

Abstract
Low-voltage-activated (T-type) calcium channels are important regulators of the transmission of nociceptive information in the primary afferent pathway and finding ligands that modulate these channels is a key focus of the drug discovery field. Recently, we characterized a set of novel compounds with mixed cannabinoid receptor/T-type channel blocking activity and examined their analgesic effects in animal models of pain. Here, we have built on these previous findings and synthesized a new series of small organic compounds. We then screened them using whole-cell voltage clamp techniques to identify the most potent T-type calcium channel inhibitors. The two most potent blockers (compounds 9 and 10) were then characterized using radioligand binding assays to determine their affinity for CB1 and CB2 receptors. The structure–activity relationship and optimization studies have led to the discovery of a new T-type calcium channel blocker, compound 9. Compound 9 was efficacious in mediating analgesia in mouse models of acute inflammatory pain and in reducing tactile allodynia in the partial nerve ligation model. This compound was shown to be ineffective in Cav3.2 T-type calcium channel null mice at therapeutically relevant concentrations, and it caused no significant motor deficits in open field tests. Taken together, our data reveal a novel class of compounds whose physiological and therapeutic actions are mediated through block of Cav3.2 calcium channels.
Keywords: hCav 3.2, T-type calcium channel, inflammatory pain, neuropathic pain, carbazole scaffold, electrophysiology
T-type calcium channels are known for regulating neuronal and cardiac pacemaker activity.1−4 They open in response to small membrane depolarizations that in turn trigger the initiation of action potentials.1,5,6 Disruption of this sensitive signaling mechanism often leads to hyperexcitability disorders such as arrhythmia, epilepsy, and pain.3,7−21 The mammalian genome expresses three different types of T-type calcium channels, Cav3.1, Cav3.2, and Cav3.3, with specific cellular functions.22 The Cav3.2 T-type channel isoform is particularly interesting due to its role in afferent pain signaling. Indeed, up-regulation of the Cav3.2 T-type channel isoform in primary afferent fibers has been linked to chronic pain disorders, whereas ablation of these channels mediates analgesia.13,14,17 The development of selective T-type channel antagonists has not been a trivial undertaking, with only a few such small organic molecules having recently been identified.15,23−33 Even more challenging is the development of Cav3 isoform selective blockers due to the large degree of sequence similarity among these three Cav3 channel family members.
Many of the known organic molecules that have been shown to modulate T-type calcium channels have structures similar to endogenous anandamide-related molecules named lipoamino acids.16,23,34 Lipoamino acids are known to interact with T-type calcium channels and several are also closely related to endocannabinoids.16,25,34,35 Therefore, it is not surprising that many of these T-type blockers also interact with cannabinoid (CB) receptors.16,25,34,35 We have previously shown that this mixed T-type/cannabinoid block has beneficial effects in inducing analgesia in animal models of inflammatory pain.16,34 However, interactions with CB receptors, particularly CB1 receptors, can have side effects that may affect mood and memory, in addition to their known psychoactive effects.36,37 Synthetic T-type calcium channel antagonists TTAP-1 based on substituted piperidines have been previously disclosed (Scheme 1).
Scheme 1. Piperidine Containing T-type Ca2+ Inhibitors TTA-P1, Dual T-type Channel Blocker/Cannabinoid Agonist NMP7, and Chemical Optimization Plan 1 To Decrease Cannabinoid Receptor Affinities.

In previous studies, we synthesized and characterized a series of novel cannabinoid ligands with the primary structure bearing a carbazole scaffold such as compound NMP7.16,34 These compounds produced mixed cannabinoid receptor/T-type channel blockers that were found to be efficacious in animal models of inflammatory and neuropathic pain. Interestingly, from structure–activity relationships (SARs), we determined that tertiary amines were important for Cav3.2 block16,23,34 and that the length of the linker attaching the tertiary amine to the carbazole scaffold affected binding to CB1 and CB2 receptors.38 Some of these compounds appeared to preferentially and potently inhibit T-type channels in vitro. The goal of this study was to optimize Cav3.2 calcium channel selectivity of our series of carbazole derivatives.
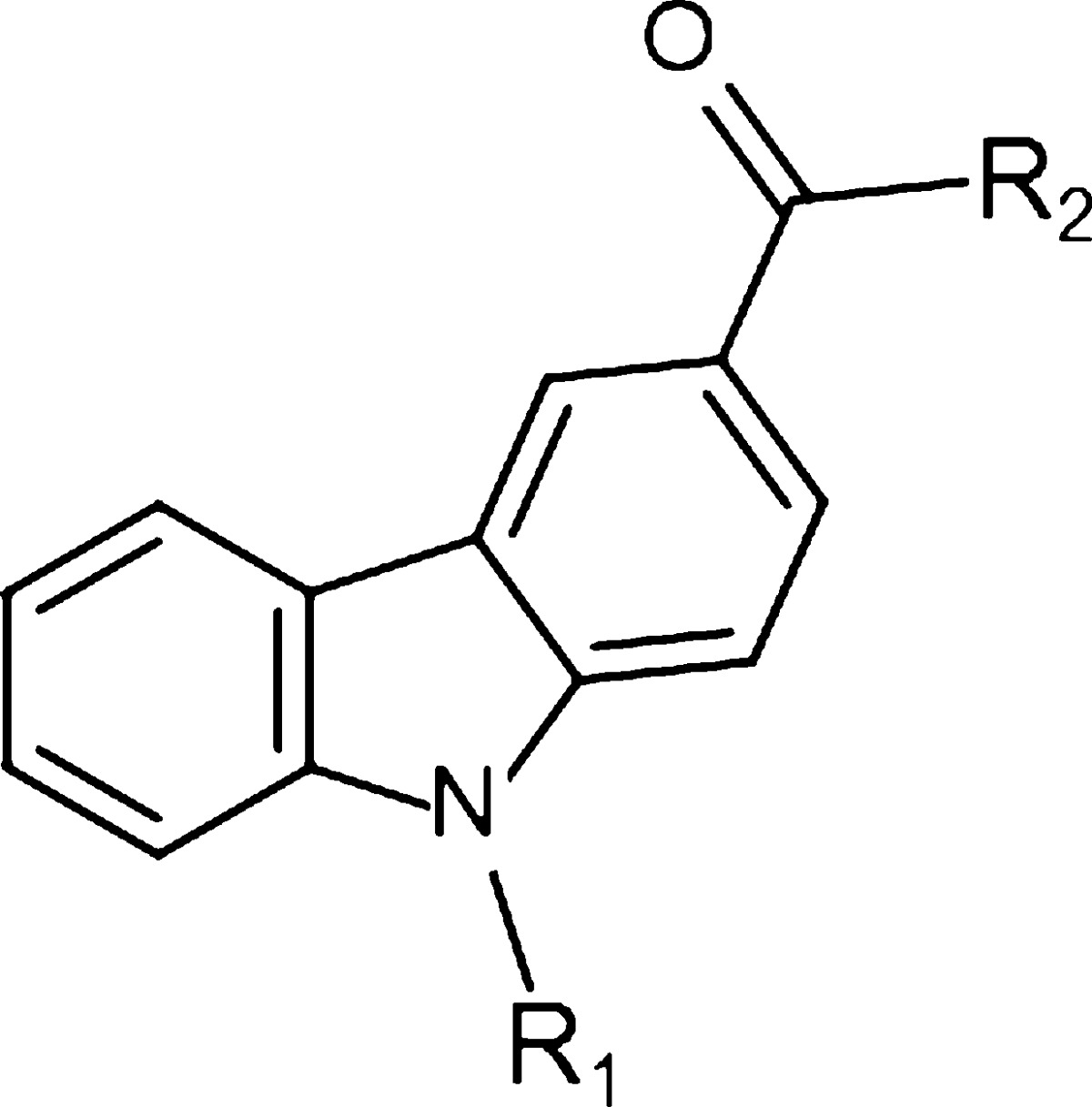
Here we used the aforementioned compounds as a starting point for the development of a new series of compounds 1 (Figure 1) based on a carbazole scaffold with an added heterocyclic bearing a tertiary amine. We modified the chain length attached to the nitrogen of the carbazole, the length of the linker between the amide bond and the heterocycle ring and introduction of a lipophilic moiety attached to the heterocycle and characterized this set of compounds in vitro for their ability to blocking transiently expressed human Cav3.2 (hCav3.2) calcium channels and tested their affinities for cannabinoid receptors. The most potent and selective compound (9) was then tested in mouse models of inflammatory and neuropathic pain, revealing potent analgesia by virtue of its Cav3.2 channel blocking ability.
Figure 1.

Percentage of whole cell current inhibition of human Cav3.2 (T-type) in response to 10 μM application of the compound series (n = 6 per compound). Note the potent and preferential block of Cav3.2 channels by compounds 9 and 10. Error bars reflect standard errors. For Cav3.2 channels, the holding and test potentials were respectively −110 and −20 mV.
Chemistry
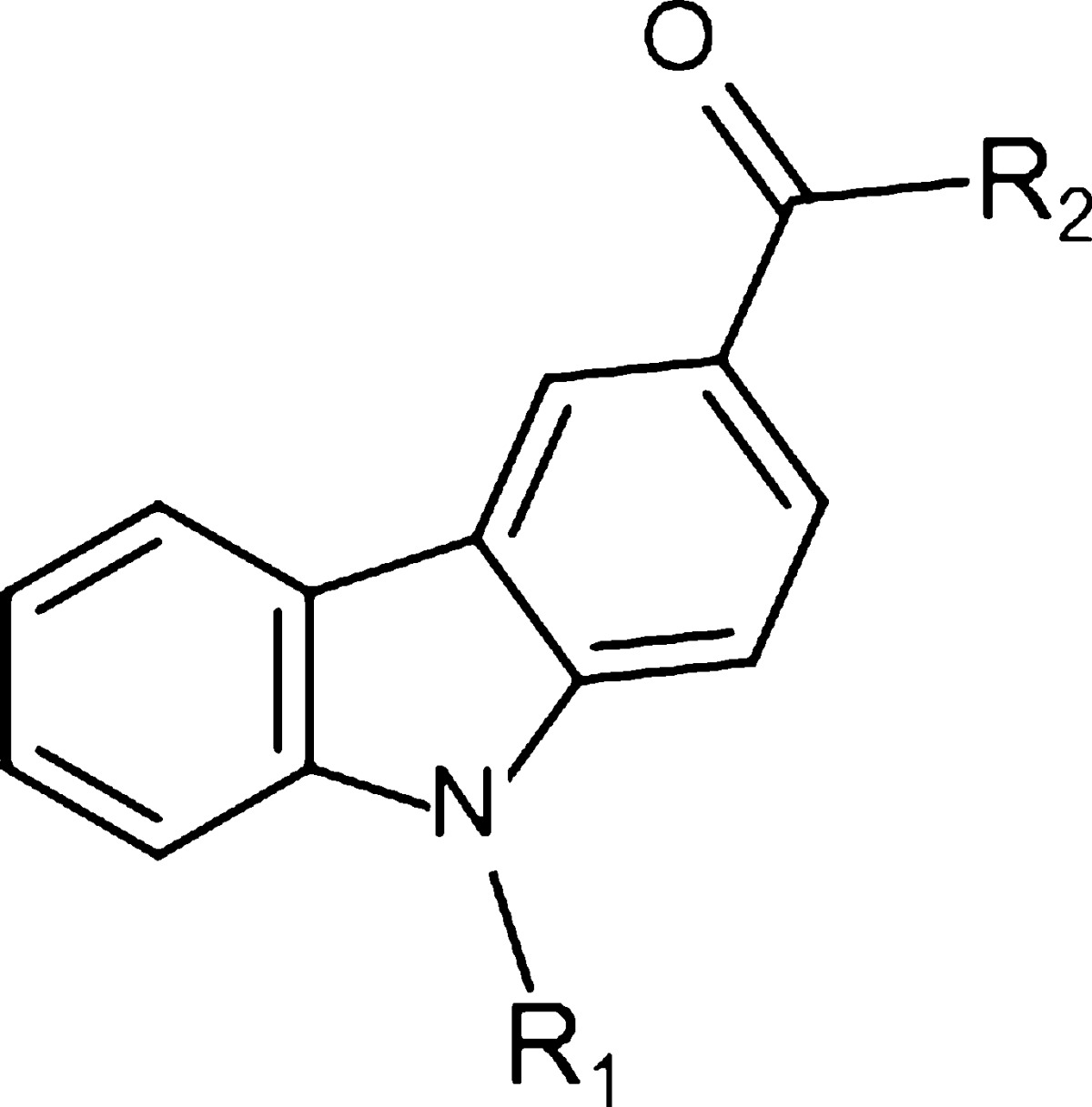
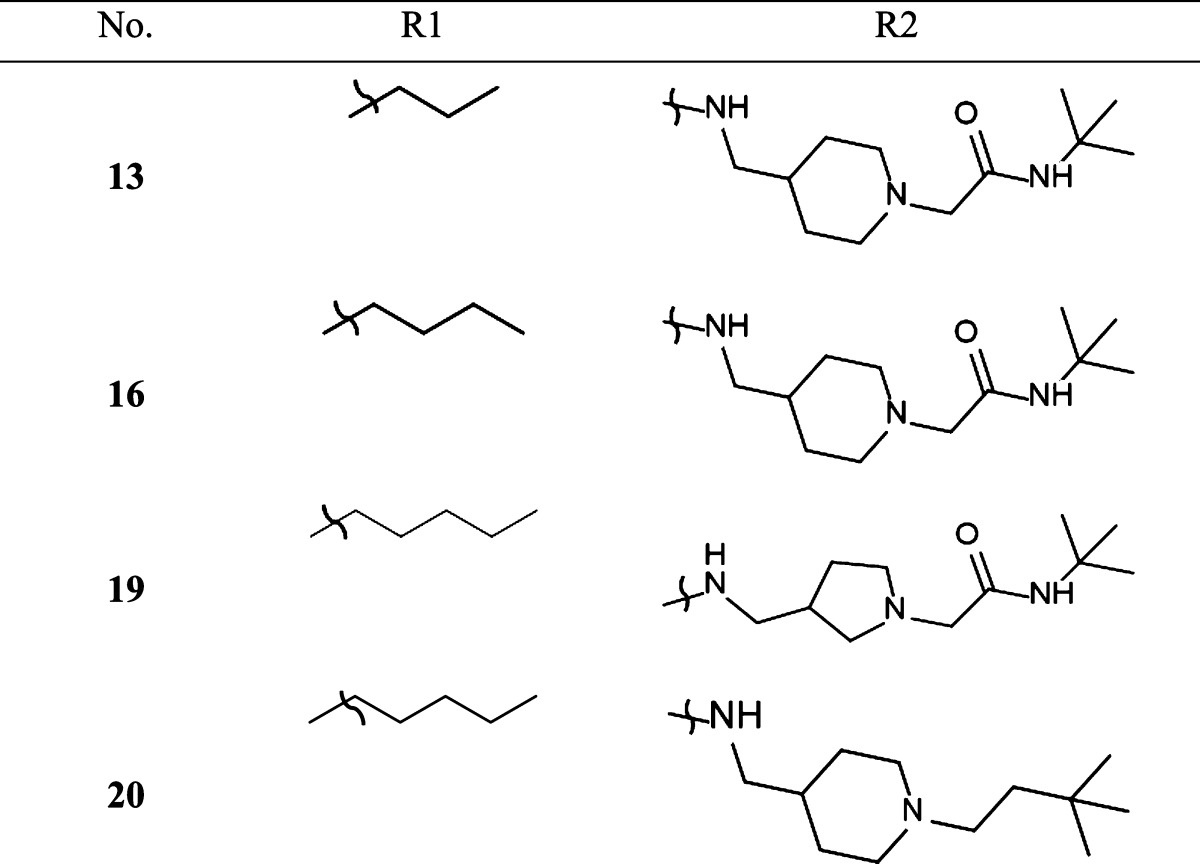
The synthesis of the carbazoles derivatives is outlined in Scheme 2. Amidation under standard peptide coupling conditions38 of N-alkylated carbazole-3-carboxylic derivatives 2 with Boc-protected amines afforded the desired amide derivatives 3 and 6. Deprotection of the Boc protecting group in the presence of TFA in dichloromethane followed by alkylation of the resultant compounds 4 and 8 with N-tert-butyl-2-chloroacetamide provided the corresponding desired compounds 9, 10, 13, 16, and 19 (Tables 1 and 2). Compound 20 was prepared by reductive amination of compound 4 using 3,3-dimethylbutyraldehyde (Table 2).
Scheme 2.

Table 1. Radioligand Competitive Binding Assays (mean ± SEM) for Carbazole-Based Analogues 7–10: Systematic Variation in the Linker Lengtha.
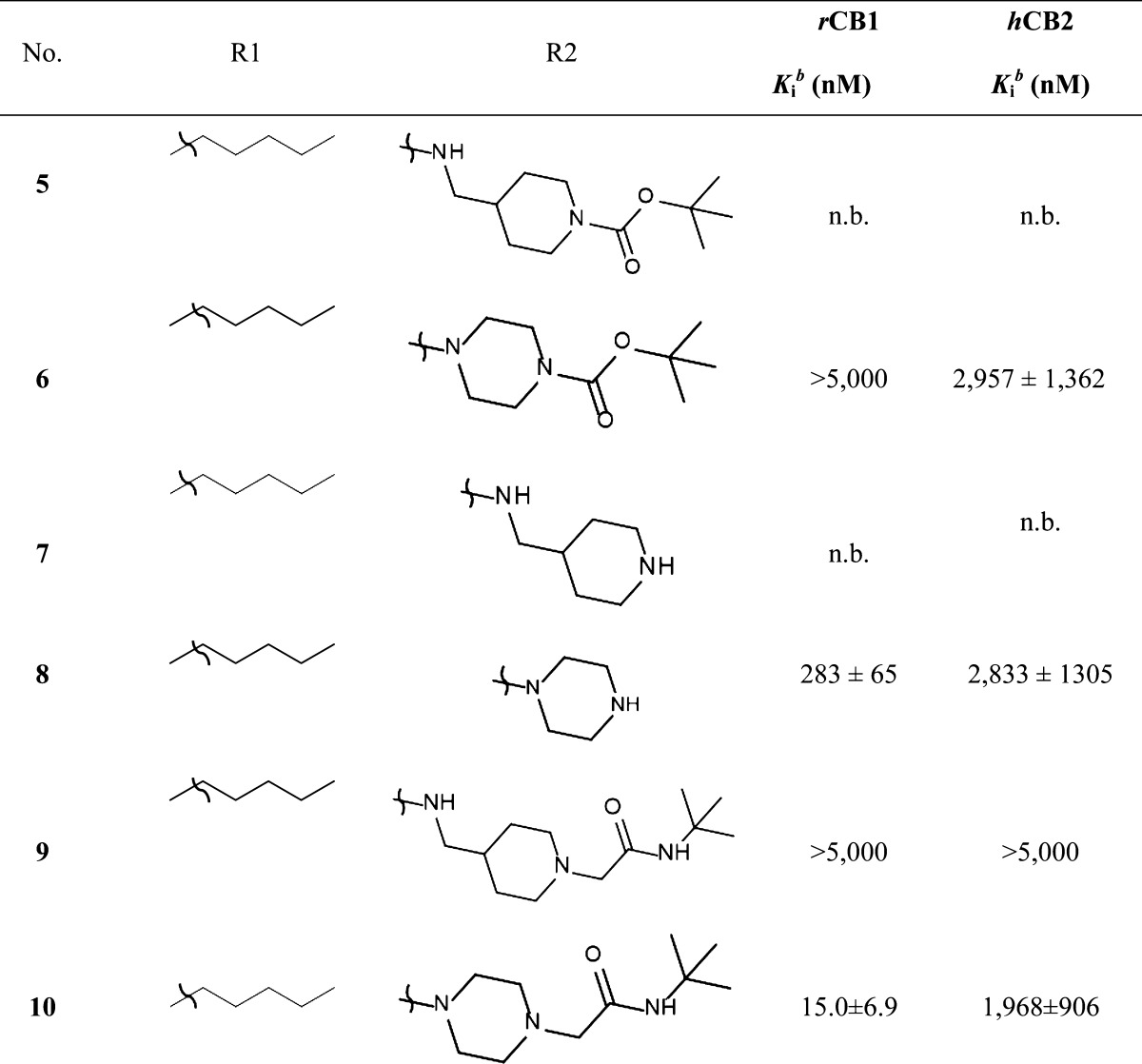
Values are means of three experiments run in triplicate with standard deviation; n.b. no binding.
Table 2. Analogues of Compound 9: Systematic Variation in N-Alkyl Chain Length and in the Region Occupied by the Heterocycle.
Results
In Vitro Characterization of the Compound Series
The entire first set of 10 compounds was screened using whole-cell voltage clamp techniques for their ability to mediate tonic block of transiently expressed hCav3.2 channels (Figure 1 and Table 3). Next, we used radioligand binding assays to assess the affinities of these compounds for both CB1 and CB2 receptors (Table 1).
Table 3. Summary of Biophysical Parameters of hCav3.2 Calcium Channel in the Absence and the Presence of Compounds 10 and 9a.
| V0.5act (mV) 3 μM | Vh (mV) 3 μM | IC50 tonic (μM) | |
|---|---|---|---|
| Wt hCav3.2 | –30.0 | –53.1 ± 1.67 | |
| compound 9 | –29.7 | –58.2 ± 1.43 | 1.48 ± 0.2 |
| compound 10 | –42.0* | –58.1 ± 1.31 | 3.68 ± 0.5 |
Note that 3 μM of compound 10 produces a significant 12 mV negative shift in the half activation potential of hCav3.2 and although there is a trend for both compounds shift the half inactivation potential of the channel, it did not reach statistical significance.
In the course of our initial exploratory work on the structure–activity relationship for this novel series of T-type channel blockers, we decided to determine the optimal linker length attached to the carbazole’s carbonyle (Table 1). We observed various degrees of inhibition of these channels, with compounds 9 and 10 being the most potent blockers of expressed hCav3.2. These two compounds mediated near complete inhibition at our standard test concentration of 10 μM (Figure 1). Interestingly, both of these compounds are very similar in structure, with both having a cyclic tertiary amine attached to the carbazole scaffold (Table 1). Previous work has indicated that this modification helps confer T-type channel blocking activity onto various organic molecules,16,28,29,34 in agreement with our data presented here. As shown in Table 1, compound 10 showed high affinity binding to CB1 receptors (15 nM), whereas its affinity for the CB2 receptor was approximately 100-fold lower (2 μM). Compound 9, however, did not bind to the two receptors with an affinity less than 5 μM for CB1 and CB2. The only difference between compounds 9 and 10 is the elongated chain attached to the nitrogen of the carbazole in 9 (Table 1). This type of modification has been shown to alter cannabinoid receptor binding,16,34,38 but it has not been demonstrated whether this modification affects the interactions of these compounds with Cav3.2. Among the first series of compounds 5–10, replacement of a piperazine moiety by a methylpiperidine moiety appeared to be the most optimal for decreasing cannabinoid receptor affinity without impacting Cav3.2 calcium channel block. This striking difference in affinity for cannabinoid receptors between compounds bearing a piperazine moiety compared to a methylpiperidine moiety underscores the importance of chain length when developing compounds that preferentially target Cav3.2 calcium channels over cannabinoid receptors. Next, we determined the optimal chain length attached to the carbazole’s endocyclic nitrogen (Table 1 and Table 2, compounds 9, 13, and 16). Among the linear N-1 alkyl chains, a pentyl chain seemed to be the most optimal for occupying its interaction site Cav3.2 channels, because systematically decreasing the length from n-pentyl in 9 negatively impacted the respective Cav3.2 blocking activities. Replacement of the piperidine ring by a pyrolidine moiety16 had a slight negative effect on Cav3.2 block, probably due to the lack of optimal ligand–receptor van der Waals contacts. Replacing the amide chain on the piperidine ring by an alkyl chain34 dramatically decreased the Cav3.2 block.
As is clearly seen in the traces in Figure 2A, the slight structural modification in 9 compared to 10 does indeed impact hCav3.2 channel inhibition. The affinity of 9 versus 10 increased more than 2-fold with the IC50 of 9 and 10 being 1.48 and 3.68 μM, respectively (Figure 2B and Table 3). In addition, compound 10 shifted the half activation potential of hCav3.2 by −12 mV (Figure 2C and Table 3). There was no significant effect on half-inactivation potential (P = 0.143) (Figure 2D and Table 3). We then tested the Cav3 channel subtype selectivity of compound 9 using a single concentration of 3 μM. This concentration blocked hCav3.2 by 69.3 ± 4% (n = 8), which was significantly (P < 0.05) greater than that of either hCav3.1 (44.5 ± 7%; n = 5) or hCav3.3 (42.5 ± 5%; n = 5). Compound 9 was thus chosen for further testing in animal models of pain.
Figure 2.

(A) Representative traces of hCav 3.2 before and after application of 3 μM compounds 10 and 9. (B) Dose–response relations for compound 9 and 10 block of hCav3.2 channels. The IC50 from the fit with the Hill equation was 1.48 and 3.68 μM, respectively (n = 6). (C) Effect of 3 μM compounds 9 and 10 on the steady state inactivation curve for Cav3.2 channels. (D) Effect of 3 μM compounds 9 and 10 on the current voltage relation for Cav3.2 channels. Note: Data in panels (B) and (C) were fitted with the Boltzmann equation, and data were obtained from 6 paired experiments.
Effects of Compound 9 in Vivo on Acute Pain
Given the Cav3.2 channel blocking property of compound 9, we hypothesized that this compound may affect pain transmission in animal models. Compound 9 was delivered by either intrathecal (i.t.) or intraperitoneal (i.p.) routes, and its effects on both the acute nociceptive and the slower inflammatory pain phases of the formalin test were evaluated. One-way ANOVA revealed that i.t. treatment of mice with compound 9 (1–10 μg/i.t., 20 min before) significantly decreased pain response time in both first (Figure 3A) and second (Figure 3B) phases (61 ± 8% and 76 ± 10% inhibition, respectively). I.p. treatment of mice with compound 9 (10–100 mg/kg, i.p., 30 min prior) also resulted in significantly (one-way ANOVA) reduced pain response time in both the first (Figure 3C) and second (Figure 3D) phases (47 ± 2% and 66 ± 48% inhibition, respectively). Importantly, systemic (via i.p.) treatment with compound 9 (30 mg/kg, i.p.) did not affect locomotor activity of mice assessed via an open-field test (Figure 4A), suggesting that the reduced response times observed in the previous formalin tests were not due to altered motor behavior. In order to investigate if the effects observed for compound 9 were specifically mediated via T-type channels, we performed a formalin test in CaV3.2 null mice that were treated either with vehicle or with compound 9 (10 μg/i.t.). The CaV3.2 null mice exhibited a lower mean response time when compared to wild-type mice, which is in agreement with previous data.14,16 As indicated in Figure 4B and C, they appear to be completely insensitive to i.t. treatment with compound 9 (10 μg i.t.) as revealed by two-way ANOVA, indicating that compound 9 mediates its analgesic effects specifically via Cav3.2 channels.
Figure 3.

(A, B) Effect of increasing doses of intrathecal compound 9 on the first and second phases of formalin-induced pain. (C, D) Effect of increasing doses of intraperitoneal compound 9 (on the first and second phases of formalin-induced pain. Each bar represents the mean ± SEM (n = 6–8), and is representative of 2 independent experiments. Asterisks denote the significance relative to the control group (***P < 0.001, one-way ANOVA followed by Dunnett’s test).
Figure 4.

(A) Effect of 30 mg/kg intraperitoneal compound 9 on locomotor activity of wild type mice in the open field test. (B, C) Comparison of effect of 10 μg/i.t. intrathecal compound 9 on the first and second phases of formalin-induced pain in wild type and Cav3.2 knockout mice, respectively. Each bar represents the mean ± SEM (n = 6–7) and is representative of 2 independent experiments. Asterisks denote the significance relative to the control group ***P < 0.001 when comparing treatment; and #P < 0.05, for comparison between genotypes (two-way ANOVA followed by Tukey’s test). Note that control mice were of the same genetic background as the Cav3.2 null mice.
Effect of Compound 9 on Chronic Neuropathic Pain
To verify whether compound 9 modulates pain transmission under neuropathic conditions, we analyzed mechanical withdrawal thresholds of mice with a partial sciatic nerve injury (PNI) and treated with compound 9 (30 mg/kg, i.p.) 14 days after nerve injury. As shown in Figure 5, sciatic nerve injury triggers mechanical hyperalgesia as confirmed by significant decrease of mechanical withdrawal thresholds when compared to baselines levels (Pre-PNI, P < 0.001). Two-way ANOVA revealed that systemic (i.p.) treatment of mice with compound 9 (30 mg/kg, i.p.) significantly attenuated the mechanical hyperalgesia induced by sciatic nerve injury when compared with the PNI + Control group for longer than 3 h after treatment. These data indicate that compound 9 treatment modulates pain transmission and mediates analgesia in this animal model of chronic neuropathic pain.
Figure 5.

Blind analyses of the time course of treatment of neuropathic mice with vehicle or compound 9. Each circle represents the mean ± SEM (n = 6), and is representative of 2 independent experiments. (*P < 0.05, ***P < 0.001, two-way ANOVA followed by Tukey’s test). The dashed line and number symbols indicate the range of data points where injured animals significantly differed from the sham treated group (P < 0.001).
Discussion
T-type calcium channels are important contributors to a range of physiological functions2−5,32,39−41 and it is well established that the Cav3.2 channel isoform plays important roles in the afferent pain pathway.12,14,15,19,42−44 Finding specific and selective blockers of these channels has proven difficult as many of the well-known T-type channel blockers such as mibefradil or ethosuximide block other channels, which can then result in side effects.12,31,45 In this study, we developed novel compounds with structures similar to some of the endogenous ligands that are known to interact with members of the T-type channel family16,23,25,34,35 and then modified them to reduce their affinity for cannabinoid receptors while attempting to increase affinity for Cav3.2 channels.
We had previously synthesized a series of compounds based on endogenous cannabinoid ligands that targeted both CB receptors and T-type calcium channels.16,34 Using data obtained from these experiments, we designed additional compounds with an extra substituted tertiary amine attached to the carbazole scaffold. We then extended the chain length attached to the carbazole to one of the compounds (compound 9) to improve its selectivity for Cav3.2 channels over CB receptors.16,34 These data show that the length of the linker between the carbazole scaffold and the heterocyclic moiety is a key drug structural determinant that can be exploited to produce better and more selective Cav3.2 channel inhibitors. This compound blocked Cav3.2 channels with approximately 2-fold higher affinity than Cav3.1 and Cav3.3 channels. At this point we do not know if compound 9 affects other molecular targets such as high voltage activated channels or sodium channels. Nonetheless, the observation that their analgesic actions were abolished upon removal of Cav3.2 channels indicates that the primary biological target for compound 9 in the context of pain signaling is Cav3.2. The potent effects of compound 9 on pain response in injured wild type animals fits with the notion that Cav3.2 channels play an important role in the afferent pain pathway17,18,20,21,42,44,46,47 and also with a number of previous studies showing that Cav3.2 channel blockers are efficacious in various pain models.12,15,20,29 Calculated physiochemical properties such as topological polar surface area (tPSA), lipophilicity (clogP), and number of hydrogen bond donor and acceptor atoms are useful indicator of druglike properties. Poor oral absorption is expected for compounds with a TPSA more than 140 Å2, with a log D > 5, more than 5 hydrogen bond donors, and more than 10 hydrogen bond acceptors. Compound 9 has a TPSA value of 66.37 Å2, a molecular weight of 490.68 g/mol, a clogP value of 4.59, and less than five hydrogen bond acceptors or donors, suggesting a reasonable probability of good oral absorption and intestinal permeability.
Compound 9 blocked Cav3.2 channels in a concentration range that was similar to that previously reported for compounds such as NMP734 and NMP181.16 These two molecules, although structurally related to compound 9 did not discriminate among the three Cav3 family members, and mediated leftward shifts in the midpoint of the steady state inactivation curve. In contrast, compound 9 did not significantly affect voltage-dependent inactivation properties of Cav3.2 channels, suggesting that this compound does not interact strongly with inactivated channels. At this point, it is not clear which structural differences among these compounds are responsible for these differential effects on channel gating. While other types of T-type calcium channel blockers such as TTA-248 or Z12321228 that are efficacious in various pain models are also known to promote voltage-dependent inactivation of Cav3.2 channels, our data showing efficacy of compound 9 in neuropathic and inflammatory pain indicate that such state-dependent inhibition is not a prerequisite for antihyperalgesic effects in vivo.
Altogether our data suggest that using a carbazole scaffold is an effective strategy for developing potent Cav3.2 calcium channel blockers for therapeutic intervention into inflammatory and neuropathic pain hypersensitivity. In addition, T-type channels are also associated with many other disorders, including epilepsy and cardiac hypertrophy;2−4,7−11,18,32,49 therefore, the novel pharmacophores that we have identified here may prove useful toward treatment of these disorders.
Methods
In Vitro Receptor Radioligand CB1 and CB2 Binding Studies
CB1 and CB2 radioligand binding data were obtained using National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) resources as described earlier.34,50−52
cDNA Constructs
Human Cav3.2 cDNA construct was kindly provided by Dr. Terrance Snutch (University of British Columbia, Vancouver, Canada).
tsA-201 Cell Culture and Transfection
Human embryonic kidney tsA-201 cells were cultured and transfected using the calcium phosphate method as described previously.53 Transfected cells were then incubated 48 h at 37 °C and 5% CO2 and then resuspended with 0.25% (w/v) trypsin-EDTA (Invitrogen) and plated on glass coverslips a minimum of 3 to 4 h before patching.
Electrophysiology
Whole-cell voltage-clamp recordings from tsA-201 cells were performed at room temperature 2–3 days after transfection. External recording solution contained (in mM): 114 CsCl, 20 BaCl2, 1 MgCl2, 10 HEPES, 10 glucose, adjusted to pH 7.4 with CsOH. Internal patch pipet solution contained the following (in mM): 126.5 CsMeSO4, 2 MgCl2, 11 EGTA, 10 HEPES adjusted to pH 7.3 with CsOH. Internal solution was supplemented with 0.6 mM GTP and 2 mM ATP and mixed thoroughly just prior to use. Liquid junction potentials for the recording solutions were left uncorrected.
Tested compounds were prepared daily from DMSO stocks diluted in external solution. Using a custom built gravity driven microperfusion system,53 diluted compounds were then applied rapidly and locally to the cells. Control vehicle experiments were performed to ensure that 0.1% DMSO had no effect on current amplitudes or on the half a ctivation and half-inactivation potentials (data not shown). Currents were measured using the whole-cell patch clamp technique and an Axopatch 200B amplifier in combination with Clampex 9.2 software (Molecular Devices, Sunnyvale, CA). After establishing whole cell configuration, cellular capacitance was minimized using the amplifier’s built-in analog compensation. Series resistance was compensated by at least 85% in all experiments. All data were digitized at 10 kHz with a Digidata 1320 interface (Molecular Devices) and filtered at 1 kHz (8-pole Bessel filter). Raw and online leak-subtracted data were both collected simultaneously. In current–voltage relation studies, the membrane potential was held at −110 mV and cells were depolarized from −80 to 20 mV in 10 mV increments. For steady-state inactivation studies, a 3.6 s conditioning prepulse of various magnitude (initial holding at −110 mV) was followed by a depolarizing pulse to −20 mV. Individual sweeps were separated by 12 s to permit recovery from inactivation between conditioning pulses. The duration of the test pulse was typically 200 ms and the current amplitude obtained from each test pulse was normalized to that observed at the holding potential of −110 mV.
Animals
During experiments, all efforts were made to minimize animal suffering according to the policies and recommendations of the International Association for the Study of Pain and all protocols used were approved by the Institutional Animal Care and Use Committee. For all experiments, either adult male C57BL/6J (wild-type) or CACNA1H knockout (Cav3.2 null) mice (20–25g) purchased from Jackson Laboratories were used. There were a maximum of five mice per cage (30 × 20 × 15 cm2), and access to food and water was unlimited. Temperature was kept at 23 ± 1 °C on a 12 h light/dark cycle (lights on at 7:00 a.m.). Intraperitoneal (i.p.) injections of drugs were a constant volume of 10 mL/kg body weight. Intrathecal (i.t.) injections used volumes of 10 μL and were performed using the method described previously54 and carried out routinely in our laboratory.16,55 All drugs were dissolved in 1% or less DMSO, whereas control animals received PBS + 1% DMSO. For each test, a different group of mice were used and only one experiment per mouse was performed. In experiments examining the action of compound 9 on PNI-induced neuropathy, the observer was blind to the conditions tested.
Formalin Test
Before experiments were performed, mice were left to acclimatize for at least 60 min. Animals were then injected intraplantarily (i.pl.) in the ventral surface of the right hindpaw with 20 μL of a formalin solution (1.25%) made up in PBS. Following i.pl. injections of formalin, individual animals were placed immediately into observation chambers and monitored from 0 to 5 min (acute nociceptive phase) and 15–30 min (inflammatory phase). The time spent licking or biting the injected paw was then considered as a nociceptive response and recorded with a chronometer. Compound 9 was delivered by i.t. (20 min prior) or by i.p. (30 min prior) and its effect on both the nociceptive and inflammatory phases of the formalin test was evaluated.
Open-Field Test
Animals received compound 9 via i.p. (30 mg/kg) route 30 min before testing, and the ambulatory behavior of the treated animals was assessed in an open-field test as described previously.56 Briefly, the apparatus consisted of a wooden box measuring 40 × 60 × 50 cm3 with a glass front wall. The floor was divided into 12 equal squares and the entire apparatus was placed in a sound free room. Animals were placed in the rear left square and left to explore freely and the number of squares crossed with all paws (crossing) in a 6 min time frame was counted. After each individual mouse session, the apparatus was then cleaned and dried with a 10% alcohol solution.
Partial Sciatic Nerve Injury (PNI)-Induced Neuropathic Pain
Before surgery, mice were anaesthetized with isoflurane (5% induction, 2.5% maintenance). Partial ligation of the sciatic nerve was performed by tying the distal 1/3 to 1/2 of the dorsal portion as previously described.57 In sham-operated mice, the sciatic nerve was exposed without ligation and all wounds were closed and treated with iodine solution. After 14 days post surgery, mice received either compound 9 (30 mg/kg, i.p.) treatment or vehicle, while sham-operated animals received only vehicle (10 mL/kg, i.p.). Mechanical hyperalgesia was then evaluated in a time-dependent manner.
Evaluation of Mechanical Hyperalgesia
Mechanical hyperalgesia responses were recorded immediately before the surgeries (baselines), 14 days after the surgeries (0), and at various time points (0.5, 1, 2, 3 h) after treatment. Measurements were made using a Dynamic Plantar Aesthesiometer (DPA, Ugo Basile, Varese, Italy). Briefly, individual animals were placed in small enclosed testing arenas (20 cm × 18.5 cm × 13 cm, length × width × height) on top of a wire mesh floor and allowed to acclimate for a period of at least 90 min. The DPA device was then positioned so that the filament was directly under the plantar surface of the ipsilateral hind paw of the animal and tested three times per session.
Data Analysis and Statistics
Data were analyzed using Clampfit 9.2 (Molecular Devices). Origin 7.5 software (Northampton, MA) was used in the preparation of all figures and curve fittings. Current–voltage relationships were fitted with the modified Boltzmann equation: I = [Gmax(Vm – Erev)]/[1 + exp((V0.5act – Vm)/ka)], where Vm is the test potential, V0.5act is the half activation potential, Erev is the reversal potential, Gmax is the maximum slope conductance, and ka reflects the slope of the activation curve. Steady-state inactivation curves were fitted using the Boltzmann equation: I =1/(1 + exp((Vm – Vh)/k)), where Vh is the half-inactivation potential and k is the slope factor. Dose–response curves were fitted with the equation y = A2 + (A1 – A2)/(1 + ([C]/IC50)n), where A1 is initial current amplitude and A2 is the current amplitude at saturating drug concentrations, [C] is the drug concentration, and n is the Hill coefficient. Statistical significance was determined by paired or unpaired Student’s t tests and one-way or repeated measures ANOVA, followed by Dunnett’s test or Tukey’s multiple comparison tests. Significant values were set as indicated in the text and figure legends. All data are given as means ± standard errors.
Chemistry
All moisture sensitive reactions were performed in an inert, dry atmosphere of argon in flame-dried glassware. Air sensitive liquids were transferred via syringe or cannula through rubber septa. Reagent grade solvents were used for extraction and flash chromatography. THF was distilled from Na/benzophenone under argon; dichloromethane (CH2Cl2) and chloroform (CHCl3) were distilled from CaH2 under argon. All other reagents and solvents which were purchased from commercial sources were used directly without further purification. The progress of reactions was checked by analytical thin-layer chromatography (Sorbent Technologies, Silica G TLC plates w/UV 254). The plates were visualized first with UV illumination followed by charring with ninhydrin (0.3% ninhydrin (w/v), 97:3 EtOH-AcOH). Flash column chromatography was performed using prepacked Biotage SNAP cartridges on a Biotage Isolera One instrument. The solvent compositions reported for all chromatographic separations are on a volume/volume (v/v) basis. 1HNMR spectra were recorded at 400 or 500 MHz and are reported in parts per million (ppm) on the δ scale relative to tetramethylsilane as an internal standard. 13CNMR spectra were recorded at 100 or 125 MHz and are reported in parts per million (ppm) on the δ scale relative to CDCl3 (δ 77.00). Melting points were determined on a Stuart melting point apparatus from Bibby Scientific Limited and are uncorrected. High Resolution mass spectrometry (HRMS) was performed on a Waters/Micromass LCT-TOF instrument. All compounds were more than 95% pure.
N-((1-(2-(tert-Butylamino)-2-oxoethyl)piperidin-4-yl)methyl)-9-pentyl-9H-carbazole-3-carboxamide (9)
Under nitrogen atmosphere, a mixture of 9-pentyl-N-(piperidin-4-ylmethyl)-9H-carbazole-3-carboxamide 7(16) (389 mg, 1.03 mmol), N-tert-butyl-2-chloroacetamide (185 mg, 1.24 mmol), potassium carbonate (427 mg, 3.09 mmol), and potassium iodide (160 mg, 0.96 mmol) in n-butanol (5 mL) was subjected to microwave irradiation at 110 °C for 3 h. The mixture was allowed to cool to room temperature, diluted with DCM (50 mL), and filtered. The organic solvents were evaporated in vacuo. The residue was purified on a Biotage KP-NH cartridge (amino-modified silica gel) using cyclohexane/EtOAc in different proportions to afford the title compound as a light yellow glass. Yield: 476 mg (94%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 1.4 Hz, 1H), 8.01 (d, J = 7.9 Hz, 1H), 7.84 (dd, J = 8.6, 1.4 Hz, 1H), 7.41 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.33 (d, J = 8.3 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.16 (ddd, J = 7.9, 7.1, 1.1 Hz, 1H), 7.00 (br s, 1H), 6.58 (t, J = 5.8 Hz, 1H), 4.19 (t, J = 7.2 Hz, 2H), 3.34 (t, J = 6.5 Hz, 2H), 2.81–2.67 (m, 4H), 2.00 (td, J = 11.7, 2.5 Hz, 2H), 1.83–1.74 (m, 2H), 1.71 (br d, J = 13.9 Hz, 2H), 1.64–1.48 (m, 1H), 1.31–1.22 (m, 15H), 0.78 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 169.84 (C=O), 168.52 (C=O), 142.33 (C), 141.15 (C), 126.44 (CH), 125.28 (C), 124.81 (CH), 122.96 (C), 122.73 (C), 120.68 (CH), 119.85 (CH), 119.68 (CH), 109.23 (CH), 108.50 (CH), 62.73 (CH2), 53.89 (CH2), 50.51 (C), 45.57(CH2), 43.37 (CH2), 36.23 (CH), 30.61 (CH2), 29.48 (CH2), 28.93 (CH3), 28.76 (CH2), 22.57 (CH2), 14.08 (CH3). ESI: m/z 491.2 (M + H)+. HRMS calcd for C30H43N4O2 (M + H)+ 491.3386, found 491.3433.
N-tert-Butyl-2-[4-(9-pentyl-9H-carbazole-3-carbonyl)piperazin-1-yl]acetamide (10)
Under argon atmosphere, to a solution of tert-butyl 4-(9-pentyl-9H-carbazole-3-carbonyl)piperazine-1-carboxylate 8(16) (360 mg, 1.03 mmol) and MeCN (10 mL) was added K2CO3 (427 mg, 3.09 mmol), KI (160 mg, 0.96 mmol), and N-tert-butyl-2-chloroacetamide (185 mg, 1.24 mmol). The reaction mixture was stirred at reflux for 3 h. The reaction mixture was cooled, diluted with DCM (30 mL), and filtered. The organic solvents were evaporated in vacuo. The organic layer was washed three times with brine (50 mL). The organic layer was then separated, dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography eluting with heptane/EtOAc (0–100%) to yield the title compound (437 mg, 92%) as yellow foam. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 1.2 Hz, 1 H), 8.09 (d, J = 7.6 Hz, 1 H), 7.38–7.55 (m, 4 H), 7.23–7.28 (m, 1 H), 6.96 (s, 1 H), 4.30 (t, J = 7.2 Hz, 2 H), 3.74 (br. s., 4 H), 2.96 (s, 2 H), 2.58 (br. s., 4 H), 1.86 (br. s., 2 H), 1.24–1.39 (m, 13 H), 0.81–1.01 (m, 3 H). 13C NMR (101 MHz, CDCl3) δ ppm 171.69 (C=O), 168.71 (C=O), 141.20 (C), 140.94 (C), 126.30 (CH), 125.60 (C), 125.19 (CH), 122.61 (C), 122.50 (C), 120.54 (CH), 120.14 (CH), 119.43 (CH), 109.07 (CH), 108.51 (CH), 77.44 (CH2), 77.33 (CH2), 77.13 (CH2), 76.81 (CH2), 62.27 (CH2), 53.48 (CH2), 50.64 (C), 43.24 (CH2), 29.38 (CH2), 28.83 (CH2), 28.66 (CH3), 22.48 (CH2), 13.98 (CH3). m/z 463.2 (M + H)+. HRMS calcd for C30H43N4O2 (M + H)+ 463.3073, found 463.2819.
tert-Butyl-4 ((9-Propyl-9H-carbazole-3-carboxamido)methyl)piperidine-1-carboxylate (11)
9-Propyl-9H-carbazole-3-carboxylic acid16 (100 mg, 0.40 mmol), HOBt (62.32 mg, 0.48 mmol), DIPEA (0.13 mL, 0.79 mmol), DMAP (58.12 mg, 0.40 mmol), and EDAC (91.22 mg, 0.40 mmol) were added upon stirring to DCM (9 mL) under nitrogen. The obtained solution was cooled down on an ice–water bath, tert-butyl 4-(aminomethyl) piperidine-1-carboxylate (119.24 mg, 0.40 mmol) was added in one portion, and the resulting reaction mixture was then allowed to warm to room temperature and stirred for 16 h. The solvent was removed in vacuo, and the obtained residue was extracted with EtOAc (50 mL). The organic layer was washed three times with brine (25 mL). The organic layer was then separated, dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography eluting with heptane/EtOAc (0–100%) to produce the title compound 11 (158.3 mg, 89%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 1.4 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.89 (dd, J = 8.7, 1.4 Hz, 1H), 7.44 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.19 (ddd, J = 8.0, 7.1, 1.1 Hz, 1H), 6.55 (t, J = 5.8 Hz, 1H), 4.26 (t, J = 7.2 Hz, 2H), 3.38 (t, J = 6.2 Hz, 2H), 3.10 (d, J = 12.1 Hz, 2H), 2.61 (td, J = 12.2, 2.5 Hz, 2H), 1.99 (s, 1H), 1.97–1.80 (m, 2H), 1.80–1.70 (m, 3H), 1.45 (s, 9H), 1.22 (qd, J = 12.8, 3.7 Hz, 2H), 0.97 (t, J = 7.4 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.53, 154.77, 142.30, 141.16, 126.42, 125.42, 124.77, 122.97, 122.74, 120.74, 119.83, 119.68, 109.22, 108.53, 79.22, 45.22, 45.39, 36.91, 31.38, 28.41, 22.58, 12.10.
9-Propyl-N-(piperidin-4-ylmethyl)-9H-carbazole-3-carboxamide (12)
Trifluoroacetic acid (5 mL) was added to a solution of 11 (150 mg, 0.33 mmol) in CH2Cl2 (5 mL), and the reaction mixture was stirred at room temperature for 4 h. The mixture was evaporated, and the residue was basified with 2 N NaOH. The solution was extracted with CH2Cl2 three times. The extracts were dried with Na2SO4 and evaporated. The crude product was purified by flash chromatography eluting with heptane/EtOAc (0–100%) to produce the title compound 12 (95.5 mg, 83%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 1.5 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.90 (dd, J = 8.7, 1.5 Hz, 1H), 7.5 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.17 (ddd, J = 8.0, 7.2, 1.2 Hz, 1H), 6.58 (t, J = 5.8 Hz, 1H), 4.25 (t, J = 7.1 Hz, 2H), 3.39 (t, J = 6.2 Hz, 2H), 3.09 (d, J = 12.1 Hz, 2H), 2.60 (td, J = 12.2, 2.5 Hz, 2H), 1.99 (s, 1H), 1.98–1.80 (m, 2H), 1.80–1.71 (m, 3H), 1.23 (qd, J = 12.8, 3.7 Hz, 2H), 0.96 (t, J = 7.5 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.51, 142.32, 141.15, 126.40, 125.41, 124.79, 122.99, 122.73, 120.73, 119.82, 119.67, 109.21, 108.50, 46.47, 46.20, 45.37, 36.91, 31.37, 22.59, 12.10.
N-((1-(2-(tert-Butylamino)-2-oxoethyl)piperidin-4-yl)methyl)-9-propyl-9H-carbazole-3-carboxamide (13)
Under argon atmosphere, to a solution of 12 (90 mg, 0.26 mmol) and MeCN (2.5 mL) was added K2CO3 (107.8 mg, 0.78 mmol), KI (43.2 mg, 0.26 mmol), and N-tert-butyl-2-chloroacetamide (47.9 mg, 0.32 mmol). The title compound was prepared according to the analogous procedure described above for compound 9 to produce the title compound 13 (121.3 mg, 82%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 1.5 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.91 (dd, J = 8.7, 1.5 Hz, 1H), 7.44 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.31 (d, J = 8.2 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.17 (ddd, J = 8.0, 7.1, 1.1 Hz, 1H), 6.98 (br s, 1H), 6.58 (t, J = 5.8 Hz, 1H), 4.26 (t, J = 7.1 Hz, 2H), 3.33 (t, J = 6.3 Hz, 2H), 2.82–2.67 (m, 4H), 2.03 (td, J = 11.8, 2.7 Hz, 2H), 1.86–1.76 (m, 2H), 1.7 (d, J = 13.7 Hz, 2H), 1.64–1.48 (m, 1H), 1.34 (s, 9H), 0.94 (t, J = 7.0 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 169.84 (C=O), 168.5 (C=O), 142.34 (C), 141.17 (C), 126.41 (CH), 126.0 (C), 124.79 (CH), 122.94 (C), 122.71 (C), 120.68 (CH), 119.84 (CH), 119.69 (CH), 109.23 (CH), 108.51 (CH), 62.74 (CH2), 53.9 (CH2), 50.52 (C), 45.58 (CH2), 45.35 (CH2), 36.22 (CH), 30.61 (CH2), 28.90 (CH3), 22.76 (CH2), 12.18 (CH3). ESI: m/z 463.3 (M + H)+. HRMS calcd for C28H40N4O2 (M + H)+ 463.3073, found 463.3089.
tert-Butyl 4-((9-Butyl-9H-carbazole-3-carboxamido)methyl)piperidine-1-carboxylate (14)
Using 9-butyl-9H-carbazole-3-carboxylic acid16 (100 mg, 0.37 mmol), HOBt (60.54 mg, 0.45 mmol), DIPEA (0.13 mL, 0.75 mmol), DMAP (54.73 mg, 0.45 mmol), tert-butyl 4-(aminomethyl) piperidine-1-carboxylate (112.22 mg, 0.45 mmol), and EDAC (85.85 mg, 0.45 mmol), the title compound was prepared according to the analogous procedure described above for compound 11, to produce the title compound 14 (147.3 mg, 85%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 1.5 Hz, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.82 (dd, J = 8.6, 1.5 Hz, 1H), 7.43 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.16 (ddd, J = 7.9, 7.1, 1.1 Hz, 1H), 6.57 (t, J = 5.8 Hz, 1H), 4.24 (t, J = 7.2 Hz, 2H), 3.37 (t, J = 6.2 Hz, 2H), 3.07 (d, J = 12.1 Hz, 2H), 2.61 (td, J = 12.2, 2.5 Hz, 2H), 2.00 (s, 1H), 1.98–1.80 (m, 2H), 1.80–1.71 (m, 3H), 1.45 (s, 9H), 1.43–1.32(m, 2H), 1.23 (qd, J = 12.8, 3.7 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.50, 154.75, 142.33, 141.16, 126.45, 125.43, 124.80, 122.97, 122.72, 120.73, 119.81, 119.68, 109.21, 108.50, 79.26, 45.47, 43.04, 36.90, 31.36, 28.41, 20.78, 14.13.
9-Butyl-N-(piperidin-4-ylmethyl)-9H-carbazole-3-carboxamide (15)
Trifluoroacetic acid (5 mL) was added to a solution of 14 (140 mg, 0.30 mmol) in CH2Cl2 (5 mL), and the title compound was prepared according to the analogous procedure described above for compound 12 to produce the title compound 15 (95.7 mg, 88%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 1.5 Hz, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.84 (dd, J = 8.7, 1.5 Hz, 1H), 7.41 (ddd, J = 8.3, 7.2, 1.1 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.17 (ddd, J = 7.9, 7.2, 1.1 Hz, 1H), 6.58 (t, J = 5.8 Hz, 1H), 4.25 (t, J = 7.2 Hz, 2H), 3.39 (t, J = 6.2 Hz, 2H), 3.09 (d, J = 12.2 Hz, 2H), 2.60 (td, J = 12.2, 2.5 Hz, 2H), 1.99 (s, 1H), 1.98–1.80 (m, 2H), 1.80–1.71 (m, 3H), 1.43–1.32(m, 2H), 1.23 (qd, J = 12.8, 3.7 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.51, 142.32, 141.15, 126.45, 125.41, 124.79, 122.99, 122.73, 120.73, 119.82, 119.67, 109.21, 108.50, 46.47, 46.20, 43.03, 36.91, 31.37, 20.79, 14.12.
N-((1-(2-(tert-Butylamino)-2-oxoethyl)piperidin-4-yl)methyl)-9-butyl-9H-carbazole-3-carboxamide (16)
Under argon atmosphere, to a solution of 15 (90 mg, 0.25 mmol) and MeCN (2.5 mL) was added K2CO3 (103.7 mg, 0.75 mmol), KI (41.5 mg, 0.25 mmol), and N-tert-butyl-2-chloroacetamide (44.9 mg, 0.30 mmol). The title compound was prepared according to the analogous procedure described above for compound 9 to produce the title compound 16 (99.9 mg, 84%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 1.5 Hz, 1H), 8.01 (d, J = 7.9 Hz, 1H), 7.8 (dd, J = 8.6, 1.5 Hz, 1H), 7.4 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.16 (ddd, J = 7.9, 7.1, 1.1 Hz, 1H), 7.01 (br s, 1H), 6.57 (t, J = 5.8 Hz, 1H), 4.24 (t, J = 7.2 Hz, 2H), 3.35 (t, J = 6.5 Hz, 2H), 2.83–2.68 (m, 4H), 2.00 (td, J = 11.7, 2.5 Hz, 2H), 1.82–1.72 (m, 2H), 1.73 (d, J = 13.8 Hz, 2H), 1.63–1.49 (m, 1H), 1.45–1.34 (m, 2H), 1.33 (s, 9H), 0.90 (t, J = 6.8 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 169.83 (C=O), 168.57 (C=O), 142.34 (C), 141.14 (C), 126.45 (CH), 125.26 (C), 124.82 (CH), 122.94 (C), 122.73 (C), 120.68 (CH), 119.85 (CH), 119.68 (CH), 109.21 (CH), 108.50 (CH), 62.71 (CH2), 53.90 (CH2), 50.50 (C), 45.59(CH2), 43.00 (CH2), 36.26 (CH), 30.66 (CH2), 29.43 (CH2), 28.92 (CH3), 20.74 (CH2), 14.10 (CH3). ESI: m/z 477.2 (M + H)+. HRMS calcd for C29H41N4O2 (M + H)+ 477.3229, found 477.3217.
tert-Butyl 4-((9-Pentyl-9H-carbazole-3-carboxamido)methyl)pyrrolidine-1-carboxylate (17)
Using 9-pentyl-9H-carbazole-3-carboxylic acid (100 mg, 0.36 mmol),16 HOBt (57.81 mg, 0.43 mmol), DIPEA (0.12 mL, 0.71 mmol), DMAP (52.29 mg, 0.43 mmol), (R)-3-(aminomethyl)-1-Boc-pyrrolidine (85.66 mg, 0.43 mmol), and EDAC (82.02 mg, 0.43 mmol), the title compound was prepared according to the analogous procedure described above for compound 11 using the amide coupling protocol, to produce the title compound 17 (141.8 mg, 86%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 1.6 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.83 (dd, J = 8.6, 1.6 Hz, 1H), 7.42 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 8.6 Hz, 1H), 7.17 (ddd, J = 8.0, 7.1, 1.1 Hz, 1H), 6.57 (t, J = 5.8 Hz, 1H), 4.16 (t, J = 7.2 Hz, 2H), 3.22–3.12 (m, 2H), 2.34–2.21 (m, 2H), 2.12–1.98 (m, 1H), 1.81–1.67 (m, 3H), 1.45 (s, 9H), 1.38–1.20 (m, 4H), 0.80 (t, J = 6.9 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.56, 154.75, 142.32, 141.13, 126.47, 125.25, 124.82, 122.92, 122.72, 120.69, 119.85, 119.68, 109.22, 108.50, 79.27, 54.28, 45.50, 39.78, 38.75, 36.26, 30.68, 29.45, 28.42, 20.75, 14.10.
9-Pentyl-N-(pyrrolidin-4-ylmethyl)-9H-carbazole-3-carboxamide (18)
Trifluoroacetic acid (5 mL) was added to a solution of compound 17 (135 mg, 0.29 mmol) in CH2Cl2 (5 mL), the title compound was prepared according to the analogous procedure described above for compound 12 to produce the title compound 18 (90.7 mg, 86%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 1.5 Hz, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.84 (dd, J = 8.6, 1.5 Hz, 1H), 7.41 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.17 (ddd, J = 7.9, 7.1, 1.1 Hz, 1H), 6.57 (t, J = 5.84 Hz, 1H), 4.16 (t, J = 7.2 Hz, 2H), 3.22–3.13 (m, 2H), 2.34–2.22 (m, 2H), 2.12–1.99 (m, 1H), 1.82–1.68 (m, 3H), 1.38–1.20 (m, 4H), 0.82 (t, J = 6.9 Hz, 3H). 13C (101 MHz, CDCl3) δ 168.55, 142.33, 141.14, 126.46, 125.24, 124.82, 122.93, 122.72, 120.69, 119.85, 119.68, 109.22, 108.50, 54.27, 45.50, 39.79, 38.76, 36.26, 30.68, 29.44, 20.76, 14.11.
N-((1-(2-(tert-Butylamino)-2-oxoethyl)pyrrolidin-4-yl)methyl)-9-pentyl-9H-carbazole-3-carboxamide (19)
Under argon atmosphere, to a solution of compound 18 (85 mg, 0.23 mmol) and MeCN (2.5 mL) was added K2CO3 (95.4 mg, 0.69 mmol), KI (38.1 mg, 0.69 mmol), and N-tert-butyl-2-chloroacetamide (41.9 mg, 0.28 mmol). The title compound was prepared according to the analogous procedure described above for compound 10 to produce the title compound 19 (96.2 mg, 88%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 1.5 Hz, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.83 (dd, J = 8.6, 1.5 Hz, 1H), 7.41 (ddd, J = 8.3, 7.1, 1.1 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.16 (ddd, J = 7.9, 7.1, 1.1 Hz, 1H), 7.01 (br s, 1H), 6.56 (t, J = 5.8 Hz, 1H), 4.16 (t, J = 7.2 Hz, 2H), 3.22–3.11 (m, 4H), 2.34–2.22 (m, 2H), 2.11–1.99 (m, 1H), 1.81–1.67 (m, 3H), 1.39–1.22 (m, 16H), 0.83 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 169.83 (C=O), 168.57 (C=O), 142.34 (C), 141.14 (C), 126.45 (CH), 125.26 (C), 124.82 (CH), 122.94 (C), 122.73 (C), 120.68 (CH), 119.85 (CH), 119.68 (CH), 109.21 (CH), 108.50 (CH), 63.41 (CH2), 60.41 (CH2), 55.56 (CH2), 50.89 (C), 47.36(CH2), 44.07 (CH2), 36.67 (CH), 30.46 (CH2), 29.53 (CH2), 28.88 (CH3), 20.58 (CH2), 14.14 (CH3). ESI: m/z 477.5 (M + H)+. HRMS calcd for C29H41N4O2 (M + H)+ 477.3229, found 477.3251.
N-{[1-(3,3-Dimethylbutyl)piperidin-4-yl]methyl}-9-pentyl-9H-carbazole-3-carboxamide (20)
9-Pentyl-9H-carbazole-3-carboxylic acid16 (100 mg, 0.27 mmol) was combined with cyanoborohydride (83.3 mg, 1.35 mmol/g) and 3,3-dimethylbutyraldehyde (0.06 mL, 0.31 mmol) and then suspended in anhydrous CH2Cl2(2 mL). The mixture was stirred at room temperature overnight. The reaction mixture was diluted with DCM (30 mL), and the organic layer was washed three times with brine (50 mL). The organic layer was then separated, dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography eluting with heptane/EtOAc (0–100%) to yield the title compound (88.4 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 8.51 (d, J = 1.4 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.86 (dd, J = 8.6, 1.4 Hz, 1H), 7.42 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.15 (ddd, J = 7.8, 7.1, 1.1 Hz, 1H), 7.01 (br s, 1H), 6.59 (t, J = 5.8 Hz, 1H), 4.20 (t, J = 7.2 Hz, 2H), 3.36 (t, J = 6.5 Hz, 2H), 2.69–2.54 (m, 4H), 2.01 (td, J = 11.7, 2.5 Hz, 2H), 1.83–1.74 (m, 4H), 1.64–1.48 (m, 1H), 1.33–1.23 (m, 8H), 0.91 (s, 9H), 0.77 (t, J = 6.9 Hz, 3H). 13C and DEPT NMR (101 MHz, CDCl3) δ 168.76 (C=O), 142.47 (C), 141.23 (C), 126.67 (CH), 125.48 (C), 124.18 (CH), 122.76 (C), 122.53 (C), 120.68 (CH), 119.85 (CH), 119.68 (CH), 109.23 (CH), 108.50 (CH), 53.89 (CH2), 52.31 (CH2), 45.67(CH2), 43.42 (CH2), 40.22 (CH2), 36.21 (CH), 30.54 (CH2), 30.12 (C), 29.42 (CH2), 29.34 (CH3), 28.71 (CH2), 22.56 (CH2), 14.03 (CH3). ESI: m/z 462.3 (M + H)+. HRMS calcd for C30H43N3O (M + H)+ 462.3484, found 462.3452.
Physicochemical Properties
Molecular properties of compound 9 were calculated using Instant JChem 6.3.3, 2014, ChemAxon, Budapest, Hungary.
Acknowledgments
Radioligand binding assays were performed by the National Institute of Mental Health’s Psychoactive Drug Screening Program Contract # HHSN-271-2008-00025-C (NIMH/PDSP). NIMH/PDSP is directed by Bryan L. Roth M.D., Ph.D. at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD. Marvin was used for drawing, displaying, and characterizing chemical structures, substructures, and reactions included in the Supporting Information, Marvin 5.11.5, 2013, ChemAxon (http://www.chemaxon.com).
Supporting Information Available
Additional characterization data; 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. P.D. designed the compounds. P.D., S.W.M., and R.R.P. synthesized the compounds. C.B. carried out all electrophysiological experiments, and V.M.G. and N.D.B. carried out all animal testing. G.W.Z. and P.D. designed and supervised the study.
This work was supported by operating grants to G.W.Z. from the Canadian Institutes of Health Research and to P.D., S.W.M., and R.R.P. from the National Institutes of Health (NIH), Grant P30-NS055022. G.W.Z. is a Canada Research Chair. C.B. holds a T. Chen Fong studentship and an Alberta Innovates – Health Solutions (AI-HS) studentship award. V.M.G. held a MITACS Elevate fellowship. N.D.B. holds an AI-HS summer studentship award.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kampa B. M.; Letzkus J. J.; Stuart G. J. (2007) Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 30, 456–463. [DOI] [PubMed] [Google Scholar]
- David L. S.; Garcia E.; Cain S. M.; Thau E.; Tyson J. R.; Snutch T. P. (2010) Splice-variant changes of the Ca(V)3.2 T-type calcium channel mediate voltage-dependent facilitation and associate with cardiac hypertrophy and development. Channels 4, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Iijima T. (2010) Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell Cardiol. 48, 65–70. [DOI] [PubMed] [Google Scholar]
- Vassort G.; Talavera K.; Alvarez J. L. (2006) Role of T-type Ca2+ channels in the heart. Cell calcium 40, 205–220. [DOI] [PubMed] [Google Scholar]
- Bender K. J.; Uebele V. N.; Renger J. J.; Trussell L. O. (2012) Control of firing patterns through modulation of axon initial segment T-type calcium channels. J. Physiol. 590, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic M. M.; Pathirathna S.; Nelson M. T.; Mancuso S.; Joksovic P. M.; Rosenberg E. R.; Bayliss D. A.; Jevtovic-Todorovic V.; Todorovic S. M. (2007) Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J. Neurosci. 27, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron S. E.; Khosravani H.; Varela D.; Bladen C.; Williams T. C.; Newman M. R.; Scheffer I. E.; Berkovic S. F.; Mulley J. C.; Zamponi G. W. (2007) Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann. Neurol. 62, 560–568. [DOI] [PubMed] [Google Scholar]
- Khosravani H. (2003) Gating Effects of Mutations in the Cav3.2 T-type Calcium Channel Associated with Childhood Absence Epilepsy. J. Biol. Chem. 279, 9681–9684. [DOI] [PubMed] [Google Scholar]
- Khosravani H.; Bladen C.; Parker D. B.; Snutch T. P.; McRory J. E.; Zamponi G. W. (2005) Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Ann. Neurol. 57, 745–749. [DOI] [PubMed] [Google Scholar]
- Powell K. L.; Cain S. M.; Ng C.; Sirdesai S.; David L. S.; Kyi M.; Garcia E.; Tyson J. R.; Reid C. A.; Bahlo M.; Foote S. J.; Snutch T. P.; O’Brien T. J. (2009) A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J. Neurosci. 29, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W.; Lory P.; Perez-Reyes E. (2010) Role of voltage-gated calcium channels in epilepsy. Pflügers Arch. 460, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton M. E.; Eberle E. L.; Shannon H. E. (2005) The antihyperalgesic effects of the T-type calcium channel blockers ethosuximide, trimethadione, and mibefradil. Eur. J. Pharmacol. 521, 79–85. [DOI] [PubMed] [Google Scholar]
- Bourinet E.; Alloui A.; Monteil A.; Barrere C.; Couette B.; Poirot O.; Pages A.; McRory J.; Snutch T. P.; Eschalier A.; Nargeot J. (2005) Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 24, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.; Na H. S.; Kim J.; Lee J.; Lee S.; Kim D.; Park J.; Chen C. C.; Campbell K. P.; Shin H. S. (2007) Attenuated pain responses in mice lacking Ca(V)3.2 T-type channels. Genes, Brain Behav. 6, 425–431. [DOI] [PubMed] [Google Scholar]
- Dogrul A.; Gardell L. R.; Ossipov M. H.; Tulunay F. C.; Lai J.; Porreca F. (2003) Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain 105, 159–168. [DOI] [PubMed] [Google Scholar]
- Gadotti V. M.; You H.; Petrov R. R.; Berger N. D.; Diaz P.; Zamponi G. W. (2013) Analgesic effect of a mixed T-type channel inhibitor/CB2 receptor agonist. Mol. Pain 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic M. M.; Pathirathna S.; Joksovic P. M.; Lee W.; Nelson M. T.; Naik A. K.; Su P.; Jevtovic-Todorovic V.; Todorovic S. M. (2008) Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J. Neurophysiol. 99, 3151–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic A.; Hwang S. M.; Scarpa J.; Hong S. J.; Todorovic S. M.; Jevtovic-Todorovic V. (2014) CaV3.2 T-Type Calcium Channels in Peripheral Sensory Neurons Are Important for Mibefradil-Induced Reversal of Hyperalgesia and Allodynia in Rats with Painful Diabetic Neuropathy. PloS One 9, e91467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch T. P.; David L. S. (2006) T-type calcium channels: an emerging therapeutic target for the treatment of pain. Drug Dev. Res. 67, 404–415. [Google Scholar]
- Todorovic S.; Jevtovic-Todorovic V. (2014) Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflügers Arch. 466, 701–706. [DOI] [PubMed] [Google Scholar]
- Zamponi G. W.; Lewis R. J.; Todorovic S. M.; Arneric S. P.; Snutch T. P. (2009) Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 60, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Barbara G.; Alloui A.; Nargeot J.; Lory P.; Eschalier A.; Bourinet E.; Chemin J. (2009) T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J. Neurosci. 29, 13106–13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladen C.; Gunduz M. G.; Simsek R.; Safak C.; Zamponi G. W. (2013) Synthesis and Evaluation of 1,4-Dihydropyridine Derivatives with Calcium Channel Blocking Activity. Pflügers Arch. 466, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Chemin J.; Monteil A.; Perez-Reyes E.; Nargeot J.; Lory P. (2001) Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 20, 7033–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y. J.; Seo H. N.; Jung S. Y.; Rhim H.; Kim J.; Choo D. J.; Lee J. Y. (2008) Synthesis and SAR study of T-type calcium channel blockers. Part II. Arch. Pharm. 341, 661–664. [DOI] [PubMed] [Google Scholar]
- Furukawa T.; Miura R.; Honda M.; Kamiya N.; Mori Y.; Takeshita S.; Isshiki T.; Nukada T. (2004) Identification of R(−)-isomer of efonidipine as a selective blocker of T-type Ca2+ channels. Br. J. Pharmacol. 143, 1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand M. E.; Smith P. L.; Bladen C.; Eduljee C.; Xie J. Y.; Chen L.; Fee-Maki M.; Doering C. J.; Mezeyova J.; Zhu Y.; Belardetti F.; Pajouhesh H.; Parker D.; Arneric S. P.; Parmar M.; Porreca F.; Tringham E.; Zamponi G. W.; Snutch T. P. (2011) A novel slow-inactivation-specific ion channel modulator attenuates neuropathic pain. Pain 152, 833–843. [DOI] [PubMed] [Google Scholar]
- Jo M. N.; Seo H. J.; Kim Y.; Seo S. H.; Rhim H.; Cho Y. S.; Cha J. H.; Koh H. Y.; Choo H.; Pae A. N. (2007) Novel T-type calcium channel blockers: dioxoquinazoline carboxamide derivatives. Bioorg. Med. Chem. 15, 365–373. [DOI] [PubMed] [Google Scholar]
- Kumar P. P.; Stotz S. C.; Paramashivappa R.; Beedle A. M.; Zamponi G. W.; Rao A. S. (2002) Synthesis and evaluation of a new class of nifedipine analogs with T-type calcium channel blocking activity. Mol. Pharmacol. 61, 649–658. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E.; Van Deusen A. L.; Vitko I. (2009) Molecular pharmacology of human Cav3.2 T-type Ca2+ channels: block by antihypertensives, antiarrhythmics, and their analogs. J. Pharmacol. Exp. Ther. 328, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E.; Kataoka K.; Dong Y. F.; Nakamura T.; Fukuda M.; Nako H.; Ogawa H.; Kim-Mitsuyama S. (2010) Benidipine, a dihydropyridine L-type/T-type calcium channel blocker, affords additive benefits for prevention of cardiorenal injury in hypertensive rats. J. Hypertens. 28, 1321–1329. [DOI] [PubMed] [Google Scholar]
- You H.; Altier C.; Zamponi G. W. (2010) CCR2 receptor ligands inhibit Cav3.2 T-type calcium channels. Mol. Pharmacol. 77, 211–217. [DOI] [PubMed] [Google Scholar]
- You H.; Gadotti V. M.; Petrov R. R.; Zamponi G. W.; Diaz P. (2011) Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol. Pain 7, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J.; Nargeot J.; Lory P. (2007) Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J. Biol. Chem. 282, 2314–2323. [DOI] [PubMed] [Google Scholar]
- Moreira F. A.; Grieb M.; Lutz B. (2009) Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. Best. Pract. Res., Clin. Endocrinol. Metab. 23, 133–144. [DOI] [PubMed] [Google Scholar]
- Witkin J. M.; Tzavara E. T.; Nomikos G. G. (2005) A role for cannabinoid CB1 receptors in mood and anxiety disorders. Behav. Pharmacol. 16, 315–331. [DOI] [PubMed] [Google Scholar]
- Petrov R. R.; Knight L.; Chen S. R.; Wager-Miller J.; McDaniel S. W.; Diaz F.; Barth F.; Pan H. L.; Mackie K.; Cavasotto C. N.; Diaz P. (2013) Mastering tricyclic ring systems for desirable functional cannabinoid activity. Eur. J. Med. Chem. 69, 881–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. L.; Bayliss D. A.; Fern R. J.; Barrett P. Q. (1999) A role for T-type Ca2+ channels in the synergistic control of aldosterone production by ANG II and K+. Am. J. Physiol. 276, F674–683. [DOI] [PubMed] [Google Scholar]
- Marger F.; Gelot A.; Alloui A.; Matricon J.; Ferrer J. F.; Barrere C.; Pizzoccaro A.; Muller E.; Nargeot J.; Snutch T. P.; Eschalier A.; Bourinet E.; Ardid D. (2011) T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. U. S. A. 108, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S.; Shioda N.; Yamamoto Y.; Tagashira H.; Fukunaga K. (2012) The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 121, 44–53. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V.; Todorovic S. M. (2006) The role of peripheral T-type calcium channels in pain transmission. Cell Calcium 40, 197–203. [DOI] [PubMed] [Google Scholar]
- Orestes P.; Osuru H. P.; McIntire W. E.; Jacus M. O.; Salajegheh R.; Jagodic M. M.; Choe W.; Lee J.; Lee S. S.; Rose K. E.; Poiro N.; Digruccio M. R.; Krishnan K.; Covey D. F.; Lee J. H.; Barrett P. Q.; Jevtovic-Todorovic V.; Todorovic S. M. (2013) Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes 62, 3828–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic S. M.; Jevtovic-Todorovic V. (2011) T-type voltage-gated calcium channels as targets for the development of novel pain therapies. Br. J. Pharmacol. 163, 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins M. E.; Horowitz B. Z.; Linden D. H.; Smith G. W.; Norton R. L.; Stump J. (1998) Life-threatening interaction of mibefradil and beta-blockers with dihydropyridine calcium channel blockers. JAMA, J. Am. Med. Assoc. 280, 157–158. [DOI] [PubMed] [Google Scholar]
- Bourinet E.; Altier C.; Hildebrand M. E.; Trang T.; Salter M. W.; Zamponi G. W. (2014) Calcium-permeable ion channels in pain signaling. Physiol. Rev. 94, 81–140. [DOI] [PubMed] [Google Scholar]
- Nelson M. T.; Joksovic P. M.; Perez-Reyes E.; Todorovic S. M. (2005) The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci. 25, 8766–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A.; Kerckhove N.; Meleine M.; Alloui A.; Barrere C.; Gelot A.; Uebele V. N.; Renger J. J.; Eschalier A.; Ardid D.; Bourinet E. (2013) State-dependent properties of a new T-type calcium channel blocker enhance Ca(V)3.2 selectivity and support analgesic effects. Pain 154, 283–293. [DOI] [PubMed] [Google Scholar]
- Simms B. A.; Zamponi G. W. (2014) Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 82, 24–45. [DOI] [PubMed] [Google Scholar]
- Armbruster B. N.; Roth B. L. (2005) Mining the receptorome. J. Biol. Chem. 280, 5129–5132. [DOI] [PubMed] [Google Scholar]
- Jensen N. H.; Roth B. L. (2008) Massively parallel screening of the receptorome. Comb. Chem. High Throughput Screening 11, 420–426. [DOI] [PubMed] [Google Scholar]
- Strachan R. T.; Ferrara G.; Roth B. L. (2006) Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discovery Today 11, 708–716. [DOI] [PubMed] [Google Scholar]
- Feng Z. P.; Doering C. J.; Winkfein R. J.; Beedle A. M.; Spafford J. D.; Zamponi G. W. (2003) Determinants of inhibition of transiently expressed voltage-gated calcium channels by omega-conotoxins GVIA and MVIIA. J. Biol. Chem. 278, 20171–20178. [DOI] [PubMed] [Google Scholar]
- Hylden J. L.; Wilcox G. L. (1980) Intrathecal morphine in mice: A new technique. Eur. J. Pharmacol. 67, 313–316. [DOI] [PubMed] [Google Scholar]
- Gadotti V. M.; Zamponi G. W. (2011) Cellular prion protein protects from inflammatory and neuropathic pain. Mol. Pain 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaster M. P.; Gadotti V. M.; Calixto J. B.; Santos A. R.; Rodrigues A. L. (2012) Depressive-like behavior induced by tumor necrosis factor-alpha in mice. Neuropharmacology 62, 419–426. [DOI] [PubMed] [Google Scholar]
- Malmberg A. B.; Basbaum A. I. (1998) Partial sciatic nerve injury in the mouse as a model of neuropathic pain: Behavioral and neuroanatomical correlates. Pain 76, 215–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.