

Abstract
Tuberous Sclerosis Complex (TSC) is an autosomal disease caused by inactivating mutations in either of the tumor suppressor genes TSC1 or TSC2. TSC-associated tumor growth is present in multiple tissues and organs including brain, kidney, liver, heart, lungs, and skin. In the kidney, TSC angiomyolipomas have aberrant vascular structures with abnormal endothelial cells, suggesting a role for endothelial mTORC1 function. In the current report, a genetically engineered mouse model (GEMM) with a conditional knockout allele of Tsc1 with a Darpp32-Cre allele displayed accelerated formation of both kidney cystadenomas and paw hemangiosarcomas. All mutant mice developed hemangiosarcomas on multiple paws by 6 weeks of age. By 16 weeks of age the average mutant hind paw was 4.0 mm in diameter, nearly double the size of control mice. Furthermore, the hemangiosarcomas and kidney cystadenomas were responsive to intraperitoneal rapamycin treatment. Immunoblotting and immunostaining for phospho-S6 (pS6) and phospho-CAD showed that that the effect of rapamycin on tumor size was through inhibition of the mTOR signaling pathway. Finally, elevated VEGF mRNA levels were also observed in hemangiosarcoma specimens. Since paw hemangiosarcomas are easily detectable and scorable for size and growth, this novel mouse model enables accelerated in vivo drug-testing for therapies of TSC related tumors.
Keywords: DARPP-32, Tsc1, tuberous sclerosis, mTOR, angiosarcoma, hemangiosarcoma
Introduction
Tuberous Sclerosis Complex (TSC) is a genetic disorder due to an inactivating mutation in either TSC1 or TSC2. The protein products of TSC1 and TSC2, hamartin and tuberin respectively, form a heterodimeric complex that inhibits the mechanistic target of rapamycin complex 1 (mTOR) pathway via RHEB-GTP modulation (1, 2). Activation of mTOR primarily regulates cell size and proliferation through promotion of translation (1). Germline TSC1 or TSC2 mutations are complemented by second hit loss of the wild type allele leading to hyperactive mTOR activity, which results in the growth of tumors in brain (subependymal nodules and giant cell astrocytomas), kidney (renal angiomylipoma), and lung (pulmonary lymphyangioleiomyomatosis) (3–5). In addition to tumor growth, a combination of intellectual disability, epilepsy, and autism occur in a majority of individuals with TSC (2). Most tumors that arise due to TSC are of a benign phenotype (4). However, renal angiomyolipomas can proliferate, and subsequent renal complications are cited as the most common cause of death in adult TSC patients. Thus, therapies that inhibit hyperactive mTOR pathway are candidates for treatment of TSC related tumors.
Rapamycin and related compounds (rapalogues) have been studied with regard to the inhibition of mTOR and treatment of TSC related tumor growth (3, 6). In multiple murine models of TSC, treatment of TSC related tumors is effective in reducing tumor size (7–9). Clinically, rapalogue treatment has been shown to result in regression of both renal angiomyolipomas (10, 11) and subependymal giant cell astrocytomas (12, 13). However rapalogue treatment does not lead to tumor elimination, and cessation of treatment results in re-growth of tumors (10, 13, 14). Other drawbacks to the use of rapamycin are side effects ranging from diarrhea, stomatitis, respiratory infection, and pyelonephritis (10). Hence identification of more efficacious therapies is desirable to further the treatment of TSC related tumor growth and subsequent clinical management.
Both cell based and animal models have been used to identify therapeutics for treatment of TSC related tumors (15). However, both Tsc1+/− and Tsc2+/− mice develop kidney cystadenomas and liver hemangiomas as their main phenotype, tumors which do not match the pathology of human TSC (16–18). Furthermore, the development of kidney and liver tumors in murine models can take from 4 to 15 months of age with varying incidence rates (17, 18). In the present study, by combining a conditional knockout allele of Tsc1 with a Darpp32-Cre allele we created a mouse that develops kidney cystadenomas and hemangiosarcomas in the extremities on an accelerated time scale. Furthermore, we tracked paw hemangiosarcoma development by simple observation during rapamycin treatment, demonstrating its use as an easily scorable measure of tumor response. Our results demonstrate the usefulness of this model for preclinical testing of novel drugs for the treatment of TSC related tumors.
Material and Methods
Breeding strategy and phenotyping
All animal procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) and Animal Resources at Children’s Hospital (ARCH). Mouse experiments were performed on a mixed strain background. Mice bearing the loxP-flanked Tsc1 allele (Tsc1c/c) (19) were bred with mice bearing the Darpp-32 Cre allele, with further matings to generate Tsc1cc Darpp-32 Cre+ male mice (20). All other non-mutant male genotypes were used as littermate controls (Tsc1cw and Tsc1ww). Genotyping for lox-p sites for Tsc1 and for the Darpp-32 Cre was performed with standard polymerase chain reaction (PCR). Primers F4530 (‘5- AGGAGGCCTCTTCTGCTACC-3’) and R4380 (‘5- CAGCTCCGACCATGAAGTG-3’) were used for Tsc1 and Cre F (‘5-GGACATGTTCAGGGATCGCCAGGCG-3’ and Cre R (‘5-GCATAACCAGTGAAACAGCATTGCTG-3’) were used for the detection of the Darpp-32 Cre.
Tumor measurements
An observer (S.G.) blind to the genotype performed paw measurements. Hind paw diameter was measured using a 0.1 mm resolution caliper in the plantar to dorsal dimension. Mean paw diameters were determined by the average of both left and right paws. Paw diameters were measured biweekly until death. Individual mice were euthanized when paw diameter of ≥ 5 mm, weight loss of ≥ 20%, or greatly extreme lethargy was seen.
Treatment protocol
All mouse treatments commenced when the diameter of rear paw tumors averaged 4.0 mm. Mice treated with rapamycin (LC Laboratories, Cat. No: R-5000) were injected intraperitoneally at 6mg/kg in 100 uL of vehicle solution (5% Tween 80, 5% Polyethyl Glycol) every other day for one month. For statistical comparisons, one and two-way ANOVAs with subsequent Bonferroni corrections, Logrank survival, and two-tailed Student T-Tests were performed using Prism (GraphPad Software, Inc., v5.0f). Alpha level for all analyses was equal to 0.05.
Histology
Standard hematoxylin and eosin (H&E) sections were prepared from mouse kidney and paws taken immediately after death of an animal. Sections were fixed in 10% formalin for 12 hours prior to staining. For cyst diameter measurements, an observer blind to the treatment and age statuses (S.L.) analyzed sections on 4X magnifications. Cyst diameter measurements were taken by identifying the largest cross sectional diameter of a defined, enclosed cyst. For cyst counts, a cyst was counted if it was enclosed and greater than 3.5 × 10−3 mm in diameter. All imaging was performed on an Olympus BX51 microscope with a Qimaging Micropublisher 3.3 RTV camera and Qimaging QCapture software (Surrey, British Columbia, Canada).
Immunoblot Analysis
Rear extremity tumor and kidney extracts were prepared in lysis buffer (20mM Tris-HCl, 140mM NaCl, 10 mM NaF, 1mM Na3VO4, 1mM EDTA) and a Dounce homogenizer. Protein concentrations were determined by Bradford Assay (Biorad Laboratories, Hercules, CA), equal concentrations were separated by electrophoresis on 10–12% Bis-Tris acrylamide gels, and then transferred onto Immobilon-P PVDF membranes (EMD Millipore, Danvers, Massachusetts). Membranes were blocked with 5% nonfat dry milk in 1X TBST (Tris Buffered Saline – 10% Triton-X) for one hour at room temperature. Primary antibodies were diluted in 5% nonfat dry milk or 5% bovine serum albumin in 1X TBST and applied to the membranes overnight at 4°C. Primary antibodies included P-CAD (Cell Signaling, Beverly, MA), CAD (Cell signaling), p-S6–240 (Cell Signaling), S6–240 (Cell Signaling), and anti-Tubulin (Abcam, Cambridge, UK). After primary antibody incubation and washing with 1X TBST, anti-mouse (Santa Cruz, Dallas, TX), or anti-rabbit (Rockland, Gilbertsville, PA) HRP- conjugated secondary antibodies were applied for one hour at room temperature in 5% nonfat dry milk solution of 1X TBST. Supersignal West Pico and Dura Chemiluminescent (Thermo Fisher Scientific) signal solutions were used to detect antibody binding and signals were collected using an ImageQuant LAS 2000 imager (GE Healthcare, Piscataway, NJ).
Immunohistochemistry
Immunohistochemical analyses were performed on paraffin embedded sections of kidney and paw after 4% paraformaldehyde fixation for 12 hours; or on frozen OCT-embedded tissues following 4% paraformaldehyde fixation. Phospho-S6 Ser 240/244 (Cell Signaling, Cat. No: 2215) was used as a primary antibody at 1:500 dilution. Secondary antibodies included Alexa Fluor 488 Anti-Goat and Anti-Rabbit used at 1:500 dilution. For X-gal staining, tissue was harvested and fixed with 4% paraformaldehyde overnight, followed by standard sucrose cryopreservation and OCT embedding. Sections were sliced and then stained in X-gal staining solution overnight at 37°C followed by counterstaining with Nuclear Fast Red as previously described (21).
qPCR
All tissue samples from kidney and paws were extracted one week after the last day of treatment in a sterile fashion and immediately frozen in liquid nitrogen. RNA was precipitated by lysing tissue using TRIzol (Invitrogen, Carlsbad, CA) and a 22-gauge syringe followed by chloroform extraction. RNA was then reverse transcribed to cDNA and amplified as previously described for qPCR (22). Amplification and quantification were run with VEGF-A, VEGF-C, VEGF-D, and GAPDH primers designed with Primer 3 and sequences were as follows: VegF-A 5′-CCGAAACCATGAACTTTCTGC-3′ (forward) and 5′-GACTTCTGCTCTCCTTCTGTC-3′ (reverse), VEGF-C 5′-GAGGTCAAGGCTTTTGAAGGC-3′ (forward) and 5′-CTGTCCTGGTATTGAGGGTGG-3′ (reverse), VEGF-D 5′-TTGAGCGATCATCCCGGTC-3′ (forward) and 5′-GCGTGAGTCCATACTGGCAAG-3′ (reverse), GAPDH 5′- GACATGCCGCCTGGAGAAAC-3′ (forward) and 5′-AGCCCAGGATGCCCTTTAGT-3′ (reverse).
Results
A new TSC mouse model with rapid development of paw hemangiosarcoma
Our intent in creating a Tsc1cc Darpp-32-Cre+ mice was to develop a unique TSC brain model with Tsc1 loss in striatal neurons (23). Although DARPP-32 (dopamine and cAMP-regulated neuronal phosphoprotein 322 or protein phosphatase 1 regulatory subunit 1B2, gene name PPP1R1B) is expressed at highest levels in brain, it is also expressed in multiple other tissues, including pancreas, prostate, lung, thymus, and bone marrow (24). To confirm successful recombination and loss of expression of Tsc1, we used a LacZ reporter allele. X-gal staining of Tsc1cc Darpp-32-Cre+ mouse tissues demonstrated expression of the cre allele in the striatum and other cortical areas (data not shown). Separated PCR experiments revealed the animals that were mutant for TSC1 and Darpp-32-Cre positive (Supplementary Figure 1). X-gal staining (blue) was also seen in renal tubules of the outer medulla and cortex as well as the paws (Figure 1A). Tsc1cc Darpp-32-Cre+ mutant mice had significantly reduced weights compared to littermate controls (Tsc1ww and Tsc1cw Darpp-32-Cre+) beginning at P25, a trend that persisted throughout adulthood (Figure 1B, P < 0.05, two-way ANOVA). Similarly, the mutant mice showed significantly reduced survival with median lifespan of 25 weeks, compared to >80 weeks for controls (Figure 1C, P < 0.001, Logrank survival).
Figure 1.
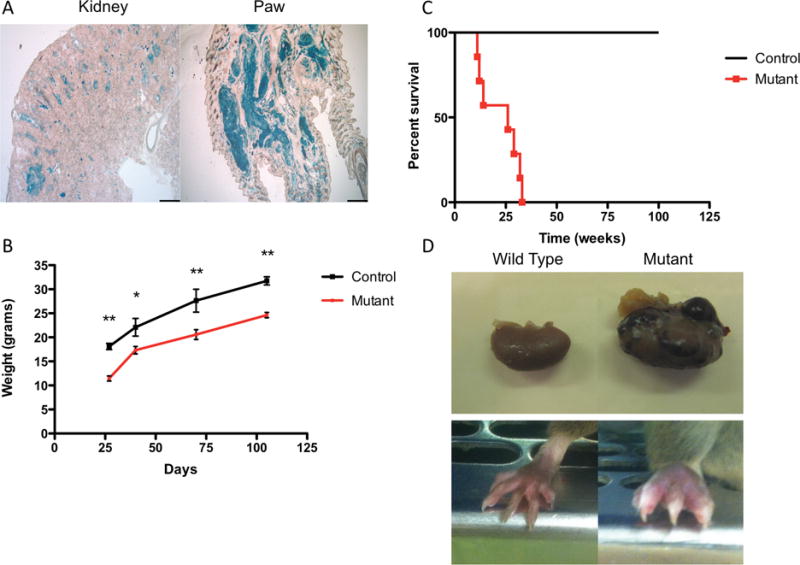
Loss of TSC1 in renal and paw tissue results in tumor formation. A) Representative images showing Lac-Z staining in 8 week old Tsc1cc Darpp-32-Cre+ kidney (left) and paw (right) tissue confirms recombination, 4X Scale bar = 500uM. Multiple tumor lesions are seen to have the blue stain. B) Tsc1cc Darpp-32-Cre+ mice show reduced mean (±SEM) weight from weaning through age 100 days. C) Tsc1cc Darpp-32-Cre+ mice have markedly reduced survival in comparison to wild type littermates. D) Gross anatomical photos taken from 17 week old Tsc1cw Darpp-32-Cre+ (left) and Tsc1cc Darpp-32-Cre+ (right) mice at sacrifice. Top highlights tumor formation in kidneys while bottom highlights increased paw size due to tumor formation. * indicates P < 0.05, ** indicates P < 0.01
Necropsy examination demonstrated that renal cystic and tumor lesions were present in all (n=11) mutant mice at the age of 8 weeks (Figure 1D). However, the most striking phenotype seen in these mice was the development of expansile tumor lesions on all four paws beginning at post-natal day 21 (Figure 1D). These progressive tumors distorted the normal anatomy of the paws. Histologically, these tumors were composed of pleiomorphic spindle cells arranged in interlacing bundles and whorling patterns. The tumors are highly vascularized and with the spindle cell shape and architecture of these vascular structures, these tumors were identified as hemangiosarcomas (Figure 2A–C, Supplementary Fig. 2A). Mitotic figures were rare. The edges of the tumors were well circumscribed but not encapsulated (Figure 2A–C). By six weeks of age all mutant mice (n=11) developed visible swelling and abnormal growth on hind paws. Similar tumor formation also occurred in the forepaws, but was of smaller extent (data not shown). Control mice (Tsc1ww and Tsc1cw Darpp-32-Cre+) showed no evidence of paw tumor formation (Figure 2D–F).
Figure 2.
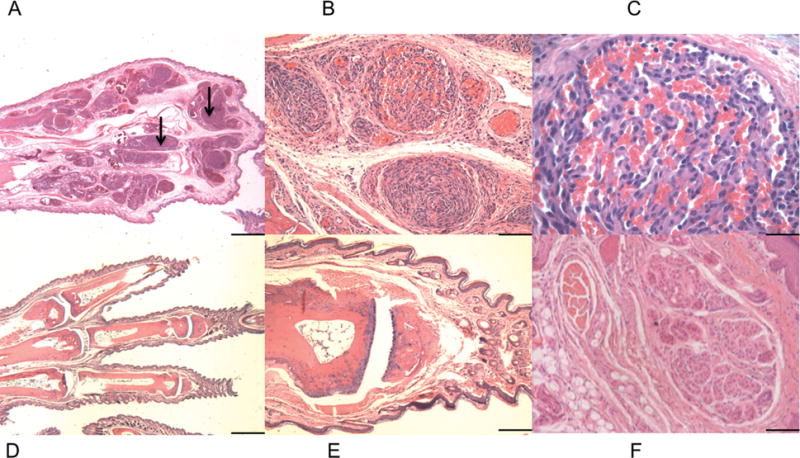
Tsc1cc DARPP Cre mouse paw pathology. A-F) Hematoxylin and Eosin staining of paw lesions show extensive clusters of aberrant vascular cells and multiple hemangiosarcomas. A–C) Paw from 8-week-old mutant mouse, at 2× (A), 20× (B), and 60× (C) magnification. Black arrows indicate paw hemangiosarcoma tissue. D–F) Paw from 8 week old wild type mouse at 2× (D), 10× (E), and 20× (F). 2× scale bar = 1 mm, 10× scale bar = 200 um, 20× scale bar = 100 um, 60× scale bar = 33 um.
Renal pathology
Pathologic evaluation demonstrated that polycystic kidney lesions were also seen at an early age in the Tsc1cc Darpp-32-Cre+ mutant mice, and were detectible in histologic sections by 8 weeks of age (Figure 3A). These lesions were cystadenomas (Figure 3A–C), similar to those described in both Tsc1+/− and Tsc2+/− mouse models (17, 18). They consisted of pure cysts with hyperplastic lining epithelial cells, partially filled cysts with papillary fronds of adenomatous growth, and solid adenomas. These lesions grew rapidly and were extensive by age 4 months, suggesting that they were a major contributor to death in these mice (Figure 3A–C, for high magnification imaging, see Supplementary Fig. 2B–C). Similar to the findings in the paws, there were no observed pathological abnormalities in the renal tissue of control mice (Figure 3D–F).
Figure 3.
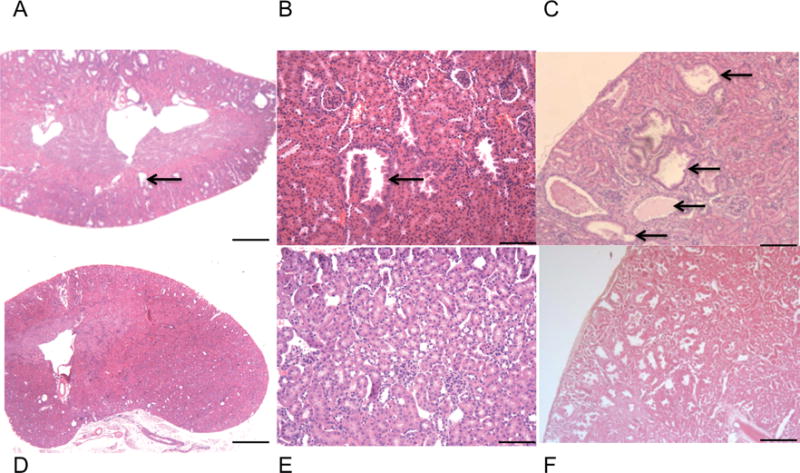
Tsc1cc DARPP Cre mouse renal pathology. A–C) Hematoxylin and Eosin staining of kidney lesions show extensive clusters of aberrant vascular cells and a variety of cystadenomas through adulthood. A) Kidney from 8-week-old mutant mouse, 2× magnification. B) Kidney from 8-week-old mutant mouse, 10× magnification. C) Kidney from 17-week-old mutant mouse, 4× magnification. Black arrows indicate cystadenomas. D) Kidney from 8-week-old wild type mouse, 2× magnification. E) Kidney from 8-week-old wild type mouse, 10× magnification. F) Kidney from 17-week-old wild type mouse, 4× magnification. 2× scale bar = 1 mm, 4× scale bar = 500um, 10× scale bar = 200 um.
Paw hemangiosarcomas provide an easily assessable tumor model of TSC related tumor growth
The paw hemangiosarcomas developed early in life (<4 months of age) and were easily assessed for size in a non-invasive, quantitative manner as shown by the difference in paw diameter between mutant and control mice (Figure 4A, P < 0.01 between mutant and control mice at all time points from P40 onwards, two-way ANOVA). We thus saw the potential for using tumor growth to assess therapeutic effectiveness. As an initial proof-of-principle, we treated these mice with rapamycin. Rapamycin was given by intraperitoneal (IP) injection at 6mg/kg three times per week for 30 days to a cohort of 11 mice beginning when the largest hemangiosarcoma reached a diameter of 4 mm (average age P102). We observed a significant reduction of 42.5% in hemangiosarcoma diameter after one month of therapy (Figure 4B, P < 0.001, one-way ANOVA). When rapamycin was discontinued after 30 days, there was significant regrowth of paw tumors (Figure 4B, P < 0.001, one-way ANOVA), consistent with observations in other TSC mouse models (25) and in TSC patients (10, 13).
Figure 4.
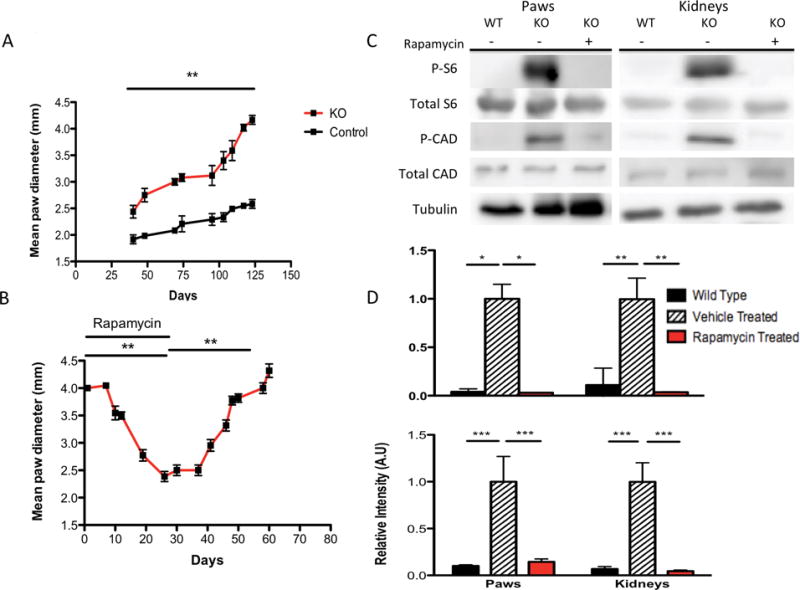
Progressive growth of paw hemangiosarcomas and response to rapamycin. A) Paw hemangiosarcomas grow progressively as seen by the mean (±SEM) paw diameter of largest hemangiosarcoma across all mice. B) Effect of rapamycin treatment on mean (±SEM) paw tumor diameter in Tsc1cc DARPP Cre mice. C) Immunoblotting of total S6-240, phospho-S6-S240, phospho-CAD, total CAD, and Tubulin expression in both paws and kidneys from wild type and Tsc1cc DARPP Cre vehicle and rapamycin treated mice. D) Normalized quantification of expression of phospho-S6-240 relative to total S6-240 (top) and phospho-CAD relative to total CAD (bottom) in both paws and kidneys from wild type and Tsc1cc DARPP Cre vehicle and rapamycin treated mice. n= 5 for phospho and total-S6-240 experimental groups, n=4 for each phospho and total-CAD experimental groups. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001.
To confirm that activation of mTORC1 was occurring in these tumors and that rapamycin was inhibiting this pathway, we examined tumor lysates by immunoblot analysis. High levels of phospho-S6 Ser240 expression were seen in kidney and paw lysates from vehicle treated mice (Figure 4C). After one week of rapamycin treatment, a significant reduction in mTORC1 activation was indicated by reduced phospho-S6 Ser40 staining relative to total S6 Ser240 staining in both kidney (Figure 4D, P < 0.01, one way ANOVA) and paw lysates (Figure 4D, P < 0.05, one way ANOVA). We also examined levels of phosphorylated and total-CAD expression and found that phospho-CAD expression was significantly elevated in vehicle treated kidney and paw lysates compared to both wild type and rapamycin treated lysates (Figure 4C and D, P < 0.001, one-way ANOVA). Histological analyses of kidneys from mutant mice revealed a significant reduction in mean cyst diameter was observed in mice treated with rapamycin (Figure 5A, P < 0.001, one-way ANOVA). Additionally, there was a significant reduction in mean renal cyst count of rapamycin treated mice (Figure 5B, P < 0.001, one-way ANOVA). However, the mean renal cyst count in rapamycin treated mice at 17 weeks was significantly greater than vehicle treated mice at 14 weeks (Figure 5B, P < 0.05, one-way ANOVA). Histological sections of vehicle treated mutant mice at 17 weeks (Figure 5C) and rapamycin treated mutant mice at 17 weeks (Figure 5D) show these reductions in cyst size and count. Immunohistochemistry analysis of paraffin embedded tissue sections of kidneys and paws confirmed the high levels of phospho-S6 in these tumors, consistent with mTORC1 activation due to loss of Tsc1, and showed that rapamycin treatment eliminated phospho-S6 expression in both kidneys and paws (Figure 6 A and B, respectively).
Figure 5.
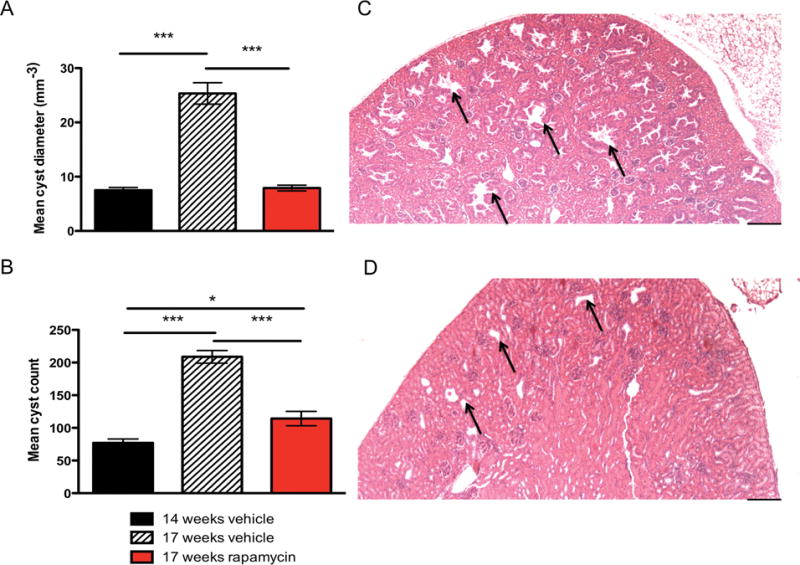
Histological analysis of kidney cyst diameter and load in Tsc1cc DARPP Cre mice. A) Mean cyst diameter in mm−3 measured in vehicle treated mice at 14 weeks, vehicle treated mice at 17 weeks, and rapamycin treated mice at 17 weeks. B) Mean cyst count measured in vehicle treated mice at 14 weeks, vehicle treated mice at 17 weeks, and rapamycin treated mice at 17 weeks. C) 4X magnification of 17-week old mutant vehicle treated mouse kidney showing extensive cyst formation with arrows highlighting the largest of the representative cysts. D) 4× magnification of 17-week old mutant rapamycin treated mouse kidney showing significantly reduced size of cysts, as highlighted by arrows. 4× scale bar = 500um. * indicates P < 0.05, *** indicates P < 0.001
Figure 6.
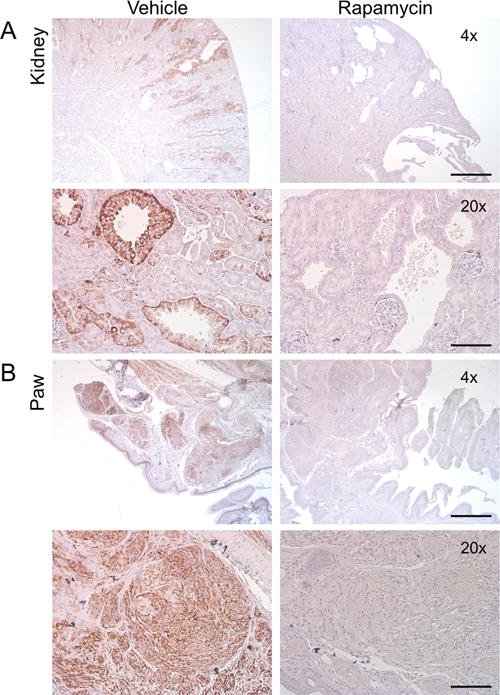
Paw hemangiosarcomas and kidney cystadenomas response to rapamycin treatment. Immunostaining of phospho-S6-S240 expression (brown) in (A) kidneys and (B) paws after vehicle or rapamycin treatment, 4× scale bar = 500um, 20× scale bar = 100 um.
VEGF mRNA expression and loss of TSC1
We had noted earlier that the paw hemangiosarcomas are deep red in appearance, suggesting that these tumors are highly vascularized. Prior findings showed that loss of Tsc1 or Tsc2 results in increased VEGF levels, which are associated with tumor development and responsive to rapamycin treatment (26, 27). Using qPCR, we observed significantly increased VEGF-A mRNA levels in mutant paws but not kidneys, compared to controls (Figure 7A, P < 0.001, Student T-test). In addition to VEGF-A mRNA levels, we also examined VEGF-C and VEGF-D mRNA levels. Compared to control mutant mice, paws from mutant mice expressed significantly highly amounts of both VEGF-C and VEGF-D mRNA expression (Figure 7B–C, P < 0.001 and 0.05 respectively, Student T-test). Furthermore, kidneys from mutant mice showed significantly elevated VEGF-D mRNA levels when compared to controls (Figure 7C, P < 0.05, Student T-Test).
Figure 7.
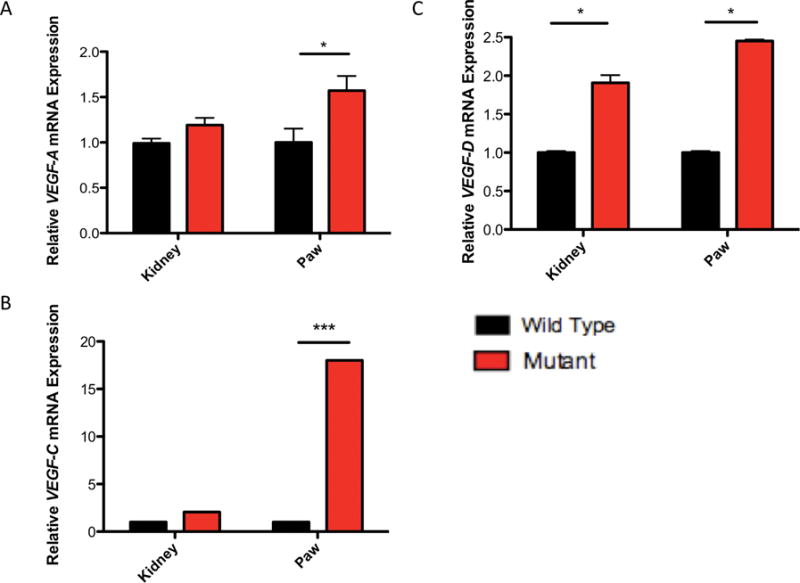
VEGF mRNA levels are elevated in Tsc1cc DARPP Cre mouse paws and kidneys. A) Relative expression of VEGF-A mRNA transcripts harvested from 17-week old control and mutant paws and kidneys. B) Relative expression of VEGF-C mRNA transcripts harvested from 17-week old control and mutant paws and kidneys. C) Relative expression of VEGF-D mRNA transcripts harvested from 17-week old control and mutant paws and kidneys. All measurements are standardized to GAPDH. * indicates P < 0.05, *** indicates P < 0.001
Discussion
In this study, we generated a new mouse tumor model in which targeted loss of Tsc1 in both the paws and the kidneys leads to rapid and progressive tumor development. In TSC patients, hamartomas appear in multiple organs due to random genetic second hits events that lead to complete loss of TSC1 or TSC2 expression and subsequent downstream activation of mTORC1. However, there are still many aspects of tumor formation in TSC that are uncertain, including the cell of origin for most TSC hamartomas, the precise molecular mechanisms of tumor progression, and the exact role of mTORC1 activation in driving tumor growth. Multiple therapeutic strategies have been identified in cell-based or biochemical studies that may provide significant control in TSC tumor growth. In this set of experiments, we took advantage of the fortuitous rapid development of paw and renal tumors in a fully viable mouse model system, Tsc1cc Darpp-32-Cre+ mice. Kidney cystadenomas and paw hemangiosarcomas were seen by P42 and P21, respectively, and enabled rapid assessment of in vivo therapies for TSC tumors. Previous mouse models of TSC related tumor growth report that extremity tumors (tail, paw, or lip) can take up to 12 months to develop (17). Furthermore, only 7% of the mutant mice in a previous model (17) developed these extremity tumors. Our conditional knockout mouse model demonstrates full penetrance of paw hemangiosarcomas by 6 weeks of life. Finally, another considerable advantage of this TSC model is that a non-invasive readout of therapeutic strategies can be performed without the need to sacrifice the mouse.
In a preliminary proof of concept drug trial, we demonstrated that rapamycin was an effective therapy for both paw hemangiosarcomas and renal cystadenomas. Most importantly, the progress of treatment could be followed through simple measurement of the paw hemangiosarcomas. Biochemically, we also provide evidence from phospho-S6 and phospho-CAD staining that this effect of rapamycin treatment is likely through inhibition of the hyperactive mTORC1 pathway in Tsc1cc Darpp-32-Cre+ mice. As it is well established that TSC tumor growth is reversed via this mechanism in both humans (10–13) and mice (7, 8), our model provides the specific development of TSC related tumors that can be assessed for responsiveness to therapeutics. Non-invasive measurement of the paw hemangiosarcomas highlights the usefulness of this model, given that the renal phenotype is also responsive to rapamycin treatment, as evidence by both the biochemical data and reduction in mean cyst diameter and apparent cyst load.
Further support for the conclusion that Tsc1cc Darpp-32-Cre+ mice present a robust model for TSC related tumor growth is evidenced by VEGF analyses. Beyond TSC related tumors, VEGF-A expression has been shown to drive tumor angiogenesis and metastasis (28). Prior research in both TSC cell based assays (27, 29, 30) and tumor tissue from patients (31) shows that hyperactivation of mTORC1 results in an increased production of hypoxia induced factor-1α (HIF-1α), a transcription factor which results in an increased expression of HIF-responsive genes such as VEGF-A (29). Furthermore, mouse models have been reported that show increased serum VEGF-A expression in Tsc1+/− mice, which was also associated with the extent of tumor development (27, 31). Interestingly a reduction in serum VEGF-A expression was observed as a result of short-term rapamycin treatment (4 days at 20 mg/kg), which also was associated with tumor reduction (27). In our model we observed an increase in VEGF-A, C, and D mRNA expression in mutant mouse paws, likely due to mTORC1 hyperactivation and subsequent tumor formation. We also observed an increase in VEGF-D mRNA levels from mutant kidneys as compared to kidneys from control mice.
Our findings with respect to VEGF mRNA levels support the conclusion that the Tsc1cc Darpp-32-Cre+ mouse is an accurate model of TSC-related tumor growth. As described, previous work has repeatedly shown that VEGF protein levels are elevated in TSC deficient mice, a phenotype that is rescued by rapamycin treatment (27, 29). These results have been examined at the protein level while the present findings begin to suggest differences in subtypes of VEGF expression at the mRNA transcript level. While we did not observe significantly increased levels of VEGF-A or C mRNA from kidneys, our analysis of VEGF-D showed the expected increase in VEGF levels from kidneys of mutant mice. VEGF-D has been proposed a potential biomarker of lymphangioleiomyomatosis severity and treatment response(32). Recent work indicates that both VEGF-A and VEGF-D can be detected in the sera of TSC patients (33). Taken together, these results indicate the similar mechanisms may be at play in tumorogenesis in Tsc1cc Darpp-32-Cre+ mouse as in patients with TSC.
The present evidence suggests that the Tsc1cc Darpp-32-Cre+ mouse can be an effective tool for the study of pharmaceutical treatments of TSC-related tumors. While there are uncertainties as to both the genetics of these tumors in terms of their abundance as well as their cell size/morphological features when compared to human tumors, this model presents a valid, quantitative, in vivo model for the testing of potential therapeutics for the treatment of TSC related tumors.
Supplementary Material
Implications.
These findings provide a strong rationale for simultaneous use of this conditional knockout mouse as an in vivo genetic model while seeking new cancer therapies for TSC related tumors.
Acknowledgments
We would like to thank Dr. Elizabeth P. Henske for assistance with treatment methods. We are grateful for the help of the Kwiatkowski lab members who aided with the use of equipment and the Sahin Lab members who contributed to the development of this project. We would also like to thank Dr. Joanne Chan for her intellectual contribution. Finally, we thank the Rodent Histopathology Core of the Dana Farber/Harvard Cancer Center, mouse histology preparation and interpretation. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516.
Financial Support: This study was funded by NIH grants R37NS031535 and P01CA120964, the Tuberous Sclerosis Alliance, Boston Children’s Hospital Translational Research Program and the Manton Family Foundation to M.S.
Footnotes
Conflict of Interest: There are no conflicts of interest to report.
Contributor Information
Jarrett D. Leech, Email: jdleech@gmail.com.
Stephen H.T. Lammers, Email: stephen.lammers@childrens.harvard.edu.
Sam Goldman, Email: segoldma@gmail.com.
Neil Auricchio, Email: neilaa@comcast.net.
Roderick T. Bronson, Email: roderick_bronson@hms.harvard.edu.
David J. Kwiatkowski, Email: dk@rics.bwh.harvard.edu.
Mustafa Sahin, Email: Mustafa.Sahin@childrens.harvard.edu.
References
- 1.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–22. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011;585:973–80. doi: 10.1016/j.febslet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auricchio N, Malinowska I, Shaw R, Manning BD, Kwiatkowski DJ. Therapeutic trial of metformin and bortezomib in a mouse model of tuberous sclerosis complex (TSC) PLoS One. 2012;7:e31900. doi: 10.1371/journal.pone.0031900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borkowska J, Schwartz RA, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011;50:13–20. doi: 10.1111/j.1365-4632.2010.04727.x. [DOI] [PubMed] [Google Scholar]
- 5.Prabhakar S, Goto J, Zhang X, Sena-Esteves M, Bronson R, Brockmann J, et al. Stochastic model of Tsc1 lesions in mouse brain. PLoS One. 2013;8:e64224. doi: 10.1371/journal.pone.0064224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Y, Kwiatkowski DJ. Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Mol Cancer Res. 2013;11:467–73. doi: 10.1158/1541-7786.MCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollizzi K, Malinowska-Kolodziej I, Stumm M, Lane H, Kwiatkowski D. Equivalent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in a mouse model of tuberous sclerosis. Mol Cancer. 2009;8:38. doi: 10.1186/1476-4598-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dabora SL, Franz DN, Ashwal S, Sagalowsky A, DiMario FJ, Jr, Miles D, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF- D levels decrease. PLoS One. 2011;6:e23379. doi: 10.1371/journal.pone.0023379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res. 2005;57:67–75. doi: 10.1203/01.PDR.0000147727.78571.07. [DOI] [PubMed] [Google Scholar]
- 10.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–24. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 12.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 13.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–8. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 14.Wienecke R, Fackler I, Linsenmaier U, Mayer K, Licht T, Kretzler M. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis. 2006;48:e27–9. doi: 10.1053/j.ajkd.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Kwiatkowski DJ. Animal models of lymphangioleiomyomatosis (LAM) and tuberous sclerosis complex (TSC) Lymphat Res Biol. 2010;8:51–7. doi: 10.1089/lrb.2009.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finlay GA, Malhowski AJ, Polizzi K, Malinowska-Kolodziej I, Kwiatkowski DJ. Renal and liver tumors in Tsc2(+/−) mice, a model of tuberous sclerosis complex, do not respond to treatment with atorvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Mol Cancer Ther. 2009;8:1799–807. doi: 10.1158/1535-7163.MCT-09-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104:687–95. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–34. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 19.Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–58. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bogush AI, McCarthy LE, Tian C, Olm V, Gieringer T, Ivkovic S, et al. DARPP-32 genomic fragments drive Cre expression in postnatal striatum. Genesis. 2005;42:37–46. doi: 10.1002/gene.20118. [DOI] [PubMed] [Google Scholar]
- 21.Kwon CH, Zhou J, Li Y, Kim KW, Hensley LL, Baker SJ, et al. Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis. 2006;44:130–5. doi: 10.1002/gene.20197. [DOI] [PubMed] [Google Scholar]
- 22.Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29:5926–37. doi: 10.1523/JNEUROSCI.0778-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiroi N, Fienberg AA, Haile CN, Alburges M, Hanson GR, Greengard P, et al. Neuronal and behavioural abnormalities in striatal function in DARPP-32-mutant mice. Eur J Neurosci. 1999;11:1114–8. doi: 10.1046/j.1460-9568.1999.00570.x. [DOI] [PubMed] [Google Scholar]
- 24.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–9. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 25.Lee N, Woodrum CL, Nobil AM, Rauktys AE, Messina MP, Dabora SL. Rapamycin weekly maintenance dosing and the potential efficacy of combination sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberous sclerosis preclinical models. BMC Pharmacol. 2009;9:8. doi: 10.1186/1471-2210-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen-Vu PA, Fackler I, Rust A, DeClue JE, Sander CA, Volkenandt M, et al. Loss of tuberin, the tuberous-sclerosis-complex-2 gene product is associated with angiogenesis. J Cutan Pathol. 2001;28:470–5. doi: 10.1034/j.1600-0560.2001.028009470.x. [DOI] [PubMed] [Google Scholar]
- 27.El-Hashemite N, Walker V, Zhang H, Kwiatkowski DJ. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003;63:5173–7. [PubMed] [Google Scholar]
- 28.Kaelin WG., Jr How oxygen makes its presence felt. Genes Dev. 2002;16:1441–5. doi: 10.1101/gad.1003602. [DOI] [PubMed] [Google Scholar]
- 29.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4:147–58. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 30.Liu MY, Poellinger L, Walker CL. Up-regulation of hypoxia-inducible factor 2alpha in renal cell carcinoma associated with loss of Tsc-2 tumor suppressor gene. Cancer Res. 2003;63:2675–80. [PubMed] [Google Scholar]
- 31.Parker WE, Orlova KA, Heuer GG, Baybis M, Aronica E, Frost M, et al. Enhanced epidermal growth factor, hepatocyte growth factor, and vascular endothelial growth factor expression in tuberous sclerosis complex. Am J Pathol. 2011;178:296–305. doi: 10.1016/j.ajpath.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young L, Lee HS, Inoue Y, Moss J, Singer LG, Strange C, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1:445–52. doi: 10.1016/S2213-2600(13)70090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siroky BJ, Yin H, Dixon BP, Reichert RJ, Hellmann AR, Ramkumar T, et al. Evidence for pericyte origin of TSC-associated renal angiomyolipomas and implications for angiotensin receptor inhibition therapy. Am J Physiol Renal Physiol. 2014;307:F560–70. doi: 10.1152/ajprenal.00569.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.