

ABSTRACT
The acyl coenzyme A (acyl-CoA) dehydrogenases (ACADs) FadE34 and CasC, encoded by the cholesterol and cholate gene clusters of Mycobacterium tuberculosis and Rhodococcus jostii RHA1, respectively, were successfully purified. Both enzymes differ from previously characterized ACADs in that they contain two fused acyl-CoA dehydrogenase domains in a single polypeptide. Site-specific mutagenesis showed that only the C-terminal ACAD domain contains the catalytic glutamate base required for enzyme activity, while the N-terminal ACAD domain contains an arginine required for ionic interactions with the pyrophosphate of the flavin adenine dinucleotide (FAD) cofactor. Therefore, the two ACAD domains must associate to form a single active site. FadE34 and CasC were not active toward the 3-carbon side chain steroid metabolite 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA (4BNC-CoA) but were active toward steroid CoA esters containing 5-carbon side chains. CasC has similar specificity constants for cholyl-CoA, deoxycholyl-CoA, and 3β-hydroxy-5-cholen-24-oyl-CoA, while FadE34 has a preference for the last compound, which has a ring structure similar to that of cholesterol metabolites. Knockout of the casC gene in R. jostii RHA1 resulted in a reduced growth on cholate as a sole carbon source and accumulation of a 5-carbon side chain cholate metabolite. FadE34 and CasC represent unique members of ACADs with primary structures and substrate specificities that are distinct from those of previously characterized ACADs.
IMPORTANCE We report here the identification and characterization of acyl-CoA dehydrogenases (ACADs) involved in the metabolism of 5-carbon side chains of cholesterol and cholate. The two homologous enzymes FadE34 and CasC, from M. tuberculosis and Rhodococcus jostii RHA1, respectively, contain two ACAD domains per polypeptide, and we show that these two domains interact to form a single active site. FadE34 and CasC are therefore representatives of a new class of ACADs with unique primary and quaternary structures. The bacterial steroid degradation pathway is important for the removal of steroid waste in the environment and for survival of the pathogen M. tuberculosis within host macrophages. FadE34 is a potential target for development of new antibiotics against tuberculosis.
INTRODUCTION
Cholesterol is a sterol that modulates membrane fluidity in eukaryotic cells and functions as a hormone precursor. In the liver, cholesterol is converted to bile acids (such as cholate and chenodeoxycholate) by epimerization of the 3β-hydroxyl group, saturation of the double bond between C-5 and C-6, introduction of hydroxyl groups at the C-7 and C-12 positions, and the shortening of the D-ring side chain from 8 carbon to 5 carbon atoms (Fig. 1) (1). Bile acids are subsequently excreted in the intestine to aid in the uptake and digestion of lipophilic nutrients. Certain actinobacteria and proteobacteria have the unique ability to grow on cholesterol and cholesterol metabolites, such as bile acids, as sole carbon and energy sources (2–4). Besides its importance in the removal of steroid waste in the environment, the bacterial steroid degradation pathway has received particular attention recently due to the fact that cholesterol is a carbon source for Mycobacterium tuberculosis within host macrophages and disruption of cholesterol degradation genes has been found to attenuate the virulence of the bacteria (5–8).
FIG 1.

Chemical structures of cholesterol (A) and cholic acid (B) and steroid compounds investigated as potential substrates for ACADs: cholyl-CoA (C), deoxycholyl-CoA (D), 3β-hydroxy-5-cholen-24-oyl-CoA (E), and 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA (4BNC-CoA) (F).
In general, the aliphatic side chain substituent on the D ring of steroids is degraded by reactions analogous to fatty acid β-oxidation reactions, involving CoA esterification followed by stepwise removal of 2 or 3 carbon units as acetyl-CoA and propionyl-CoA (9). Due to differences in side chain lengths, cholesterol and cholate side chains will undergo 3 and 2 rounds of β-oxidation reactions, respectively. While some bacteria, such as M. tuberculosis, can grow only on cholesterol and not bile acids, the soil actinomycete Rhodococcus jostii RHA1 is able to grow on both cholesterol and cholate (2, 10). R. jostii RHA1 also has separate paralogous genes for degradation of the D-ring aliphatic side chains of these two types of steroids (10).
Acyl coenzyme A (acyl-CoA) dehydrogenases (ACADs) are key enzymes involved in β-oxidation reactions. They are classified based on their substrate specificities (11, 12). Those involved in fatty acid β-oxidation are the short-, medium-, long-, and very-long-chain acyl-CoA dehydrogenases (SCAD, MCAD, LCAD, and VLCAD, respectively). The other members are involved in amino acid oxidation pathways and are named isovaleryl-CoA dehydrogenase (i3VD, also known as IVD, and i2VD, also known as short branched-chain acyl-CoA dehydrogenase), isobutyryl-CoA dehydrogenase (IBH), and glutaryl-CoA dehydrogenase. The sequence identities among these members range from 35 to 45%. Structurally, the ACAD members display similar folds, suggesting a common evolutionary origin. With the exception of VLCAD, which is a homodimer and membrane bound, all of the above-mentioned ACADs are soluble homotetramers. Each subunit contains a glutamate base that abstracts a proton from the Cα of the CoA ester substrate, with the transfer of a hydride from Cβ to the isoalloxazine of flavin adenine dinucleotide (FAD) cofactor, leading to the formation of a double bond between Cα and Cβ. In each active site, the isoalloxazine ring of the FAD is bound by one subunit, but the adenosine of the FAD extends toward the opposite subunit. Classical ACADs contains two symmetrical active sites with two FAD molecules bound per dimer.
Recently, an ACAD from M. tuberculosis involved in dehydrogenation of cholesterol metabolites with a 3-carbon side chain on the D ring was discovered that is encoded by two genes, fadE28 and fadE29, in tandem within an operon. The FadE28-FadE29 complex was found to adopt a unique α2β2 quaternary structure (13). Although FadE28 and FadE29 (also known as ChsE1 and ChsE2) are homologous to classical ACADs, only FadE29 contains a catalytic glutamate, indicating that there is only 1 active site per FadE28-FadE29 dimer. Subsequently, other ACADs encoded within the M. tuberculosis cholesterol degradation gene cluster (FadE23, -24, -26, -27, -31, -32, and -33) were found to be heteromeric ACADs, similar to FadE28-FadE29 (14). FadE26-FadE27 was shown to utilize the 5-carbon side chain substrate 3β-hydroxy-5-cholen-24-oyl-CoA, although specificity constants or specific activities of this enzyme for this substrate and other potential substrates have not been reported (14). The substrate specificities of the other M. tuberculosis ACADs and their roles in cholesterol degradation have not been determined. Heteromeric ACADs have also been shown recently to be involved in dehydrogenation of a 3-carbon side chain derived from the AB ring catabolism of cholate in Comamonas testosteroni TA441 (15).
Another unusual ACAD that is essential for growth on cholesterol in M. tuberculosis is FadE34 (16), which has approximately twice the number of amino acid residues as other previously characterized ACADs. Homologues of FadE34 are found in the cholesterol and cholate degradation clusters of R. jostii RHA1 (Ro04483 and Ro05816 [CasC], respectively) (10). Here we show from sequence and biochemical analyses that FadE34 and CasC contain two ACAD domains per polypeptide, of which only the C-terminal ACAD domains contain the catalytic glutamates. Kinetic analysis reveals that CasC and FadE34 have distinct substrate specificities from FadE28-FadE29, displaying activities toward steroid substrates containing 5-carbon but not 3-carbon side chains.
MATERIALS AND METHODS
Chemicals.
Cholic acid, deoxycholic acid, hexanoyl-CoA, propionyl-CoA, FAD, and ferrocenium hexafluorophosphate were from Sigma-Aldrich (Oakville, ON, Canada). 4-Pregnen-3-one-20β-carboxylic acid was from Steraloids Inc. (Newport, RI). 3β-Hydroxy-5-cholen-24-oic acid was from TCI America (Portland, OR). CoA was from BioShop Canada Inc. (Burlington, ON, Canada). Restriction enzymes and Pfu polymerase were from Thermo Scientific (Ottawa, ON, Canada). T4 DNA ligase was from New England BioLabs (Pickering, ON, Canada). Nickel-nitrilotriacetic acid (Ni2+-NTA) Superflow resin was from Qiagen (Mississauga, ON, Canada). All other chemicals were obtained from Fisher Scientific (Nepean, ON, Canada) or Sigma-Aldrich unless otherwise stated.
Bacterial strains and plasmids.
R. jostii RHA1 was obtained from Lindsay Eltis (Department of Microbiology and Immunology, University of British Columbia, BC, Canada). pCR-Blunt II-TOPO vectors and Escherichia coli DH5α were purchased from Invitrogen (Oakville, ON, Canada). M. tuberculosis H37Rv genomic DNA was a gift from Marcel Behr (McGill University, Montreal, QC, Canada).
DNA manipulation.
DNA was purified, digested, and ligated using standard protocols. The genes casC (ro05816), casI (ro05822), casG (ro05820), fadE28, and fadE29 were PCR amplified from genomic DNA with the primers listed in Table S1 in the supplemental material. Due to difficulties in direct PCR amplification of fadE34 (rv3573c), two sets of nested primers were used in the PCRs. The fadE34, casG, and fadE28 genes were inserted into plasmid pET28a (Novagen), casC and casI were inserted into plasmid pTIPQC1-His (17, 18), and fadE29 was inserted into pBTLT7 (19) using primer-introduced NdeI and HindIII restriction sites. Site-specific mutagenesis was performed according to the modified QuikChange method (20) using primers listed in Table S2 in the supplemental material. To create an in-frame deletion of casC, a second SacI restriction site was introduced by site-specific mutagenesis replacing A to G at nucleotide position 1765. Digestion of the resultant plasmid with SacI followed by religation led to an in-frame deletion of a 1,465-bp fragment in the middle of the casC gene. This gene was inserted into plasmid pK18mobsacB and transformed into E. coli S17-λpir for biparental mating with R. jostii RHA1 using a previously described method (21). R. jostii RHA1 ΔcasC knockout mutant was confirmed by PCR of genomic DNA. Cloned genes and gene mutations were confirmed by DNA sequencing at Laboratory Services (University of Guelph).
Sequence alignment.
Multiple-sequence alignments were generated with ClustalX using default parameters (22) and visualized using ESPript 3 (23).
Protein expression and purification.
Recombinant E. coli BL21(λDE3) containing casG, fadE34, or fadE28-fadE29 was grown at 37°C in 4 liters of LB medium supplemented with kanamycin or kanamycin and tetracycline. At mid-log phase (optical density at 600 nm [OD600] of 0.4 to 0.6), expression of the recombinant proteins was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were grown for a further 24 h at 37°C for the bacteria containing fadE34 and fadE28-fadE29, while casG was expressed at 15°C. R. jostii RHA1 cells containing casI or casC in pTIPQC1-His were grown at 30°C in 4 liters of LB medium supplemented with chloramphenicol. At mid-log phase (OD600 = 0.4 to 0.6), expression of recombinant proteins was induced with 1 μg/ml of thiostrepton. Cells were grown for a further 24 h at 30°C. All cells were harvested by centrifugation at 4,503 × g for 8 min.
The harvested cell pellets were resuspended in 20 mM HEPES (pH 8.0). E. coli cells were lysed by passing through a French press 3 times at 10,000 lb/in2, and R. jostii cells were lysed by passing them through a French press 5 times at a pressure of 13,000 lb/in2. E. coli lysates were centrifuged at 39,191 × g for 30 min, while R. jostii lysates were centrifuged 3 times at 39,191 × g for 10 min, with the removal of cellular debris at each step. The supernatant was passed through a 0.45-μm filter and incubated for 1 h at 4°C with Ni2+-NTA resin and wash buffer containing 50 mM sodium phosphate buffer, 300 mM sodium chloride, and 20 mM imidazole (pH 8.0). The mixture was poured into a gravity column and washed with wash buffer. The His-tagged proteins were eluted with elution buffer containing 50 mM sodium phosphate buffer, 300 mM sodium chloride, and 150 mM imidazole (pH 8.0). A 1 mM concentration of FAD was added to the eluted ACADs, and the buffer was exchanged into 20 mM HEPES (pH 8.0) by dilution in a stirred cell equipped with a YM10 filter (Amicon). Purified enzymes were stored at −80°C.
For preparation of deflavinated FadE28-FadE29 complex, the protocol described above for purification of His-tagged proteins was followed, with the following changes. The crude extract containing the ACAD was incubated overnight at 4°C with Ni2+-NTA resin in deflavination wash buffer containing 50 mM sodium phosphate, 300 mM sodium chloride, 20 mM imidazole, and 1 M potassium chloride (pH 8.0). FAD was not added to fractions containing the ACAD after elution.
Determination of protein concentrations, purities, and molecular masses.
Protein concentrations were determined by the Bradford assay using bovine serum albumin as the standard (24). Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to assess the purity of the enzymes. The native molecular weights of purified FadE34 and CasC were estimated using gel filtration on a calibrated HiLoad 26/60 Superdex 200 column (GE Healthcare). The proteins used for calibration were horse heart cytochrome c (12.4 kDa), bovine erythrocyte carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), yeast alcohol dehydrogenase (150 kDa), and sweet potato β-amylase (200 kDa). The void volume was determined using blue dextran (2,000 kDa). Proteins were eluted at 2 ml/min in 20 mM HEPES (pH 7.5) containing 150 mM sodium chloride at 25°C. The concentrations of FAD bound to the purified acyl-CoA dehydrogenases were determined spectrophotometrically, as described previously (25), and reported as an average of duplicates.
Substrate synthesis.
3-Oxo-23,24-bisnorchol-4-en-22-oyl-CoA (4BNC-CoA) was synthesized using acyl-CoA synthetase CasI, as described previously (26). CoA esters of cholic acid, deoxycholic acid, and 3β-hydroxy-5-cholen-24-oic acid were synthesized using CasG (10, 27). Briefly, reaction mixtures were incubated overnight at 22°C and contained 1.0 mM cholic acid or deoxycholic acid, 1.0 mM CoA, 5.0 mM magnesium chloride, 2.5 mM ATP, and 1.5 μM CasG dissolved in a total of 10 ml of 100 mM HEPES (pH 7.5). 3β-Hydroxy-5-cholen-24-oyl-CoA was synthesized similarly using CasG; however, due to limited solubility, the final reaction mixture contained 0.2 mM 3β-hydroxy-5-cholen-24-oic acid. The reactions were stopped via acidification to pH 4, and reaction products were centrifuged at 17,000 × g for 5 min. Products were purified via high-performance liquid chromatography (HPLC) using an ÄKTA Explorer 100 (Amersham Pharmacia Biotech, Baie d'Urfé, QC, Canada) equipped with a Discovery C18 column (15 cm by 4.6 mm column; 5-μm particles) (Sigma-Aldrich) and a gradient of 0 to 80% acetonitrile. 4BNC-CoA was eluted at 32% acetonitrile, deoxycholyl-CoA was eluted at 42% acetonitrile, cholyl-CoA was eluted at 43% acetonitrile, and 3β-hydroxy-5-cholen-24-oyl-CoA was eluted at 45% acetonitrile. The eluates were collected, evaporated, and lyophilized. HPLC-purified steroid CoA esters were subjected to liquid chromatography-mass spectrometry (LC-MS) to further confirm the identities of these compounds (4BNC-CoA, m/z 1,094.3456 ([M+H]+); deoxycholyl-CoA, m/z 1,142.4038 ([M+H]+), cholyl-CoA, m/z 1,158.398 ([M+H]+); and 3β-hydroxy-5-cholen-24-oyl-CoA, m/z 1,124.3921 ([M+H]+).
The concentration of 4BNC-CoA was determined using the extinction coefficient 17,200 M−1 cm−1 at 248 nm (26). The concentrations for the CoA esters of cholic acid, deoxycholic acid, and 3β-hydroxy-5-cholen-24-oic acid were determined by an endpoint acyl-CoA dehydrogenase assay assuming a 1:2 ratio of substrate to reduced ferrocenium hexafluorophosphate (ε300 = 4,300 M−1 cm−1) (28).
Steady-state kinetic assays.
All assays were performed at least in duplicate in a total volume of 1 ml of TAPS [N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid] buffer (pH 8.5) at 25°C, using a Varian Cary 3 spectrophotometer equipped with a temperature-controlled cuvette holder. Dehydrogenase activity was measured using ferrocenium hexafluorophosphate as the artificial electron acceptor (ε300 = 4,300 M−1 cm−1) at a ratio of 2:1 for reduced ferrocenium molecules to substrate dehydrogenation (28). Assay mixtures contained 100 mM TAPS buffer (pH 8.5), 250 μM ferrocenium hexafluorophosphate, and variable substrate concentrations as described previously (13). Data were fitted to the Michaelis-Menten equation by nonlinear regression using the software GraphPad Prism.
Characterization of a ΔcasC R. jostii RHA1 mutant.
The R. jostii RHA1 wild type, a ΔcasC knockout, and a knockout complemented with a casC gene in plasmid pTIPQC1-His were grown in 100 ml of M9 minimal medium supplemented with trace elements, vitamin B1, and 20 mM pyruvate for 4 days at 30°C. A 250-ml volume of each starter culture was inoculated into the same medium containing either 1 mM cholic acid or 20 mM pyruvate as the sole carbon source (29). For the complemented strain and the wild type containing pTIPQC1 plasmid, the media were also supplemented with chloramphenicol (34 μg/ml) and thiostrepton (2.5 μg/ml). Cell densities at various time points over 2 weeks were estimated by absorbance measurements at 600 nm. Aliquots of culture supernatant were removed at late log phase, and 5α-cholestane (0.25 mM) was added as an internal standard. Ethyl acetate extracts of acidified culture supernatant for gas chromatography-mass spectrometry (GC-MS) were then derivatized with bis(trimethylsilyl)trifluoroacetamide-trimethylchlorosilane as previously described (30) and analyzed on an Agilent 7890A gas chromatograph interfaced with an Agilent 59756 MS detector (DB-5MS column [30 m by 0.25 mm by 0.25 μm] containing a built-in 10-m Duraguard precolumn). Liquid chromatography-mass spectrometry analyses of underivatized samples were performed on an Agilent 1200 HPLC liquid chromatograph interfaced with an Agilent UHD 6530 Q-Tof mass spectrometer at the Mass Spectrometry Facility of the Advanced Analysis Centre, University of Guelph. A C18 column (Agilent Poroshell 120; 150 mm by 4.6 mm by 2.7 μm) was used for chromatographic separation with the following solvents: water with 0.1% formic acid (solvent A) and acetonitrile with 0.1 formic acid (solvent B). The mobile phase gradient was as follows: initial conditions were 2% solvent B for 1 min, increasing to 100% B in 19 min, followed by column wash at 100% solvent B for 3.5 min and 10 min of reequilibration. The first 2 and last 5 min of gradient were sent to waste and not the spectrometer. The flow rate was maintained at 0.4 ml/min. The mass spectrometer electrospray capillary voltage was maintained at 4.0 kV and the drying gas temperature at 250°C with a flow rate of 8 liters/min. Nebulizer pressure was 30 lb/in2, and the fragmentor was set to 160. Nitrogen was used both as nebulizing and drying gas and as collision-induced gas. The mass-to-charge ratio was scanned across the m/z range of 50 to 1,400 m/z in 4-GHz (extended dynamic range) positive-ion auto-tandem MS (MS/MS) mode. Two precursor ions per cycle were selected for fragmentation. The instrument was externally calibrated with ESI TuneMix (Agilent). The sample injection volume was 50 μl. Chromatograms were analyzed within Agilent Qualitative Analysis software B 06.0, finding compounds by the Molecular Feature algorithm and generating possible compound formulas with Molecular Formula Generator. Formulas including the elements C, H, O, and N and fragmentation data for each compound were generated.
RESULTS
Sequence analysis of FadE34 and CasC.
The amino acid sequences of FadE34 and CasC were analyzed using INTERPRO (31); the results showed that both proteins have two putative ACAD domains separated by a short linker. These ACAD domains span from amino acid residues 5 to 343 and 367 to 709 for FadE34 and residues 2 to 364 and 386 to 732 for CasC. These separate ACAD domains, designated the N-terminal ACAD domain and C-terminal ACAD domain, respectively, for each protein were aligned with FadE28, FadE29, FadE26, FadE27, and classical ACADs that utilize aliphatic substrates (see Fig. S1 in the supplemental material). Sequence similarities of the proteins in the alignment are indicated in Table S3 in the supplemental material. The catalytic glutamate in human IVD and rat LCAD are conserved in the C-terminal ACAD domains of CasC and FadE34 (see Fig. S1). In addition, an arginine that interacts with CoA of the substrate and several amino acid side chains that form hydrogen bond interactions with the isoalloxazine and adenosine ribose of FAD in classical ACADs are also conserved in the C-terminal ACAD domains of CasC and FadE34 (Table 1). There is also a conserved arginine residue in the N-terminal ACAD domains of CasC and FadE34. The corresponding arginine in the three-dimensional structure of IVD (PDB accession number 1IVH) interacts with the pyrophosphate moiety of the FAD molecule (32).
TABLE 1.
Amino acid residues involved in catalysis or forming of side chain interactions with FAD or CoA in classical ACADs and their corresponding amino acids in steroid-degrading ACADsa
| ACAD | Catalytic glutamate | Interaction with isoalloxazine of FAD | Interaction with CoA of substrate | Interaction with adenosine ribose of FAD | Interaction with pyrophosphate of FAD |
|---|---|---|---|---|---|
| MCAD_Pig | E376A | T136A, T168A | R256A | T378A, Q380A | S142A, R281B |
| SCAD_Melsdenii | E367A | T129A, T162A | R247A | T369A, E371A | T135A, R272B |
| IBH_Human | E376A | T139A, S171A | R255A | S378A, E380A | S145A, R280B |
| LCAD_Rat | E291A | T173A, T205A | R292A | T414A, E295A | S179A, R317B |
| IVD_Human | E254A | S136A, T168A | R255A | T377A, E379A | S142A, R280B |
| FadE26_Mtb | E247b G378c | S130, S162 | R248 | T380, E382 | T136, K272 |
| FadE29_Mtb | E241b G365c | T126, T158 | R242 | V367, E369 | T132, P265 |
| CasCCterm_Rjostii | E598b G330c | S482, S514 | R599 | T717, N334 | S488, P623 |
| FadE34Cterm_Mtb | E581b G326c | S466, S498 | R582 | T694, Q330 | S472, E606 |
| FadE27_Mtb | S225b G378c | D126, G153 | T226 | T358, Q360 | G132, R251 |
| FadE28_Mtb | A201b G323c | N107, G134 | L202 | P325, H327 | L113, R227 |
| CasCNterm_Rjostii | A213b L348c | Q121, -d | A214 | T351, S353 | L127, R239 |
| FadE34Nterm_Mtb | A210b L329c | D126, -d | A211 | S336, R338 | V129, R236 |
ACADs in the comparison are short-chain acyl-CoA dehydrogenase from Megasphaera elsdenii (SCAD_Melsdenii), pig medium-chain acyl-CoA dehydrogenase (MCAD_Pig), human isovaleryl-CoA dehydrogenase (IVD_Human), rat long-chain acyl-CoA dehydrogenase (LCAD_Rat), human isobutyryl-CoA dehydrogenase (IBH_Human), FadE26 (FadE26_Mtb), FadE27 (FadE27_Mtb), FadE28 (FadE28_Mtb), FadE29 (FadE29_Mtb), the N-terminal ACAD domain of FadE34 (FadE34Nterm_Mtb), the C-terminal ACAD domain of FadE34 (FadE34Cterm_Mtb) from M. tuberculosis H37Rv, and the N-terminal ACAD domain of CasC (CasCNterm_Rjostii) and the C-terminal ACAD domain of CasC (CasCCterm_Rjostii) from R. jostii RHA1. The functional active site of classical ACADs is a dimer with two symmetrical active sites. For simplicity, subscript A and subscript B indicate residue contribution from two different subunits in the functional dimer of classical ACADs. There are two possible positions for the catalytic glutamate in the primary sequences of classical ACADs. Corresponding residues in steroid-degrading ACADs that are identical or similar to classical ACADs are underlined. FadE26, FadE29, and the C-terminal ACAD domains in FadE34 and CasC contain the catalytic glutamate in a position similar to that in LCAD and IVH.
Residue aligned with catalytic glutamate of LCAD and IVH.
Residue that aligned with catalytic glutamate of SCAD, MCAD, and IBH.
Hyphen corresponds to a gap in the sequence alignment.
Expression and purification of recombinant proteins.
The FadE28-FadE29 complex was purified from recombinant E. coli with yields comparable to those previously reported (33) (see Fig. S2 in the supplemental material). fadE34 was PCR amplified from M. tuberculosis H37Rv genomic DNA and inserted into the vector pET28a for expression in E. coli BL21(DE3). The enzyme was purified by Ni2+-NTA chromatography with a yield of 4 mg per liter of culture (see Fig. S2). Attempts to express casC in E. coli led to formation of inclusion bodies. The gene can, however, be overexpressed in R. jostii RHA1 using the rhodococcal expression vector pTIPQC1-His. CasC was purified with a yield of 4 mg per liter of culture. By gel filtration, the estimated native molecular mass of FadE34 was 172.25 kDa and that of CasC was 175.02 kDa, consistent with the expected sizes of homodimers. Although each homodimer will have four ACAD domains, the stoichiometries of FAD determined from two separate purifications were 2.1 and 2.0 per FadE34 dimer and 2.4 and 2.3 per CasC dimer.
Specificities of FadE34, CasC, and FadE28-FadE29.
Substrate specificities of FadE34, FadE28-FadE29, and CasC were tested by steady-state kinetics. FadE34 and CasC were not active toward the 3-carbon side chain 4BNC-CoA but were active toward the steroid CoA esters containing 5-carbon side chains: cholyl-CoA, deoxycholyl-CoA, and 3β-hydroxy-5-cholen-24-oyl-CoA. The enzymes obey Michaelis-Menten kinetics, and a representative plot of initial velocity versus cholyl-CoA concentration for CasC is shown in Fig. 2. CasC has similar specificity constants toward the substrates cholyl-CoA, deoxycholyl-CoA, and 3β-hydroxy-5-cholen-24-oyl-CoA (Table 2). These three substrates differ only by the ring structure, with the 3β-hydroxy-5-cholen-24-oyl-CoA being a cholesterol metabolite. FadE34, on the other hand, has specificity constants at least 2 orders of magnitude higher for 3β-hydroxy-5-cholen-24-oyl-CoA than for cholyl-CoA and deoxycholyl-CoA. Interestingly, FadE34 and CasC displayed small but detectable activity toward hexanoyl-CoA (0.015 s−1 and 0.024 s−1, respectively) but not with propionyl-CoA. FadE28-FadE29, on the other hand, has no detectable activity toward 5-carbon side chain steroid metabolites but was active toward 4BNC-CoA, with a Km value of 6.3 ± 0.88 μM and a kcat value of 2.2 ± 0.19 s−1.
FIG 2.

Steady-state kinetic analysis of CasC with cholyl-CoA. The initial velocity is shown as a function of cholyl-CoA concentration. The line represents a best fit of the Michaelis-Menten equation to the data.
TABLE 2.
Steady-state kinetic parameters of acyl-CoA dehydrogenasesa
| Substrate | Enzyme | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| Cholyl-CoA | FadE34 | 47 ± 5.8 | 0.22 ± 0.0095 | (4.8 ± 0.20) × 103 |
| CasC | 5.7 ± 0.69 | 16 ± 0.61 | (2.8 ± 0.11) × 106 | |
| Deoxycholyl-CoA | FadE34 | 27 ± 3.5 | 0.44 ± 0.019 | (1.6 ± 0.069) × 104 |
| CasC | 2.2 ± 0.35 | 9.9 ± 0.60 | (4.4 ± 0.23) × 106 | |
| 3β-Hydroxy-5-cholen-24-oyl-CoA | FadE34 | 2.5 ± 0.79 | 3.7 ± 0.40 | (1.5 ± 0.16) × 106 |
| CasC | 3.0 ± 0.76 | 10 ± 1.0 | (3.5 ± 0.34) × 106 |
Acyl-CoA dehydrogenase assays were performed at 25°C and included 250 μM ferrocenium hexafluorophosphate in 100 mM TAPS buffer (pH 8.5).
Site-specific mutagenesis.
The putative catalytic glutamates in the C-terminal ACAD domains of CasC and FadE34 were replaced with glutamines by site-specific mutagenesis. Purified FadE34E581Q and CasCE598Q were yellow, indicating that the FAD cofactor remained bound in these variants. However, the variant enzymes displayed less than 1% activity with cholyl-CoA as the substrate compared to their corresponding wild-type enzymes. The FadE28-FadE29E241Q variant had less than 1% of the wild-type activity toward 4BNC-CoA, similar to previously reported results (13).
The putative arginine residues that interact with the pyrophosphate of FAD in FadE28 and the N-terminal ACAD domains of FadE34 and CasC were replaced with alanines using site-specific mutagenesis. All variant enzymes bound to the Ni-NTA column and were eluted with the same concentration of imidazole as the wild-type enzyme. Purified variant enzymes migrated similarly to the wild-type enzyme on SDS-PAGE (see Fig. S2 in the supplemental material). FadE34R236A, CasCR239A, and FadE28R227A-FadE29 complex were, however, colorless after purification, indicating the loss of bound FAD in these enzymes. FadE34R236A and CasCR239A had <2% of the wild-type activity using cholyl-CoA as the substrate, even after the addition of 1 mM exogenous FAD to the enzyme assay. In contrast, while purified FadE28R227A-FadE29 complex is inactive with 4BNC-CoA, addition of 1 mM exogenous FAD in the assay led to the recovery of 93% of the wild-type activity. With the addition of 1 mM exogenous FAD in the assay, the FadE28R227A-FadE29 variant showed a catalytic efficiency (kcat/Km) of (1.95 ± 0.065) × 105 M−1 s−1 for 4BNC-CoA, comparable to the value obtained for the wild-type enzyme (3.5 × 105 M−1s−1). Using a fixed concentration of 8.6 μM 4BNC-CoA, the apparent Km for FAD in the FadE28R227A-FadE29 variant was determined to be 1,000-fold higher than for the wild-type deflavinated enzyme (Km,app of 1.20 ± 0.16 mM, versus Km, app of 1.24 ± 0.16 μM).
Analysis of R. jostii RHA1 ΔcasC mutant.
The gene encoding CasC in the R. jostii RHA1 genome was knocked out by allelic replacement of the wild-type gene with casC containing an in-frame deletion of 1,465 bp. The ΔcasC mutant, when grown in minimal medium with pyruvate as the sole carbon source, had growth rates and final OD600 values that were similar to those of the wild type grown in the same medium. However, the mutant displayed a significant lag phase, of approximately 100 h, when grown in cholate as a sole carbon source. The ΔcasC mutant also had an increased doubling time (51 h, compared to 4.4 h in the wild type) and produced only 66% of the final biomass during stationary phase compared to the wild type (Fig. 3). The mutant containing an intact casC gene on a plasmid (pTIPQC1-His casC) had a doubling time of 16 h and reached an optical density similar to that of the wild type during stationary phase. The lag phase observed in the complemented mutant can be partially attributed to the presence of chloramphenicol and thiostrepton in the medium required for plasmid maintenance and the induction of casC expression on the plasmid. Wild-type R. jostii RHA1 transformed with pTIPQC1 and grown in the presence of chloramphenicol and thiostrepton also displayed a long lag phase (Fig. 3).
FIG 3.

Growth curves of wild-type and ΔcasC R. jostii RHA1 strains on cholate. Cells of wild-type R. jostii RHA1 (open squares), the wild type containing plasmid pTIPQC1 (triangles), the ΔcasC mutant (circles), and a ΔcasC strain containing the casC gene in plasmid pTIPQC1-His (solid squares) were grown in mineral medium supplemented with 1 mM cholate at 30°C. Growth curves represent the averages of two independent experiments with similar results.
The metabolites found in the culture supernatant of the wild type and ΔcasC complemented strain grown in cholate, as analyzed by GC-MS, had retention times and MS spectra that correspond to previously described trimethylsilyl (TMS) derivatives of cholate metabolites 3,7(R),12(S)-trihydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5 (10)-trien-22-oate (THSBNC), 1β(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aα-methylhexahydro-5-indanone (HHIDP), and 1-ylidene(2′-propanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aα-methylhexahydro-5-indanone (YHHIDP) (Table 3) (30). All these metabolites contain an isopropanoate side chain attached to the cyclopentane ring. The ΔcasC mutant culture supernatant contained a small amount of THSBNC but not HHIDP or YHHIDP. Instead, the mutant accumulated a unique major metabolite with the following GC-MS profile: a retention time (Rt) of 17.9 min and MS (70 eV, electron ionization) data of m/z 556.4 (0.1%), 541.4 (12.7%), 466 (8.0%), 451 (8.6%), 376 (23%), 361 (18%), 233 (33%), 217 (26%), 189 (13.8%), 147 (25%), and 73 (100%). The fragmentation pattern was similar to that of TMS derivatives of HHIDP except that the molecular ion [M]+ and fragments [M-15]+ (-CH3) and [M-90]+ (-TMSOH) are higher by 28 mass units than HHIDP. This difference corresponds to the mass of an ethylene (C2H2) and is consistent with a 5-carbon side chain cholate metabolite, 1β(2′-pentanoate)-3aα-H-4α(3″(R)-hydroxy-3″-propanoate)-7aα-methylhexahydro-5-indanone (2′-pentanoate HHIDP) (Fig. 4). Underivatized ethyl acetate extracts of culture supernatants were also subjected to LC-MS analyses. The major peak in the ΔcasC mutant sample corresponded to an M+H of 341.1956, and the Molecular Formula Generator program assigned an empirical formula of C18H28O6 with an error of 1 ppm. This is in agreement with the predicted formula for 2′-pentanoate HHIDP. This metabolite was not detected in the wild-type culture supernatant.
TABLE 3.
Comparison of metabolites from cholate-grown culture supernatants of wild-type, ΔcasC, and ΔcasC/pTIPQcHiscasC R. jostii strainsa
| Compound | GC retention time (min) | m/z of parental ion | Relative abundance (%) |
||
|---|---|---|---|---|---|
| Wild type | ΔcasC/pTIPQcHiscasC complemented strain | ΔcasC mutant | |||
| THSBNC | 20.3 | 678 | 37 | 111 | 11.6 |
| HHIDP | 15.6 | 528 | 3 | 7 | ND |
| YHHIDP | 15.9 | 526 | 3 | 6 | ND |
| 2′-Pentanoate HHIDP | 17.9 | 556 | ND | ND | 128 |
Metabolites were derivatized with trimethylsilane and analyzed by GC-MS as described in Materials and Methods. The amount of each metabolite is presented relative to the added internal 5α-cholestane standard (0.25 mM; retention time on GC = 17.8 min). ND, not detected.
FIG 4.
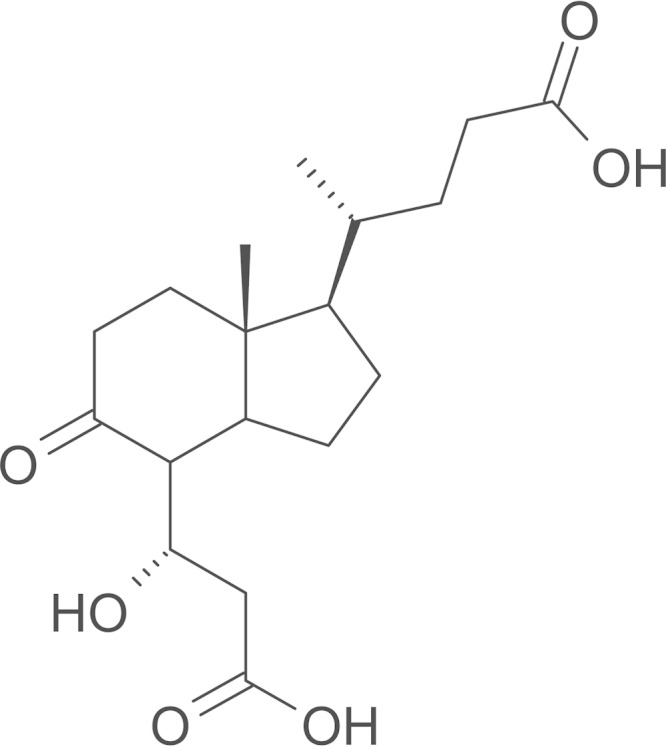
Structure of 2′-pentanoic acid HHIDP.
DISCUSSION
Previously, the acyl-CoA dehydrogenase FadE28-FadE29 in M. tuberculosis has been shown to be active toward cholesterol metabolites containing a 3-carbon isopropyl side chain at the D ring (13). We show here that FadE34 in M. tuberculosis and its homologue in the cholate degradation pathway of R. jostii RHA1, CasC, have the ability to catalyze the dehydrogenation of 5-carbon but not 3-carbon CoA ester substituents on the D ring, providing evidence that ACADs that metabolize steroid side chains have distinct chain length specificities.
FadE34 and CasC were found to have primary structures made up of a fusion of two ACAD domains. The N-terminal ACAD domain is similar to FadE28, while the C-terminal domain is similar to FadE29 and contains the catalytic glutamate. There is only one FAD molecule bound per polypeptide in FadE34 and CasC, and replacement of the catalytic glutamate in the C-terminal ACAD domain in both of these enzymes resulted in a significant reduction in activity. Comparable reduction in ACAD activity was observed when the catalytic glutamate of human LCAD was replaced with glutamine (34). Replacement of the arginine residue predicted to interact with the pyrophosphate of FAD in FadE28 and the N-terminal ACAD domains of FadE34 and CasC with alanines disrupted FAD binding. Therefore, the N- and C-terminal domains of FadE34 and CasC must interact to form a single active site. Together, these results suggest that despite FadE34 and CasC having four ACAD domains in the oligomeric state, these unique ACADs have half the number of FAD molecules and active sites of ACADs that utilize aliphatic substrates. It is reasonable to assume that ACADs involved in steroid side chain degradation evolved from a gene duplication event followed by the loss of a catalytic site in one of the domains or subunits, possibly to facilitate binding of the large steroid ring substrates. In CasC and FadE34, the genes are duplicated in frame, resulting in a polypeptide with two ACAD domains.
Kinetic assays were performed on CasC and FadE34 with several CoA ester substrates. The results indicate that these ACADs have high specificity constants for steroid CoA esters containing five-carbon side chains. CasC, however, has broader substrate specificity than FadE34, since it can utilize cholyl-CoA, deoxycholyl-CoA, and 3β-hydroxy-5-cholen-24-oyl-CoA with similar catalytic efficiencies. Sequence comparisons between CasC and FadE34 show that the N-terminal ACAD domains of the enzymes are less similar than the C-terminal ACAD domains (52% versus 69%). Although the N-terminal ACAD domain is not involved in catalysis, since this domain lacks the catalytic glutamate, whether it has a bearing on substrate specificity of these ACADs requires further investigation.
The R. jostii RHA1 ΔcasC mutant had decreased growth compared to that of the wild type. GC-MS and LC-MS analyses of culture supernatant metabolites revealed that the mutant accumulated a cholate metabolite that is consistent with an impaired ability to process the 5-carbon side chain. This result also shows that the A ring of cholate can be metabolized even if the side chain on the D ring remains intact. A small amount of the cholate metabolite with a 3-carbon side chain, THSBNC, was found in the growth medium of the ΔcasC mutant, which may be due to the presence of an orthologue of fadE34, Ro04483, within the cholesterol degradation cluster in R. jostii RHA1 that is constitutively expressed (10). This gene may have partially compensated for the loss of casC function in the mutant. On the other hand, although there are other ACADs encoded within the cholesterol gene clusters of M. tuberculosis, fadE34 was found to be essential for in vitro growth of M. tuberculosis on cholesterol based on transposon mutagenesis (16), implying that other genes in the genome cannot replace its function in M. tuberculosis.
In the cholate side chain degradation gene cluster of R. jostii RHA1 there are 2 additional genes encoding ACADs, casL and casN (10). CasL and CasN are homologues of FadE28 and FadE29, respectively, from M. tuberculosis (32% sequence identity between FadE28 and CasL; 47% sequence identity between FadE29 and CasN). Unlike fadE28 and fadE29 and other previously reported genes for heteromeric ACADs from M. tuberculosis, casL and casN are not adjacent in the genome but are instead separated by a gene encoding a putative hydratase (casM). Whether CasL and CasN form a heterocomplex and are involved in degradation of the 3-carbon side chain metabolite of cholate after the first round of β-oxidation remains to be determined.
Genes encoding proteins with two ACAD domains are present in other steroid-degrading Actinobacteria but are not prevalent in Proteobacteria (see Fig. S3 in the supplemental material). There is one gene encoding a protein with 2 ACAD domains in Comamonas testosteroni CNB-2, but other bacteria that can grow on cholate, such as Pseudomonas sp. strain CHOL1 (3), do not appear to have similar genes. Their genomes do, however, contain a number of homologues of fadE28 or fadE29. Future studies of ACADs from Proteobacteria should clarify their role in cholate catabolism and provide a basis for comparison with the Actinobacteria enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by National Science and Engineering Research Council of Canada grant 238284 (to S. Y. K. Seah).
We thank Dyanne Brewer from the University of Guelph Advanced Analysis Centre for assistance with GC-MS and LC-MS experiments.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02420-14.
REFERENCES
- 1.Russell DW. 2003. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 2.Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holert J, Alam I, Larsen M, Antunes A, Bajic VB, Stingl U, Philipp B. 2013. Genome sequence of Pseudomonas sp. strain Chol1, a model organism for the degradation of bile salts and other steroid compounds. Genome Announc 1(1):e00014-12. doi: 10.1128/genomeA.00014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horinouchi M, Hayashi T, Kudo T. 2012. Steroid degradation in Comamonas testosteroni. J Steroid Biochem Mol Biol 129:4–14. doi: 10.1016/j.jsbmb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Brzostek A, Pawelczyk J, Rumijowska-Galewicz A, Dziadek B, Dziadek J. 2009. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol 191:6584–6591. doi: 10.1128/JB.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langenhoff A, Inderfurth N, Veuskens T, Schraa G, Blokland M, Kujawa-Roeleveld K, Rijnaarts H. 2013. Microbial removal of the pharmaceutical compounds ibuprofen and diclofenac from wastewater. Biomed Res Int 2013:325806. doi: 10.1155/2013/325806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunau WH, Dommes V, Schulz H. 1995. Beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res 34:267–342. doi: 10.1016/0163-7827(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 10.Mohn WW, Wilbrink MH, Casabon I, Stewart GR, Liu J, van der Geize R, Eltis LD. 2012. A gene cluster encoding cholate catabolism in Rhodococcus spp. J Bacteriol 194:6712–6719. doi: 10.1128/JB.01169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghisla S, Thorpe C. 2004. Acyl-CoA dehydrogenases. A mechanistic overview. Eur J Biochem 271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim JJ, Miura R. 2004. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur J Biochem 271:483–493. doi: 10.1046/j.1432-1033.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- 13.Thomas ST, Sampson NS. 2013. Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain. Biochemistry 52:2895–2904. doi: 10.1021/bi4002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wipperman MF, Yang M, Thomas ST, Sampson NS. 2013. Shrinking the FadE proteome of Mycobacterium tuberculosis: insights into cholesterol metabolism through identification of an alpha2beta2 heterotetrameric acyl coenzyme A dehydrogenase family. J Bacteriol 195:4331–4341. doi: 10.1128/JB.00502-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horinouchi M, Hayashi T, Koshino H, Malon M, Hirota H, Kudo T. 2014. Identification of 9alpha-hydroxy-17-oxo-1,2,3,4,10,19-hexanorandrostan-5-oic acid in steroid degradation by Comamonas testosteroni TA441 and its conversion to the corresponding 6-en-5-oyl coenzyme A (CoA) involving open reading frame 28 (ORF28)- and ORF30-encoded acyl-CoA dehydrogenases. J Bacteriol 196:3598–3608. doi: 10.1128/JB.01878-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakashima N, Tamura T. 2004. Isolation and characterization of a rolling-circle-type plasmid from Rhodococcus erythropolis and application of the plasmid to multiple-recombinant-protein expression. Appl Environ Microbiol 70:5557–5568. doi: 10.1128/AEM.70.9.5557-5568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carere J, McKenna SE, Kimber MS, Seah SY. 2013. Characterization of an aldolase-dehydrogenase complex from the cholesterol degradation pathway of Mycobacterium tuberculosis. Biochemistry 52:3502–3511. doi: 10.1021/bi400351h. [DOI] [PubMed] [Google Scholar]
- 19.Baker P, Pan D, Carere J, Rossi A, Wang W, Seah SY. 2009. Characterization of an aldolase-dehydrogenase complex that exhibits substrate channeling in the polychlorinated biphenyls degradation pathway. Biochemistry 48:6551–6558. doi: 10.1021/bi9006644. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Naismith JH. 2008. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TD. 2011. Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 50:5096–5107. doi: 10.1021/bi101892z. [DOI] [PubMed] [Google Scholar]
- 22.Sievers F, Higgins DG. 2014. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Aliverti A, Curti B, Vanoni MA. 1999. Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol Biol 131:9–23. [DOI] [PubMed] [Google Scholar]
- 26.Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. 2011. Activity of 3-ketosteroid 9alpha-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem 286:40717–40724. doi: 10.1074/jbc.M111.289975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casabon I, Swain K, Crowe AM, Eltis LD, Mohn WW. 2014. Actinobacterial acyl coenzyme A synthetases involved in steroid side-chain catabolism. J Bacteriol 196:579–587. doi: 10.1128/JB.01012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehman TC, Thorpe C. 1990. Alternate electron acceptors for medium-chain acyl-CoA dehydrogenase: use of ferricenium salts. Biochemistry 29:10594–10602. doi: 10.1021/bi00499a004. [DOI] [PubMed] [Google Scholar]
- 29.Casabon I, Crowe AM, Liu J, Eltis LD. 2013. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol Microbiol 87:269–283. doi: 10.1111/mmi.12095. [DOI] [PubMed] [Google Scholar]
- 30.Swain K, Casabon I, Eltis LD, Mohn WW. 2012. Two transporters essential for reassimilation of novel cholate metabolites by Rhodococcus jostii RHA1. J Bacteriol 194:6720–6727. doi: 10.1128/JB.01167-12; doi: 10.1128/JB.01167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiffany KA, Roberts DL, Wang M, Paschke R, Mohsen AW, Vockley J, Kim JJ. 1997. Structure of human isovaleryl-CoA dehydrogenase at 2.6 A resolution: structural basis for substrate specificity. Biochemistry 36:8455–8464. doi: 10.1021/bi970422u. [DOI] [PubMed] [Google Scholar]
- 33.Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. 2011. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem 286:43668–43678. doi: 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Djordjevic S, Dong Y, Paschke R, Frerman FE, Strauss AW, Kim JJ. 1994. Identification of the catalytic base in long chain acyl-CoA dehydrogenase. Biochemistry 33:4258–4264. doi: 10.1021/bi00180a021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.