

Abstract
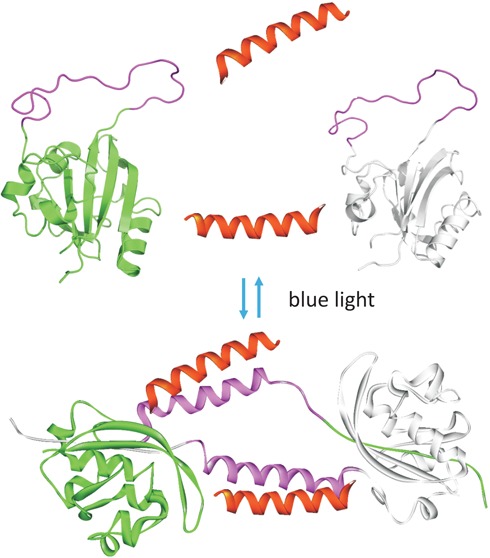
The design of new optogenetic tools for controlling protein function would be facilitated by the development of protein scaffolds that undergo large, well-defined structural changes upon exposure to light. Domain swapping, a process in which a structural element of a monomeric protein is replaced by the same element of another copy of the same protein, leads to a well-defined change in protein structure. We observe domain swapping in a variant of the blue light photoreceptor photoactive yellow protein in which a surface loop is replaced by a well-characterized protein–protein interaction motif, the E-helix. In the domain-swapped dimer, the E-helix sequence specifically binds a partner K-helix sequence, whereas in the monomeric form of the protein, the E-helix sequence is unable to fold into a binding-competent conformation and no interaction with the K-helix is seen. Blue light irradiation decreases the extent of domain swapping (from Kd = 10 μM to Kd = 300 μM) and dramatically enhances the rate, from weeks to <1 min. Blue light-induced domain swapping thus provides a novel mechanism for controlling of protein–protein interactions in which light alters both the stability and the kinetic accessibility of binding-competent states.
Photoswitchable proteins can elicit a change in biological activity in response to light.1 Just as fluorescent proteins have been employed as tools to monitor cellular events, photoswitchable proteins offer a means to control them. Channelrhodopsins, a family of naturally occurring or engineered light-switchable ion channels,2 are gaining widespread use in neuroscience because they allow precise spatiotemporal control of action potential firing in neurons. There are many processes in biology that exhibit complex spatial and temporal patterns akin to electrical signaling in the nervous system, but for which no known photoswitchable effectors analogous to the channelrhodopsins exist. Structure-based design of novel photoswitchable effectors would be greatly facilitated by the development of protein scaffolds that undergo large, well-defined structural changes upon illumination.
Domain swapping, defined by Eisenberg as the process whereby a secondary or tertiary structural element of a monomeric protein is replaced by the same element of another copy of the same protein,3−5 must produce a large structural change in the hinge region, the part of the structure that must reorient to alter the oligomeric state of the protein. In addition, domain swapping may be coupled to complete folding–unfolding transitions of protein structure.6 Consistent with the large and well-defined structural changes associated with domain swapping, the process is associated with radical changes in protein activity. For example, domain swapping can effectively turn on and off protein splicing,7 membrane interactions,8 an ability to mediate stable cell junctions,9 and the process of amyloid fibril formation.10
Here we report domain swapping in a circularly permuted variant of photoactive yellow protein (PYP). PYP is well-suited for photoswitching applications because it is small and water-soluble, folds reversibly, and undergoes a large change in conformational dynamics upon conversion to the light state.11,12 Our intention was to couple the light-driven conformational change in PYP to a functional change in a target protein sequence. Loh and colleagues have described how protein function can be controlled by coupling folding of a target protein to unfolding of a control protein by setting up an antagonistic folding–unfolding equilibrium between the two proteins.6,13 This type of mutually exclusive folding can be achieved by inserting the target protein sequence into a surface loop of the control protein so that the attachment points are too close together for the target protein to adopt its normally folded conformation unless the control protein unfolds, or unless it undergoes a domain swap.6 To make folding and unfolding of the control protein light-dependent, we required a loop in the control protein for which the ends were much less constrained in the light state than in the dark state. Analysis of the changes in conformational dynamics of PYP between light and dark states suggests that none of the native loop constraints change dramatically.14 We therefore designed a circular permutant cPYP in which the original N- and C-termini were joined in a new surface loop and new N- and C-termini were created at positions 115 and 114 of the original sequence.15 This protein underwent a normal photocycle, and the constraints on the ends of the newly created loop were found to change substantially between light and dark forms.15
In this study, a cPYP variant was created (designated c-E-helix-PYP) in which a heterodimeric coiled-coil-forming sequence (E-helix)16,17 [(IAALEKE)2IAALE] is inserted at the newly created surface loop. The E-helix sequence was designed by Hodges and colleagues on the basis of extensive data on sequence properties governing coiled-coil formation.17−19 The E-helix sequence binds with high specificity and affinity to a complementary K-helix peptide [Ac-WG(IAALKEK)2IAALK-NH2] but must adopt a helical conformation to do so. The distance between the loop ends in the dark-state folded structure of cPYP is too short to permit the E-helix insert to adopt an α-helical conformation. Thus, we expected this state to show weak affinity for the K-helix. Our original expectation was that the enhanced flexibility of the light state of PYP would then permit the E-helix to adopt a conformation competent for K-helix binding. Instead, we discovered that c-E-helix-PYP undergoes domain swapping that leads to the presence of both monomeric and dimeric forms of the protein. The two forms interconvert extremely slowly (weeks) in the dark but do so rapidly (<1 min) under blue light illumination. Domain swapping produces a change in the hinge region of the protein, so that only the domain-swapped dimeric state can bind the K-helix peptide. This novel type of light-driven protein conformational change produces essentially complete on–off control because the high thermal barrier to interconversion means the active (binding-competent) state cannot be accessed without exposure to light.
Materials and Methods
Gene Synthesis
DNA encoding c-E-helix-PYP (codon optimized
for Escherichia coli) was synthesized by BioBasic
Inc. (Toronto, ON) and inserted into the pET24b(+) vector using Ndel
and HindIII restriction sites. The expressed sequence was
Protein Expression and Purification
The expression and purification protocol for c-E-helix-PYP was adapted from the work of Devanathan et al.20 DNA (0.2 ng) was transformed into BL21*(pLysS) competent cells and plated onto agar plates containing 30 μg/mL kanamycin. The following day, a single colony was used to inoculate 25 mL of Luria-Bertani broth (LB) that had been supplemented with kanamycin (30 μg/mL). The 25 mL overnight culture was used to inoculate 1 L of LB supplemented with 30 μg/mL kanamycin. Cells were grown at 37 °C to an OD600 of 0.6 and then induced with 0.5 mM IPTG. The temperature was adjusted to 18 °C, and the cells were grown for a further 1.5 h. At this point, 25 mg of activated chromophore dissolved in 1 mL of ethanol was added to the medium. The synthesis of the activated chromophore, 4-hydroxycinnamic acid S-thiophenyl ester, was conducted as detailed by Changenet-Barret et al.,21 except that the product was not recrystallized. The cells were then grown for 14 h before centrifugation to separate the medium from the protein-containing cell pellet.
The pellet was resuspended in lysis buffer [50 mM sodium phosphate, 300 mM sodium chloride, and 5 mM magnesium chloride (pH 8.0)] and frozen at −20 °C until it was purified. The resuspended cell pellet was sonicated in pulses on ice for 5 min and then centrifuged at 12K rpm for 1 h to separate the supernatant from the pellet. The pellet was then resuspended in lysis buffer containing 6 M urea and subsequently centrifuged at 12K rpm for an additional 1 h. The protein-containing supernatant was then applied to a Ni2+-NTA column that had been equilibrated with lysis buffer containing 6 M urea. The resin was washed with 10 column volumes (CV) of lysis buffer containing 6 M urea. The resin was subsequently washed with lysis buffer containing decreasing concentrations of urea, followed by high-salt buffer (i.e., lysis buffer supplemented with 2 M NaCl). The resin was then washed with 5 CV of lysis buffer supplemented with 5 mM imidazole to elute nonspecifically bound proteins. The protein was eluted by increasing the concentration of imidazole to 200 mM.
The eluted protein was dialyzed extensively against 40 mM Tris-OAc, 1 mM EDTA, and 100 mM NaCl (pH 7.5) [1× TAE and 100 mM NaCl (pH 7.5)] at 4 °C. The dialyzed protein was concentrated to ∼1.5 mL using an Amicon ultracentrifugal device [10000 Da NMWL (nominal molecular weight limit)] (Millipore). The protein was then applied to a Superdex 75 10/300 GL column (GE Healthcare) running in 1× TAE, 100 mM NaCl buffer (pH 7.5). This permitted physical separation of the dimer and monomer for subsequent experiments. UV–vis absorbance spectroscopy was used to determine which eluted fractions had the highest ratios of absorbance at 446 nm to that at 278 nm. A value of ∼2 was typical for both dimeric and monomeric c-E-helix-PYP. The identity of the sample was then confirmed by electrospray ionization mass spectrometry (ESI-MS). Additional dimer was produced as needed by incubating 500 μM c-E-helix-PYP in the dark for 18 h [1× TAE, 100 mM NaCl, and 1.5 M Gdn (pH 7.5, 30 °C)], followed by size exclusion chromatography as described above. The monomer could be produced by blue light irradiation of 10 μM protein [1× TAE and 100 mM NaCl (pH 7.5, 30 °C)], followed by size exclusion chromatography as described above.
UV–Vis Spectra and Thermal Relaxation Kinetics
UV–vis spectra and kinetic measurements were performed using a PerkinElmer Lambda 35 or 25 spectrophotometer or using a diode array UV–vis spectrophotometer (Ocean Optics Inc., USB4000), in each case coupled to a temperature-controlled cuvette holder (Quantum Northwest, Inc.). Protein concentrations were determined using an extinction coefficient at λmax (∼446 nm) of 45 × 103 M–1 cm–1. Thermal relaxation experiments were conducted at 10 °C to prevent interconversion between monomeric and dimeric species. Irradiation of the protein samples was conducted using a Luxeon III Star Royal Blue Lambertian light emitting diode (455 nm) operating at approximately 700 mA (∼50 mW/cm2). Changes in the absorbance spectrum at 446 nm were monitored to determine the rate constants for thermal relaxation. Monomer data were fit to a single-exponential function. Dimer data were fit to a double-exponential function, or in the case where kinetics were measured as the K-peptide was added, global fitting was performed using a triple-exponential function (see Figure S9 of the Supporting Information). All fitting was performed using IgorPro (Wavemetrics).
CD Measurements
CD experiments were conducted on an Olis RSM100 CD spectrometer. Dimeric c-E-helix-PYP was prepared at a concentration of 12 μM in 5 mM sodium phosphate (pH 7.5). Monomeric c-E-helix-PYP was produced by illuminating the dimer sample with blue light. This allowed for a direct comparison of CD traces without any changes in protein concentration. A cylindrical quartz cell with a path length of 0.1 cm was used for all measurements. Samples were fully dark adapted before they were scanned in the far-UV region from 260 to 190 nm at 30 °C. The integration time was 1 s, and three scans were averaged to give a final spectrum. The spectra were converted to mean residue ellipticities and smoothed using a binomial algorithm provided in IgorPro (Wavemetrics).
Sedimentation Equilibrium
Samples of purified monomeric and dimeric c-E-helixPYP were prepared at concentrations of 5.5, 11.1, and 22.2 μM in 1× TAE, 100 mM NaCl buffer (pH 7.5) and analyzed by analytical ultracentrifugation in sedimentation equilibrium mode (AUC facility, Department of Biochemistry, University of Toronto). The absorbance at 446 nm was monitored at 4 °C and speeds of 16000, 19000, and 22000 rpm.
Nuclear Magnetic Resonance (NMR)
Labeled c-E-helix-PYP (15N, 19F, and 15N and 19F) was prepared as follows. An expression plasmid was transformed into BL21*(pLysS) cells and plated on LB-Agar containing 30 μg/mL kanamyacin. A single colony was selected from this plate and transferred to a 50 mL LB culture containing 30 μg/mL kanamycin. The following day, 6 mL of the overnight culture was centrifuged for 15 min at 4000 rpm, and the pellet was resuspended in 50 mL of M9 medium (not isotope-labeled). The 50 mL culture was grown at 37 °C and 250 rpm until it had reached the midlog growth phase (OD600 ∼ 0.5–0.7), after which it was centrifuged as described above. The pellet was used to inoculate 1 L of 99% 15N-enriched M9 minimal medium, supplemented with 0.3% d-glucose, 0.1% 15NH4Cl (Cambridge Isotope Laboratories, Inc.), 30 mg/L kanamycin, 10 mg/L thiamine, 10 mg/L biotin, 1 mM MgSO4, 1 mM CaCl2, and 1% Bioexpress cell growth 15N medium (Cambridge Isotope Laboratories, Inc.), and the culture was subsequently grown at 37 °C and 250 rpm until it had again reached the midlog phase. Protein expression was induced by the addition of 0.5 mM IPTG. The temperature was adjusted to 18 °C, and the cells were grown for a further 1.5 h, after which 25 mg of activated chromophore21 dissolved in 1 mL of ethanol was added. The culture was then grown overnight. The following day, the cells were harvested by centrifugation at 4000 rpm and 4 °C for 1 h. To incorporate 5-fluorotrytophan into c-E-helix-PYP, 60 mg/L 5-fluoro-l,d-Trp (Aldrich) was introduced into the bacterial culture 1 h prior to induction.22 Here, the activated chromophore was added at the time of induction of protein expression by addition of 0.5 mM IPTG, and growth was halted 2 h after induction. The protein was purified in the same manner as unlabeled samples (as described above). The expected masses of the proteins were confirmed by ESI-MS.
NMR experiments were conducted at CSICOMP (Department of Chemistry, University of Toronto) on an Agilent DD2 700 MHz spectrometer equipped with an HFCN cold probe. Spectra were acquired with a 15N NH HSQC-TROSY Watergate pulse sequence from the Varian “Biopack” library. All spectra were acquired at 37 °C with 2048 points spanning 8928.6 Hz in the 1H dimension and 256 increments spanning 2202 Hz in the 15N dimension. Sample concentrations varied from 200 to 400 μM, and the number of transients collected was varied from 8 to 80 depending on the concentration available. Size exclusion chromatography was used following acquisition to confirm that monomer–dimer interconversion did not occur.
Spectra were processed using the NMRPipe23 processing suite. FID signals were zero filled to double the original data size and apodized using a sine window function prior to Fourier transformation. In the indirect dimension, linear prediction was applied to double the original data size. Analysis was aided by NMRViewJ (One Moon Scientific).
Peptide Synthesis
Automated Fmoc-SPPS was conducted on a CEM Microwave Peptide Synthesizer (CEM Corp.) using Rink Amide MBHA resin (0.4 Loading, AnaSpec, Inc.) at 0.1 mmol scale. Coupling was performed using HBTU [O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate] (AnaSpec, Inc.), DIPEA (N,N-diisopropylethylamine) (Sigma-Aldrich Inc.), and a 5-fold molar excess of amino acid (AnaSpec, Inc.). Peptides were purified by high-performance liquid chromatography (HPLC) (Zorbax Rx-C8 semipreparative column) using a linear gradient from 5 to 65% acetonitrile/H2O (with 0.1% trifluoroacetic acid) over 30 min. The molecular composition of the peptides was confirmed by ESI-MS, and lyophilized peptides were used for subsequent reactions. 5-Iodoacetamide fluorescein (5-IAF) was purchased from AnaSpec, Inc. K-Helix-F labeling was conducted by reacting 1.1 mM 5-IAF with 1 mM peptide in 5 mM phosphate buffer (pH 8.0) followed by HPLC purification.
Native Polyacrylamide Gel Electrophoresis (PAGE) Analysis
Native PAGE was conducted with 12.5% polyacrylamide gels. A running buffer of 25 mM Tris with 192 mM glycine (pH 8.3) was used. Gel images were quantified using ImageJ (http://imagej.nih.gov/ij/index.html). Fluorescently labeled complexes were electrophoresed under the same conditions. They were then visualized with a Bio-Rad PharosFX Plus Molecular Imager equipped with an external 488 nm laser and a 530 nm band-pass filter. Equilibrium constants were determined using c-E-helix-PYP at varying concentrations that were incubated under constant blue light illumination, or in the dark with 1 M Gdn HCl. Guanidinium was removed by rapidly diluting the samples with native buffer [1× TAE and 100 mM NaCl (pH 7.5)] and then concentrating the sample in an Amicon centrifugal filter (10000 Da NMWL) (Millipore). The rate of monomer formation under illumination was determined using 20 μM dimeric c-E-helix-PYP that was irradiated with a mounted high-power light-emitting diode (LED) (455 nm) purchased from ThorLabs. The light intensity was controlled with a variable current LED driver, and the fraction of cis-isomerized protein was monitored with a diode array UV–vis spectrophotometer (Ocean Optics Inc., USB4000).
Results
c-E-Helix-PYP Exists as a Slowly Exchanging Monomer–Dimer Pair
The circularly permuted form of PYP containing the E-helix sequence as a loop insert (c-E-helix-PYP) was expressed in bacteria and purified under denaturing conditions. After Ni2+-NTA chromatography, c-E-helix-PYP was applied to a gel filtration column. In contrast to other c-PYP constructs we have investigated, which elute as a single peak, we found that c-E-helix-PYP eluted as two well-separated peaks from the gel filtration column (Figure 1a). We identified these two peaks as monomeric and dimeric forms of c-E-helix-PYP by sedimentation equilibrium ultracentrifugation (Figure 1b). Both monomeric and dimeric forms behave as a single species by ESI-MS under denaturing conditions and have a characteristic PYP UV–visible spectrum (Figure 1c), consistent with both species being holoproteins. Because the PYP chromophore is covalently linked to the only cysteine residue, the dimeric form cannot be the result of intermolecular disulfide bonding.
Figure 1.
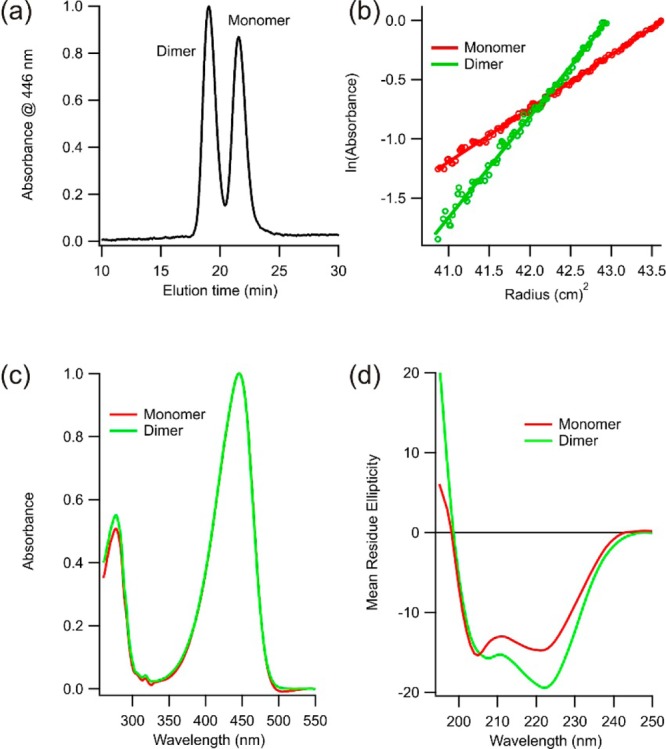
(a) Size exclusion (Sephadex G75) chromatogram of c-E-helix-PYP following purification by Ni2+-NTA chromatography. (b) Sedimentation equilibrium data confirming the two peaks in panel a correspond to monomeric and dimeric species of c-E-helix-PYP. (c) Electronic absorption spectra of the dark-adapted monomer and dimer. (d) Far-UV circular dichroism spectra of the dark-adapted monomer and dimer.
Under standard buffer conditions (Tris-acetate, pH 7.5, and 100 mM NaCl), we found that monomer–dimer exchange occurred on a very slow time scale; no appreciable exchange was seen in 1 week at 30 °C when the sample was dark-adapted (Figure S1 of the Supporting Information). However, the monomeric form could be produced by diluting the protein under denaturing conditions and then removing the denaturant. Likewise, the dimeric form could be produced by concentrating the protein in the presence of denaturant.
Characterization of the Monomer and Dimer
Because of the slow rate of monomer–dimer exchange, we were able to use biophysical methods to characterize the monomer and dimer separately. The UV–visible absorption spectra of monomeric and dimeric c-E-helix-PYP are essentially superimposable (Figure 1c), indicating that both species have a properly folded, wild-type-like chromophore binding pocket. Far-UV circular dichroism, however, indicates that the dimer has more helical content (Figure 1d).
We recorded 15NH HSQC-TROSY spectra of dark-adapted monomeric and dimeric forms of c-E-helix-PYP. Monomeric c-E-helix-PYP shares a large number [∼100 (Table S1 of the Supporting Information)] of overlapping resonances with c-PYP15 (Figure S2 and Table S1 of the Supporting Information). Because of this, we were able to assign most of the c-E-helix-PYP resonances (except those of the E-helix insert) by inspection. We also found that dimeric c-E-helix-PYP showed significant overlap with cPYP (Figure S3 and Table S1 of the Supporting Information). This indicates that dimerization of c-E-helix-PYP occurs with relatively small changes to the environments of the assigned residues, which consist mostly of those in the PYP core (Figures S2–S4 of the Supporting Information). There are differences in the spectra of the monomer and dimer, but these occur mostly for cross-peaks that have no corresponding cross-peak in cPYP. Also considering the CD data (Figure 1d), we attribute this to a conformational change in the E-helix loop insert upon dimerization.
We considered whether formation of the dimer by c-E-helix-PYP may be occurring via interaction of two E-helices. Although homodimerization of E-helices in solution has been observed, the interaction is very weak for E-helices of this length (three heptads).16,24 Moreover, we found that adding the complementary K-helix peptide to a solution of dimeric c-E-helix-PYP did not lead to monomerization (Figure S1 of the Supporting Information) despite the fact that E-helix and K-helix peptides alone interact with high affinity (Kd = 70 nM).16,24 Instead, the very slow rate of monomer–dimer exchange is consistent with domain swapping in which a large number of intermolecular contacts are present at the dimerization interface. Domain swapping results in very little structural rearrangement outside the hinge region, as intramolecular contacts in the natively folded monomer become intermolecular contacts in the dimer.3−5 Thus, domain swapping would explain the very high degree of similarity of the monomer–dimer UV–vis spectra and monomer–dimer NMR spectra.
For domain swapping to occur, a hinge loop in the monomer must become disengaged to allow for reciprocal exchange. Previous reports show that domain swapping is sensitive to mutations in the hinge region.25,26 cPYP does not domain swap,15 and it differs from c-E-helix-PYP only in the loop connecting the original N- and C-termini. We therefore hypothesized that the hinge region was the E-helix sequence. This hypothesis predicts that dimer formation occurs via swapping of the N-terminal β-strand between monomers (Figure 2d). Such swapping would be expected to result in a change in conformation of the E-helix linker, which would be consistent with the differences observed in monomer–dimer HSQC spectra and CD spectra (vide supra).
Figure 2.

(a) Region of a 15NH HSQC-TROSY spectrum of the c-E-helix-PYP monomer (red) and the monomer containing 5-F-Trp (blue). Residue numbers are indicated. (b) Model of the c-E-helix-PYP monomer with residues whose NH chemical shifts are affected by fluorine substitution (except those on the same strand as 5-F-Trp) colored blue. Residues shown in the HSQC detail (a) are labeled and shown as sticks. The fluorine atom is shown as a green sphere on a stick representation of the 5-F-Trp side chain. The E-helix sequence is colored magenta. (c) Same region as in panel a of a 15NH HSQC-TROSY spectrum showing the c-E-helix-PYP dimer labeled with 15N only (green) and the heterodimer sample (blue). The heterodimer sample is a mixture of three molecular species: (1) dimers made from two 15N-labeled monomers, (2) dimers made from two 19F-labeled monomers, and (3) dimers made from one 15N-labeled monomer and one 19F-labeled monomer. Species (2) is invisible in the NH HSQC spectrum, but both species 1 and 3 are present. The same subset of resonances is affected by fluorine substitution as for the monomer. (d) Model of the c-E-helix-PYP domain-swapped dimer with one monomer colored white and the other green. The E-helix sequence is colored magenta. Residues affected by fluorine substitution are colored blue.
A Fluorotryptophan Probe for N-Terminal Strand Swapping
c-E-Helix-PYP has a single tryptophan residue, which is located on the N-terminal β-strand that we propose is swapping (Figure 2). We therefore made use of a 5-fluorotryophan (5-F-Trp) probe to test this hypothesis. We first produced 15N-labeled monomeric c-E-helix-PYP containing 5-F-Trp. We found that introduction of this probe did not affect the protein globally [the majority of HN chemical shifts were unaffected (Figure S6 of the Supporting Information)] but caused specific chemical shift perturbations at residues that are near the fluorine atom in space (Table S2 of the Supporting Information and Figure 2a). These residues are on the N-terminal strand and adjacent strands in the central β-sheet (Figure 2b).
We then produced dimeric c-E-helix-PYP from an equal mixture of 15N-only-labeled and 5-F-Trp-only-labeled monomers. This mixture is expected to contain differently labeled c-E-helix-PYP dimers, i.e., 25% 15N/15N dimers without 5-F-Trp, 25% 5-F-Trp/5-F-Trp dimers without 15N (which do not contribute to the NH HSQC spectra), and 50% 5-F-Trp/15N dimers. If N-terminal strand swapping is occurring, we expect the 5-F-Trp probe of one monomer to be inserted between 15N-labeled β-strands of the other. We should then expect similar chemical shift perturbations as seen in the doubly labeled (15N and 5-F-Trp) monomer. Consistent with the N-terminal strand swapping hypothesis, the 15N/5-F-Trp dimer produced these same chemical shift perturbations (Figure 2c and Table S2 of the Supporting Information). These data also indicate that the β-strands adjacent to the swapped strand cannot be involved in swapping. If these strands were also exchanged between monomers, their 15NH chemical shifts would not be perturbed by the 19F label.
Blue Light Accelerates Domain Swapping and Promotes Monomerization
To estimate the equilibrium constant for dimer formation, we required conditions that increased the rate of exchange between monomeric and dimeric states so that equilibrium could be reached in a reasonable period of time. It was found that increasing the temperature to 30 °C and adding 1 M GdnHCl to the solution permitted equilibration between states to occur overnight (∼14 h). These conditions do not cause complete denaturation of the protein as evidenced by the persistence of a normal UV–vis absorbance trace (Figure S7 of the Supporting Information), but presumably, they induce some fraction of the sample to transiently unfold. Native PAGE permitted separation of the monomer and the dimer species, and quantification of band intensities permitted calculation of the dimerization affinity (Figure 3). Using a range of total protein concentrations, an average value for Kd(dark) of 10 μM was obtained (Figure S8 of the Supporting Information). Because an increased temperature and 1 M GdnHCl would be expected to decrease the helical content of the E-helix insert, and domain swapping is expected to be promoted by the incompatibility of the folded E-helix insert with the monomer state, we would expect these solution conditions to decrease the propensity of c-E-helix-PYP to form a dimer compared to those seen under native conditions.
Figure 3.
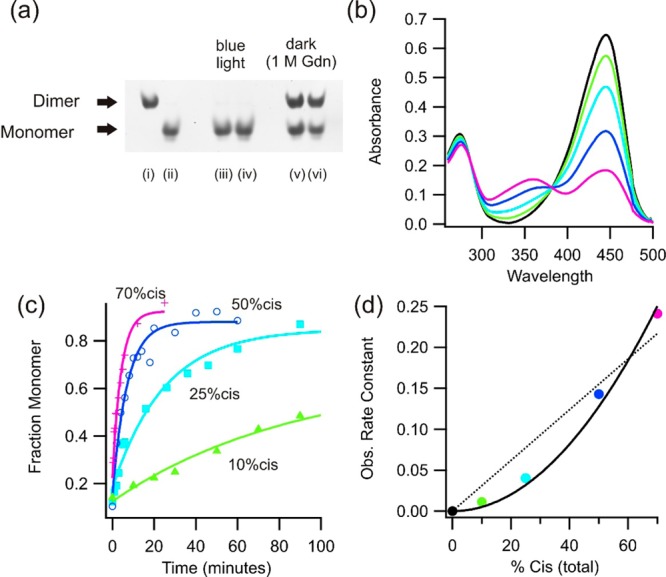
(a) Analysis of monomer–dimer equilibration using native PAGE (20 μM, 1× TAE, 100 mM NaCl, pH 7.5, 30 °C). Pure dimeric (i) and monomeric (ii) samples are shown before equilibration. Irradiation of a dimeric (iii) or monomeric (iv) sample produces >98% monomer. Dark equilibration overnight in the presence of 1 M GdnHCl from either the dimer (v) or monomer (vi) produces ∼60% dimer. (b) Steady-state UV–vis spectra of c-E-helix-PYP obtained with different intensities of blue light. (c) Observed fraction of monomer produced over time (as measured using native PAGE) from a dimer sample (as in panel a) for the different illumination intensities shown in panel b. (d) Monomerization rate constant (calculated from the data in panel c) as a function of the total percent of cis isomer. The rate constant is clearly not linearly dependent on the percent cis (···) but is a function of the percent cis isomer squared (—).
Both monomeric and dimeric forms of c-E-helix-PYP undergo typical photocycles as measured by UV–vis spectroscopy (Figure 3 and Figure S9 of the Supporting Information). Blue light irradiation causes the intensity of the absorption band at 446 nm to decrease and that of a new band at 350 nm (corresponding to the cis, protonated chromophore) to increase. When blue light irradiation ceases, a thermal process restores the trans, dark-adapted state.27 For both forms of c-E-helix-PYP, the thermal back reaction has a half-life on the order of a few seconds (Figure S9 of the Supporting Information). The light state of PYP exhibits greatly altered protein conformational dynamics relative to those of the dark state.11 Previous work suggests that irradiation produces a partially unfolded state that can be mimicked by addition of denaturant.28,29 By varying the intensity of blue light to produce different steady-state fractions of cis isomerized protein, we found that the rate of dimer–monomer exchange was dependent on the square of the total percent cis protein (Figure 3b–d). This finding is consistent with monomerization occurring from dimers containing two cis chromophores. With sufficient light intensity to produce a 70% total cis content, the dimer could be fully converted to the monomer in <1 min, in the absence of GdnHCl (Figure 3c). Thus, the barrier to strand swapping, which presumably requires the breakage of numerous H-bonds between the N-terminal strand and the rest of the β-sheet, is greatly reduced when both monomers in the domain-swapped dimer are in their light states.
Blue light greatly accelerates the rate of domain swapping, and native PAGE analysis of blue light-irradiated c-E-helix-PYP, at 30 °C (in the absence of GdnHCl), showed the protein had a greatly decreased tendency to form domain-swapped dimers [Kd(light) ∼ 300 μM (Figure 3 and Figure S8 of the Supporting Information)]. For example, at a total protein concentration of 20 μM, equilibration in the dark (with 1 M GdnHCl) leads to more than 50% of the protein being in the dimer fraction, while equilibration under blue light leads to >98% monomer (Figure 3).
Blue Light Induces a Functional Change in c-E-Helix-PYP
As noted above, the E-helix insert is a capable of forming a heterodimeric coiled coil with a complementary K-helix peptide.17 In c-E-helix-PYP, the E-helix apparently functions as the hinge loop sequence and is expected to undergo a change in conformation upon oligomerization.3 CD spectra suggest that the E-helix becomes more helical in the dimeric form (Figure 1d). We investigated the ability of monomeric and dimeric forms of c-E-helix-PYP to interact with the K-helix peptide. Addition of 1 equiv of the K-helix peptide to a solution of dimeric c-E-helix-PYP leads to an ∼15-fold slowing of thermal recovery, but no effect is seen with the monomer (Figure S9 of the Supporting Information), suggesting that the K-peptide interacts with only the domain-swapped dimeric form of the protein. A K-helix peptide containing two Leu-to-Ala mutations (Ac-WGIAAAKEKIAALKEKIAAAK-NH2) that shows a much weaker affinity for the E-helix sequence17,30 does not have this effect, indicating the interaction is occurring via formation of a coiled coil (Figure S9 of the Supporting Information). To visualize K-helix binding directly, we synthesized a fluorescein-labeled K-helix peptide [K-helix-F; Ac-CAAA(IAALKEK)2IAALKGW-NH2]. Figure 4b shows c-E-helix-PYP–K-helix-F complexes analyzed by native PAGE. Strong fluorescence is associated with dimeric c-E-helix-PYP when K-helix-F is added, but not with the monomeric state (Figure 4b), even at >100-fold greater protein concentrations.
Figure 4.

Native PAGE analysis of c-E-helix-PYP–K-helix-F complexes. Coomassie staining (left) showing the migration pattern of the dimer and monomer. Fluorescence image (right) (488 nm excitation, 530 nm emission) showing that fluorescein-labeled K-peptide interacts with only the dimeric form of the protein. Protein concentrations were (i) 18.7, (ii) 7.5, (iii) 0.75, and (iv) 0.4 μM.
Discussion
In principle, photoswitchable effectors can be designed by fusing a photoswitchable protein to a target protein of interest.1 The photoswitchable protein undergoes a change in conformation or dynamics upon illumination, which can in turn affect the conformational dynamics, and thereby activity, of the protein of interest. This strategy for engineering photocontrol has been successful using several natural photoreceptors, including LOV domains,31−35 phytochromes,36 cryptochromes,37 and photoactive yellow protein (PYP).38−40 To produce a large change in activity, the photoswitchable protein must fully inhibit the target protein in one state and release it in the other (usually the light state). Most photoswitches are based on constraints that make one state closed or incompatible with binding and one state binding-competent.1,33 For example, the J-α-helix of the Avena sativa LOV domain detaches from the core of the LOV protein upon illumination, and this conformational change has been used to control interaction with partner proteins. A problem with this strategy is that the binding-competent dark state may be accessed thermally.41 Indeed, Yao et al. have used NMR relaxation dispersion measurements to measure the degree to which the detached J-α-helix state is accessed thermally and thereby to estimate the available free energy for light-driven allosteric regulation by A. sativa LOV.42
Light-driven domain swapping provides a solution to the problem of dark-state thermal activation. In this scenario, two protein conformational states that cannot interconvert thermally at room temperature can be induced to interconvert by blue light. When one state can interact with a partner protein and the other cannot, essentially on and off switching can result. The behavior of the system described here is shown schematically in Figure 5.
Figure 5.
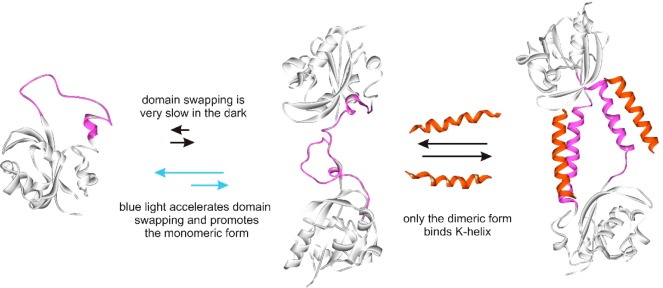
Schematic diagram showing domain swapping and K-helix binding of c-E-helix-PYP.
Coupling domain swapping to photoisomerization in c-E-helix-PYP is based on observations made by Loh and colleagues, who found that insertion of a structured domain into a surface loop of a control protein can lead to domain swapping of the control protein when the locations of loop ends are incompatible with the folded state of the control protein.6,13 In a circular permutant of PYP, the locations of the loop ends are light-dependent.15 The light state of PYP has characteristics of a molten globule,28 so that a folded target can be accommodated in a surface loop relatively easily. Thus, the light state of c-E-helix-PYP has a tendency to domain swap that is weakened compared to that of the dark state. While the light state may be able to bind a K-helix, as we originally intended, it is short-lived (∼1 s); thus, optical control of K-helix binding is seen primarily through the light-promoted conversion of the domain-swapped dimer to monomer. Slowing the photocycle with a targeted mutation may lead to photocontrol of K-helix binding by a monomeric form of the protein.
Because the mechanism for photocontrol of function occurring here depends on the compatibility of the insert sequence with the constraints of the cPYP surface loop, but not on the sequence of the insert directly, a wide variety of protein–protein interactions could in principle be photocontrolled in this manner.
In addition to altering the relative stability of monomeric and domain-swapped dimeric forms, light has a dramatic effect on the kinetics of domain swapping, decreasing the half-life from weeks to minutes. Depending on the intended application for a specific photoswitchable effector, more rapid domain swapping in the dark and/or an increased dark-state dimerization affinity may be desired. Both these characteristics have been found to be sensitive to point mutations in proteins that undergo domain swapping and so could likely be introduced in this system.5
Interestingly, recent structural studies of red and far-red light signaling by bacteriophytochromes suggest natural systems may have evolved an analogous mechanism for tight optical control of transcriptional processes. Bellini and Papiz43 have reported that a bacteriophytochrome from Rhodopseudomonas palustris (RpBphP1), when irradiated with 760 nm light, undergoes protomer swapping with a transcriptional regulatory domain RpPpsR2 to form a heterodimeric complex. The swapping involves numerous intermolecular contacts and does not appear to occur in the dark, suggesting light switching involves lowering of the activation barrier for swapping. Light-induced domain swapping may thus provide a general mechanism for optical switching of protein function where a very low background signal is required.
Supporting Information Available
Native PAGE analysis of equilibria, 15NH HSQC spectra, NMR chemical shifts, and denaturation data and kinetic data of PYP switching rates in the presence and absence of K-helix peptide. This material is available free of charge via the Internet at http://pubs.acs.org.
This work has been supported by the Natural Sciences and Engineering Research Council of Canada and the National Institutes of Health (Grant R01MH086379). NMR instrumentation at the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers was supported by CFI (19119). J.M.R. was supported by an Ontario Graduate Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Moglich A.; Moffat K. (2011) Engineered photoreceptors as novel optogenetic tools. Photochem. Photobiol. Sci. 9, 1286–1300. [DOI] [PubMed] [Google Scholar]
- Hegemann P.; Moglich A. (2011) Channelrhodopsin engineering and exploration of new optogenetic tools. Nat. Methods 8, 39–42. [DOI] [PubMed] [Google Scholar]
- Gronenborn A. M. (2009) Protein acrobatics in pairs: Dimerization via domain swapping. Curr. Opin. Struct. Biol. 19, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Eisenberg D. (2002) 3D domain swapping: As domains continue to swap. Protein Sci. 11, 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F.; Schymkowitz J.; Itzhaki L. S. (2012) Implications of 3D domain swapping for protein folding, misfolding and function. Adv. Exp. Med. Biol. 747, 137–152. [DOI] [PubMed] [Google Scholar]
- Ha J. H.; Karchin J. M.; Walker-Kopp N.; Huang L. S.; Berry E. A.; Loh S. N. (2012) Engineering domain-swapped binding interfaces by mutually exclusive folding. J. Mol. Biol. 416, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranko A. S.; Oeemig J. S.; Kajander T.; Iwai H. (2013) Intermolecular domain swapping induces intein-mediated protein alternative splicing. Nat. Chem. Biol. 9, 616–622. [DOI] [PubMed] [Google Scholar]
- Junedi S.; Yasuhara K.; Nagao S.; Kikuchi J.; Hirota S. (2014) Morphological change of cell membrane by interaction with domain-swapped cytochrome C oligomers. ChemBioChem 15, 517–521. [DOI] [PubMed] [Google Scholar]
- Li Y.; Altorelli N. L.; Bahna F.; Honig B.; Shapiro L.; Palmer A. G. III (2013) Mechanism of E-cadherin dimerization probed by NMR relaxation dispersion. Proc. Natl. Acad. Sci. U.S.A. 110, 16462–16467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel P. C. (2012) Domain swapping and amyloid fibril conformation. Prion 6, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P. L.; Lovett J. E.; Carl P. J.; Cammarata M.; Lee J. H.; Jung Y. O.; Ihee H.; Timmel C. R.; van Thor J. J. (2011) The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J. Am. Chem. Soc. 133, 9395–9404. [DOI] [PubMed] [Google Scholar]
- Kumauchi M.; Hara M. T.; Stalcup P.; Xie A.; Hoff W. D. (2008) Identification of six new photoactive yellow proteins: Diversity and structure-function relationships in a bacterial blue light photoreceptor. Photochem. Photobiol. 84, 956–969. [DOI] [PubMed] [Google Scholar]
- Cutler T. A.; Mills B. M.; Lubin D. J.; Chong L. T.; Loh S. N. (2009) Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 386, 854–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C.; Houben K.; Derix N. M.; Marks D.; van der Horst M. A.; Hellingwerf K. J.; Boelens R.; Kaptein R.; van Nuland N. A. (2005) The solution structure of a transient photoreceptor intermediate: Delta25 photoactive yellow protein. Structure 13, 953–962. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Burns D. C.; Al-Abdul-Wahid M. S.; Woolley G. A. (2013) A circularly permuted photoactive yellow protein as a scaffold for photoswitch design. Biochemistry 52, 3320–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litowski J. R.; Hodges R. S. (2001) Designing heterodimeric two-stranded α-helical coiled-coils: The effect of chain length on protein folding, stability and specificity. J. Pept. Res. 58, 477–492. [DOI] [PubMed] [Google Scholar]
- Litowski J. R.; Hodges R. S. (2002) Designing heterodimeric two-stranded α-helical coiled-coils. Effects of hydrophobicity and α-helical propensity on protein folding, stability, and specificity. J. Biol. Chem. 277, 37272–37279. [DOI] [PubMed] [Google Scholar]
- Grigoryan G.; Reinke A. W.; Keating A. E. (2009) Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 458, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason J. M.; Schmitz M. A.; Muller K. M.; Arndt K. M. (2006) Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc. Natl. Acad. Sci. U.S.A. 103, 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanathan S.; Genick U. K.; Getzoff E. D.; Meyer T. E.; Cusanovich M. A.; Tollin G. (1997) Preparation and properties of a 3,4-dihydroxycinnamic acid chromophore variant of the photoactive yellow protein. Arch. Biochem. Biophys. 340, 83–89. [DOI] [PubMed] [Google Scholar]
- Changenet-Barret P.; Espagne A.; Katsonis N.; Charier S.; Baudin J. B.; Jullien L.; Plaza P.; Martin M. M. (2002) Excited-state relaxation dynamics of a PYP chromophore model in solution: Influence of the thioester group. Chem. Phys. Lett. 365, 285–291. [Google Scholar]
- Evanics F.; Bezsonova I.; Marsh J.; Kitevski J. L.; Forman-Kay J. D.; Prosser R. S. (2006) Tryptophan solvent exposure in folded and unfolded states of an SH3 domain by 19F and 1H NMR. Biochemistry 45, 14120–14128. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Su J. Y.; Hodges R. S.; Kay C. M. (1994) Effect of chain length on the formation and stability of synthetic α-helical coiled coils. Biochemistry 33, 15501–15510. [DOI] [PubMed] [Google Scholar]
- O’Neill J. W.; Kim D. E.; Johnsen K.; Baker D.; Zhang K. Y. (2001) Single-site mutations induce 3D domain swapping in the B1 domain of protein L from Peptostreptococcus magnus. Structure 9, 1017–1027. [DOI] [PubMed] [Google Scholar]
- Rousseau F.; Schymkowitz J. W.; Wilkinson H. R.; Itzhaki L. S. (2001) Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc. Natl. Acad. Sci. U.S.A. 98, 5596–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kort R.; Vonk H.; Xu X.; Hoff W. D.; Crielaard W.; Hellingwerf K. J. (1996) Evidence for trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 382, 73–78. [DOI] [PubMed] [Google Scholar]
- Lee B. C.; Croonquist P. A.; Sosnick T. R.; Hoff W. D. (2001) PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J. Biol. Chem. 276, 20821–20823. [DOI] [PubMed] [Google Scholar]
- Lee B. C.; Pandit A.; Croonquist P. A.; Hoff W. D. (2001) Folding and signaling share the same pathway in a photoreceptor. Proc. Natl. Acad. Sci. U.S.A. 98, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N. E.; Kay C. M.; Hodges R. S. (1992) Synthetic model proteins: The relative contribution of leucine residues at the nonequivalent positions of the 3–4 hydrophobic repeat to the stability of the two-stranded α-helical coiled-coil. Biochemistry 31, 5739–5746. [DOI] [PubMed] [Google Scholar]
- Strickland D.; Moffat K.; Sosnick T. R. (2008) Light-activated DNA binding in a designed allosteric protein. Proc. Natl. Acad. Sci. U.S.A. 105, 10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland D.; Lin Y.; Wagner E.; Hope C. M.; Zayner J.; Antoniou C.; Sosnick T. R.; Weiss E. L.; Glotzer M. (2012) TULIPs: Tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 9, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lungu O. I.; Hallett R. A.; Choi E. J.; Aiken M. J.; Hahn K. M.; Kuhlman B. (2012) Designing photoswitchable peptides using the AsLOV2 domain. Chem. Biol. 19, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta-Mena L. B.; Reade A.; Mallory M. J.; Glantz S.; Weiner O. D.; Lynch K. W.; Gardner K. H. (2014) An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol. 10, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. I.; Frey D.; Lungu O. I.; Jaehrig A.; Schlichting I.; Kuhlman B.; Hahn K. M. (2009) A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Sato S.; Huq E.; Tepperman J. M.; Quail P. H. (2002) A light-switchable gene promoter system. Nat. Biotechnol. 20, 1041–1044. [DOI] [PubMed] [Google Scholar]
- Kennedy M. J.; Hughes R. M.; Peteya L. A.; Schwartz J. W.; Ehlers M. D.; Tucker C. L. (2010) Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan S. A.; Al-Abdul-Wahid S.; Woolley G. A. (2010) Structure-based design of a photocontrolled DNA binding protein. J. Mol. Biol. 399, 94–112. [DOI] [PubMed] [Google Scholar]
- Morgan S. A.; Woolley G. A. (2010) A photoswitchable DNA-binding protein based on a truncated GCN4-photoactive yellow protein chimera. Photochem. Photobiol. Sci. 9, 1320–1326. [DOI] [PubMed] [Google Scholar]
- Ui M.; Tanaka Y.; Araki Y.; Wada T.; Takei T.; Tsumoto K.; Endo S.; Kinbara K. (2012) Application of photoactive yellow protein as a photoresponsive module for controlling hemolytic activity of staphylococcal α-hemolysin. Chem. Commun. 48, 4737–4739. [DOI] [PubMed] [Google Scholar]
- Strickland D.; Yao X.; Gawlak G.; Rosen M. K.; Gardner K. H.; Sosnick T. R. (2010) Rationally improving LOV domain-based photoswitches. Nat. Methods 7, 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Rosen M. K.; Gardner K. H. (2008) Estimation of the available free energy in a LOV2-J α photoswitch. Nat. Chem. Biol. 4, 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini D.; Papiz M. Z. (2012) Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Structure 20, 1436–1446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.