex in inducing fear generalization, however, are still under investigation. Some studies have related this maladaptive outcome to deficits in pattern separation, which, in turn, may involve the adult hippocampal neurogenesis (Kheirbek et al., 2012). Furthermore, both the medial prefrontal cortex and hippocampus belong to a neural circuit related to memory specificity and generalization (Xu and Südhof, 2013). Based on these findings, a question to be addressed in future studies is whether noradrenergic hyperactivation through β-adrenoceptors in the hippocampus, and/or its network, could form an overconsolidated fear memory and maintain a generalized fear expression over time. It has been shown that propranolol has limited efficacy in preventing the development of PTSD in humans when given after the traumatic event (Pitman et al., 2002; Stein et al., 2007; Hoge et al., 2012). At least one key aspect, namely the interval between trauma exposure and administration of this drug (immediately vs. ≥ 6h), may account for this apparent divergence. Indeed, it is able to attenuate the PTSD outcome when administrated at the time of discharging a highly stressful stimulus capable of inducing PTSD features in humans (Bhuvaneswar et al., 2014).
A relative resistance to fear extinction following yohimbine-induced generalized fear memory formation is also shown herein. Indeed, despite using a protocol of extinction able to mitigate a contextual fear memory in rats (Stern et al., 2013), animals kept expressing significant amounts of conditioned and generalized fear. This result agrees with that seen in healthy humans after enhancing noradrenergic activity during aversive memory consolidation with yohimbine (Soeter and Kindt, 2012). As it also corresponds to results reported in PTSD patients (Milad et al., 2009; Cain et al., 2012), the current experimental design may be suitable to investigate pharmacological interventions able to maximize the effectiveness of the extinction approach, which in turn mimics the exposure therapy in humans, a key behavioral intervention for PTSD (Daskalakis et al., 2013). It may also allow further investigation of the noradrenergic system’s role in brain regions, such as the amygdala, in which a dysfunctional activation has been associated with impaired extinction (Milad et al., 2009; Debiec et al., 2011).
Whereas suppressing the expression of an aversive memory by extinction has often been considered to exert only a temporary effect (Quirk and Mueller, 2008; Maren et al., 2013), interventions affecting reconsolidation tend to induce a more permanent outcome (Alberini, 2011). For instance, the disrupting effects of clonidine (0.3mg/kg) and cannabidiol (10mg/kg) on the reconsolidation of conditioned fear memories are long lasting in rats (Gamache et al., 2012; Stern et al., 2012; Gazarini et al., 2013). Fear memories consolidated under enhanced noradrenergic activity, however, were unaffected by the same dose of these drugs. A potential explanation for this finding is that a generalized fear memory might be less prone to labilization than is necessary to trigger the reconsolidation process on its brief retrieval and reactivation. Corroborating this assumption, administrating D-cycloserine prior to memory retrieval allowed the disrupting effects of clonidine and cannabidiol on reconsolidation, in view of the subsequent attenuation of both conditioned and generalized fear. It has been shown that administering the same D-cycloserine dose used here potentiates the reconsolidation of an auditory fear memory in rats (Lee et al., 2006). At least two aspects, namely the baseline levels of conditioned freezing (80 vs. 40%) and nature (abnormal vs. normal) of the aversive memory may account for this divergence. Of note, the latter argument may also explain, at least in part, the divergent results reported by studies investigating the efficacy of potential reconsolidation blockers (Pitman et al., 2002; Vaiva et al., 2003; Stein et al., 2007; Muravieva and Alberini, 2010; Debiec et al., 2011; Hoge et al., 2012; Soeter and Kindt, 2012, 2013). Our results also highlight two other aspects. First, generalized fear expression, and probably other outcomes related to a PTSD-like memory, can be ameliorated by associating pharmacological interventions that potentiate memory labilization and disrupt its subsequent reconsolidation. Second, the mechanism of action of a drug able to disrupt the reconsolidation of a generalized fear memory may be unrelated to the neurochemical mechanism responsible for inducing it, as both clonidine and cannabidiol were effective.
Not only the inability to restrict fear and increased systolic blood pressure to the appropriate context, but also a relative resistance to behavioral (extinction training) and pharmacological (use of reconsolidation blockers) interventions were shown by combining classical conditioning with a single and systemic administration of yohimbine during contextual fear memory consolidation in rats. With a slight change in conditioning parameters (i.e., pairing Context A with 1 instead of 3 footshocks as used here), this experimental approach has also been found to allow the induction of fear sensitization (Gazarini et al., 2013), which is associated with PTSD (Anisman, 2011) and noradrenergic-mediated signaling mechanisms (Olson et al., 2011). Therefore, it seems more relevant than conducting the contextual fear conditioning alone to both model a reliable PTSD-like phenotype and to discover/develop potential interventions for its effective attenuation, such as the dual-step pharmacological approach as demonstrated herein. Another applicability of the current experimental design may be for the investigation of an approach combining pharmacological and behavioral interventions able to maximize the effectiveness of the fear extinction. Moreover, as generalized fear expression was concurrent with the formation of a persistent and extinction-resistant fear memory, it may also arise from overconsolidation induced by enhanced noradrenergic activity, which could extend the original formulation of the PTSD overconsolidation hypothesis (Pitman, 1989).
Statement of Interest
None.
Acknowledgments
This work was supported by Brazilian grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). We thank Professors AP Carobrez, RDS Prediger, and JE da Silva Santos for kindly donating some drugs used herein.
References
- Adamec R, Muir C, Grimes M, Pearcey K. (2007). Involvement of noradrenergic and corticoid receptors in the consolidation of the lasting anxiogenic effects of predator stress. Behav Brain Res 179:192–207. [DOI] [PubMed] [Google Scholar]
- Alberini CM. (2011). The role of reconsolidation and the dynamic process of long-term memory formation and storage. Front Behav Neurosci 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisman H. (2011). Sensitization in relation to posttraumatic stress disorder. Biol Psychiatry 70:404–405. [DOI] [PubMed] [Google Scholar]
- Bhuvaneswar CG, Ruskin JN, Katzman AR, Wood N, Pitman RK. (2014). Pilot study of the effect of lipophilic vs. hydrophilic beta-adrenergic blockers being taken at time of intracardiac defibrillator discharge on subsequent PTSD symptoms. Neurobiol Learn Mem. 112:248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos SG, Giachero M, Maldonado H, Molina VA. (2010). Previous stress attenuates the susceptibility to Midazolam’s disruptive effect on fear memory reconsolidation: influence of pre-reactivation D-cycloserine administration. Neuropsychopharmacology 35:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, Maynard GD, Kehne JH. (2012). Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin Investig Drugs 21:1323–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS. (2012). Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Phil Trans R Soc B 367:3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalakis NP, Yehuda R, Diamond DM. (2013). Animal models in translational studies of PTSD. Psychoneuroendocrinology 38:1895–1911. [DOI] [PubMed] [Google Scholar]
- Debiec J. (2012). Memory reconsolidation processes and posttraumatic stress disorder: promises and challenges of translational research. Biol Psychiatry 71:284–285. [DOI] [PubMed] [Google Scholar]
- Debiec J, Bush DE, LeDoux JE. (2011). Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats--a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety 28:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudai Y. (2004). The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol 55:51–86. [DOI] [PubMed] [Google Scholar]
- Dudai Y. (2012). The restless engram: consolidations never end. Annu Rev Neurosci 35:227–247. [DOI] [PubMed] [Google Scholar]
- Ehlers A, Hackmann A, Michael T. (2004). Intrusive re-experiencing in post-traumatic stress disorder: phenomenology, theory, and therapy. Memory 12:403–415. [DOI] [PubMed] [Google Scholar]
- Elzinga BM, Bremner JD. (2002). Are the neural substrates of memory the final common pathway in posttraumatic stress disorder (PTSD)? J Affect Disord 70:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamache K, Pitman RK, Nader K. (2012). Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology 37:2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazarini L, Stern CA, Carobrez AP, Bertoglio LJ. (2013). Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1- and β-adrenergic receptors. Learn Mem 20:210–219. [DOI] [PubMed] [Google Scholar]
- Hoge EA, Worthington JJ, Nagurney JT, Chang Y, Kay EB, Feterowski CM, Katzman AR, Goetz JM, Rosasco ML, Lasko NB, Zusman RM, Pollack MH, Orr SP, Pitman RK. (2012). Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci Ther 18:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JW, Pfister SL, Biediger AM, Herzig TC, Keeton TK. (1988). The pharmacokinetic properties of yohimbine in the conscious rat. Naunyn Schmiedebergs Arch Pharmacol 337:583–587. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Aston-Jones G. (1995). Extranuclear dendrites of locus coeruleus neurons: activation by glutamate and modulation of activity by alpha adrenoceptors. J Neurophysiol 74:2427–2436. [DOI] [PubMed] [Google Scholar]
- Ji JZ, Wang XM, Li BM. (2003). Deficit in long-term contextual fear memory induced by blockade of beta-adrenoceptors in hippocampal CA1 region. Eur J Neurosci 17:1947–1952. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Fennell JE, Keyes M, Fiallos AM, Myers KM, Davis M, Duncan EJ. (2009). Posttraumatic stress disorder may be associated with impaired fear inhibition: relation to symptom severity. Psychiatry Res 167:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirbek MA, Klemenhagen KC, Sahay A, Hen R. (2012). Neurogenesis and generalization: a new approach to stratify and treat anxiety disorders. Nat Neurosci 15:1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon JA, Carobrez AP. (2009). Olfactory fear conditioning paradigm in rats: effects of midazolam, propranolol or scopolamine. Neurobiol Learn Mem 91:32–40. [DOI] [PubMed] [Google Scholar]
- LaLumiere RT, Buen TV, McGaugh JL. (2003). Post-training intra-basolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J Neurosci 23:6754–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci 26:10051–10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon I, Abelson JL, Flagel SB, Raz J, Young EA. (1999). Neuroendocrine and psychophysiologic responses in PTSD: a symptom provocation study. Neuropsychopharmacology 21:40–50. [DOI] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. (2013). The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14:417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66:1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravieva EV, Alberini CM. (2010). Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem 17:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. [DOI] [PubMed] [Google Scholar]
- Olson VG, Rockett HR, Reh RK, Redila VA, Tran PM, Venkov HA, Defino MC, Hague C, Peskind ER, Szot P, Raskind MA. (2011). The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry 70:441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Ressler KJ. (2013). Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci 16:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry BD, Giller EL, Jr, Southwick SM. (1987). Altered platelet alpha 2-adrenergic binding sites in posttraumatic stress disorder. Am J Psych 144:1511–1512. [DOI] [PubMed] [Google Scholar]
- Pitman R, Sanders K, Zusman R, Healy A, Cheema F, Lasko N. (2002). Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry 51:189–192. [DOI] [PubMed] [Google Scholar]
- Pitman RK. (1989). Post-traumatic stress disorder, hormones, and memory. Biol Psychiatry 26:221–223. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. (2012). Biological studies of post-traumatic stress disorder. Nat Rev Neurosci 13:769–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyslawski J, Roullet P, Sara SJ. (1999). Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci 19:6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. (2008). Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33:56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ. (2000). Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 7:73–84. [DOI] [PubMed] [Google Scholar]
- Sara SJ. (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223. [DOI] [PubMed] [Google Scholar]
- Schwabe L, Nader K, Wolf OT, Beaudry T, Pruessner JC. (2012). Neural signature of reconsolidation impairments by propranolol in humans. Biol Psychiatry 71:380–386. [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M. (2012). Stimulation of the noradrenergic system during memory formation impairs extinction learning but not the disruption of reconsolidation. Neuropsychopharmacology 37:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeter M, Kindt M. (2013). High trait anxiety: a challenge for disrupting fear memory reconsolidation. PLOS ONE 8:e75239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Kerridge C, Dimsdale JE, Hoyt DB. (2007). Pharmacotherapy to prevent PTSD: Results from a randomized controlled proof‐of‐concept trial in physically injured patients. J Trauma Stress 20:923–932. [DOI] [PubMed] [Google Scholar]
- Stern CA, Gazarini L, Takahashi RN, Guimarães FS, Bertoglio LJ. (2012). On disruption of fear memory by reconsolidation blockade: evidence from cannabidiol treatment. Neuropsychopharmacology 37:2132–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern CA, Gazarini L, Vanvossen AC, Hames MS, Bertoglio LJ. (2013). Activity in prelimbic cortex subserves fear memory reconsolidation over time. Learn Mem 21:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel S, Feenstra MG, Sara SJ. (2004). Noradrenergic action in prefrontal cortex in the late stage of memory consolidation. Learn Mem 11:453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A, Marmar CR. (2003). Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol Psychiatry 54:947–949. [DOI] [PubMed] [Google Scholar]
- Xu W, Südhof TC. (2013). A neural circuit for memory specificity and generalization. Science 339:1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, McFarlane AC, Shalev AY. (1998). Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biol Psychiatry 44:1305–1313. [DOI] [PubMed] [Google Scholar]
- Zoladz PR, Diamond DM. (2013). Current status on behavioral and biological markers of PTSD: a search for clarity in a conflicting literature. Neurosci Biobehav Rev 37:860–895. [DOI] [PubMed] [Google Scholar]