

Abstract
Glutathione (GSH) plays an important role in maintaining redox homeostasis inside cells. Currently, there are no methods available to quantitatively assess the GSH concentration in live cells. Live cell fluorescence imaging revolutionized the field of cell biology and has become an indispensable tool in current biological studies. In order to minimize the disturbance to the biological system in live cell imaging, the probe concentration needs to be significantly lower than the analyte concentration. Because of this, any irreversible reaction-based GSH probe can only provide qualitative results within a short reaction time and will exhibit maximum response regardless of the GSH concentration if the reaction is completed. A reversible reaction-based probe with an appropriate equilibrium constant allows measurement of an analyte at much higher concentrations and, thus, is a prerequisite for GSH quantification inside cells. In this contribution, we report the first fluorescent probe—ThiolQuant Green (TQ Green)—for quantitative imaging of GSH in live cells. Due to the reversible nature of the reaction between the probe and GSH, we are able to quantify mM concentrations of GSH with TQ Green concentrations as low as 20 nM. Furthermore, the GSH concentrations measured using TQ Green in 3T3-L1, HeLa, HepG2, PANC-1, and PANC-28 cells are reproducible and well correlated with the values obtained from cell lysates. TQ Green imaging can also resolve the changes in GSH concentration in PANC-1 cells upon diethylmaleate (DEM) treatment. In addition, TQ Green can be conveniently applied in fluorescence activated cell sorting (FACS) to measure GSH level changes. Through this study, we not only demonstrate the importance of reaction reversibility in designing quantitative reaction-based fluorescent probes but also provide a practical tool to facilitate redox biology studies.
Glutathione (GSH) is the most abundant nonprotein thiol in mammalian cells and plays an important role in maintaining redox homeostasis inside cells.1,2 Variations in intracellular GSH concentration have been linked to many pathological processes including cancer, aging, and diabetes.3 In order to understand the influence of GSH in these processes, it is necessary to precisely measure the GSH concentration in live cells. In this contribution, we report the first quantitative fluorescent probe for determination of GSH levels in live cells.
Currently, there are no methods available to quantitatively assess the GSH concentration in live cells. Although many GSH responsive chromogenic and fluorogenic reagents have been developed, quantification using these reagents can only be performed on cell lysates.4 Additionally, despite the fact that myriad GSH fluorescent probes are reported for live cell imaging, none of these probes can provide meaningful quantitation of intracellular GSH concentrations.5−16 Redox-sensitive green fluorescent proteins (roGFPs) remain one of the most popular GSH probes for live cell imaging. However, they can only monitor the ratios of GSH to the oxidized form GSSG, not absolute concentrations.17,18 Additionally, the conventional roGFPs lack specificity and respond slowly to changes in redox potential. Therefore, the most widely used probe for studying redox biology is the fusion of human glutaredoxin-1 (Grx1) to roGFP2.18,19 However, it is well-known that Grx1 is a key player in maintaining redox homeostasis.20,21 The main disadvantage of Grx1-roGFP2 as a redox probe is that overexpression of this protein may change the redox status of the probed cells. In contrast, small molecule probes are advantageous in this regard and are less likely to change the cellular redox status.
In order to minimize the disturbance on the biological system in live cell imaging, the probe concentration needs to be significantly lower than the concentration of analyte. Because of this, any irreversible reaction-based GSH probe will exhibit the maximum response regardless of the GSH concentration.8,9,22 This problem is not limited to GSH but is also true for the detection of other molecules in live cells (e.g., nitric oxide,23,24 hydrogen peroxide,25,26 and hydrogen sulfide27−32). To overcome this issue, a reversible reaction-based probe with an appropriate equilibrium constant (Keq) must be used.33−40 This approach allows one to measure analyte at concentrations significantly higher than the concentration of the probe and thus is a prerequisite for GSH quantification inside cells.
In addition to a reversible reaction, a preferable probe for GSH quantification in cells needs to have (a) ratiometric fluorescent readouts, (b) a suitable wavelength for imaging, and (c) membrane permeability. In a complex intracellular environment, it is difficult to accurately control probe concentrations. Ratiometric fluorescent probes are molecules that exhibit shifts in their spectra upon interaction with analytes. They are preferred in live cell imaging because they allow quantitative measurements of analyte concentrations independent of the probe concentration. In addition, the excitation wavelengths of the probe and its product with the analyte should match the commonly used laser wavelengths for confocal microscopes. Moreover, the probe molecule needs to have a reasonable aqueous solubility to maintain an adequate concentration inside cells while still possessing sufficient membrane permeability. Herein, we report the first quantitative fluorescent probe—ThiolQuant Green (TQ Green)—to determine GSH levels in live cells and demonstrate that TQ Green can be used not only in high resolution confocal microscope imaging but also in bulk measurements using fluorescence activated cell sorting (FACS).
Results and Discussion
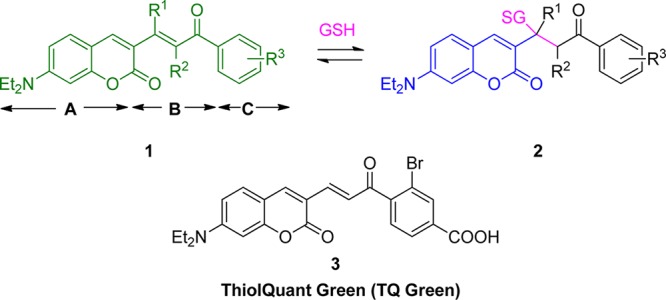
In this study, we exploited the reversible Michael addition reaction to design a ratiometric probe that can quantify intracellular GSH concentration.41,42 It should be noted that despite the attempts to use Michael addition reaction for GSH probes, an inappropriate Keq of these probes accounts for the failure of GSH quantification.22 In this study, analyte GSH is the Michael donor and the probe is the Michael acceptor. We chose a modular design for the GSH ratiometric probes (Scheme 1). Module A is protected 7-amino coumarin, whose fluorescence (Fl) property is suitable for confocal experiments. Module C is a modulator with an aromatic structure to extend the absorption (Abs) wavelength of module A, and module B is the reaction center of the Michael acceptor that connects modules A and C. Upon reaction with GSH, the extended conjugation of 1 is interrupted to form 2, causing a hypsochromic shift in both Abs and Fl. Quantification of the Fl emission ratios of 1 and 2 allows us to deduce the GSH concentration, following previously reported procedures for Ca2+ ratiometric probes.43 The Keq with GSH and the time to reach equilibrium can be adjusted by altering the R1, R2, and R3 substituents. After a few iterations (Supporting Information (SI), Table S1), we developed probe 3, designated as TQ Green. TQ Green has an appropriate Keq with GSH and meets all the GSH probe design criteria outlined in the introduction section.
Scheme 1. Modular Design of GSH Probes and Structure of TQ Green.
TQ Green displays ratiometric changes in spectroscopic properties upon reaction with GSH in a phosphate-buffered saline at pH 7.4 (PBS). TQ Green absorbs at 479 nm and fluoresces at 590 nm (λex = 488 nm, Figure 1). The extinction coefficients of TQ Green and TQ Green-GSH (the adduct between TQ Green and GSH) at their maximum absorption wavelengths are (2.3 ± 0.2) × 104 M–1·cm–1 (λmax = 479 nm, Table 1) and (1.6 ± 0.2) × 104 M–1·cm–1 (λmax = 406 nm, Table 1), respectively. Upon reacting with GSH, the Abs and Fl peaks shift hypsochromically to 406 and 463 nm (λex = 405 nm), respectively. The Abs of TQ Green and TQ Green-GSH are close to 488 and 405 nm, respectively, which are two commonly used laser wavelengths for confocal microscopes. For all the following experiments, excitation and absorption wavelengths 488 and 405 nm were used.
Figure 1.
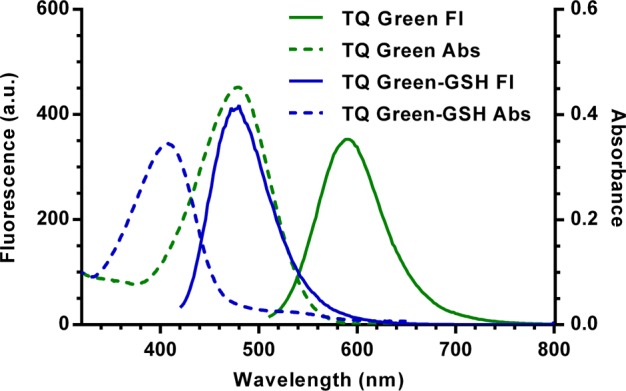
UV–vis and fluorescence spectra of TQ Green (λex = 488 nm) and TQ Green-GSH (λex = 405 nm).
Table 1. Summary of Physical Chemical Properties of TQ Green and TQ Green-GSH.
| TQ Green | TQ Green-GSH | |
|---|---|---|
| abs max wavelength | 479 nm | 406 nm |
| Fl emission max wavelength | 590 nm (λex = 488 nm) | 463 nm (λex = 405 nm) |
| extinction coefficient | (2.3 ± 0.2) × 104 M–1·cm–1 (at λmax = 479 nm) | (1.6 ± 0.2) × 104 M–1·cm–1 (at λmax = 406 nm) |
| quantum yield (PBS) | 0.0094 ± 0.0004 (λex = 479 nm) | 0.0059 ± 0.0003 (λex = 406 nm) |
| quantum yield (methanol) | 0.16 ± 0.05 (λex = 488 nm) | NAa |
| log D (pH = 7.4) | 0.70 | NAb |
| pseudo first-order constant | kobs = (5.98 ± 0.03) × 10–3 s–1 ; t1/2 = 116 s (concentration: TQ Green at 20 μM; GSH at 40 mM) | |
| second-order rate constant | 0.150 ± 0.001 M–1·s–1 | |
| equilibrium constants | Kd′ = 14.8 mM (based on UV–vis), Kd = 1.6 mM | |
TQ Green-GSH is not soluble in methanol.
TQ Green-GSH, GSH are not soluble in octanol thus not suitable for standard log D measurement; the estimated value based on HPLC result is < −1.0.
The reaction between TQ Green and GSH is reversible. To demonstrate this reversibility, three experiments were performed. First, when incubating TQ Green (20 μM) with excessive amounts of GSH (40 mM) in PBS, the Abs at 488 nm decreased with a concurrent increase at 405 nm following pseudo-first-order kinetics (kobs = (5.98 ± 0.03) × 10–3 s–1) and a half-life of 116 s based on a global fitting for the decay and growth at 488 and 405 nm, respectively (Table 1 and Figure 2a). The second-order rate constant between TQ Green and GSH is 0.150 ± 0.001 M–1·s–1. There were no appreciable Abs changes observed after 20 min. Following a pause for another 80 min to ensure that the equilibrium between TQ Green and GSH was fully established, we added two equivalents of an irreversible Michael acceptor (5,6-dihydro-2H-pyran-2-one) to deplete all the GSH in solution (Figure 2a). We observed almost full restoration of the absorbance values at 405 and 488 nm, demonstrating the reversibility of the reaction between TQ Green and GSH.
Figure 2.
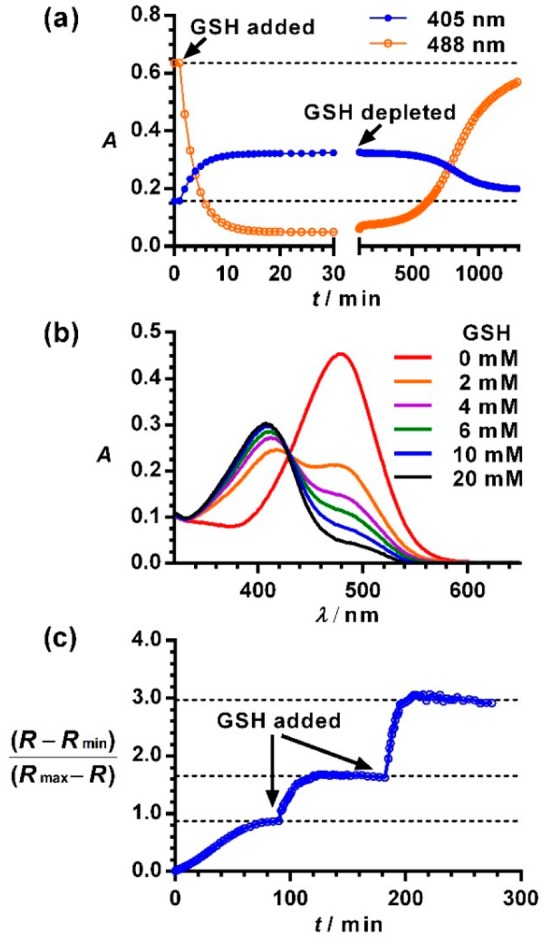
Reversibility of the reaction between TQ Green and GSH. (a) Recovery of reacted TQ Green by depleting GSH. (b) Concentration dependent ratiometric spectra of TQ Green in PBS under anaerobic conditions for 18 h. (c) Responsiveness of TQ Green to the concentration changes of GSH. Rmin and Rmax were measured at 0 and 80 mM of GSH, respectively.
In the second experiment, we allowed TQ Green (16 μM) to react with different concentrations of GSH (0–20 mM) under anaerobic conditions (GSH is easily oxidized in air, Figure S1) for 1, 18, and 72 h. No significant changes in Abs or Fl were observed at these three time points, indicating reaction equilibria were fully established 1 h after reaction. An increase in concentration of GSH resulted in a decrease of Abs at 488 nm with a concurrent increase at 405 nm with an isosbestic point at 426 nm (Figure 2b). If the reaction between TQ Green and GSH were irreversible, TQ Green would be completely consumed, allowing enough reaction time, and would not behave in a concentration dependent manner.
In the third experiment, GSH was added to a solution of TQ Green in three portions with 90 min intervals that served to ensure that equilibrium was fully established. R is defined as the ratio of the signal intensities (Abs or Fl) between TQ Green-GSH and TQ Green. Rmin and Rmax correspond to the R values at zero and saturated GSH concentrations (80 mM), respectively. (R – Rmin)/(Rmax – R), which is proportional to the GSH concentration (vide infra), plateaued within 90 min after the addition of GSH (Figure 2c). The introduction of additional GSH caused (R – Rmin)/(Rmax – R) changes and re-establishment of the reaction equilibrium. This experiment demonstrated that unlike previously reported GSH probes,7−9 TQ Green can respond to the changes in GSH concentrations. Overall, the three experiments demonstrated the reversible nature of the reaction between TQ Green and GSH as well as the effectiveness and necessity for developing reversible reaction-based small molecule probes.
We also determined Keq between TQ Green and GSH and the relationship of the ratiometric changes as a function of GSH concentration. Kd′ is the apparent dissociation constant for the reaction between TQ Green and GSH. The following equation can be derived (refer to the SI for equation deduction and discussion):
Plotting (R – Rmin)/(Rmax – R) as a function of GSH concentration afforded a superb linear relationship with r2 = 0.999 (Figure 3 and Figure S2). The reciprocal of the slope affords Kd′ as 14.8 mM. It is assumed that at 80 mM of GSH, TQ Green is fully converted to TQ Green-GSH. Comparing the absorption values of TQ Green and TQ Green-GSH at 488 nm, we can calculate εCouBro,488nm/εCouBro–GSH,488nm = 9.2. On the basis of the relationship that Kd′ = KdεCouBro,488nm/εCouBro–GSH,488nm (refer to the SI for details), we can deduce that Kd = 1.6 mM (Figure S3).
Figure 3.

Linear relationship between (R – Rmin)/(Rmax – R) and GSH concentration. The reciprocal of the slope is the apparent dissociation constant Kd′. R is based on UV–vis absorption measurements.
Meanwhile, TQ Green showed good specificity toward GSH under physiological conditions. Free cysteine and the surface exposed cysteine residues on proteins inside cells could potentially compete with GSH in TQ Green reactions. It is known that in contrast to the 1–10 mM concentrations of GSH inside cells, cysteine concentrations are in the range of 0.1–1 mM, approximately an order of magnitude lower than GSH levels.2,44 Assuming cysteine and GSH have similar reactivities, the presence of cysteine will introduce an error no more than 10%. At a 0.1 mM concentration of cysteine, we did not observe appreciable reactions between TQ Green and cysteine within 2 h (Figure 4). Proteins containing free thiol groups may also react with TQ Green to affect the measurement of GSH levels. On average, the protein concentration inside cells is 3 × 106 molecules/μm3, which corresponds to a concentration of 5 mM.45 Assuming there is one free thiol on each protein molecule, a 5 mM bovine serum albumin (BSA) solution should reasonably mimic intracellular proteins because (a) a BSA molecule has a free thiol on its surface and (b) the molecular weight of BSA (65 kDa) is close to the average molecular weight of intracellular proteins (∼50 kDa).45 As shown in Figure 4, TQ Green undergoes little reaction with 5 mM of BSA within the experimental time scale. Through these experiments and analyses, we can conclude that TQ Green has good specificity toward GSH under physiological conditions.
Figure 4.
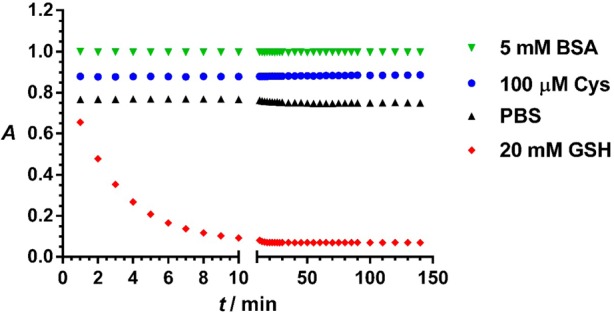
Reaction specificity of TQ Green and GSH under physiological concentrations. For clarity, data points for the TQ Green reaction with BSA, cysteine, and PBS were offset by 0.1 unit from each other on the y axis. Data points represent the absorbance of reaction mixtures of TQ Green (32 μM) with cysteine (100 μM, blue), BSA (5 mM, green), GSH (20 mM red), and water (black) in PBS (pH 7.4) at 479 nm, the maximum absorption wavelength for TQ Green.
In order to allow TQ Green to efficiently penetrate into cells for GSH measurements, we converted the carboxylic acid group to an acetoxymethyl (AM) ester (Figure S4). Once inside cells, the AM ester will be readily hydrolyzed by esterases to regenerate TQ Green (Figure 5). To verify this, we incubated TQ Green-AM (Scheme 2, 40 μM) in PBS for 2 h and in a 500 times diluted HeLa cell lysate for 10 h at 37 °C (Note: the dilution factor for the cell lysate is calculated based on the fact that ∼400 000 HeLa cells, which have a total volume of ∼2 μL, were lysed in 1 mL of lysis buffer, resulting in 500 times dilution of cellular components, Table 2). A mixture of TQ Green and TQ Green-AM standard samples and the reaction mixtures after incubation in PBS and in cell lysate were separated using high performance liquid chromatography (HPLC) with a tandem of UV–vis and mass spectrometer (MS) detectors. We observed that TQ Green-AM ester is pretty resistant to hydrolysis in PBS. In contrast, TQ Green-AM ester was completely converted to a mixture of TQ Green and TQ Green-GSH adduct after 10 h of incubation in a diluted cell lysate based on both UV–vis and MS analyses (Figure 5). Assuming esterase hydrolysis follows pseudo-first order kinetics and the concentration of TQ Green-AM is no more than 1 μM (loading concentration), TQ Green-AM will be completely hydrolyzed to TQ Green within 1 min once inside cells. This experiment demonstrated that TQ Green-AM is a TQ Green precursor that can be efficiently regenerated inside cells. This is also consistent with the fact that AM esters are widely used in many molecular probes, such as calcium probe Fura-243 and H2S probe SF7.46
Figure 5.
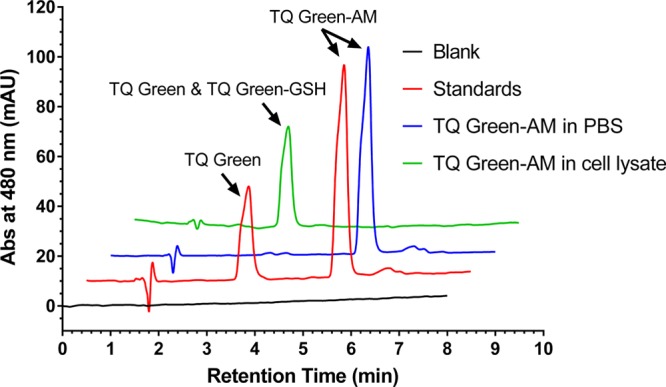
Regeneration of TQ Green from TQ Green-AM under intracellular environment. TQ Green-AM (40 μM) was incubated in PBS for 2 h and in a 500 times diluted HeLa cell lysate for 10 h at 37 °C. The reaction products were analyzed by HPLC with a tandem of UV–vis and MS detectors. Analytical standards TQ Green and TQ Green-AM (red trace) were used to determine the retention time of the corresponding compounds. TQ Green-AM did not show appreciable hydrolysis in PBS within 2 h (blue trace), indicating that TQ Green-AM stays intact before entering cells under the live imaging conditions. TQ Green-AM was completely hydrolyzed after 10 h of incubation in 500 times diluted cell lysate (green trace), indicating that TQ Green-AM can be completely converted into TQ Green within ∼1 min under an intracellular environment. Note, under the elution conditions used, TQ Green and TQ Green-GSH cannot be separated. But their identities were confirmed by MS. All the traces were offset by 0.5 min on the x axis and 10 mAU on the y axis from each other for clarity.
Scheme 2. Structure of TQ Green-AM Ester.

Table 2. Quantification of GSH Levels in Different Cell Lines Using Live Imaging Based and Lysate Based Methodsa.
| imaging
based |
lysate based |
||||
|---|---|---|---|---|---|
| CGSH (mM) | Nb | GSH per cell (fmol)c | cell volume (μm3)d | CGSH (mM) | |
| 3T3-L1 | 4.6 ± 0.9 | 41 | 10.3 ± 0.4 | 4500 ± 700 | 2.3 ± 0.4 |
| HepG2 | 6.2 ± 1.9 | 105 | 16.2 ± 1.5 | 3100 ± 500 | 5.2 ± 0.8 |
| HeLa | 4.6 ± 0.8 | 151 | 13.2 ± 0.9 | 2700 ± 400 | 4.9 ± 0.8 |
| PANC-1 | 6.4 ± 1.8 | 89 | 32.7 ± 3.1 | 4800 ± 700 | 6.8 ± 1.1 |
| PANC-28 | 6.6 ± 1.7 | 80 | 26.7 ± 2.7 | 4900 ± 800 | 5.4 ± 0.9 |
All the errors represent standard deviations.
N is the number of cells used for quantification.
Amount of GSH per cell.
Cell volume measured by packed cell volume tubes. The values shown are the average of three measurements. The errors for GSH levels in the lysate measurement mainly originate from the cell volume measurements using packed cell volume tubes.
TQ Green can establish equilibrium with GSH within 30 min inside cells. As shown in Figure 2, the half-life of TQ Green in 40 mM GSH solution is 116 s (∼2 min) at 25 °C. In a 10 mM GSH environment, the TQ Green half-life will be ∼8 min assuming pseudo-first-order kinetics. Therefore, it takes about three half-lives (∼24 min) to get ∼90% consumption of TQ Green at 25 °C. As a rule of thumb, based on the Arrhenius equation, reaction rates generally double for every 10 °C increase in temperature. At 37 °C, the intracellular environment, TQ Green should be able to establish equilibrium with GSH within 15–30 min, which is the incubation time used for all the following imaging experiments in this study.
To determine the subcellular distribution of TQ Green, we costained HeLa cells with probes specific to different organelles, including mitochondria, endoplasmic reticulum (ER), lysosomes, and endosomes (Figure 6). TQ Green displays an extranuclear distribution pattern, which suggests that TQ Green may have preference for specific organelles. Further analyses revealed that TQ Green mainly colocalizes with mitochondria and ER, and with lysosomes and endosomes to a much lesser extent (Figure 6). We infer that TQ Green that did not colocalize with any of the organelle specific markers tested resides in cytoplasm. The goal of our current study is to determine the cytoplasmic GSH concentrations in live cells. Therefore, reduced distribution of TQ Green in the nucleus does not affect our quantification of cytoplasmic GSH. The colocalization of TQ Green with ER and mitochondria is of specific interest for the future development of organelle specific probes.
Figure 6.

Subcellular distribution of TQ Green. HeLa cells were costained with TQ Green (green) and different organelle specific probes, including MitoTracker Red, ER-Tracker Red, LysoTracker Red, and mRFP-Rab5 fusion protein (endosome localized red fluorescent protein (RFP)). Orange color in the overlay column indicates colocalization. It should be noted that due to the transfection efficiency, some of the cells did not express mRFP-Rab5.
Despite the fact that TQ Green colocalizes with ER and mitochondria, it is unclear whether TQ Green resides in lipid membranes or an aqueous environment, which is critical to quantitative analyses because fluorophores tend to have different quantum yields in hydrophilic and hydrophobic environments. We obtained the quantum yields for TQ Green and TQ Green-GSH in PBS as 0.0094 ± 0.0004 and 0.0059 ± 0.0003, respectively (Table 1, refer to the SI for a detailed explanation). In contrast, the quantum yield for TQ Green increased significantly to 0.16 ± 0.05 in an organic solvent (Note: TQ Green-GSH is not soluble in organic solvents; therefore, the quantum yield of TQ Green-GSH in organic solvents is unavailable). To further understand TQ Green distribution between hydrophobic and hydrophilic environments, we measured the log D values of TQ Green and TQ Green-GSH using an octanol–PBS (pH 7.4) biphasic system (Table 1). We found that TQ Green has a log D value of 0.7, indicating a preference for hydrophobic environments and, thus, the potential enrichment of TQ Green in intracellular lipid membranes. In addition, we discovered that the absorbance maximum of TQ Green shifts from 480 to 455 nm when changing the solvents from PBS to octanol. We hypothesized that if TQ Green is distributed in lipid membranes, the absorbance maximum and fluorescence intensity will change. We used liposomes to mimic the lipid membranes and found that an increasing concentration of liposomes indeed blue-shifts the absorption of TQ Green and enhance its fluorescence (data not shown). Consistent with the log D measurements, this experiment also supports the hypothesis that TQ Green accumulates in lipid membranes. Considering the high quantum yield of TQ Green in hydrophobic solvents, lipid enrichment of TQ Green seemingly complicates intracellular GSH quantification. After careful analysis, we found that if thermodynamic equilibria completely establish between the distributions of TQ Green and TQ Green-GSH in lipid membranes and aqueous environment, and between TQ Green and GSH, the fluorescence intensity ratios of TQ Green and TQ Green-GSH are still proportional to GSH concentrations (please refer to the SI for detailed explanation). However, the Kd′ values may be different under calibration conditions and inside cells, which may introduce a systemic error (vide infra).
In order to quantify intracellular GSH concentration, a calibration curve for TQ Green in known concentrations of GSH was established using a confocal microscope (Figure 7, refer to the SI for details). A fixed excitation laser energy was appropriately chosen to ensure that the Fl signals of TQ Green in different concentrations of GSH fit into the dynamic range of the microscope. A calibration curve with a reasonable linearity (r2 = 0.97) was generated using known concentrations of GSH solutions (Figures 7, S5, and S6). It should be noted that with the settings of our confocal microscope, we found that R is in a reasonable linear relationship with GSH concentrations. This is because Kd′ is an instrument dependent parameter and R is proportional to GSH concentrations if Kd′ is much larger than 10 mM (refer to the SI for details). Therefore, for all the cell imaging studies, R is plotted against GSH concentrations in standard curves and quantification, instead of (R – Rmin)/(Rmax – R).
Figure 7.

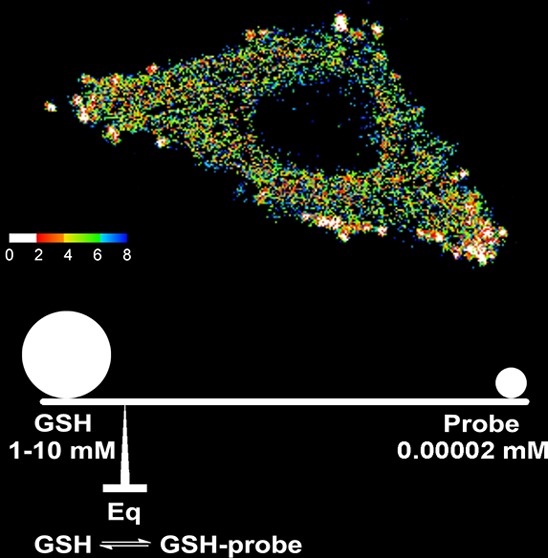
Measurements of GSH levels in HeLa cells based on ratiometric fluorescence imaging. (a) Representative images of HeLa cells treated with TQ Green-AM. The ratiometric image represents the distribution of GSH levels (the calibration bar represents the ratiometric reading instead of GSH concentration). (b) Standard curve of R, the fluorescence intensity ratio between 405 and 488 nm excitation, as a function of GSH concentration produced using the same instrument setting as the live cell imaging experiment. The data point in red represents the GSH concentration in HeLa cells based on statistical analyses of >40 cells. Error bars represent standard deviations.
On the basis of this calibration curve, we first determined the GSH concentration to be 4.6 ± 0.8 mM in HeLa cells (Figure 7), which is consistent with previously reported values.47 A representative image is shown in Figure 7 to demonstrate intracellular distribution of GSH levels. We further expand the live imaging measurements in several other cell lines, including 3T3-L1, HepG2, PANC-1, and PANC-28 cells (Table 2). For comparison, we also measured the total amount of GSH in these cell lines using their cell lysate following a well-established protocol.48 To further convert the amount of GSH into concentrations, we measured the corresponding cell volumes using packed cell volume (PCV) tubes. As shown in Figure 8, the concentrations measured using TQ Green live imaging are well correlated with the values obtained from bulk lysate measurements.
Figure 8.

Correlation between the GSH concentrations measured in live cells and in lysates. The y axis represents concentrations derived from live imaging, while the x axis represents concentrations determined using cell lysate. All imaging results are from statistical analysis of >40 cells. All assay results are from >3 replicates under the same conditions. Error bars represent standard deviations. The slope of the correlation line (the dash line) is 1.1.
It is worth noting that the correlation line (dashed line in Figure 8) has a slope of 1.1, indicating that the GSH concentrations from live imaging are ∼10% higher than the values measured using lysates. This systemic error may originate from a loss of lysate during the cell homogenization process. Alternatively, it is also possible that a systemic error arose because the calibration curve was generated using TQ Green adsorbed onto the surface of polystyrene beads in PBS, which is different from the intracellular environment. In addition, intracellular cysteine may also react with TQ Green to afford an overestimated GSH concentration using the imaging method (vide supra). This systemic error will be investigated in our future studies. It should be noted that the GSH level in 3T3-L1 cells based on TQ Green imaging (4.6 ± 0.9 mM) deviates the most from the bulk lysate measurement (2.3 ± 0.4 mM). We noticed that unlike the spherical cancer cells, 3T3-L1 cells are stellate, which prevents tight packing in the cell volume measurement. Therefore, we suspect that the inconsistency in GSH levels between TQ Green imaging and bulk lysate measurement in 3T3-L1 is due to an overestimation of the 3T3-L1 cell volume, and thus an underestimation of GSH concentration in the lysate. The live imaging method also results in relatively large standard deviations. This is mainly because the imaging method measures GSH concentrations in individual cells and the heterogeneity of the cells broadens the distribution of the measurements. However, this can be advantageous if single cell behavior is the area of interest. Overall, TQ Green enables quantitative measurements of GSH levels in live cells.
To further demonstrate the ability of TQ Green to monitor GSH level changes and the reproducibility of live imaging based measurements, we treated PANC-1 cells with 50 μM diethyl maleate (DEM) for 24 h to decrease the GSH levels. We also treated PANC-1 cells with DEM for a very short time to stimulate the uptake of cysteine, thus increasing the GSH levels.49 After removal of DEM, the cells were further incubated under normal conditions for an additional 22 h before imaging. As shown in Figure 9, we observed a 27% decrease and 28% increase in GSH levels with GSH inhibition and stimulation experiments, respectively (P < 0.001). Importantly, the GSH level obtained in DEM nontreated cells after 24 h is essentially the same as the value measured at time zero (P = 0.63), indicating excellent reproducibility of live imaging based GSH quantification.
Figure 9.
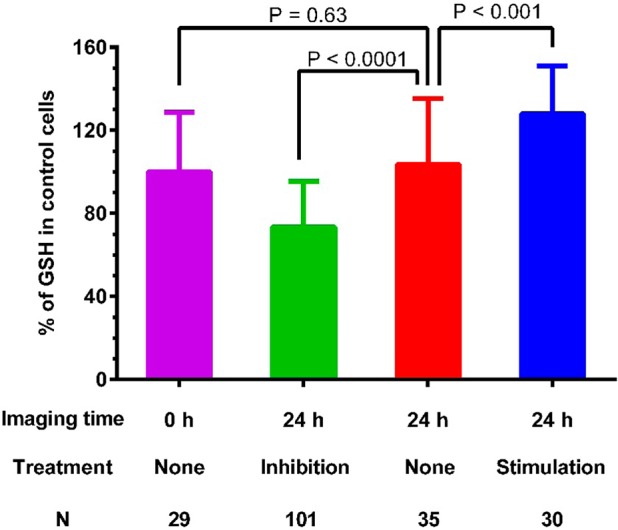
Detection of GSH level changes in PANC-1 cells using TQ Green live imaging. PANC-1 cells were treated with diethyl maleate (50 μM) for 24 and 2 h to inhibit and stimulate GSH levels, respectively. All the cells were imaged 24 h after starting the experiment. Results are statistical analyses of >25 cells. P values shown are based on unpaired student t tests. Error bars represent standard deviations. N is the number of cells analyzed.
The GSH levels are generally cell cycle dependent.50 We observed the same GSH levels in PANC-1 cells at 0 and 24 h time points (Figure 9). The cells used in this experiment were not synchronized. Therefore, the GSH levels reflect an average of cells in different cell cycles. Because of the heterogeneity of the cells, we are unable to determine the concentration changes as a function of time if no stimulating or inhibiting signals are introduced. In our future studies, we plan to follow individual cells over a long period of time to monitor the GSH concentration changes in different cell cycles.
Not only is TQ Green capable of quantifying GSH levels in high resolution confocal microscope experiments, but it is also suitable for FACS based bulk cell measurements. PANC-1 cells were treated with DEM under both inhibition and stimulation conditions as described in Figure 9. After 24 h, cells were further incubated with TQ Green-AM for 30 min and washed with trypan blue to quench the fluorescence that originated from surface bound TQ Green. The fluorescence intensities of each individual cell were quantified using FACS with both 405 and 488 nm excitations. As shown in Figure 10c, the fluorescence ratio of 405 and 488 nm, which is positively correlated with GSH levels, changes accordingly upon different cell treatment conditions. It should be noted that in the FACS histograms (Figure 10a and b), the 488 nm channel changed significantly under different cell treatment conditions, while the 405 nm channel remained essentially the same. This is because the fluorescence intensities of TQ Green and TQ Green-GSH differ significantly with excitation at 488 nm but have very similar fluorescence intensities with excitation at 405 nm (Figure S7). When TQ Green-GSH is formed at the expense of TQ Green, the loss of TQ Green fluorescence at 405 nm excitation coincides with the gain of TQ Green-GSH fluorescence, which makes the 405 nm fluorescence remain unchanged.
Figure 10.

Quantification of GSH levels using fluorescence activated cell sorting (FACS). PANC-1 cells were treated with diethyl maleate (50 μM) for 24 and 2 h to inhibit and stimulate GSH levels, respectively. The GSH levels of the cells were measured 24 h after starting the experiment using FACS. Red, green, and blue traces and bars represent inhibition, stimulation, and control conditions. (a,b) Histograms of the 405 nm (Pacific Blue) and 488 nm (FITC) channels are shown. (c) Fluorescence intensity ratios of 405 and 488 nm as a function of treatment conditions. Results are statistical analyses of >4000 cells. P values shown are based on unpaired student t tests. Error bars, representing SEM, are too small to show clearly.
In summary, we demonstrated the importance of reaction reversibility in designing quantitative reaction-based fluorescent probes. We developed the first quantitative imaging of intracellular GSH concentration using a reversible reaction-based ratiometric fluorescent probe. We successfully applied the probe to measure the intracellular GSH concentrations and found that the imaging based measurements are well-correlated with lysate based bulk measurements. In addition, we showed that this live imaging method has excellent reproducibility and is able to detect GSH level changes in cells following the stimulation and inhibition effect of DEM. Furthermore, TQ Green is also suitable for GSH measurements using FACS. It should be noted that the reverse reaction between GSH and TQ Green is sluggish (Figure 2a). For this reason, TQ Green is suitable for one-point measurement or monitoring increases in GSH levels but is unable to respond quickly to any decreases in GSH concentrations. Another caveat is that TQ Green does not distribute exclusively in cytosol and also penetrates into ER and mitochondria, which were reported to have different GSH levels from the cytosol.19 Therefore, the imaging analyses of TQ Green should be considered as a measurement of the global GSH level. Our future work will focus on developing GSH probes with fast kinetics in both forward and reverse reactions in order to quantitatively monitor the dynamics of intracellular GSH and increasing the subcellular specificity of this probe. We believe that this study will not only provide a convenient tool for redox biology studies but also guide future development of reaction-based fluorescent probes for quantitative imaging in live cells.
Methods
Materials
All the chemicals were purchased from Sigma-Aldrich and Alfa Aesar unless otherwise specified. All solvents and reagents were used as obtained without further purification. Polystyrene beads (4.5 μm, catalog # 17135–5) were purchased from Polysciences Inc. All the organelle specific dyes were purchased from Thermo Fisher Scientific Inc.
Instrumentation
NMR spectra were recorded on a Varian NMR (1H at 400 MHz) spectrometer. Chemical shifts (δ) were given in parts per million with reference to solvent signals [1H NMR: CDCl3 (7.26), CD3OD (3.31)]. UV–vis measurements were performed with a 2 × 10 mm quartz cuvette in a Cary 60 UV–vis spectrometer. Fluorescence measurements were performed with the same quartz cuvette in a Cary Eclipse fluorescence spectrophotometer with an excitation slit of 5 nm, an emission slit of 10 nm, and PMT at 650 V. Flash chromatography was performed on a Teledyne ISCO CombiFlashRf200. HPLC measurements were performed on Agilent Infinity 1200 HPLC with inline diode array and ESI-MS detectors. An Olympus FV1000 laser scanning confocal microscope system was used for cell imaging. ESI mass spectrometry was measured on a BrukerMS microTOF ESI, at the Shared Equipment Authority at Rice University. Flow cytometry was performed on BD LSR II Flow Cytometer at BCM core facilities.
Chemical Synthesis and Characterization
Refer to the SI for details.
Determination of Equilibrium Constant of TQ Green and GSH Reaction
TQ Green was dissolved in PBS buffer (10 mM, pH 7.4) containing 1% DMSO with a concentration at 32 μM. GSH was dissolved in the same PBS buffer with a concentration from 0 to 80 mM. The above solutions were mixed at 1:1 ratio, with the exception of the solution containing 80 mM of GSH and TQ Green, which was prepared by directly mixing TQ Green stock solution in DMSO with a 80 mM of GSH solution at a ratio of 1:100. All solutions were protected with nitrogen and stored in a glovebox to prevent any oxygen entering the solution. Samples of all solutions were taken out at 1, 18, 72, and 144 h after mixing. UV–vis and fluorescence were measured for all solutions.
Cell Culture and Treatment for Imaging
All cell lines used in this study were purchased from American Type Culture Collection (ATCC) and grown in DMEM (Gibco, 11965) media supplemented with 10% FBS and 1% 1003 Pen Strep (Gibco). Cells were cultured under a controlled atmosphere (37 °C, 5% CO2). Glass dishes were used for the cell culture according to confocal scanning requirements. Cells were treated with TQ Green-AM (20 nM to 1 μM with 0.0025–1% DMSO in PBS) for 30 min, followed by two washing steps with trypan blue and PBS prior to imaging. Fluorescent images were acquired with a 405 nm laser/430–470 nm emission filter and 488 nm laser/575–620 nm emission filter. All the microscope settings were kept consistent in each experiment.
Calibration for Confocal Microscopy
GSH solutions (0–80 mM in PBS (10 mM, pH 7.4)) were prepared and mixed with TQ Green solution (5 μM final concentration). The above solutions were further mixed with a suspension containing 4.5 μm polystyrene beads. Cover glasses were used to hold the solutions for confocal microscopy. The same microscope settings were adopted from prior experiments.
Subcellular Colocalization Imaging
HeLa cells were cultured on glass bottom dishes as described before. For endosome labeling, cells were transfected with pCMV:mRFP-Rab5 plasmid 24 h prior to imaging; for mitochondria labeling, cells were treated with 100 nM MitoTracker Red CMXRos (Thermo Fisher Scientific Inc., catalog# M-7512) in PBS 30 min prior to imaging; for ER labeling, cells were treated with 1 μM ER-Tracker Red CMXRos (Thermo Fisher Scientific Inc., catalog# E34250) in PBS 30 min prior to imaging; for lysosome labeling, cells were treated with 50 nM LysoTracker Red DND-99 (Thermo Fisher Scientific Inc., catalog# L-7528) in PBS 120 min prior to imaging. All cells were also costained with 1 μM TQ Green-AM in PBS 30 min prior to imaging. Fluorescent images were acquired with a 405 nm laser/430–470 nm filter, 488 nm laser/505–545 nm filter, and 559 nm laser/575–620 nm filter.
Glutathione Reductase Assay and Cell Volume Measurement
The procedure was adopted from the literature with minor modification.48 All cells were grown on six-well plates until the cell number reached about 5 × 105 in each well before harvesting. Cells were washed with cold PBS buffer twice and digested by 0.25 mL of trypsin under RT (treatment time varies, usually around 5 min). Then, 1 mL of fresh medium was added to neutralize, and the solution was immediately transferred to a cold 1.5 mL Eppendorf tube. A small sample was used for cell counting every time. The sample was centrifuged at 1000g for 5 min at 4 °C, and the supernatant was discarded. The cell pellet was then washed with PBS and centrifuged again under the same conditions. The supernatant was removed, and cell-lysis buffer (1 mL 0.1% Triton-X and 0.6% sulfosalicylic acid in EDTA added PBS buffer) was added. Cells were homogenized using a Teflon pestle at 4 °C. The suspension was centrifuged at 3000g for 4 min at 4 °C, and the supernatant was transferred to a new cold Eppendorf tube, which is ready for assay measurement.
In a 96-well microtiter plate, 20 μL of cell lysate samples were placed in each well. Freshly made solutions of 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) and glutathione reductase (GR) with concentrations of 0.33 g/L and 1.67 units/mL, respectively, were added to the same well to make the final volume 140 μL. After about 30 s, 60 μL of β-NADPH (0.67 g/L) was added, and the mixture was immediately measured for absorbance at 412 nm every 30 s for 2 min. The slope of the absorbance changes was proportional to the GSH concentration. A standard curve with known GSH concentrations was used to calibrate all the results.
Cell volumes were measured by centrifuging the cell suspension (∼4 × 105 cells per sample) at 2500g for 1 min in packed cell volume (PCV) tubes (Sigma-Aldrich, cat. no. Z760986).
Measurement of GSH Concentration Changes in PANC-1 Cells
PANC-1 cells were incubated with DEM (50 μM) for 2 h for stimulation of cysteine uptake. After replacement with fresh medium, cells were cultured under normal conditions for another 22 h. A separate dish of PANC-1 cells was incubated with DEM (50 μM) as an inhibitor for 24 h. TQ Green-AM (1 μM with 1% DMSO) was used to stain the cells for 30 min, followed by two washing steps with trypan blue and PBS prior to measurement. The same imaging procedures as above were performed. GSH concentrations were calculated based on the calibration curve. FACS was performed from harvested cells after the same treatment as mentioned above; fluorescent intensities were recorded with 405 nm laser/420–460 nm filter (pacific blue channel) and 488 nm laser/515–545 nm filter (FITC channel). Data were processed with FlowJo.
Acknowledgments
This work is supported in part by the Cancer Prevention and Research Institute of Texas (CPRIT R1104) and the Welch Foundation (Q-1798) to J.W., National Institute of Health (R01AG045183) to M.C.W., Texas Advanced Computing Center (TACC), the Center for Drug Discovery at Baylor College of Medicine, the Dan L. Duncan Cancer Center, the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom, the John S. Dunn Gulf Coast Consortium for Chemical Genomics, and the Shared Equipment Authority instrumentation at Rice University. M.Z. thanks the support from the Integrative Molecular and Biomedical Sciences Graduate Program Training Grant (NIH 5 T32 GM008231). We also thank Dr. S. Ho for sharing pCMV:mRFP-Rab5 plasmid. We also appreciate the discussion and suggestion from the reviewers, Drs. T. Wensel and T. Palzkill at Baylor College of Medicine and Dr. M. Janes and his molecular probe team at Thermo Fisher Scientific Inc.
Supporting Information Available
Experimental details on synthesis and cell imaging studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Balaban R. S.; Nemoto S.; Finkel T. (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495. [DOI] [PubMed] [Google Scholar]
- Banerjee R. (2012) Redox outside the box: linking extracellular redox remodeling with intracellular redox metabolism. J. Biol. Chem. 287, 4397–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaestro B. M.; Malone M. (1995) Glutathione in health and disease: pharmacotherapeutic issues. Ann. Pharmacother. 29, 1263–1273. [DOI] [PubMed] [Google Scholar]
- Winther J. R.; Thorpe C. (2014) Quantification of thiols and disulfides. Biochim. Biophys. Acta 1840, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Kwon Y.; Kim D.; Lee D.; Kim G.; Hu Y.; Ryu J. H.; Yoon J. (2014) Cyanine-based fluorescent probe for highly selective detection of glutathione in cell cultures and live mouse tissues. J. Am. Chem. Soc. 136, 5351–5358. [DOI] [PubMed] [Google Scholar]
- Jung H. S.; Chen X.; Kim J. S.; Yoon J. (2013) Recent progress in luminescent and colorimetric chemosensors for detection of thiols. Chem. Soc. Rev. 42, 6019–6031. [DOI] [PubMed] [Google Scholar]
- Acharya J. R.; Zhang H.; Li X.; Nesterov E. E. (2009) Chemically controlled amplified ratiometric fluorescence in surface-immobilized end-capped oligo(p-phenylene ethynylene)s. J. Am. Chem. Soc. 131, 880–881. [DOI] [PubMed] [Google Scholar]
- Niu L. Y.; Guan Y. S.; Chen Y. Z.; Wu L. Z.; Tung C. H.; Yang Q. Z. (2012) BODIPY-based ratiometric fluorescent sensor for highly selective detection of glutathione over cysteine and homocysteine. J. Am. Chem. Soc. 134, 18928–18931. [DOI] [PubMed] [Google Scholar]
- Lim C. S.; Masanta G.; Kim H. J.; Han J. H.; Kim H. M.; Cho B. R. (2011) Ratiometric detection of mitochondrial thiols with a two-photon fluorescent probe. J. Am. Chem. Soc. 133, 11132–11135. [DOI] [PubMed] [Google Scholar]
- Yi L.; Li H.; Sun L.; Liu L.; Zhang C.; Xi Z. (2009) A highly sensitive fluorescence probe for fast thiol-quantification assay of glutathione reductase. Angew. Chem., Int. Ed. 48, 4034–4037. [DOI] [PubMed] [Google Scholar]
- Shao N.; Jin J.; Wang H.; Zheng J.; Yang R.; Chan W.; Abliz Z. (2010) Design of bis-spiropyran ligands as dipolar molecule receptors and application to in vivo glutathione fluorescent probes. J. Am. Chem. Soc. 132, 725–736. [DOI] [PubMed] [Google Scholar]
- Sun Y. Q.; Liu J.; Zhang H.; Huo Y.; Lv X.; Shi Y.; Guo W. (2014) A Mitochondria-Targetable Fluorescent Probe for Dual-Channel NO Imaging Assisted by Intracellular Cysteine and Glutathione. J. Am. Chem. Soc. 136, 12520–12523. [DOI] [PubMed] [Google Scholar]
- Yu F.; Li P.; Wang B.; Han K. (2013) Reversible near-infrared fluorescent probe introducing tellurium to mimetic glutathione peroxidase for monitoring the redox cycles between peroxynitrite and glutathione in vivo. J. Am. Chem. Soc. 135, 7674–7680. [DOI] [PubMed] [Google Scholar]
- McMahon B. K.; Gunnlaugsson T. (2012) Selective detection of the reduced form of glutathione (GSH) over the oxidized (GSSG) form using a combination of glutathione reductase and a Tb(III)-cyclen maleimide based lanthanide luminescent ‘switch on’ assay. J. Am. Chem. Soc. 134, 10725–10728. [DOI] [PubMed] [Google Scholar]
- Ahn Y. H.; Lee J. S.; Chang Y. T. (2007) Combinatorial rosamine library and application to in vivo glutathione probe. J. Am. Chem. Soc. 129, 4510–4511. [DOI] [PubMed] [Google Scholar]
- Wang K.; Peng H.; Wang B. (2014) Recent Advances in Thiol and Sulfide Reactive Probes. J. Cell. Biochem. 115, 1077–1022. [DOI] [PubMed] [Google Scholar]
- Hanson G. T.; Aggeler R.; Oglesbee D.; Cannon M.; Capaldi R. A.; Tsien R. Y.; Remington S. J. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044–13053. [DOI] [PubMed] [Google Scholar]
- Gutscher M.; Pauleau A. L.; Marty L.; Brach T.; Wabnitz G. H.; Samstag Y.; Meyer A. J.; Dick T. P. (2008) Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 5, 553–559. [DOI] [PubMed] [Google Scholar]
- Montero D.; Tachibana C.; Rahr Winther J.; Appenzeller-Herzog C. (2013) Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 1, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaert N. L.; van der Vliet A.; Guala A. S.; McGovern T.; Hristova M.; Pantano C.; Heintz N. H.; Heim J.; Ho Y. S.; Matthews D. E.; Wouters E. F.; Janssen-Heininger Y. M. (2006) Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. U.S.A. 103, 13086–13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B.; Ezerina D.; Amoako T. N.; Riemer J.; Seedorf M.; Dick T. P. (2013) Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 9, 119–125. [DOI] [PubMed] [Google Scholar]
- Kim G. J.; Lee K.; Kwon H.; Kim H. J. (2011) Ratiometric fluorescence imaging of cellular glutathione. Org. Lett. 13, 2799–2801. [DOI] [PubMed] [Google Scholar]
- Nagano T.; Yoshimura T. (2002) Bioimaging of nitric oxide. Chem. Rev. 102, 1235–1270. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Lin W.; Xie Y.; Chen B.; Song J. (2011) Development of a ratiometric fluorescent sensor for ratiometric imaging of endogenously produced nitric oxide in macrophage cells. Chem. Commun. 47, 9372–9374. [DOI] [PubMed] [Google Scholar]
- Srikun D.; Miller E. W.; Domaille D. W.; Chang C. J. (2008) An ICT-based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells. J. Am. Chem. Soc. 130, 4596–4597. [DOI] [PubMed] [Google Scholar]
- Albers A. E.; Okreglak V. S.; Chang C. J. (2006) A FRET-based approach to ratiometric fluorescence detection of hydrogen peroxide. J. Am. Chem. Soc. 128, 9640–9641. [DOI] [PubMed] [Google Scholar]
- Bae S. K.; Heo C. H.; Choi D. J.; Sen D.; Joe E. H.; Cho B. R.; Kim H. M. (2013) A ratiometric two-photon fluorescent probe reveals reduction in mitochondrial H2S production in Parkinson’s disease gene knockout astrocytes. J. Am. Chem. Soc. 135, 9915–9923. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhu C.; Yang Z.; Chen J.; He Y.; Jiao Y.; He W.; Qiu L.; Cen J.; Guo Z. (2013) A ratiometric fluorescent probe for rapid detection of hydrogen sulfide in mitochondria. Angew. Chem., Int. Ed. 52, 1688–1691. [DOI] [PubMed] [Google Scholar]
- Wan Q.; Song Y.; Li Z.; Gao X.; Ma H. (2013) In vivo monitoring of hydrogen sulfide using a cresyl violet-based ratiometric fluorescence probe. Chem. Commun. 49, 502–504. [DOI] [PubMed] [Google Scholar]
- Wang B.; Li P.; Yu F.; Chen J.; Qu Z.; Han K. (2013) A near-infrared reversible and ratiometric fluorescent probe based on Se-BODIPY for the redox cycle mediated by hypobromous acid and hydrogen sulfide in living cells. Chem. Commun. 49, 5790–5792. [DOI] [PubMed] [Google Scholar]
- Wu M. Y.; Li K.; Hou J. T.; Huang Z.; Yu X. Q. (2012) A selective colorimetric and ratiometric fluorescent probe for hydrogen sulfide. Org. Biomol. Chem. 10, 8342–8347. [DOI] [PubMed] [Google Scholar]
- Yu F.; Li P.; Song P.; Wang B.; Zhao J.; Han K. (2012) An ICT-based strategy to a colorimetric and ratiometric fluorescence probe for hydrogen sulfide in living cells. Chem. Commun. 48, 2852–2854. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Murphy C. S.; Kuang G. C.; Hazelwood K. L.; Constantino M. H.; Davidson M. W.; Zhu L. (2009) A fluorescent heteroditopic ligand responding to free zinc ion over six orders of magnitude concentration range. Chem. Commun. 7408–7410. [DOI] [PubMed] [Google Scholar]
- Sreenath K.; Allen J. R.; Davidson M. W.; Zhu L. (2011) A FRET-based indicator for imaging mitochondrial zinc ions. Chem. Commun. 47, 11730–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan E. M.; Jaworski J.; Okamoto K.; Hayashi Y.; Sheng M.; Lippard S. J. (2005) QZ1 and QZ2: rapid, reversible quinoline-derivatized fluoresceins for sensing biological Zn(II). J. Am. Chem. Soc. 127, 16812–16823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan E. M.; Lippard S. J. (2009) Small-molecule fluorescent sensors for investigating zinc metalloneurochemistry. Acc. Chem. Res. 42, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomat E.; Nolan E. M.; Jaworski J.; Lippard S. J. (2008) Organelle-specific zinc detection using zinpyr-labeled fusion proteins in live cells. J. Am. Chem. Soc. 130, 15776–15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert A. R. (2014) Designing reaction-based fluorescent probes for selective hydrogen sulfide detection. J. Inorg. Biochem. 133, 136–142. [DOI] [PubMed] [Google Scholar]
- Paige J. S.; Nguyen-Duc T.; Song W.; Jaffrey S. R. (2012) Fluorescence imaging of cellular metabolites with RNA. Science 335, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack R. L.; Jaffrey S. R. (2013) New approaches for sensing metabolites and proteins in live cells using RNA. Curr. Opin. Chem. Biol. 17, 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B.; Greaney M. F. (2005) Reversible Michael addition of thiols as a new tool for dynamic combinatorial chemistry. Chem. Commun. 886–888. [DOI] [PubMed] [Google Scholar]
- Johansson M. H. (2012) Reversible Michael additions: covalent inhibitors and prodrugs. Mini Rev. Med. Chem. 12, 1330–1344. [DOI] [PubMed] [Google Scholar]
- Higley M. J.; Sabatini B. L. (2008) Calcium signaling in dendrites and spines: practical and functional considerations. Neuron 59, 902–913. [DOI] [PubMed] [Google Scholar]
- Jones D. P.; Go Y. M.; Anderson C. L.; Ziegler T. R.; Kinkade J. M. Jr.; Kirlin W. G. (2004) Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 18, 1246–1248. [DOI] [PubMed] [Google Scholar]
- Milo R.; Jorgensen P.; Moran U.; Weber G.; Springer M. (2010) BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin V. S.; Lippert A. R.; Chang C. J. (2013) Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc. Natl. Acad. Sci. U.S.A. 110, 7131–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schans G. P.; Vos O.; Roos-Verheij W.; Lohman P. (1986) The influence of oxygen on the induction of radiation damage in DNA in mammalian cells after sensitization by intracellular glutathione depletion. Int. J. Radiat. Biol. 50, 453–465. [DOI] [PubMed] [Google Scholar]
- Rahman I.; Kode A.; Biswas S. K. (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 1, 3159–3165. [DOI] [PubMed] [Google Scholar]
- Bannai S. (1984) Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J. Biol. Chem. 259, 2435–2440. [PubMed] [Google Scholar]
- Poot M.; Kavanagh T. J.; Kang H. C.; Haugland R. P.; Rabinovitch P. S. (1991) Flow cytometric analysis of cell cycle-dependent changes in cell thiol level by combining a new laser dye with Hoechst 33342. Cytometry 12, 184–187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.