

Abstract
With the advent of antiretroviral therapy that can control virus replication below the detection levels of conventional assays, a new clinical landscape of AIDS emerged, in which non-AIDS complications prevail over AIDS-defining conditions. These comorbidities are diverse and affect multiple organs, thus resulting in cardiovascular, kidney, neurocognitive and liver disease, osteopenia/osteoporosis, and cancers. A common feature of these conditions is that they are generally associated with accelerated aging. The mechanism behind these comorbidities is chronic excessive inflammation induced by HIV infection, which persists under antiretroviral therapy. Progressive simian immunodeficiency virus (SIV) infection of nonhuman primates (NHPs) closely reproduces these comorbidities and offers a simplified system in which most of the traditional human risk factors for comorbidities (i.e., smoking, hyperlipidemia) are absent. Additionally, experimental conditions can be properly controlled during a shorter course of disease for SIV infection. As such, NHPs can be employed to characterize new paradigms of AIDS pathogenesis and to test the efficacy of interventions aimed at alleviating non-AIDS-related comorbidities.
Keywords: Human immunodeficiency virus, Simian immunodeficiency virus, Chronic inflammation, Hypercoagulation, HIV-associated neurocognitive disease (HAND), Microbial translocation, Aging, Dysbiosis, Chronic obstructive pulmonary disease (COPD), HIV-associated nephropathy (HIVAN)
Introduction: the New Clinical Landscape of AIDS
The advent of antiretroviral therapies (ART) is one of the most prominent accomplishments of modern medicine, which dramatically changed the clinical landscape of human immunodeficiency virus (HIV) infection. With therapies, the life expectancy of a motivated patient has increased to 59 years, albeit not yet fully restored [1]. ART has other multiple limitations: (a) it requires life-long adherence preventing effective treatment from being delivered in a sustainable way to all in need; (b) it is associated with short- and long-term toxicity; (c) it does not completely restore immune integrity; and (d) it is not curative and does not eradicate HIV-1 [1]. Additionally, patients that are effectively treated have a higher risk of non-AIDS-related overall morbidity and mortality compared to uninfected individuals of the same age [2••]. Non-AIDS comorbidities are diverse and affect multiple organs thus resulting in cardiovascular (CV), kidney, neurocognitive and liver disease, osteopenia/osteoporosis, and cancers. A common feature of these conditions is that they are generally associated with aging. Indeed, HIV-infected patients experience frailty and other geriatric signs earlier than the general population [2••].
Several pathogenic mechanisms are at the origin of non-AIDS comorbidities observed in the HIV-infected patient on ART, such as higher frequency of risk factors (substance abuse, diet, obesity, hypertension), drug side effects and toxicity, and, most importantly, virus-induced immune dysfunction and chronic inflammation. In infected patients in whom HIV-1 replication is controlled with ART, the failure of immune recovery is associated with high levels of T cell activation [3], inflammatory monocyte activation [4], and increased levels of inflammatory biomarkers [5, 6, 4]. Furthermore, the biomarkers of hypercoagulability are also increased in chronically infected patients. The elevations of inflammatory and coagulation biomarkers are independent predictors of significant increases in morbidity and mortality in HIV-infected patients compared to the general population [6, 4]. These features are so consistently observed in HIV-infected patients that it was recently proposed that they can comprise a new HIV-associated syndrome [7•].
Understanding how chronic inflammation and hypercoagulation occur in HIV-infected subjects may generate new therapeutic paradigms to control end-organ diseases. These new approaches must be tested preclinically in animal models. Here, we discuss the utility of nonhuman primate (NHP) models for the study of HIV-related comorbidities and of therapeutic strategies aimed at controlling them.
NHP Models for AIDS Research
NHP models are instrumental for studies of pathogenesis, transmission and prevention of HIV infection. Both HIVs have a simian origin, with HIV-1 originating from chimpanzees/gorillas and HIV-2 originating from multiple cross-species transmissions from sooty mangabeys [8]. As both chimpanzees and sooty mangabeys are highly endangered and therefore cannot be used for controlled studies, alternative models are used that mainly rely on the use of Asian macaques [9]. Macaques are not naturally infected with simian immunodeficiency virus (SIV) in the wild, but upon experimental infection with SIVs, they develop a clinical syndrome that is remarkably similar to AIDS in humans [9]. SIV infection in macaques recapitulates the most important features of HIV infection: (i) the development of a chronic progressive infection that eventually progresses to simian AIDS (characterized by opportunistic infections, cancers, and neuropathology-neuro-AIDS); (ii) the occurrence of a controlled infection in a minority of macaques harboring specific MHC class I alleles, similar to human elite controllers; (iii) similar viral replication and CD4+ T cell depletion patterns and alteration of the same memory T cell subsets; (iv) the failure of immune responses to contain the virus; (v) the similar pathogenic consequences of viral replication, with mucosal dysfunction, microbial translocation, and persistent immune activation and inflammation; and (vi) the persistence of the virus in cellular reservoirs even after complete suppression with antiretroviral therapy [9, 10].
As such, it is considered that macaques infected with SIV represent one of the best animal models for a human disease in the history of modern medicine [9, 10]. The only major limitation to the use of this model is that HIV-1 does not infect macaques because of multiple host restriction factors [11]. This limitation has largely been surpassed by using SIV for macaque infection, as multiple SIV strains infect macaques. SIVsmm/SIVmac, which are derived from sooty mangabeys, similar to HIV-2 [12] are the most widely used. SIVagm strains, which are derived from African green monkeys (AGMs) [8], are used for infecting pigtailed macaques (PTMs), in which upon passage induces a progressive infection [13, 14], and rhesus macaques, in which SIVagm is functionally cured [15]. Furthermore, artificial chimeras including HIV-1 genes in a backbone of SIVmac, called simian-human immunodeficiency viruses (SHIVs), have been constructed for vaccine and therapeutic studies [16]. More recently, HIV-1-like chimeras were constructed with only minimal SIV content included only to counteract the host restriction factors [17].
Over the years, the use of the NHP model for AIDS permitted major advances in the field of HIV transmission, pathogenesis, prevention, and therapy [9, 10]. Comparative studies between pathogenic SIV infections in macaques and nonpathogenic SIV infections in their natural hosts (i.e., sooty mangabeys and AGMs) decisively contributed to the new paradigms of HIV pathogenesis in which chronic immune activation, dysfunction of the lymphoid tissues, and preferential depletion/preservation of different CD4+ T cell subsets are key contributors to AIDS progression [18, 9]. In this context, one of the key questions in the field is whether or not NHP models will be useful to study the HIV comorbidities that form the current clinical landscape of AIDS. In this review, we will provide compelling evidence that NHP models can and should be employed to model HIV comorbidities and new therapeutic approaches.
Use of NHPs to Study Immune Activation and Inflammation Associated With Chronic SIV Infection
Chronic immune activation and inflammation are the best predictors of HIV disease progression to AIDS [19]. In HIV-infected patients, immune activation is characterized by increased T cell turnover, high frequency of T cells that express an activated phenotype, polyclonal innate cell (B cells, monocytes/macrophages/myeloid, and plasmacytoid dendritic cells) activation, high levels of plasma cytokines and chemokines, and increased levels of inflammatory mediators [20, 2••].
The causes of chronic immune activation and inflammation in HIV-infected patients are multiple and intricate, therefore difficult to tackle: (i) the virus itself induces immune activation directly, with the levels of the “set-point” immune activation being at least partially correlated to those of virus replication [20], (ii) coinfection with other viral pathogens, including different herpesviruses [21], (iii) HIV-induced alterations of the mucosal barrier that results in the chronic translocation of the microbial products from the intestinal lumen to systemic circulation [22–25], and (iv) dysfunctional immunoregulatory factors [2••]. The chronic inflammation caused by the intricate action of these factors will in turn produce the early fibrosis of lymphoid tissues resulting in CD4+ T cell regenerative failure and disease progression to AIDS [26]. Inflammation is partially reversed by ART, but reversion is incomplete, and residual inflammation is responsible for the reported comorbidities.
Over the years, the use of NHP models was instrumental for generating this paradigm of HIV infection, in which inflammation is at the core of HIV pathogenesis [2••, 7•, 8–27]. Very strong support for the role of chronic immune activation and inflammation in the pathogenesis of AIDS came from the field of SIV infection in their natural hosts. In these species, which do not develop AIDS despite high levels of virus replication, an acute increase in the levels of immune activation and inflammation [18–28] is resolved to preinfection levels during chronic infection [29]. Furthermore, chronic SIV infection in natural hosts does not undermine the integrity of mucosal barrier, and consequently, there is no increase in microbial translocation [30, 31]. Therefore, there is a general consensus that lack of disease progression in natural hosts is due to their ability to avoid the negative consequences of SIV-related inflammatory responses. We confirmed the impact of chronic immune activation and inflammation on driving disease progression by administering Ontak (an IL-2 coupled with a diphtheria toxin, which targets CD25, a molecule expressed on Treg surface) to chronically SIVagm-infected AGMs. Ontak administration resulted in the increased levels of immune activation and inflammation and consequently enhanced viral replication and mucosal CD4+ T cell depletion [32]. In other sets of experiments, we mimicked microbial translocation by administering lipopolysaccharide (LPS) to chronically SIVsab-infected AGMs, both in single dose [32] and in prolonged administration [33••]. In both experiments, LPS increased the levels of systemic immune activation and inflammation and boosted viral replication [33••, 32]. Finally, in an unpublished work, we demonstrated that alcohol-induced mucosal alterations (and microbial translocation) resulted in increased levels of systemic immune activation and inflammation in chronically infected AGMs. Altogether, these experiments provided strong support to the fact that immune activation/inflammation are key factors of disease progression. Moreover, these studies validate microbial translocation as a major driver of immune activation.
Conversely, macaques infected with SIV recapitulate all the biological features of chronic HIV infection, and thus present with increased levels of chronic immune activation, inflammation, and microbial translocation [34, 14, 33••, 31]. The macaque model is therefore ideally suited for testing therapeutic strategies aimed at breaking the vicious circle of HIV pathogenesis and contribute to the normalization of chronic inflammation and immune activation, which may complement ART in HIV-infected patients [35••, 36••, 37].
Use of NHPs to Model HIV-related Aging
With more than half of HIV-infected patients in the USA anticipated to be over 50 years of age by 2015, the overall risk of noninfectious complications will be significantly higher and will become an important challenge for the management of chronic HIV infection [38]. Chronic inflammatory disease becomes apparent by the fifth decade of life, coinciding with the end of most procreation and raising a family [39]. Certain chronic infections (such as the cytomegalovirus infection) which have no discernible effect on young and middle-aged individuals are clearly associated with complications of aging and mediating diseases in individuals at risk [40]. Furthermore, T cell activation does not appear to have a direct effect on HIV disease progression in young adults but has a discernible effect in patients over 50 years [2••].
Interestingly, the inflammatory and coagulation abnormalities, as well as the levels of associated biomarkers (i.e., D-dimer, IL-6, C-reactive protein, sCD14) predictive for HIV disease progression and mortality in middle-aged HIV-infected patients, are strikingly similar to those observed in much older uninfected subjects, in which the same markers are known to be predictive of morbidity and mortality [41].
In older HIV-infected patients, a higher risk of death is related to a higher incidence of noninfectious complications, including CV disease and other chronic diseases related to aging [42]. Furthermore, treatment initiation at an older age is consistently associated with a less effective CD4+ T cell restoration and a higher risk for development of non-AIDS related complications [42]. This prompted the US Department of Health and Human Services (DHHS) to recommend the initiation of ART in all HIV-infected patients >50 years of age, regardless of their CD4+ T cell counts (http://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/10) [43]. However, ART alone does not eliminate the increased risk of death due to non-AIDS comorbidities in elderly patients, which is currently a major clinical problem for the management of HIV infection. Yet, to date, there is no efficient therapeutic solution to this situation.
In macaques, aging is associated with similar immune senescence, with a switch toward a proinflammatory status [44]. While there is a reduction in the overall levels of white blood cells (WBCs) [45], some cell subtypes (i.e., lymphocytes, monocytes, and polymorphonuclear cells) increase with age. One study reported no significant decline in NK and T cells but a significant decline of B cells in aged macaques [46]. Changes in the macaque T cell subsets include significant declines in CD4+ and CD8+ naïve T cells and increases of the CD4+ and CD8+ effector memory T cells occurring with age [46].
Interestingly, an age-related expression of IL-8 and IL-6 was reported to occur in airway epithelium after LPS exposure in macaques confirming the age-related proinflammatory status [47].
This overall proinflammatory switch characteristic of aged macaques might increase the levels of target cells for the virus and thus result in a more pathogenic infection. In other virus infections, such as influenza or SARS, higher levels of viral replication were reported to occur in aged animals [48].
These increases in the proinflammatory status are shared between pathogenic and nonpathogenic models of SIV infection. In our unpublished studies, we observed that the age-related alterations in immune cell populations were similar between uninfected macaques and AGMs. We also documented age-related increases in the levels of D-dimer in wild AGMs [49]. Interestingly, while SIV infection is generally nonpathogenic in natural hosts, we and others have described a handful of AIDS cases in AGMs, mandrills, sooty mangabeys, and black mangabeys [50], the vast majority of which occurred in older NHPs. Another interesting observation was that all these animals had increased persistent levels of immune activation (i.e., significantly increased Ki-67 expression in the lymph nodes) [50]. This was surprising, as it is well established that the levels of immune activation markers do not significantly change from the baseline in chronically SIV-infected NHPs that are natural hosts [50]. Finally, a common pathological feature in the old African NHPs that progressed to AIDS was a condition characterized by the severe accumulation of infected macrophages in tissues known as “giant cell disease” [50]. Significant increases in neopterin, an inflammatory marker related to monocyte activation, were also observed in the AIDS cases occurring in the elder NHP hosts. Altogether, these findings suggest that increased levels of immune activation and inflammation (particularly macrophage activation) that occur in older animals may drive progression to AIDS even in species that are generally resistant to disease progression.
In summary, similar to humans [38], NHPs develop chronic systemic inflammation with age. While this age-related inflammatory status results in the development of well-documented degenerative effects in elderly patients [38], the impact of age-related inflammation on HIV/SIV pathogenesis is not well studied.
Use of NHPs to Model and Control Gastrointestinal (GI) Tract and Liver Dysfunction
The GI tract is one of the systems most affected by HIV infection. The intestine contains the largest number of CD4+ T lymphocytes [22]. Even more important, the predominant CD4+ T cell subset in the gut is the effector memory cell, which expresses high levels of CCR5, the main HIV co-receptor [22]. As such, the intestine is the main site of HIV replication and CD4+ T cell depletion [22] early in the disease course. In addition to these direct effects, HIV exerts a major indirect effect in the gut by inducing epithelial cell activation and apoptosis [22]. Altogether, these processes result in a loss of mucosal integrity and translocation of microbial products from the intestinal lumen to systemic circulation [22]. Antiretroviral therapy may alleviate these effects, but control of microbial translocation is incomplete with ART [22] and is a leading cause of residual inflammation, which in turn results in incomplete immune restoration, hypercoagulation, and comorbidities. The role of microbial translocation in inducing inflammation and hypercoagulability was directly validated in NHP models [51•, 36••].
Intestinal permeability is also increased in multiple pathogenic conditions, such as inflammatory bowel disease, pancreatitis, graft-versus-host disease, excessive alcohol consumption, as well as obesity and diabetes or with age [25, 22]. The extensive studies of this process show that microbial products can stimulate the innate and adaptive effectors both locally and systemically. In addition to these effects on the immune system, microbial products may also stimulate nonimmune cells (i.e., endothelial cells of the CV system) [52]. In HIV-infected patients, microbial translocation has characteristic features: thus, generalized immune activation is associated with preferential loss of cell subsets involved in maintaining epithelial integrity at mucosal surfaces and antimicrobial immunity, such as the CD4+ T cells secreting IL-17 and IL-22 [53, 54]. These features were established by comparative studies of the pathogenesis of SIV infection in progressive and nonprogressive animal models.
Studies have also shown that HIV may induce gut dysbiosis, with an enrichment of microbial species that can catabolize tryptophan through the kynurenine pathway, and thus promote the loss of Th17 cells [55–57]. It was also reported that pathogenic, but not nonpathogenic, nonprogressive SIV infection also altered the enteric viral communities [58•]. Furthermore, dysbiosis may increase the production of trimethylamine-N-oxide, a potent proatherogenic factor [59], which would provide a mechanistic pathway between microbial translocation and the high incidence of CV disease in HIV-infected people [60].
In addition to gut leakage, microbial translocation may occur through inefficient metabolism of microbial products in the liver. Liver dysfunction is frequent in HIV-infected patients and may play a significant role in the pathogenesis of other comorbidities. Hepatic dysfunction may alter multiple critical coagulation factors which are produced by the liver, which may explain some of the coagulation abnormalities associated with HIV infection. It was indeed shown that HIV replication leads to short-term alterations of both pro- and anticoagulant factors [61]. To date, however, the effect of liver dysfunction on the overall outcome of HIV infection is not known. In the SIVsab PTM model, we observed frequent inflammatory infiltrates with mononuclear cells in the hepatic sinusoids or in the portal spaces and around the central vein. Sometimes, these infiltrates are severe (Fig. 1a) bridging several portal spaces, similar to the lesions observed in chronic active hepatitis. As a result, in some of the animals, we observed hepatic fibrosis that moderately alters the hepatic architecture (Fig. 1b). The effect of these modifications on NHP liver function is not yet known.
Fig. 1.
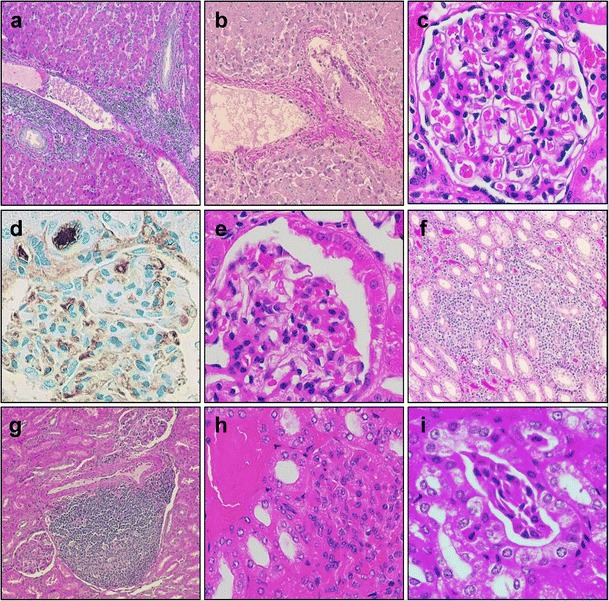
Liver and kidney lesions detected in SIVsab-infected NHPs. a Severe bridging mononuclear infiltrate peri portal and around the central veins (SIVsab/PTM, HE); b hepatic fibrosis (SIVsab/PTM, HE); c no kidney lessons are detected in SIVagm-infected African green monkeys; d microthrombi in the glomerular capillaries and afferent arterioles (SIVsab/PTM, IHC for fibrinogen); e parietal epithelial cell hyperplasia associated with collapsed capillaries (a condition involved in the development of focal, segmental glomerulosclerosis) (SIVsab/PTM, HE); f interstitial nephritis—diffuse mononuclear infiltration associated with hyaline casts in the renal tubules (SIVsab/PTM, HE); g interstitial pseudogranulomatous nephritis (SIVsab/PTM, HE)—interstitial nephritis with focal collections of lymphocytes with a germinal center-like area surrounded by other mononuclear cells; h interstitial nephritis (detail—dilated tubules with hyaline casts); i interstitial nephritis (detail—dilated tubules with cellular casts—adherent desquamated epithelial cells of the tubule lining (SIVsab/PTM, HE)
There are multiple mechanisms of liver dysfunction in HIV-infected patients: (i) effects of HIV infection: direct infection of Kupffer and stellate cells; indirect-through microbial translocation, chronic inflammation, and coagulopathy [2••]; (ii) action of cofactors, such as chronic alcohol consumption or coinfection with hepatitis C virus [62]; and (iii) ART toxicity [61].
Due to the key role played by the gastrointestinal tract in the pathogenesis of AIDS, multiple therapeutic approaches aimed at controlling immune activation and inflammation, microbial translocation, and dysbiosis are currently being evaluated, including in NHP models. Prebiotics and probiotics, to alter the bowel flora and reduce the levels of potentially pathogenic bacteria, have shown promising results in both NHP models [35••] and humans [63]. The administration of bovine colostrum, which binds LPS and may prevent translocation, had no clinical effect in HIV-infected subjects [64]. Sevelamer administration to acutely SIV-infected NHPs improved the levels of immune activation and coagulation [36••]; in chronically HIV-infected patients, sevelamer did not reduce microbial translocation but had a statistical effect on soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol [65]. In SIV-infected macaques, treatment with IL-21 resulted in a better preservation of the intestinal Th17 cells and reduced microbial translocation [66]. None of these studies provided a clear clinical improvement, but they collectively provided strong support for pursuing interventions aimed at alleviating the GI alterations in order to improve the clinical outcome of HIV infection.
Use of NHPs to Model CV Comorbidities
Accumulating evidence suggests that HIV-infected individuals have a higher rate of CV events versus uninfected persons. The proportion of deaths from heart disease doubled between 1996 and 2006, and health insurance claims data indicated a dramatic increase of acute myocardial infarction (MI) and coronary disease incidence in HIV-infected patients (sevenfold higher incidence compared to general population). CV conditions associated with HIV infection also include hypertension, metabolic disease (hyperglycemia, lipid disorders), and accelerated atherosclerosis leading to premature coronary artery disease. Interestingly, the increased risk of MI in patients with HIV infection versus age-matched uninfected patients is observed even after adjusting for Framingham risk factors and other comorbidities, suggesting that traditional models for estimating CV risk may underestimate risk in HIV patients and that there may be pathways in the pathogenesis of CV disease that are specific to HIV-infected patients [67].
The etiology, pathophysiology, and determinants of CV disease in HIV-infected patients are poorly understood, and the relative contributions of HIV itself, conventional risk factors, and ART to CV risk are unknown, thus constraining strategies to mitigate HIV-related CV disease. ART itself may contribute to CV disease (directly or through effects on lipid profiles); however, the discontinuation of ART appears to increase CV risk, implicating viral replication in the pathogenesis of CV disease. Furthermore, studies in NHPs clearly showed that CV disease occurs in the absence of ART thus reinforcing an independent role of SIV infection in the development of CV disease [33••]. Although traditional risk factors (smoking, hypertension, dyslipidemia) are highly prevalent in HIV-infected subjects, evidence strongly suggests that chronic inflammation is the main factor behind CV disease, as indicated by elevations in soluble and cellular biomarkers of inflammation, endothelial dysfunction, and hypercoagulation, processes which contribute to the pathogenesis of “conventional” CV disease.
The abnormal levels of coagulation factors have been consistently observed in HIV-infected patients for over 20 years [68, 69]. D-dimer (produced during clot lysis) has been associated with mortality and CV disease in HIV-infected patients [70, 6] and predicts venous thromboembolic disease in these patients [71]. D-dimer increases and hypercoagulation occur in both untreated patients [72, 6] as well as in patients receiving ART (albeit administration of ART results in a partial reduction in D-dimer levels [73, 74]). With regard to the sources of alteration in coagulation status, a general consensus is emerging in the field in support of a role of chronic inflammation characteristic to chronic HIV infection [70, 75]. Activated coagulation on the other side has the capacity to enhance inflammation thus creating a vicious circle of inflammation/hypercoagulation.
Our studies in NHPs established a strong correlation between chronic inflammation, monocyte activation, and hypercoagulation and HIV disease progression. In these studies, we compared pathogenic and nonpathogenic SIV infections (which, in spite of comparable levels of viral replication, have different levels of chronic immune activation and inflammation). These studies have shown that in AGMs and sooty mangabeys, the resolution of immune activation and inflammation are associated with normal levels of coagulation, while in pathogenic SIV infection of the PTMs, chronic inflammation and disease progression are associated with high levels of hypercoagulopathy [33••]. Both the increased levels of D-dimer (DD) and thrombin-antithrombin complex (TAT) strongly predicted disease progression and death in NHPs [33••], similar to HIV-infected patients [33••].
Also, our experimental studies demonstrated that interventions aimed at controlling persistent inflammation also lowered the biomarkers of hypercoagulation [33••, 34, 35••, 36••] in SIV-infected NHPs thus reinforcing a causative relationship between these two parameters.
The major limitation for addressing CV complications in HIV-infected patients is the interference of a plethora of confounding factors that cannot be dissected. First, CV disease may be due either to HIV infection or to ART itself [76, 6]. Hence, ART may have a dual effect on CV complications: either reducing the risk of CV events by reducing inflammation and coagulation or by increasing the CV risk through adverse metabolic effects (lipid changes) and vascular toxic effects. While the effects of the virus and treatments can be theoretically dissociated, this task is difficult to achieve, since most of HIV-infected patients are treated. Furthermore, the nature of the risk factors of CV disease (i.e., diet, alcohol and cigarette usage, sedentarism, obesity) further complicates these studies making patient matching between groups very difficult. It is thus not surprising that to date, our understanding of the relationship between HIV infection, ART, and CV disease is unclear and incomplete, which is a critical barrier toward identifying interventions to reduce CV disease-related mortality in HIV patients with HIV.
Therefore, the use of the NHP models, which minimize the confounding factors, can provide an excellent model for the study of the interrelation between immune activation and inflammation, hypercoagulation, and CV disease in HIV-1 infected subjects.
Besides hypercoagulation, progressive SIV infection in the macaque model faithfully reproduces the large array of CV abnormalities described in HIV patients. We analyzed multiple tissues from SIV/PTMs and SIV/AGMs to identify and compare CV lesions between pathogenic and nonpathogenic SIV infections. In contrast to the natural hosts which do not present with CV lesions (Fig. 2a) the SIV/PTM model closely reproduces the spectrum of CV abnormalities reported in HIV-infected patients, i.e., thrombotic microangiopathy (TMA), arteriopathy, myocardial hypertrophy and fibrosis, atherosclerosis (ATS), infarction, and myocarditis [77–80]. Many of these pathological conditions were previously reported in persistent progressive SIV/HIV-2 infected rhesus macaques (RMs) and PTMs [81–84].
Fig. 2.
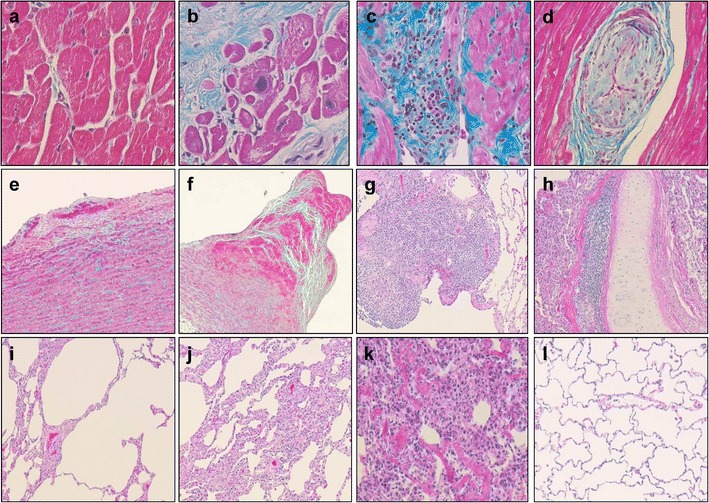
Cardiovascular and lung lesions detected in the SIVsab-infected NHPs. a Lack of mononuclear infiltration, fibrosis, and microthrombosis in the chronically SIVsab-infected AGM lung (SIVsab/AGM, collagen staining); b myocardial fibrosis with subsequent myocardial hypertrophy (SIVsab/PTM, collagen staining); c massive mononuclear cell infiltrates in the myocardium (SIVsab/PTM, collagen staining); d arteritis—small artery with thickened wall and obstructed lumen (SIVsab/PTM, collagen staining); e incipient atherosclerosis plaque (SIVsab/PTM, HE); f advanced fibrotic plaques complicated with thrombosis (SIVsab/PTM, HE); g massive mononuclear infiltration in the lung parenchyma (SIVsab/PTM, HE); h mononuclear infiltration in the large bronchi wall (SIVsab/PTM, HE); i localized emphysema characterized by destruction and dilatation of the airways distal to terminal bronchioles (SIVsab/PTM, HE); j thickened alveolar walls infiltrated with mononuclear cells and numerous thrombi in capillaries (SIVsab/PTM, HE); k disruption of the lung architecture, hyaline membranes, and fibrosis (SIVsab/PTM, HE); l lack of mononuclear infiltration, fibrosis, and microthrombosis in the chronically SIVsab-infected AGM lung (SIVsab/AGM, HE)
Most frequently and in agreement with their hypercoagulable status described above, SIVsab/PTMs have extensive in situ coagulopathies: similar to the renal TMA described in HIV-infected patients, numerous thrombi are accumulating in the glomerular capillary loops (Fig. 1d), afferent and efferent arterioles (Fig. 1d), and small arteries in the kidneys of SIV-infected PTMs. TMA can also be detected in the small vessels in the intestine, lung, and brain [33••], thus explaining the frequent neurological disease observed in PTMs [10]. Microthrombosis may play a significant role in tissue fibrosis development and thus may contribute to lower immune restoration at a certain tissue site or to end-stage organ disease development.
Similar to HIV-infected patients, SIV-infected macaques present with myocardial hypertrophy (enlarged myocytes with irregular nuclei), fibrosis, with increased collagen deposition (Fig. 2b) that replaces either small groups of drop-out myocytes or larger areas of infarction and myocarditis, with infiltration with mononuclear cells (Fig. 2c), and myocytolysis. Arteriopathy characterized by thickened arterial wall, infiltrated with mononuclear cells and obstructed lumen (Fig. 2d) was also detected in several organs such as the heart and kidney. Finally, although atherosclerosis (ATS) lesions are very rare in SIV-negative PTMs, fatty streak (the first visible lesions in the development of ATS) composed by foamy macrophages were found to accumulate in the tunica intima, underneath the aorta endothelium in several SIV-infected PTMs (Fig. 2e) [33••]. In rare cases, we also detected fibrotic ATS plaques complicated with thrombosis (Fig. 2f).
To conclude, our studies in pathogenic and nonpathogenic NHP models of AIDS clearly showed that CV disease is specifically associated to pathogenic SIV infection and absent in nonpathogenic SIV infection. SIV-associated comorbidities occur in the absence of conventional risk factors for CV disease and in the absence of ART and can be linked to persistent inflammation characteristic to pathogenic HIV/SIV infection. Therefore, our studies point to a causal relationship between the two conditions and suggest that strategies targeting persistent inflammation may alleviate CV disease. Therefore, the animal model proves instrumental for the study of CV comorbidities.
Use of NHPs to Model Respiratory Comorbidities
In the past, one of the hallmarks of AIDS clinical presentation was Pneumocystis carinii (now jiroveci) and bacterial pneumonia. With the advent of ART and the improvement of the immune status in patients receiving ART, infectious complications of AIDS are now better controlled. Currently, noninfectious lung diseases, like chronic obstructive pulmonary disease (COPD), are emerging as key clinical conditions [85].
COPD, an impairment of expiratory airflow limitation, is one of the leading causes of global death and of global years lived with disability. The prevalence of COPD in HIV-infected patients ranges between 3 and 23 % in cross-sectional studies. In prospective studies, the identified prevalence of COPD is 10 % which increases to 19 % at 4.4 years of follow-up, suggesting that higher life expectancy of HIV-infected patients creates the premise for COPD to emerge as a major comorbidity [85]. This is not unexpected, as the frequency of COPD increases with age and often presents in later adulthood even in the noninfected individuals. Therefore, it is expected that COPD prevalence will dramatically increase in HIV-infected patients whose average age is significantly increasing [2••].
To date, it is not known how HIV increases the risk for COPD. Its main triggers are chronic inflammation, respiratory infections, oxidative stress, and ART. The role of bacterial pneumonia in COPD is not clear, some studies reporting a clear association between the two conditions while others finding no such association. Altered respiratory microbiome is also postulated to be responsible for the HIV-associated COPD and such studies are currently in progress. P. jiroveci colonization of the lung is frequent in patients with AIDS, and in uninfected patients, more severe forms of COPD have high frequency of colonization [2••]. While these results are strictly observational and not causal, P. jiroveci can induce matrix metalloproteinase activity thus causing lung tissue destruction which contribute to the pathogenesis of COPD in HIV-infected patients [85].
It was also reported that the CD8+ T cells accumulated in the lungs of COPD patients interact with macrophages and neutrophils, releasing chemokines, cytokines, and growth factors in the lungs, thus contributing to chronic inflammation. Thus, the levels of CD8+ T cells in bronchoalveolar lavages correlate to the degree of viral RNA and both correlate with the severity of alveolitis [85].
NHP models of pathogenic SIV infection closely recapitulate pulmonary lesions observed in HIV-infected patients. In PTMs infected with SIVsab, we observed a large range of pulmonary lesions (Fig. 2). In earlier stages of infection, most frequently, massive mononuclear infiltrates may be seen in the lung parenchyma (Fig. 2g) or in the vicinity of the large bronchi (Fig. 2h). With disease progression, localized emphysema (Fig. 2i) and most frequently thickened alveolar walls (Fig. 2j) due to mononuclear infiltration can be seen. The complete disruption of the lung architecture, hyaline membranes depositions, and consequent lung fibrosis is also a characteristic of late chronic SIV infection in PTMs (Fig. 2k). In high contrast, the natural host maintains a healthy lung architecture during chronic SIVsab infection (Fig. 2l). The prospective studies of COPD in SIV-infected monkeys with progressive disease have yet to be completed. The animal model permits us to refine the system for risk factors, as one of the major trigger of COPD and potentially confounding factor of such studies, cigarette smoking, is not present in the model. Also, NHP studies will enable a more invasive approach that permits clear conclusions regarding the mechanisms and possible interventions aimed at preventing respiratory comorbidities in HIV-infected patients.
Use of NHPs to Model Kidney Comorbidities
Although kidney disease has not been recognized as a priority noncommunicable disease (NCD) by the 2011 United Nations General Assembly, the meeting recognized the linkage between NCDs and HIV/AIDS and called for an integrated model of response to these two conditions [86].
Acute kidney injury defined by a sudden loss of kidney function (hours to days) and characterized by the accumulation of urea and creatinine, decreased urine output, or both is common in patients with HIV. Acute kidney injury has been shown to predispose to end-stage renal disease and increased mortality in HIV patients [87]. Chronic kidney disease defined by either structural or functional kidney damage (abnormal urinalysis, imaging studies, or histology) with a duration of at least 3 months with or without a decreased glomerular filtration rate or decreased kidney function, with or without evidence of kidney damage, [87] was also described in HIV patients. Chronic kidney disease can be caused, accelerated, or complicated by HIV.
HIV-associated nephropathy (HIVAN) is most frequently characterized by collapsing focal glomerulosclerosis with microcystic tubular dilatation [88]. In addition, other histopathological lesions associated with kidney disease in HIV patients are immune complex glomerulonephritis with “lupus-like” features, IgA nephropathy, cryoglobulinemia, and amyloidosis. HIVAN is common in HIV-infected patients with less than 200 CD4+ T cells/μl and associated with significant proteinuria. Untreated, HIVAN rapidly progresses to end-stage renal disease and death (50 % mortality at 4.47 months in patients that do not receive ART) [87].
The mechanisms responsible for kidney disease development are poorly understood. Both HIV infection and immune suppression in general are considered risk factors for renal impairment. While the pathway by which a low CD4+ T cell count impacts renal function independent of HIV viremia or opportunistic infections is not known, there is a general consensus in the field that the main mechanism behind this association is persistent inflammation [89]. The preservation of the immune function through early initiation of ART seems to be essential to minimize the risk of renal disease in HIV-infected patients [89].
The complex interactions between HIV infection and kidney disease development can be dissected by using NHP progressive models. Natural hosts do not develop kidney impairment (Fig. 1c), while the clinical presentation of kidney disease associated with progressive SIV infection in macaque recapitulates the features of HIV infection. A clinical syndrome equivalent to HIVAN was described in macaques infected with SIV, characterized by focal and segmental glomerulosclerosis and collapsing glomerulopathy [90]. We have recently confirmed similar features in SIVsab-infected PTM, our model of pathogenic SIV infection, in which we identified histological lesions similar to HIVAN, i.e., the hyperplasia of the epithelial lining of the Bowman capsule (Fig. 1e) associated with collapsing/glomerulosclerosis glomerulopathy (Fig. 1e), diffuse (Fig. 1f) or pseudogranulomatous (Fig. 1g) interstitial nephritis associated with dilated microtubules containing either hyaline (Fig. 1h) or cellular casts (Fig. 1i). As described for HIV patients, the SIVsab-infected PTMs presented with SIV-arteriopathy in the kidney and other organs, i.e., thickened wall arteries and vessel occlusion (Fig. 2) [33••]. Other SHIV-infected macaques presented with nephrotic syndrome, characterized by peripheral edema, hypoalbuminemia, and proteinuria [91]. A case of immunoglobulin-A nephropathy with crescentic glomerulonephritis was described in a PTM coinfected with SIVmac and Mycobacterium tuberculosis [92]. Finally, we previously described a high frequency of thrombotic microangiopathy (TMA) in the SIVsab-infected PTMs [33••], a condition described in HIV patients which is associated with a poor prognostic and accelerated death [67]. TMA is characterized by the presence of numerous microthrombi in the glomerulus capillaries [33••] and in the afferent and efferent arterioles (Fig. 1d).
Considering that the NHP kidney presents similar histopathologies as HIV-infected patients, the NHP appears perfectly suited to model and understand the pathogenesis of renal comorbidities. Furthermore, the NHP model can be used to assess the safety, efficacy, and impact that complementary therapies aimed at controlling the deleterious consequences of persistent immune activation and inflammation can have on the kidneys.
Use of NHPs to Model HIV-associated Neurocognitive Disorders
Neuropathogenesis of HIV infection dramatically changed after the advent of ART. Nowadays, most of the neuro-AIDS features are due to persistent low level of HIV, chronic inflammation, potential drug toxicity, and accelerated aging (which is associated with neurodegeneration) [93]. In patients receiving ART, associated neuropathology ranges from asymptomatic neurocognitive impairment (ANI) and mild neurocognitive disorder (MND), to HIV-associated dementia (HAD) [94]. Altogether, these clinical conditions are called HIV-associated neurocognitive disorder (HAND).
Prior to the general introduction of ART, HAD was dominating the clinical spectrum of HAND. HAD is a progressive disabling subcortical dementia that presents with progressive loss of attention and concentration, motor impairment, and various behavioral components that lead to death in less than a year [93]. HAD associates pathologic changes in the brain including generalized atrophy, changes in white matter causing a leukoencephalopathy, microglial nodules characteristic to viral encephalitis, and multinucleated giant cells. Interestingly, in untreated patients, the severity of dementia is more closely associated with inflammatory response markers than with plasma viral load.
In the ART era, however, HAND is driven by ANI and MND [94]. These clinical entities are defined by neuropsychometric testing abnormalities with or without functional impairment in activities of daily living. There are authors that question the utility of ANI due the fact that functional impairment is imprecise and therefore may represent a confounding factor [93].
In addition to the clinical signs, HAND is predicted by several biomarkers: the levels of CD4+ T cells at the nadir, the levels of sCD14 and of sCD163, as well as those of viral DNA in the circulating monocytes [93]. Cardiovascular biomarkers, such as D-dimer, as well as metabolic risk factors were also reported to be predictive of HAND. In the cerebrospinal fluid, markers associated with chronic inflammation and neuronal injury are predictive for HAND [94].
In longitudinal studies, the prevalence of HAND ranges from 18 to 25 %. The risk of HAND is increased by severe comorbidities (co-infections, drug abuse, other neurological conditions) or failure of ART [94]. Other studies could not document the significant deterioration of the neurocognitive impairment in patients on ART. Neurocognitive deterioration persists in some HIV patients in spite of effective ART [94].
Due to the nature of the lesions behind HAND, invasive studies are not possible in humans, and the only available data can be collected at the necropsy. Therefore, animal models that permit more invasive approaches and even animal sacrifice for the study of the magnitude of the lesions in relation with the stage of disease progression are critical for the study of HAND [95]. Multiple animal models have been developed over time, and, similar to other comorbidities, SIV-infected PTMs appear to be the model of choice for the study of neurocognitive comorbidities [95]. They have the advantage not only of a more condensed duration of progression but also of a higher prevalence of neurocognitive disorders.
In macaques, the SIV invasion of the brain occurs early in infection and is mediated by infected mononuclear cells and T cells that cross the blood brain barrier [96]. Virus replication in the brain macrophages correlates with virus ability to replicate productively in cultured macrophages derived from peripheral blood of RMs with encephalopathy. As such, studies in the NHP models confirmed that only the SIV strains able to replicate in macrophages can cause encephalitis [96].
The study of the early stages of the neurocognitive disorders in macaques can permit understanding the cellular and molecular mechanisms of disease progression and to understand the role of viral and host factor functions and interactions during both active and latent infection. Furthermore, the study of NHP models will help with a better understanding of the issues related to ARV penetrability in CNS. The consequences of drug abuse on neurocognition and encephalitis can also be modeled in NHPs. To date, modeling HIV infection in macaques permitted to establish both the efficacy and the limitations of therapies targeting immune activation in the CNS.
Use of NHPs to Model HIV-associated Osteopenia/Osteoporosis
Bone disorders are common in HIV-infected patients with >50 % of HIV-infected adults being osteopenic (presenting with low bone mineral density), with 15 % more being osteoporotic (associating to low bone mineral density distorted micro-architecture and high risk for fracture) [97]. The pathophysiology underlying HIV-associated bone abnormalities is not completely understood [98]. Among the factors contributing to bone abnormalities are (i) comorbidities (i.e., kidney disease, hypogonadism, and hypovitaminosis D) that predispose to bone catabolism; (ii) the virus, which may contribute by altering immune B cell function and enhancing bone resorption [97]; (iii) antiretroviral therapy, through immune reconstitution, suppressive effects on bone cells, and alteration of vitamin D metabolism; and (iv) chronic inflammation that fuels all the other factors [99]. Understanding the mechanisms of the bone disorder is a subject of intense research and can largely benefit from studies in animal models, because it is generally acknowledged that lifestyle factors (absent in the animal models) may at least contribute to the maintenance of these alterations [99].
To date, no studies have modeled bone pathology in either pathogenic or nonpathogenic SIV infections. Yet, macaques have been used in the past for the study of metabolic disorders [9, 10] and may constitute models for osteopathy associated with HIV infection.
Conclusion
NHP models can and should be employed for the study of non-AIDS comorbidities associated with HIV infection. As presented here, NHP models recapitulate most of the key features of these comorbidities. NHPs have the advantage of a simplified system in which most of the risk factors for comorbidities are absent and experimental conditions (i.e., timing of infection, stage of acute or chronic SIV infection, certain diets, etc.) can be properly controlled. Similar to other areas of AIDS research, such as pathogenesis, cure or vaccine research, and pre- and postexposure prophylaxis [100], NHP models may be instrumental for the study of non-AIDS comorbidities to characterize and better define the new landscape of AIDS pathogenesis and to test the efficacy of interventions aimed to alleviate non-AIDS-related comorbidities.
Acknowledgments
We thank Daniel Douek, Jason Brenchley, and Jake Estes for the helpful discussion. We would like to thank the multiple groups of scientists that had seminal contributions to this field, which could not be cited here due to restrictions in the number of cited references. This work was supported by the funds from grants RO1 AI064066 (IP) [National Institute of Allergy and Infectious Diseases (NIAID)] RO1 HL117715 (IP) from the National Heart, Lung and Blood Institute and RO1 RR025781 (CA and IP) from the National Center for Research Resources (NCRR). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Ivona Pandrea, Russell Tracy, and Cristian Apetrei report that they have grants from NIH.
Alan Landay, Cara Wilson, and Jennifer Stock declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on HIV Pathogenesis and Treatment
Contributor Information
Ivona Pandrea, Phone: +412-624-3242, Email: pandrea@pitt.edu.
Alan Landay, Email: alanday@rush.edu.
Cara Wilson, Email: cara.wilson@ucdenver.edu.
Jennifer Stock, Phone: +412-624-3242, Email: jls329@pitt.edu.
Russell Tracy, Email: Russell.Tracy@uvm.edu.
Cristian Apetrei, Phone: +412-624-3235, Email: apetreic@pitt.edu.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Fauci AS, Folkers GK, Dieffenbach CW. HIV-AIDS: much accomplished, much to do. Nat Immunol. 2013;14(11):1104–1107. doi: 10.1038/ni.2735. [DOI] [PubMed] [Google Scholar]
- 2.••.Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. 2013;39(4):633–645. doi: 10.1016/j.immuni.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J. Infect. Dis. 2011;204(8):1217–1226. doi: 10.1093/infdis/jir507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 2011;203(6):780–790. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 2003;187(10):1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 6.Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.•.Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. 2013;119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VandeWoude S, Apetrei C. Going wild: lessons from T-lymphotropic naturally occurring lentiviruses. Clin Microbiol Rev. 2006;19:728–762. doi: 10.1128/CMR.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans DT, Silvestri G. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS. 2013 doi: 10.1097/COH.0b013e328361cee8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lifson JD, Haigwood NL. Lessons in nonhuman primate models for AIDS vaccine research: from minefields to milestones. Cold Spring Harb Perspect Med. 2012;2(6):a007310. doi: 10.1101/cshperspect.a007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanco-Melo D, Venkatesh S, Bieniasz PD. Intrinsic cellular defenses against human immunodeficiency viruses. Immunity. 2012;37(3):399–411. doi: 10.1016/j.immuni.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, Hardcastle J, et al. Molecular epidemiology of SIVsm in US Primate Centers unravels the origin of SIVmac and SIVstm. J Virol. 2005;79(14):8991–9005. doi: 10.1128/JVI.79.14.8991-9005.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirsch VM, Dapolito G, Johnson PR, Elkins WR, London WT, Montali RJ, et al. Induction of AIDS by simian immunodeficiency virus from an African green monkey: species-specific variation in pathogenicity correlates with the extent of in vivo replication. J Virol. 1995;69(2):955–967. doi: 10.1128/jvi.69.2.955-967.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandell DT, Kristoff J, Gaufin T, Gautam R, Ma D, Sandler N, et al. Pathogenic features associated with increased virulence upon simian immunodeficiency virus cross-species transmission from natural hosts. J Virol. 2014;88(12):6778–6792. doi: 10.1128/JVI.03785-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, et al. Functional cure of SIVagm infection in rhesus macaques results in complete recovery of CD4+ T cells and is reverted by CD8+ cell depletion. PLoS Pathog. 2011;7(8):e1002170. doi: 10.1371/journal.ppat.1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Prete GQ, Ailers B, Moldt B, Keele BF, Estes JD, Rodriguez A, et al. Selection of unadapted, pathogenic SHIVs encoding newly transmitted HIV-1 envelope proteins. Cell Host Microbe. 2014;16(3):412–418. doi: 10.1016/j.chom.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatziioannou T, Del Prete GQ, Keele BF, Estes JD, McNatt MW, Bitzegeio J, et al. HIV-1-induced AIDS in monkeys. Science. 2014;344(6190):1401–1405. doi: 10.1126/science.1250761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandrea I, Apetrei C. Where the wild things are: pathogenesis of SIV infection in African nonhuman primate hosts. Curr HIV/AIDS Rep. 2010;7(1):28–36. doi: 10.1007/s11904-009-0034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2008 [DOI] [PMC free article] [PubMed]
- 20.Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104(4):942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 21.Doisne JM, Urrutia A, Lacabaratz-Porret C, Goujard C, Meyer L, Chaix ML, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. 2004;173(4):2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- 22.Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. 2012;30:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7(3):235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 24.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 25.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10(9):655–666. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 26.Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest. 2011;121(3):998–1008. doi: 10.1172/JCI45157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev. 2013;254(1):326–342. doi: 10.1111/imr.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wijewardana V, Kristoff J, Xu C, Ma D, Haret-Richter G, Stock JL, et al. Kinetics of myeloid dendritic cell trafficking and activation: impact on progressive, nonprogressive and controlled SIV infections. PLoS Pathog. 2013;9(10):e1003600. doi: 10.1371/journal.ppat.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandrea I, Sodora DL, Silvestri G, Apetrei C. Into the wild: simian immunodeficiency virus (SIV) infection in natural hosts. Trends Immunol. 2008;29(9):419–428. doi: 10.1016/j.it.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon S, Klatt NR, Milush JM, Engram J, Dunham RM, Paiardini M, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free SIV-infected sooty mangabeys. J Immunol. 2007;179:3026–3034. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol. 2007;179(5):3035–3046. doi: 10.4049/jimmunol.179.5.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, et al. Cutting edge: experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol. 2008;181(10):6687–6691. doi: 10.4049/jimmunol.181.10.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.••.Pandrea I, Cornell E, Wilson C, Ribeiro RM, Ma D, Kristoff J, et al. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood. 2012;120:1357–1366. doi: 10.1182/blood-2012-03-414706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klatt NR, Harris LD, Vinton CL, Sung H, Briant JA, Tabb B et al. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation, and IL-17 production in the absence of SIV infection. Mucosal Immunol. 2010:in press. doi:10.1038/mi.2010.14. [DOI] [PMC free article] [PubMed]
- 35.••.Klatt NR, Canary LA, Sun X, Vinton CL, Funderburg NT, Morcock DR, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J Clin Invest. 2013;123(2):903–907. doi: 10.1172/JCI66227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.••.Kristoff J, Haret-Richter G, Ma D, Ribeiro RM, Xu C, Cornell E, et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J Clin Invest. 2014;124(6):2802–2806. doi: 10.1172/JCI75090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandrea I, Kristoff J, Xu C, Ma D, Stock JL, Ross S et al. Combination antibiotic and antiinflammatory therapy reduces acute inflammation and hypercoagulation in SIV-infected pigtailed macaques. PLoS pathogens. 2015;11(in revision.). [DOI] [PMC free article] [PubMed]
- 38.High KP, Brennan-Ing M, Clifford DB, Cohen MH, Currier J, Deeks SG, et al. HIV and aging: state of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J Acquir Immune Defic Syndr. 2012;60(1):S1–S18. doi: 10.1097/QAI.0b013e31825a3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capri M, Salvioli S, Monti D, Caruso C, Candore G, Vasto S, et al. Human longevity within an evolutionary perspective: the peculiar paradigm of a post-reproductive genetics. Exp Gerontol. 2008;43(2):53–60. doi: 10.1016/j.exger.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev. 2011;10(3):319–329. doi: 10.1016/j.arr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Althoff KN, Justice AC, Gange SJ, Deeks SG, Saag MS, Silverberg MJ, et al. Virologic and immunologic response to HAART, by age and regimen class. AIDS. 2010;24(16):2469–2479. doi: 10.1097/QAD.0b013e32833e6d14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, Cahn P, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA : J. Am. Med. Assoc. 2012;308(4):387–402. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 44.Willette AA, Coe CL, Birdsill AC, Bendlin BB, Colman RJ, Alexander AL, et al. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. Age. 2013;35(6):2215–2227. doi: 10.1007/s11357-013-9518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haberthur K, Engelman F, Barron A, Messaoudi I. Immune senescence in aged nonhuman primates. Exp Gerontol. 2010;45(9):655–661. doi: 10.1016/j.exger.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Didier ES, Sugimoto C, Bowers LC, Khan IA, Kuroda MJ. Immune correlates of aging in outdoor-housed captive rhesus macaques (Macaca mulatta) Immun. Ageing. 2012;9(1):25. doi: 10.1186/1742-4933-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maniar-Hew K, Clay CC, Postlethwait EM, Evans MJ, Fontaine JH, Miller LA. Innate immune response to LPS in airway epithelium is dependent on chronological age and antecedent exposures. Am J Respir Cell Mol Biol. 2013;49(5):710–720. doi: 10.1165/rcmb.2012-0321OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Josset L, Engelmann F, Haberthur K, Kelly S, Park B, Kawoaka Y, et al. Increased viral loads and exacerbated innate host responses in aged macaques infected with the 2009 pandemic H1N1 influenza A virus. J Virol. 2012;86(20):11115–11127. doi: 10.1128/JVI.01571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma D, Jasinska AJ, Feyertag F, Wijewardana V, Kristoff J, He T, et al. Factors associated with siman immunodeficiency virus transmission in a natural African nonhuman primate host in the wild. J Virol. 2014;88(10):5687–5705. doi: 10.1128/JVI.03606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandrea I, Silvestri G, Apetrei C. AIDS in African nonhuman primate hosts of SIVs: a new paradigm of SIV infection. Curr HIV Res. 2009;6:57–72. doi: 10.2174/157016209787048456. [DOI] [PubMed] [Google Scholar]
- 51.•.Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6(8):e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 53.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112(7):2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009;5(2):e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5(193):193ra91. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steele AK, Lee EJ, Vestal B, Hecht D, Dong Z, Rapaport E, et al. Contribution of intestinal barrier damage, microbial translocation and HIV-1 infection status to an inflammaging signature. PLoS One. 2014;9(5):e97171. doi: 10.1371/journal.pone.0097171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dillon SM, Lee EJ, Kotter CV, Austin GL, Dong Z, Hecht DK, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7(4):983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.•.Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151(2):253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. 2013;173(8):614–622. doi: 10.1001/jamainternmed.2013.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Justice AC, Freiberg MS, Tracy R, Kuller L, Tate JP, Goetz MB, et al. Does an index composed of clinical data reflect effects of inflammation, coagulation, and monocyte activation on mortality among those aging with HIV? Clin. Infect. Dis : Off Pub Infect Dis Soc Am. 2012;54(7):984–994. doi: 10.1093/cid/cir989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pandrea I, Amedee A, Bagby G, Nelson S. Alcohol’s role in HIV transmission and disease progression. Alcohol Res Health. 2010;33:203–218. [PMC free article] [PubMed] [Google Scholar]
- 63.Gori A, Rizzardini G, Van’t Land B, Amor KB, van Schaik J, Torti C, et al. Specific prebiotics modulate gut microbiota and immune activation in HAART-naive HIV-infected adults: results of the “COPA” pilot randomized trial. Mucosal Immunol. 2011;4(5):554–563. doi: 10.1038/mi.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Byakwaga H, Kelly M, Purcell DF, French MA, Amin J, Lewin SR, et al. Intensification of antiretroviral therapy with raltegravir or addition of hyperimmune bovine colostrum in HIV-infected patients with suboptimal CD4+ T-cell response: a randomized controlled trial. J Infect Dis. 2011;204(10):1532–1540. doi: 10.1093/infdis/jir559. [DOI] [PubMed] [Google Scholar]
- 65.Sandler NG, Zhang X, Bosch RJ, Funderburg NT, Choi AI, Robinson JK, et al. Sevelamer does not decrease lipopolysaccharide or soluble CD14 levels but decreases soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol levels in individuals with untreated HIV infection. J Infect Dis. 2014;210(10):1549–1554. doi: 10.1093/infdis/jiu305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pallikkuth S, Micci L, Ende ZS, Iriele RI, Cervasi B, Lawson B, et al. Maintenance of intestinal Th17 cells and reduced microbial translocation in SIV-infected rhesus macaques treated with interleukin (IL)-21. PLoS Pathog. 2013;9(7):e1003471. doi: 10.1371/journal.ppat.1003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Palella FJ, Jr, Phair JP. Cardiovascular disease in HIV infection. Curr Opin HIV AIDS. 2011;6(4):266–271. doi: 10.1097/COH.0b013e328347876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bissuel F, Berruyer M, Causse X, Dechavanne M, Trepo C. Acquired protein S deficiency: correlation with advanced disease in HIV-1-infected patients. J Acquir Immune Defic Syndr. 1992;5(5):484–489. doi: 10.1097/00126334-199205000-00009. [DOI] [PubMed] [Google Scholar]
- 69.Shen YM, Frenkel EP. Thrombosis and a hypercoagulable state in HIV-infected patients. Clin. Appl. Thromb. Hemost : Off J Intern Acad Clin Appl Thromb. Hemost. 2004;10(3):277–280. doi: 10.1177/107602960401000311. [DOI] [PubMed] [Google Scholar]
- 70.Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One. 2012;7(9):e44454. doi: 10.1371/journal.pone.0044454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Musselwhite LW, Sheikh V, Norton TD, Rupert A, Porter BO, Penzak SR, et al. Markers of endothelial dysfunction, coagulation and tissue fibrosis independently predict venous thromboembolism in HIV. AIDS. 2011;25(6):787–795. doi: 10.1097/QAD.0b013e3283453fcb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calmy A, Gayet-Ageron A, Montecucco F, Nguyen A, Mach F, Burger F, et al. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS. 2009;23(8):929–939. doi: 10.1097/QAD.0b013e32832995fa. [DOI] [PubMed] [Google Scholar]
- 73.Hatano H, Strain MC, Scherzer R, Bacchetti P, Wentworth D, Hoh R, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: a randomized, placebo-controlled trial. J Infect Dis. 2013;208(9):1436–1442. doi: 10.1093/infdis/jit453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palella FJ, Jr, Gange SJ, Benning L, Jacobson L, Kaplan RC, Landay AL, et al. Inflammatory biomarkers and abacavir use in the Women’s Interagency HIV Study and the Multicenter AIDS Cohort Study. AIDS. 2010;24(11):1657–1665. doi: 10.1097/QAD.0b013e3283389dfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010;115(2):161–167. doi: 10.1182/blood-2009-03-210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 77.Butt AA, Chang CC, Kuller L, Goetz MB, Leaf D, Rimland D, et al. Risk of heart failure with human immunodeficiency virus in the absence of prior diagnosis of coronary heart disease. Arch Intern Med. 2011;171(8):737–743. doi: 10.1001/archinternmed.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen IS, Anderson DW, Virmani R, Reen BM, Macher AM, Sennesh J, et al. Congestive cardiomyopathy in association with the acquired immunodeficiency syndrome. N Engl J Med. 1986;315(10):628–630. doi: 10.1056/NEJM198609043151007. [DOI] [PubMed] [Google Scholar]
- 79.De Castro S, D’Amati G, Gallo P, Cartoni D, Santopadre P, Vullo V, et al. Frequency of development of acute global left ventricular dysfunction in human immunodeficiency virus infection. J Am Coll Cardiol. 1994;24(4):1018–1024. doi: 10.1016/0735-1097(94)90864-8. [DOI] [PubMed] [Google Scholar]
- 80.Lipshultz SE, Chanock S, Sanders SP, Colan SD, Perez-Atayde A, McIntosh K. Cardiovascular manifestations of human immunodeficiency virus infection in infants and children. Am J Cardiol. 1989;63(20):1489–1497. doi: 10.1016/0002-9149(89)90014-3. [DOI] [PubMed] [Google Scholar]
- 81.Chalifoux LV, Simon MA, Pauley DR, MacKey JJ, Wyand MS, Ringler DJ. Arteriopathy in macaques infected with simian immunodeficiency virus. Lab Investig. 1992;67(3):338–349. [PubMed] [Google Scholar]
- 82.Eitner F, Cui Y, Hudkins KL, Schmidt A, Birkebak T, Agy MB, et al. Thrombotic microangiopathy in the HIV-2-infected macaque. Am J Pathol. 1999;155(2):649–661. doi: 10.1016/S0002-9440(10)65161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shannon RP, Simon MA, Mathier MA, Geng YJ, Mankad S, Lackner AA. Dilated cardiomyopathy associated with simian AIDS in nonhuman primates. Circulation. 2000;101(2):185–193. doi: 10.1161/01.CIR.101.2.185. [DOI] [PubMed] [Google Scholar]
- 84.Yanai T, Lackner AA, Sakai H, Masegi T, Simon MA. Systemic arteriopathy in SIV-infected rhesus macaques (Macaca mulatta) J Med Primatol. 2006;35(2):106–112. doi: 10.1111/j.1600-0684.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 85.Kunisaki KM. Will expanded ART use reduce the burden of HIV-associated chronic lung disease? Curr Opin HIV AIDS. 2014;9:27–33. doi: 10.1097/COH.0000000000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Assembly. UNG. Political declaration of the high-level meeting of the general assembly on the prevention and control of non-communicable diseases. 66th session of the United Nations General Assembly; New York, NY2011. p. September 2011.
- 87.Kalyesubula R, Wearne N, Semitala FC, Bowa K. HIV-associated renal and genitourinary comorbidities in Africa. J Acquir Immune Defic Syndr. 2014;67(Suppl 1):S68–S78. doi: 10.1097/QAI.0000000000000259. [DOI] [PubMed] [Google Scholar]
- 88.D’Agati V, Suh JI, Carbone L, Cheng JT, Appel G. Pathology of HIV-associated nephropathy: a detailed morphologic and comparative study. Kidney Int. 1989;35(6):1358–1370. doi: 10.1038/ki.1989.135. [DOI] [PubMed] [Google Scholar]
- 89.Ryom L, Mocroft A, Lundgren JD. Antiretroviral therapy, immune suppression and renal impairment in HIV-positive persons. Curr Opin HIV AIDS. 2014;9(1):41–47. doi: 10.1097/COH.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 90.Stephens EB, Tian C, Dalton SB, Gattone VH., 2nd Simian-human immunodeficiency virus-associated nephropathy in macaques. AIDS Res Hum Retrovir. 2000;16(13):1295–1306. doi: 10.1089/08892220050117050. [DOI] [PubMed] [Google Scholar]
- 91.Clarke CL, Eckhaus MA, Zerfas PM, Elkins WR. Peripheral edema with hypoalbuminemia in a nonhuman primate infected with simian-human immunodeficiency virus: a case report. J Am Assoc Lab Anim Sci: JAALAS. 2008;47(1):42–48. [PMC free article] [PubMed] [Google Scholar]
- 92.Borda JT, Pauley DR, MacKey JJ, Alvarez X, Simon MA, Klumpp SA. Immunoglobulin-A nephropathy with crescentic glomerulonephritis in a pigtailed macaque (Macaca nemestrina) Vet Pathol. 2004;41(1):44–49. doi: 10.1354/vp.41-1-44. [DOI] [PubMed] [Google Scholar]
- 93.Johnson TP, Nath A. New insights into immune reconstitution inflammatory syndrome of the central nervous system. Curr Opin HIV AIDS. 2014;9(6):572–578. doi: 10.1097/COH.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Winston A, Vera JH. Can antiretroviral therapy prevent HIV-associated cognitive disorders? Curr Opin HIV AIDS. 2014;9(1):11–16. doi: 10.1097/COH.0000000000000016. [DOI] [PubMed] [Google Scholar]
- 95.Williams R, Bokhari S, Silverstein P, Pinson D, Kumar A, Buch S. Nonhuman primate models of NeuroAIDS. J Neurol. 2008;14(4):292–300. doi: 10.1080/13550280802074539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zink MC, Laast VA, Helke KL, Brice AK, Barber SA, Clements JE, et al. From mice to macaques—animal models of HIV nervous system disease. Curr HIV Res. 2006;4(3):293–305. doi: 10.2174/157016206777709410. [DOI] [PubMed] [Google Scholar]
- 97.Ali MK, Magee MJ, Dave JA, Ofotokun I, Tungsiripat M, Jones TK, et al. HIV and metabolic, body, and bone disorders: what we know from low- and middle-income countries. J Acquir Immune Defic Syndr. 2014;67(Suppl 1):S27–S39. doi: 10.1097/QAI.0000000000000256. [DOI] [PubMed] [Google Scholar]
- 98.Mallon PW. Aging with HIV: osteoporosis and fractures. Curr Opin HIV AIDS. 2014;9(4):428–435. doi: 10.1097/COH.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 99.Cotter AG, Mallon PW. The effects of untreated and treated HIV infection on bone disease. Curr Opin HIV AIDS. 2014;9(1):17–26. doi: 10.1097/COH.0000000000000028. [DOI] [PubMed] [Google Scholar]
- 100.Apetrei C, Pandrea I, Mellors JW. Nonhuman primate models for HIV cure research. PLoS Pathog. 2012;8(8):e1002892. doi: 10.1371/journal.ppat.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]