

Abstract
Histone dacetylases (HDACs) are a group of enzymes that remove acetyl groups from histones and regulate expression of tumor suppressor genes. They are implicated in many human diseases, especially cancer, making them a promising therapeutic target for treatment of the latter by developing a wide variety of inhibitors. HDAC inhibitors interfere with HDAC activity and regulate biological events, such as cell cycle, differentiation and apoptosis in cancer cells. As a result, HDAC inhibitor-based therapies have gained much attention for cancer treatment. To date, the FDA has approved three HDAC inhibitors for cutaneous/peripheral T-cell lymphoma and many more HDAC inhibitors are in different stages of clinical development for the treatment of hematological malignancies as well as solid tumors. In the intensifying efforts to discover new, hopefully more therapeutically efficacious HDAC inhibitors, molecular modeling-based rational drug design has played an important role in identifying potential inhibitors that vary in molecular structures and properties. In this review, we summarize four major structural classes of HDAC inhibitors that are in clinical trials and different computer modeling tools available for their structural modifications as a guide to discover additional HDAC inhibitors with greater therapeutic utility.
Keywords: HDAC inhibitors, cancer, molecular modeling, clinical trials
1. Background
Cancer is a disease driven by genetic and genomic alterations such as amplifications, translocations, deletions, and point mutations. However, cancer development is also tied to epigenetic changes due to modifications such as DNA methylation and post-translational histone acetylations that can alter DNA accessibilities and chromatin structures without alterations in the DNA sequence. The basic unit of chromatin is the nucleosome, which comprises 147 base pairs of DNA superhelix wrapped around a histone core consisting of two copies each of the core histones [1]. Histones are the primary protein components of chromatin of five classes (H1, H2A, H2B, H3 and H4). H1 is a linker histone and the remaining are the core histones. The core plays an important role in establishing interactions between the nucleosomes and within the nucleosome particle itself [2,3]. The N-terminal tails of core histones are flexible and unstructured, but the rest are predominantly globular and well structured. Depending on the epigenetic modifications that occur in DNA and in histone tails, chromatin can adopt different conformational changes that control the activation or repression of gene transcription.
There are at least eight distinct types of histone post-translational modifications, namely acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deamination and proline isomerization. It can be viewed as a regulatory code that resides in the pattern of post-translational modifications for which the histone amino terminal tails are the target. The N-ε-lysine acetylation and deacetylation of histone are controlled by two groups of enzymes: histone acetyltransferase (HAT) and histone deacetylase (HDAC). The balance between acetylation and deacetylation of histones or the reverse activities of HATs and HDACs regulate gene expression through chromatin modifications [4,5]. Histone acetylation by HAT plays a key role in transcriptional activation, whereas deacetylation of histones promotes transcriptional repression and silencing of genes. An excessive level of histone acetylation induces apoptotic cell death, whereas excessive level of histone deacetylation has been linked to cancer pathologies by promoting the repression of tumor regulatory genes. Disruption of HAT and HDAC activities has been associated with the development of a wide variety of human cancers [5]. HDAC inhibitors cause an increase of the acetylated level of histones, which in turn promote the re-expression of the silenced regulatory genes in cancer cells and reverse the malignant phenotype. Due to this effect, HDAC inhibitors have recently emerged as potential cancer therapeutic agents.
2. Classification of HDAC Family
In the human genome, eighteen HDAC family members have been identified and are grouped into four classes based on their homology to yeast HDACs. Classes I, II and IV are Zn2+-dependent metalloproteins, whereas Class III is a nicotinamide adenine dinucleotide (NAD+)-dependent enzyme. Class I family of HDACs consists of HDAC1, 2, 3 and 8 proteins sharing sequence homology with yeast reduced potassium dependency-3 (Rpd3), and are mainly located in the nucleus of the cells [6,7]. Class II family HDACs are homologous to the yeast histone deacetylases 1 (Hda1) and are further divided into two subgroups, Class IIA (HDAC4, 5, 7 and 9) and Class IIB (HDAC6 and 10). Unlike Class I family HDACs, Class II family HDACS are primarily localized in the cytoplasm; however depending upon the phosphorylation status they can be shuttled between the cytoplasm and nucleus [8,9]. HDAC11 is the only member of Class IV family localized in the nucleus. It has a unique structure but shares some of the sequences of Class I and II enzymes. HDAC11 has been implicated in the regulation of interleukin-10 expression [10,11], OX40L surface expression [12] and expression of the DNA replication licensing factor Cdt1 [13]. Class III family comprise of seven members and they share sequence homology with yeast silent information regulator-2 (Sir2) protein. Hence Class III family HDACs are also known as sirtuins (SIRTs), and the seven members of this family are SIRT1 through SIRT7. SIRTs are located in three important cellular compartments: nucleus, cytoplasm and mitochondrion [14]. Phylogenetically SIRTs are further divided into four classes (SIRT1, SIRT2 and SIRT3 belong to Class I, a sole member of SIRT4 to Class II, SIRT5 to Class III, and SIRT6 and SIRT7 to Class IV) [14,15]. Sirtuins have emerged as potential therapeutic targets for the treatment of various diseases, such as cancer, cardiovascular, aging and neurodegenerative related diseases [16,17,18,19]. A recent review has summarized the possibility of sirtuins, especially SIRT1 and SIRT2, for cancer therapy agents [20]. Table 1 summarizes the classification, cellular localization, protein size, some biological implications and crystal structure availability of HDACs. This review focuses on recent development of inhibitors of metal-dependent “classical” HDACs (Classes I, II, and IV) that are in clinical trials as anti-cancer agents, and different computer modeling tools for the development of HDAC inhibitors.
Table 1.
Histone deacetylase enzymes: classification, amino acid size, cellular localization, physiological functions and crystal structure availability.
| Metal Dependent | |||||
|---|---|---|---|---|---|
| Class | Members | Size (aa) | Cellular Localization | Physiological Function | X-ray Crystal |
| I | HDAC1 | 483 | Nucleus | Cell survival and proliferation | Yes |
| HDAC2 | 488 | Nucleus | Cell proliferation, Insulin resistance | Yes (core domain) | |
| HDAC3 | 428 | Nucleus | Cell survival and proliferation | Yes | |
| HDAC8 | 377 | Nucleus | Cell proliferation | Yes | |
| IIA | HDAC4 | 1084 | Nucleus/Cytoplasm | Regulation of skeletogenesis and gluconeogenesis | Yes (catalytic & glutamine rich domains) |
| HDAC5 | 1122 | Nucleus/Cytoplasm | Cardiovascular growth and function, gluconeogenesis, cardiac myocytes and endothelial cell function | No | |
| HDAC7 | 912 | Nucleus/Cytoplasm | Thymocyte differentiation, endothelial function, glucogenesis | Yes (catalytic domain) | |
| HDAC9 | 1069 | Nucleus/Cytoplasm | Homologous recombination, thymocyte differentiation, cardiovascular growth and function | No (structure is known for aa 138–158) | |
| IIB | HDAC6 | 1215 | Cytoplasm | Cell motility, control of cytoskeletal dynamics | Yes (zinc finger and ubiquitin binding domains) |
| HDAC10 | 669 | Cytoplasm | Homologous recombination, Autophagy mediated cell- survival | No | |
| IV | HDA11 | 347 | Nucleus | Immunomodulators-DNA replication | No |
| NAD+ Dependent | |||||
| III | SIRT 1 | 747 | Nucleus, Cytoplasm | Aging, redox regulation, cell survival, autoimmune system regulation | Yes (catalytic domain) |
| SIRT 2 | 389 | Nucleus | Cell survival-cell migration and invasion | Yes | |
| SIRT 3 | 399 | Mitochondria | Urea Cycle, Redox balance, ATP regulation, metabolism, apoptosis and cell signaling | Yes | |
| SIRT 4 | 314 | Mitochondria | Energy metabolism, ATP regulation, metabolism, apoptosis and cell signaling | No | |
| SIRT 5 | 310 | Mitochondria | Urea cycle, Energy metabolism, ATP regulation, metabolism, apoptosis and cell signaling | Yes | |
| SIRT 6 | 355 | Nucleus | Metabolic regulation | Yes | |
| SIRT 7 | 400 | Nucleus | Apoptosis | No | |
3. Histone Deacetylases and Cancer
HDACs play a major role in the epigenetic regulation of gene expression through their effects on the compact chromatin structure. In recent years, HDACs have become promising therapeutic targets with the potential to reverse the aberrant epigenetic states associated with cancer. Alterations in acetylation levels and overexpression of various HDACs in many cancer cell lines and tumor tissues have been reported [21]. Characterization of post-translational modifications to histone H4 in a comprehensive panel of normal tissues, cancer cell lines and primary tumors suggests that global loss of monoacetylation at Lys16 of histone H4 is a common hallmark of human cancer cells, implicating a critical role of HDAC activity in establishing tumor phenotypes [22]. In cancer pathological conditions where the classical HDACs are overexpressed, inhibitors of HDACs were found to be effective in reversing the malignant phenotype of transformed cells and have subsequently emerged as promising cancer therapeutic agents. HDAC inhibitors have the potential to disrupt multiple signaling pathways to inhibit tumor growth and induce apoptosis. HDAC inhibitors can not only target histones but have the ability to influence a variety of processes such as cell cycle arrest, angiogenesis, immune modulation and apoptosis by targeting nonhistone proteins [21,23]. Several nonhistone proteins have been identified as HDAC substrates with diverse biological functions and they include, transcription factors (E2F, p53, c-Myc, NF-κB), hypoxia-inducible factor 1 alpha (HIF-1α), estrogen receptor (ER α), androgen receptor (AR), MyoD, Chaperons (HSP90), signaling mediators (Stat3, Smad7), DNA repair proteins (Ku70), α-tubulin, β-catenin, retinoblastoma protein (pRb) and many others [24,25].
Thus the disruption of multiple pathways by HDAC inhibitors and their lack of enzyme specificity cause additional complication to rational drug design for a specific disease state. In clinical studies several classes of HDAC inhibitors demonstrated potent anticancer activities with remarkable tumor specificity, such as cutaneous T-cell lymphoma and peripheral T-cell lymphoma [26,27,28,29].
To date, three HDAC inhibitors have been approved for cancer therapy by the US Food and Drug Administration (FDA). The first drug, Vorinostat (SAHA, Zolina), developed by Merck & Co. Inc. was approved in October 2006 for use in patients with cutaneous T-Cell Lymphoma (CTCL), a rare type of non-Hodgkin’s lymphoma of the skin. Vorinostat is structurally related to trichostatin A (TSA), a hydroxamic acid-containing natural product that was found to possess HDAC inhibitor activity and originally used as an antifungal drug. The second drug, romidepsin (Istodax, FK228, FR901228, depsipeptide), developed by Gloucester Pharmaceuticals (acquired by Celgene in 2009) was approved at the end of 2009, also for the treatment of T-cell lymphoma. Romidepsin is a unique natural product isolated from the cultures of Chromobacterium violaceum, a Gram negative bacterium isolated from a Japanese soil sample [30]. In June 2011, romidepsin was also approved for peripheral T-cell lymphoma (PTCL) in patients who have received at least one prior therapy. The third drug, belinostat (Beleodaq, PXD-101), developed by Spectrum Pharmaceuticals was approved on July 3, 2014 for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL) [31].
Over the past several years, a number of small molecule HDAC inhibitors have been subjected to clinical trials for various types of cancers. Based on their distinct chemical structure, these inhibitors can be grouped into four different classes, comprising hydroxamic acids, benzamides, cyclic peptides and short-chain fatty acids [32]. Vorinostat and belinostat belong to the hydroxamic acid class, and romidepsin is a member of the cyclic peptide class. The most widely explored class of HDAC inhibitors that have entered pre-clinical or clinical studies as anti-cancer agents are the hydroxamic acid-based compounds. Besides Vorinostat and belinostat, some of the novel hydroxamic acid based HDACi that are in different stages of clinical studies are abexinostat (PCI-24781), pracinostat (SB939), resminostat (RAS2410, 4SC-201), givinostat (ITF2357), quisinostat (JNJ-26481585), panobinostat (LBH589) and CUDC-101 [33]. Interestingly, HDAC inhibitors share common structural features so that they can properly interact with different portions of the catalytic channel of the enzyme. HDAC inhibitors generally consist of three parts in chemical structure with distinct pharmacophore features: (1) a zinc chelating group; (2) a spacer group; which is generally hydrophobic and (3) an enzyme binding group that confers specificity and is generally aromatic in character [34]. A range of natural and synthetic HDAC inhibitors have been characterized for their antitumor activities. Although not fully understood, the clinical activities of these compounds are thought to be mediated in part by the induction of histone acetylation where the chromatin configuration adopts a permissive or more open form for potential reactivation of aberrantly suppressed genes, leading to inhibition of cell proliferation, cell differentiation and apoptosis [35]. In the following sections, we describe the clinical development of different classes of HDAC inhibitors.
4. FDA Approved Drugs
To date, only three HDAC inhibitors have been approved by the FDA for the treatments of CTCL (vorinostat (SAHA) and romidepsin (Istodax)) and PTCL (belinostat (Beleodaq) and romidepsin). Currently all three drugs are being further evaluated for other diseases as well as in other hematological malignancies and solid tumors, either as a single agent or in combination with other drugs. The following subsections summarize the research done with these three drugs for various diseases.
4.1. Vorinostat
FDA approval of this hydroxamic acid based drug for the treatment of cutaneous manifestation of CCTL in patients with progressive, persistent or recurrent disease was based on Phase II clinical trials that enrolled 74 patients who had stage IB or higher CTCL. The objective response rate determined by direct evidence of clinical benefit was 30% [26]. For hematological malignancies, vorinostat can be given orally with a maximum tolerated dose of 400 mg once daily or 200 mg twice daily, but the dose level can be increased up to 600 mg in solid tumors [36]. Preclinical studies involving vorinostat have demonstrated its use as a potent radiosensitizer in human glioblastoma cell lines [37]. Vorinostat in combination with temozolomide and radiotherapy are currently in an ongoing clinical trial (NCT00731731) for treating patients with newly diagnosed glioblastoma multiforme (GBM). GBM is the most common and aggressive malignant brain tumor with very poor prognosis. Vorinostat showed potent apoptotic and anti-proliferative effect in both type I and type II human endometrial cancers by modifying the expression of specific genes related to the insulin-like growth factor-I (IGF-I) receptor signaling pathway [38]. In type I cell lines, vorinostat increased the IGF-IR phosphorylation, up-regulated PTEN and p21 expression, and reduced p53 and cyclin D1 levels. In type II cell lines, vorinostat up-regulated IGF-IR and p21 expression, and down-regulated the expression of total AKT, PTEN and cyclin D1. Interestingly, vorinostat hyperacetylated histone H3 in both type I and type II endometrial cancer cell lines, implying the role of histone H3 in endometrial cancer. Endometrial cancer is the most common gynecologic cancer that begins in the endometrium, the inner lining of the uterus, and these are classified into Type I and Type II groups, with type I being the most frequent [39,40]. In murine and human lung cancer cell lines and genetically engineered mouse lung cancer models, Vorinostat reduced cancer cell growth, cyclin D1 and cyclin E expressions, but increased p27 expression, histone acetylation and apoptosis [41]. Under hypoxia, radiosensitization by vorinostat in combination with capecitabine decreased colonogenicity in vitro, and inhibited tumor growth in vivo in xenograft models of colorectal carcinoma [42]. Currently vorinostat in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) that exhibits poor prognosis by itself is in clinical trials for treating patients with untreated PTCL [43]. Vorinostat has also been found to be a potent agent in the treatment of gastrointestinal (GI) cancer [44]. Vorinostat has also been implicated in having an effect on other types of cancers, such as brain metastasis, refractory colorectal, advanced solid tumors, melanoma, pancreatic, lung cancer and multiple myeloma. In terms of its target, vorinostat inhibits Class I, II and IV HDAC proteins, but not the NAD+-dependent Class III HDAC [45,46,47].
4.2. Romidepsin (Depsipeptide, ISTODAX)
The second HDAC inhibitor approved for the treatment of CTCL was based on two large phase II studies: a multi-institutional study based at the NCI in the US (71 patients), and an international study (96 patients) [27,28]. The treatment schedule was identical across both studies and the overall response rate was 34% in both studies. Romidepsin also induced complete and durable responses in patients with relapsed or refractory PTCL across all major PTCL subtypes, regardless of the number or types of prior therapies, with an objective response rate of 25%, which led to the approval of single agent romidepsin for the treatment of relapsed or refractory PTCL in the US [48]. Similarly, a phase II trial enrolling 47 patients with PTCL of various subtypes including PTCL NOS, angioimmunoblastic, ALK-negative anaplastic large cell lymphoma, and enteropathy-associated T-cell lymphoma also showed an overall response rate of 38% [49]. Romidepsin was also implicated in inhibiting the growth of non-small cell lung cancer (NSCLC) cells. A recent study concluded that romidepsin and bortezomib cooperatively inhibit A549 NSCLC cell proliferation by altering the histone acetylation status, expression of cell cycle regulators and matrix metalloproteinases [50]. Investigation of romidepsin for the treatment of inflammatory breast cancer (IBC), the most metastatic variant of locally advanced breast cancer, revealed that it potentially induced destruction of IBC tumor emboli and lymphatic vascular architecture [51]. Romidepsin, either as a single agent, or in combination with paclitaxel, effectively eliminated both primary tumors and metastatic lesions at multiple sites formed by the SUM149 IBC cell line in the Mary-X preclinical model [51]. A combination of depsipeptide and gemcitabine was tested in patients with advanced solid tumors including pancreatic, breast, NSCLC and ovarian and the study identified a dose level of 12 mg/m2 romidepsin and 88 mg/m2 gemcitabine for phase II trial [52]. In another phase I trial, romidepsin was evaluated in patients with advanced cancers including patients with thyroid cancer and identified tolerable doses for the treatment [53]. According to clinicaltrials.gov, romidepsin is currently being evaluated in nearly 30 studies, either as a single agent or in combination with other drugs for treating mainly T-cell lymphoma.
4.3. Belinostat (Beleodaq)
Approval of the third pan-HDAC inhibitor, belinostat was based on a multi-center, single arm BELIEF trial of 120 evaluable patients with PTCL that was refractory or had relapsed after prior treatment [54]. Among patients with histologically confirmed PTCL (n = 120), the overall response rate was 25.8%. Similar to other two FDA approved drugs, belinostat was also tested in Phase I and Phase II clinical trials for both solid and hematological cancers. For example, the response rate of belinostat was tested for a second line therapy in 13 patients with recurrent or refractory malignant pleural mesothelioma and identified two patients with stable disease [55]. A Phase II trial of belinostat in women with platinum resistant epithelial cancer (OEC) and micropapillary (LMP) ovarian tumors showed good drug tolerance in both patient groups [56]. Belinostat was also tested in patients with recurrent or refractory advanced thymic epithelial tumors and the response rate was 8% among the thymoma patients but found no response among thymic carcinoma patients [57]. A phase II multicenter study was undertaken to estimate the efficacy of belinostat for the treatment of myelodysplastic syndrome (MDS), a cancer in which the bone marrow does not make enough healthy blood cells [58]. However, this study met the stopping rule in the first stage of enrollment itself, hence the trial was closed to further accrual.
A Phase II study involving 29 women with recurrent or persistent platinum-resistant ovarian cancer was also conducted to evaluate the impact of belinostat in combination with carboplatin [59]. The overall response rate was 7.4% and the addition of belinostat to carboplatin had little activity in a platinum-resistant ovarian cancer patients. Phase II clinical activity of belinostat was also tested in combination with carboplatin and paclitaxel by enrolling 35 women with previously treated ovarian cancer [60]. Combination of these three drugs were reasonably well tolerated with an overall response rate of 43% and demonstrated clinical benefits in patients with OEC. In patients with relapsed or refractory acute myeloid leukemia (AML), the effect of belinostat was studied in a phase II clinical study and it was found that the effect of belinostast as a single agent is minimal in AML patients [61]. A phase 1/II trial of belinostat in combination with cisplatin (P), doxorubicin (A), and cyclophosphamide (C) in thymic epithelial tumors (TETs) showed that belinostat in combination with PAC was active and feasible in TETs [62]. A preclinical study of belinostat in three hepatocellular carcinoma cell lines (PLC/PRF/5, Hep3B and HepG2) showed that it can inhibit cell growth in a dose dependent manner and induce histone acetylation in all three cell lines [63]. Antileukemia activity of this compound as a single drug and in combination with all-trans-retinoic acid was characterized in promyelocytic leukemia HL-60 and NB4 cell lines, where belinostat can exert dose-dependent growth inhibitory or proapoptotic effects promoting cell cycle arrest at the G0/G1 or the S transition phase [64].
While three hydroxamic acid derivatives as HDAC inhibitors have been clinically approved, the indication is mainly CTCL, not any solid tumor form. So far, few ongoing clinical trials are designed to combat solid tumors, and the ones that have been completed had very limited therapeutic outcome with regard to using the HDAC inhibitors for treatment of nonhematological cancers. This persistent gap limits the utility of HDAC inhibitors, but more importantly, it calls for the discovery of more selective inhibitors that are also pharmaceutically more robust.
The exact reasons why HDAC inhibitors are more effective in hematological malignancies than in solid tumors are not well understood, but some observations suggest that the inhibitors having gone through clinical trials so far may not be sufficiently stable to reach solid tumor sites, and that they may not be target specific for solid tumors.
5. Different Classes of HDAC Inhibitors
5.1. Hydroxamic Acid Derivatives
Some of the initial clinical studies established that hydroxamic acid-based compound vorinostat is well tolerated in patients with CTCL, and observed promising anti-cancer activities in different types of cancer, such as diffuse large B-cell lymphoma, Hodgkin lymphoma, and other haematological and solid tumors [36,65,66,67]. Vorinostat was also found to inhibit tumor growth in rodent models of a variety of cancers (prostate cancer, leukemia, breast cancer, glioma, and lung cancer) [68]. Given the diverse anti-cancer activities of vorinostat, much effort has been made to explore hydroxamic acid derivatives as potential treatment for various cancers. Indeed, over the past several years, many hydroxamic acid derivative based HDACis have entered pre-clinical or clinical studies as anti-cancer agents with promising results, including abexinostat, pracinostat, resminostat, givinostat, panobinostat, and CUDC-101. These HDAC inhibitors are described below in more detail.
5.1.1. Abexinostat (PCI-24781)
Abexinostat is a novel hydroxamate-based HDACi that showed broad spectrum anticancer activities in preclinical studies. As a single agent and in combination with the proteasome inhibitor bortezomib, abexinostat was tested in neuroblastoma cell lines [69]. Western blotting analysis showed the cleavage of caspase-3 and PARP, indicating apoptosis as a primary mechanism of action. Further studies with xenograft mouse models indicated increased survival among animals treated with a combination of abexinostat and bortezomib. This oral pan-HDACi was evaluated in patients with advanced solid tumors in two single agent phase I studies (PCYC-402 and CL1-78454-002), resulting in an optimal schedule for allowing higher doses in the next stage of the trials in solid tumors [70]. The effect of abexinostat, alone or in combination with conventional chemotherapy agents, was also tested in vivo in human soft tissue sarcoma (STS) models [71]. As a single-agent abexinostat showed modest effects on STS growth and metastasis, but marked inhibition effect was observed in combination with chemotherapy. In a phase I study, pazopanib (PAZ: a tyrosine kinase inhibitor approved for use in renal cell carcinoma) in combination with abexinostat was tested in patients with metastatic solid tumors, and the results presented at the ACSO annual meeting 2014 showed partial tumor response and disease stabilization. Further studies are being done and this trial is currently recruiting patients (clinical trial information, NCT01543763). Similarly a phase I study was done with abexinostat in combination with cisplatin in patients with advanced keratinizing nasopharyngeal carcinoma (NPC), leading to the identification of optimal doses for combination therapy (clinical trial information: ISRCTN96922360).
5.1.2. Pracinostat (SB939)
Pracinostat is another hydroxamate-based HDAC inhibitor for which clinical trials have been carried out. A phase II study tested the activity and tolerability of pracinostat in patients with intermediate or high risk myelofibrosis (MF) where pracinostat was shown to have clinical benefit and modest activity in patients with MF [72]. In another phase II study, pracinostat was tested in advanced solid tumor patients [73]. The drug was well tolerated, but there was no clear relationship between the acetylated histone H3 changes and dose level or antitumor response. Pracinostat was also found to be well tolerated in children with refractory solid tumors [74].
5.1.3. Resminostat
Resminostat was evaluated in a pharmacokinetics and pharmacodynamics phase I study for patients with advanced solid tumors, yielding a recommended phase II dose of 600 mg/day [75]. Low micro- molar concentrations of resminostat abrogated cell growth and strongly induced apoptosis in multiple myeloma (MM) cell lines [76]. Synergistic effects were observed when it was used in combination with melphalan, bortezomib and S-2209 [76]. In a Phase II SAPHIRE trial, resminostat was also tested in relapsed or refractory Hodgkin Lymphoma (HL) [77,78]. Assessment of disease status was carried out by computed tomography in combination with positron emission tomography (PET/CT). This study achieved clear objective responses in relapsed/refractory HL patients and showed excellent safety profiles in heavily pre-treated patient population. Resminostat was also tried in patients with advanced hepatocellular carcinoma (HCC), either alone or in combination with sorafenib [79]. The combination treatment provided a substantial overall survival (OS) benefit (median OS of 8.1 months) for advanced HCC patients who had developed progressive tumor disease under first-line sorafenib therapy. Resminostat is also in clinical trials for treating advanced colorectal carcinoma (NCT01277406).
5.1.4. Givinostat
Givinostat is a hydroxamic acid-containing HDAC inhibitor which has shown clinical benefits in patients with Hodgkin’s lymphoma, chronic lymphocytic leukemia and multiple myeloma. A phase II study was conducted to evaluate the safety and efficacy of givinostat in patients with JAK2V617F positive myeloproliferative neoplasms (MPN), a type of blood cancer [80]. Complete and partial responses were documented suggesting givinostat as a promising drug for further clinical investigation in patients with MPN. An in vitro study determined if givinostat and hydroxyurea induce synergistic cytotoxicity in JAK2V617F cells [81]. At low doses, both givinostat and hydroxyurea potentiated the pro-apoptotic effects of each other in the JAK2V617F HEL and UKE1 cell lines. As a single agent, givinostat and hydroxyurea induced 6.8%–20.8%, and 20.4%–42.4% cell death, respectively, whereas in combination of these two drugs the cell death was 35.8%–75.3%. This study suggested that a combined treatment with givinostat and hydroxyurea is a potential strategy for the management of JAK2V617F myeloproliferative neoplasms. A phase I safety and pharmacokinetic trial in healthy males was also done with givinostat and identified the safe therapeutic dosing of givinostat [82]. In another multicenter, open-label phase II trial, patients with polycythemia vera (PV), unresponsive to the maximum tolerated doses (TMD) of hydroxycarbamide (HC), were treated with givinostat in combination with TMD of HC [83]. Complete or partial response was observed in 55% and 50% of the patients who received 50 or 100 mg of givinostat, respectively. This study showed that the combined use of givinostat and HC was safe and well tolerated, and clinically effective in HC-responsive PV patients.
5.1.5. Panobinostat (LB589)
This hydroxamate-based panobinostat showed activity in clinical trials with different solid and heamatological cancers. The antitumor activity of panobinostat in patients with previously treated small-cell lung cancer (SCLC) was tested in a multicenter, nonrandomized phase II trial [84]. Although panobinostat was well tolerated and induced tumor shrinkage and sustained stable disease in SCLC, this study was prematurely closed because of a lack of activity. A phase I study investigated the effect of panobinostat in patients with primary myelofibrosis (PMF), post-essential thrombocythemia myelofibrosis (post-ET MF) and post-polycythemia vera myelofibrosis (post-PV MF) [85]. Panobinostat was well tolerated in MF patients with clinical improvements indicated by 100% reduction in palpable splenomegaly, and stable disease or near complete remission was observed in some patients. A phase I trial of panobinostat in 14 patients with advanced solid tumors was conducted in three cohorts. Even though stable disease (for ≥4 months) was observed in six patients, complete or partial responses were not observed in this study [86].
In another phase I trial in patients with advanced solid tumors or cutaneous T-cell lymphoma, good tolerance to panobinostat was observed when administered orally thrice in a week [87]. A multicenter, international Phase II study examined the safety and activity of panobinostat in 129 patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation, and observed tumor reductions in 74% of the patients with a 1 year survival rate of 78% [88]. In another phase II trial, panobinostat as a single agent was tested in red blood cell transfusion-dependent low or intermediate-1 risk MDS patients, but did not demonstrate a meaningful clinical activity [89]. A phase II trial of panobinostat in patients with low or intermediate-1 risk MDS observed only limited activity [90].
5.1.6 CUDC-101
A recent study has shown simultaneous inhibition of HDAC and receptor tyrosine kinases (epidermal growth factor receptor—EGFR—and human epidermal growth factor receptor 2—HER2) in cancer cells, and displayed antiproliferative and proapoptotic activities in vitro as well as in drug-resistant in vivo tumor models [91]. Hence it has the potential to improve the treatment of heterogeneous and drug resistant tumors that cannot be controlled with singe-target agents. This synergistic inhibition was also tested in patients with advanced solid tumor using CUDC-101, and the drug was found to induce histone H3 acetylation in some of the patients. This study recommended a dose of 275 mg/m2 CUDC-101 for further clinical testing [92]. Then a phase 1b (expansion) was conducted to further evaluate the safety and tolerability of CUDC-101 in patients with diverse cancers (advanced breast, gastric, head and neck, NSCLC or liver cancer), The drug was found to be well tolerated in these patients and exhibited antitumor activity [93,94]. Table 2 summarizes all the hydroxamic acid based HDAC inhibitors as potential therapeutics for various cancers that were in clinical trials.
Table 2.
Hydroxamic acid based HDAC inhibitors in clinical trials.
| Hydroxamic Acid Based HDAC Inhibitors (HDACi) | HDAC Specificity (Class) | In Vitro Potency | Combination | Cancer Types | Reference |
|---|---|---|---|---|---|
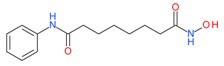 Vorinostat (SAHA) |
I and II | nM | Temozolomide + radiation | Glioblastoma Multiforme (GBM) | [95] |
| CHOP | Peripheral T-cell lymphoma (PTCL) | [43] | |||
| - | Gastrointestinal(GI) | [44] | |||
| Whole brain radiation | Brain metastasis | [37] | |||
| 5-fluorouracil/leucovorin(5FU/LV) | Refractory colorectal and solid tumors | [96,97] | |||
| Hydroxychloroquine | Advanced solid tumors | [98] | |||
| Marizomib | Melanoma, Pancreatic and Lung cancer | [99] | |||
| Bortezomib | Multiple myeloma | [100] | |||
| 5-fluorouracil | Metastatic colorectal | [101] | |||
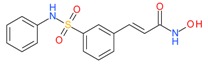 Belinostat (Beleodaq) |
I and II | μM | - | Malignant pleural mesothelioma | [55] |
| - | Epithelial & microcapillary ovarian cancers | [56] | |||
| - | Thymic epithelial tumor(TETs) | [57] | |||
| - | Myelodysplastic syndrom (MDS) | [58] | |||
| Carboplatin | Platinum resistant ovarian cancer | [59] | |||
| Carboplatin + Paclitaxel | Ovarian cancer | [60] | |||
| - | Acute myeloid leukemia (AML) | [61] | |||
| Cisplatin + doxorubicin + cyclophosphamide | Thymic epithelial tumors | [62] | |||
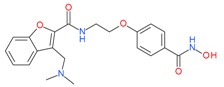 Abexinostat(PCI-24781) |
I and II | nM | - | Advanced solid tumors | [70] |
| Pazopanib | Metastatic solid tumor | [95] | |||
| Cisplatin+radiation | Nasopharyngeal carcinoma (NPC) | [102] | |||
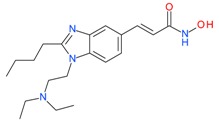 Pracinostat (SB939) |
I, II and IV | μM | - | Myelofibrosis(MF) | [72] |
| - | Advanced solid tumors | [73] | |||
| - | Refractory solid tumors | [74] | |||
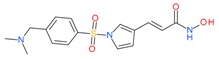 Resminostat |
I and II | μM | - | Advanced solid tumors | [75] |
| - | Relapsed/refractory Hogdkin Lymphoma (HL) | [77,78] | |||
| or Sorafenib | Advanced hepatocellular carcinoma (HCC) | [79] | |||
| - | Colorectal carcinoma | [95] | |||
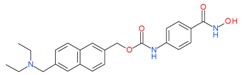 Givinostat (ITF-2357) |
I and II | nM | - | Myeloproliferative neoplasms(MPN) | [80] |
| Hydroxycarbamide | Polycythemia vera | [83] | |||
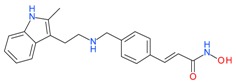 Panobinostat |
I and II | μM | - | Small cell lung cancer (SCLC) | [84] |
| - | Myelofibrosis(MF) | [85] | |||
| - | Advanced solid tumors | [86] | |||
| - | Cutaneous T-cell lymphoma | [87] | |||
| - | Relapsed/refractory hogdkins lymphoma | [88] | |||
| - | Myelodysplastic syndrome (MDS) | [89] | |||
 CUDC-101 |
I and II | nM | - | Advanced solid tumors | [92–94] |
5.2. Benzamide Derivatives
Benzamide containing HDAC inhibitors are another class of compounds that showed both in vitro and in vivo anticancer activities. Among them mocetinostat (MGCD0103) and entinostat (MS-275) are two examples of benzamide derivatives that had been taken to clinical trials.
5.2.1. Mocetinostat (MGCD0103)
This benzamide derivative HDAC inhibitor is selective for both Class I and IV histone deacetylases [103]. A phase 1 trial of mocetinostat in patients with leukemia or myelodysplastic syndromes (MDS) showed the drug was safe and exhibited antileukemia activity in these patients [104]. Three patients achieved a complete bone marrow response (blasts ≤ 5%) too. Mocetinostat was also tested in pancreatic cancer cell lines, and found dose-dependent growth arrest, cell death and cell cycle arrest. This effect was found to be enhanced when treated in combination with MC1568 (Class IIA selective HDACi) or tubastatin A (HDAC6 selective inhibitor) [105]. A phase II clinical trial in patients with chronic lymphocytic leukemia (CLL) also demonstrated efficacy with manageable side effects profile [106]. In patients with advanced solid tumors, mocetinostat inhibited HDAC activity and induced Histone H3 acetylation in peripheral white blood cells from these patients, and the trial identified a dose level of 45 mg/m2/day for Phase II studies [107]. The safety and efficacy of this compound was tested in patients with relapsed classical Hodgkin’s Lymphoma during a phase II clinical trial [108]. Even though the treatment showed promising clinical activity with manageable toxicity in patients with relapsed classical Hodgkin’s lymphoma, four patients died during the study, of which two might have been treatment-related deaths. As a result, this study has been terminated (clinical trial identifier: NCT00358982).
5.2.2. Entinostat
Many clinical studies have investigated the activity of entinostat in many cancer cells, which include non-small cell lung cancer, breast cancer, lymphoblastic leukemia, renal cell cancer, colon cancer, metastatic melanoma and more. It is a Class I selective HDAC inhibitor and is well tolerated either as a single agent or in combination with other drugs [109]. For example, in a phase I trial, entinostat in combination with 13-cis-retinoic acid (CRA) was tested to determine the safety, tolerability, and the pharmacokinetic/pharmacodynamic profiles of entinostat and CRA in advanced solid tumors. While objective responses were not achieved, the combination drug was well tolerated and prolonged stable disease occurred in patients with prostate, pancreatic, and kidney cancer. In a randomized phase II trial to evaluate the effect of erlotinib with or without entinostat in advanced state NSCLC patients [110]. No improved outcome of patients in the overall study population was observed when compared with erlotinib monotherapy. Similarly, a placebo controlled randomized phase II study evaluated the effect of entinostat alone or combined with the aromatase inhibitor exemestane in breast cancer patients [111]. This study showed that a combination therapy of entinostat and exemestane is well tolerated and demonstrated clinical activity in patients with ER+ advanced breast cancer. Another phase I trial tested entinostat in patients with refractory solid tumors and lymphomas [112]. Prolonged disease stabilization was seen in some patients, and the drug was well tolerated and demonstrated antitumor activity.
5.3. Short Chain Fatty Acids
These compounds represent another class of HDAC inhibitor with simple structures that showed clinical potential in various studies. Valproic acid and phenylbutyrate are two well characterized compounds that belong to this class of compounds. They both display HDAC inhibition for Class I and IIa HDACs, but they tend to be less potent in inhibiting the HDAC activity than the hydroxamic acid based HDAC inhibitors.
Valproic Acid
This compound has entered in clinical trials as a single agent as well as in combination with other drugs [113]. In a phase I study, valproic acid (VPA) was tested in pediatric patients with refractory solid or central nervous system (CNS) tumors [113]. Increased histone acetylation in peripheral blood mononuclear cells was documented in 50% of patients studied, and the drug was well tolerated when administered three times daily to maintain a through concentration. In a pilot phase II study, VPA was also tested for the treatment of neuroendocrine tumors (NETs) and also to determine whether VPA can induce Notch 1 signaling in vivo [114]. Overall treatment with VPA was well tolerated in patients with NETs and was found to activate Notch1 signaling in vivo, suggesting its role in treating patients with low grade neuroendocrine carcinoma.
VPA was also tested in combination with other drugs for the treatment of various cancers [115,116,117,118]. For example, a phase I study of the combination of bevacizumab (anti-angiogenic agent) and VPA was conducted in patients with advanced cancers, and demonstrated that the combination of bevacizumab and VPA is safe in patients with colorectal, prostate, and gastroesophageal cancers with ≥ 6 months of stable disease [115]. VPA was also tested in advanced stage NSCLC patients in combination with 5-aza-2'-deoxycytidine (decitabine). This combination therapy was found to be effective in reactivating hypermethylated genes as demonstrated by re-expressing fetal Hb, but was limited by unacceptable neurological toxicity at a relatively low dosage [116]. VPA in combination with S-1, an oral fluoropyrimidine derivative consisting of 5-fluorouracil, was tested in pancreatobiliary tract cancers and showed manageable safety and preliminary antitumor activity in these patients [117]. Table 3 summarizes the benzamide, short chain fatty acid, and cyclic peptide HDAC inhibitors and their respective activities against various cancers tested in clinical trials.
Table 3.
Benzamide, short chain fatty acid and cyclic peptide based HDAC inhibitors in clinical trials.
| HDACi | HDAC Specificity (Class) | In Vitro Potency | Combination | Cancer Types | Reference |
|---|---|---|---|---|---|
| Benzamide Based HDAC Inhibitors (HDACi) | |||||
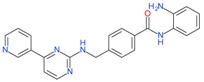 Mocetinostat (MGCD0103) |
I and IV | μM | - | Leukemia | [104] |
| - | Myelodysplastic syndrome (MDS) | [104] | |||
| - | Chronic lymphocytic leukemia (CLL) | [106] | |||
| - | Advanced solid tumors | [107] | |||
| - | Relapsed Hodgkin’s lymphoma | [108] | |||
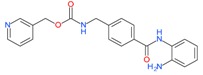 Entinostat (MS-275) |
I | μM | 13-cis retinoic acid(CRA) | Advanced solid tumors | [109] |
| Erlotinib | NSCLC | [110] | |||
| Exemestane | Breast cancer | [111] | |||
| - | Refractory solid tumors and lymphoma | [112] | |||
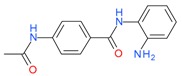 Tacedinaline (CI994) |
I | μM | - | Advanced solid tumor | [119] |
| Short Chain Fatty Acid Based HDAC Inhibitors (HDACi) | |||||
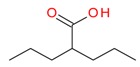 Valproic acid |
I | mM | - | Refractory solid or central nervous system (CNS) tumors | [113] |
| - | Neuroendocrine tumors(NET) | [114] | |||
| Bevacizumab | Colorectal, Prostate, Breast, melanoma | [115] | |||
| Decitabine | NSCLC | [116] | |||
| S-1 | Pancreatobiliary | [117] | |||
| Hydralazine | Solid cancer | [118] | |||
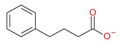 Phenylbutyrate |
I and II | mM | - | Refractory solid tumor or lymphoma | [95] |
| - | Recurrent brain tumor | [95] | |||
| Azacitidine | Acute myeloid leukemia or MDS | [95] | |||
| Azacitidine | Prostate cancer | [95] | |||
| Azacitidine | NSCLC | [95] | |||
| Cyclic Peptide Based HDAC Inhibitors (HDACi) | |||||
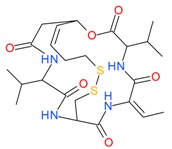 Romidepsin (Depsipeptide) |
I | nM | - | Relapsed or refractory PTCL | [48,49] |
| Bortezomib | NSCLC | [50] | |||
| Abraxane | Inflammatory breast cancer | [95] | |||
| Gemcitabine | Pancreatic, Breast, NSCLC, Ovarian | [52] | |||
| - | Thyroid cancer | [53] | |||
6. Natural HDAC Inhibitors
A large number of HDAC inhibitors are of natural origin. Hydroxamic acid-based trichostatin A (TSA) was one of first natural hydroxamate compounds isolated from the actinomycete Streptomyces hygroscopicus that was found to inhibit HDACs at IC50 less than 10 nM, with over 300-fold selectivity against Class IIa HDACs [120,121]. Another natural hydroxamate found to have anti-proliferative effects against various human tumor cells is amamistatin, isolated from Nocardia asteroides [122,123]. Short-chain fatty acids, such as sodium butyrate, the byproduct of anaerobic microbial fermentation inside the gastrointestinal tract, have been found to inhibit different classes of HDAC [124]. Both TSA and sodium butyrate downregulate the expression of Bcl-2 and induce apoptosis in lymphoma cells [125]. Short chain fatty acids like butyrate and propionate have been shown to increase apoptosis of neutrophils through HDAC inhibition [126]. Both propionate and butyrate based compounds are being tested in clinical trials for many diseases. Natural cyclopeptide FR235222 isolated from the fermentation broth of Acremonium sp. caused accumulation of acetylated histone H4, inhibition of human leukemia cell (U937) proliferation, and cell cycle arrest in the G1 phase via p21 [127]. Other natural cyclopeptides that have been demonstrated to act as HDAC inhibitors are chlamydocin from Diheterospora chlamydosporia [128], apicidin from Fusarium sp. [129] and azumamide A-E from the marine sponge Mycale izuensis [130,131], and the microbial metabolite trapoxin [132]. In addition to romidepsin, some other natural products that belong to the depsipeptide class with antitumor activities are largazole from cyanobacterium Symploca sp [133] spiruchostatin from Pseudomonas [134] and burkholdacs and thailandepsin from Burkholderia thailandensis [135]. They all exhibited prominent antitumor activity against various mammalian solid tumors. Several analogues of chlamydocin, largazole and apicidin also demonstrated anticancer activities in various cancers. Stilbene-based HDAC inhibitors such as resveratrol from red grapes have demonstrated promising activities for the prevention and treatment of cancer [136]. Resveratrol and its analogue piceatannol from blueberries are also known to be SIRT1 activators. Similarly several organosulfur compounds such as diallyl disulfide and allyl mercaptan from garlic [137,138] and sulforaphane from broccoli sprouts [139] inhibit HDAC activity in various cancer cells including colon, prostate and breast cancer cells.
Various other natural products from different sources are also found to inhibit HDAC activity. Two dimensional drawing of all the compounds discussed here are depicted in Figure 1.
Figure 1.

Natural product HDAC inhibitors and their sources.
7. Miscellaneous
7.1. Thioester Based HDACi
Thioesters are used for prodrug strategies. Largazole is a depsipeptide with a thioester moiety purified from marine cyanobacteria and it is a Class I selective HDAC inhibitor. Largazole upon protein-assisted hydrolysis liberates the bioactive largazole thiol. Disulfide prodrug strategy to modulate largazole-based compounds resulted in enzymatic activities comparable to the natural product largazole [140]. KD5170, a mercaptoketone-based Class I and II HDAC inhibitor which is another thioester prodrug demonstrated broad spectrum cytotoxicity against a range of human tumor-derived cell lines. In the proposed mechanism of action, the thioester prodrug undergoes hydrolysis to generate mercaptoketone that coordinates Zn2+ in a bidentate or monodentate fashion in the active site of HDACs [141,142]. Similarly, thioester derivatives of the natural product psammaplin A, a prodrug requiring reduction of its disulfide to the corresponding thiol monomer for the potential inhibition of HDACs, exhibited both potent cytotoxicity and enzymatic inhibitory activity against recombinant HDAC1 [143]. Among the three thioester compounds that contain an oxime or methyloxime or ketone moiety on the linker that connects the cap group, the ketone containing compound was found to be highly potent against recombinant HDAC1, displaying an IC50 of 5 nm. Preliminary investigation discounted the hydrolysis of thioester under the buffered conditions of the assay and direct cleavage of the acetyl group by the deacetylase enzyme. So in this case, rather than acting as a prodrug, the authors state that it is highly plausible that the thioacetate group can function as a potent zinc-binding group.
7.2. Epoxide Based HDACi
Epoxides are another known group of inhibitors of zinc dependent HDAC enzymes. Epoxide bearing natural compounds such as trapoxins and depudecin are reported to form covalent bonds with HDACs [144]. The HDAC activity of these compounds occur at micromolar to nanomolar concentrations [145,146]. Depudecin is a microbial metabolite containing two epoxide groups, whereas trapoxin has only one epoxide group. 1-Alaninechlamydocin isolated from Tolypocladium sp. showed potent antiproliferative/cytotoxic activities in human pancreatic cancer cell lines MIA PaCa-2 at low nanomolar concentrations and induced G2/M cell cycle arrest and apoptosis [147]. It exhibited comparable potency to the cyclic epoxytetrapeptide HDAC inhibitor trapoxin A, but greater potency than SAHA and apicidin in pancreatic carcinoma cell line MIA PaCa-2.
7.3. Electrophilic Ketone Based HDACi
Trifluoromethyl ketones are known to be readily hydrated and have been reported as potent inhibitors of aspartyl, cysteine and serine proteases, as well as zinc dependent enzymes. Trifluoromethyl ketones attached to aromatic amides showed micromolar inhibitory activities as HDAC inhibitors in breast and fibrosarcoma cell lines [148]. Similarly cyclic tetrapeptides containing trifluoromethyl and pentafluoromethyl ketones as zinc binding functional groups were also found to be potent HDAC inhibitors with promising anticancer activities [149]. Fluorinated ketones are considerably more electrophilic because of the presence of strong electron withdrawing effect of the fluoride. Therefore trifluoromethyl ketones are readily hydrated in aqueous media at physiological pH. Trifluoromethyl ketones containing a thiophene linker have been reported as Class IIa selective HDAC inhibitors. A recent study demonstrated that the trifluoromethyl ketone moiety served as a potent zinc binding group [150]. The study also identified silanediol as a zinc binding group with potential for future development of non-hydroxamate Class I and Class IIb HDAC inhibitors. Figure 2 shows structures of some of the thioester and epoxide compounds that are discussed here.
Figure 2.

Structures of thioester- and epoxide-based HDAC inhibitors.
8. Toxicity in Clinical Trials
As with any class of anticancer agents, HDAC inhibitors are also associated with toxicities. The most common grade 3 and 4 adverse events observed with the use of HDAC inhibitors were thrombocytopenia, neutropenia, anemia, fatigue and diarrhea [48,56,74,86,108,109]. In some cases, HDAC-induced thrombocytopenia can be rapidly reversible upon withdrawal of the drug [151]. Nausea, vomiting, anorexia, constipation and dehydration were also seen in patients receiving HDAC inhibitors. Deaths have been reported in clinical studies involving HDAC inhibitors. For example, when mocetinostat was tested in patients with relapsed Hodgkin’s lymphoma four patients died, of which two were treatment-related deaths [108]. Similarly deaths were also reported in clinical trials involving vorinostat [66], givinostat [152] and many other HDAC inhibitors. Thus more studies are needed to determine the toxicity of HDAC inhibitors before a clinical trial can be done, to minimize the cytotoxic effects in patients.
9. Basic Structure of Zinc Binding HDAC Inhibitors
As discussed here, a number of structurally distinct classes of HDAC inhibitors (hydroxamic acid, benzamide, cyclic peptide, short chain fatty acid) have been tested in clinical trials. Interestingly, most of the zinc-dependent HDAC inhibitors have common pharmacophores consisting of three distinct domains: (1) cap group or a surface recognition unit, usually a hydrophobic and aromatic group, which interacts with the rim of the binding pocket; (2) zinc binding domain (ZBD), such as the hydroxamic acid, carboxylic acid or benzamide groups, which coordinates to the active site of Zn2+ ion; and (3) a saturated or unsaturated linker domain with linear or cyclic structure, that connects the cap group to the ZBD [153]. Crystallographic analyses of HDAC in complex with hydroxamate compounds have revealed that the capping group is solvent exposed and interacts with the amino acids near the entrance of the active site, whereas the metal binding moiety resides in the interior of the protein and form complexes with the metal ion [34,154,155,156]. The linker serves to position the capping group and the metal binding domain appropriately for providing high affinity interactions with the proteins. Figure 3 shows the pharmacophoric summary and structure of a few selected HDAC inhibitors.
Figure 3.

Structure of representative HDAC inhibitors and their pharmacophores. The cap group, linker and the zinc binding domain (ZBD) are colored green, red and blue, respectively.
Variations in any or all three domains have variably contributed to the potency and selectivity in various HDAC inhibitors. In the case of the metal binding moiety, the functional groups contain hydroxamic acid, benzamides, thiols, ketones or epoxides. Comparing clinically important three drugs, vorinostat, entinostat and valproic acid that contain a hydroxamate, benzamide and a carboxylate metal binding moiety, respectively, a drastic change in the IC50 value was observed, when the hydroxamate (110–370 nM) [157,158] was changed to a benzamide (2 μM) [159] or a carboxylate (50 μM) [160]. Thus the presence of a carboxylate acid or a benzamide resulted in reduced inhibitory activity, perhaps due to their weaker metal-binding capacity than a hydroxamate group. Other studies also confirmed that hydroxamic acid is generally a more potent HDAC inhibitor than carboxylic acid [161]. Modification of the linker group, with different chain length, saturated or unsaturated hydrocarbons, including cyclic hydrocarbons have also displaced variations in the inhibitory activity. As a result, HDAC inhibitor design has involved these three modifications, as evident from several articles and reviews [33,162,163]. Thus finding the optimal structural requirements for HDAC inhibition is essential for developing more potent and specific inhibitors of different isoforms of HDAC.
10. Mechanism of action of HDAC inhibitors
HDAC inhibitors increase the level of histone acetylation and the mechanism for their antiproliferative effect is clearly associated with inhibition of HDAC activity. However, this effect alone is not sufficient to confer activity, because several trials have demonstrated increased histone acetylation in tumor samples despite little clinical effect [107,164]. HDACs can not only act on and modify histones, but also have many different cellular substrates and target proteins, including proteins that are involved in tumor progression, cell cycle control, apoptosis, angiogenesis and cell invasion. Thus HDAC inhibitors exert multiple cellular effects and the mechanism of action includes cell cycle arrest, activation of apoptotic pathway, induction of autophagy, reactive oxygen species generation, and angiogenesis.
HDACi mediated tumor cell death is mainly due to induction of apoptosis, which occurs through intrinsic (mitochondrial) or extrinsic (death receptor) pathways, both of which lead to caspase activation and cell death. Extrinsic pathway is initiated by binding of ligands, such as Fas ligand (FasL), tumor necrosis factor (TNF) and TNF-related apoptosis-inducing ligand (TRAIL) to their respective cell surface death receptors (DR), whereas intrinsic pathways are activated by disruption of mitochondrial membranes by cellular stresses such as chemotherapy, ionizing radiation, and withdrawal of growth factors [165]. Suberic bishydroxamate induces apoptosis in melanoma cells by the upregulation of Bim, Bax, Bak, while down regulating the expression of anti-apoptotic X-linked inhibitor of apoptosis, B-cell lymphoma-extra-large (Bcl-xL) and myeloid cell leukemia 1 (Mcl-1) [166]. Vorinostat treatment caused the general transcriptional induction of BH3-only pro-apoptotic protein encoding genes (Bad, Bim, Bix, Noxa), the multi-domain pro-apoptotic gene BAK1 and genes encoding death effector components downstream of mitochondrial damage (Diablo, Apaf1, Casp9, HtrA2 and CytC) in transformed fibroblasts [167]. Besides, the pro-survival genes, such as Bcl2A1, Bcl2L1 (encoding Bcl-xL) and Bcl2L2 (encoding Bcl-w), were concomitantly repressed in these cells. HDACi upregulated the expression of pro-apoptotic proteins Bmf, Bid, and Bim that belong to the Bcl2 family, and down regulated the expression of the anti-apoptotic proteins of the Bcl2 family such as Bcl2 and Bcl-x [21].
HDACi can also induce cell cycle arrest at G1/S or G2/M transition, leading to differentiation and/or apoptosis. HDACi-mediated increase in CDK inhibitor p21WAF1/CIP1 expression leads to cell cycle arrest at G1/S [168]. Silencing of HDAC3 has been found to induce the expression of p21WAF1/CIP1 and cell cycle arrest in the G2/M phase in colon cancer cells [169]. Vorinostat was found to promote cell cycle arrest at G1/S and G2/M and subsequent apoptosis of leukemic K562, HL60 and THP1 cells [170].
In another mechanism of action, HDACi can block tumor angiogenesis by inhibition of hypoxia inducible factors (HIF). Hypoxia upregulates gene expression of VEGF by stabilizing the transcription factor HIF 1α and tumor suppressor gene Von Hippel Lindau (VHL) degrades HIF 1α. Under hypoxic conditions trichostatin A (TSA) has been shown to upregulate VHL and p53 while downregulating VEGF and HIF 1α to block angiogenesis [171]. HDACi also contribute to the anti-angiogenic pathway by disrupting Hsp90 mediated chaperone function and exposing HIF 1α to proteosomal degradation [172].
HDAC inhibitors indirectly damage DNA by inducing changes in chromatin conformation upon histone acetylation that might expose the DNA to UV rays, ionizing radiation, ROS and chemotherapeutic genotoxic chemicals. This complex biochemical reaction can eventually lead to double strand breaks (DSBs) in DNA. The pan HDACi, vorinostat was shown to induce DSBs in normal (HFS) and cancer (LNCaP, A549) cells [173]. Normal cells in contrast to cancer cells repair the DSBs despite continued culture with vorinostat, whereas in transformed cells the level of biomarker of DBSs in DNA, phosphorylated histone variant γH2AX, increased with continued culture with vorinostat. DSBs are repaired by two independent pathways, homologous recombination (HR) and non-homologous end joining (NHEJ). HDACi can downregulate the levels of DNA repair proteins, such as Ku70 and Ku86 that are involved in NHEJ pathway [174,175]. Similarly HDACi suppressed the gene expression of DNA repair proteins like RAD51, BRCA1 and BRCA2 [176].
Generation of reactive oxygen species (ROS) is another key event in HDACi induced cell death, causing DNA damage. Free radical scavengers like N-acetylcysteine reduce ROS generation which in turn abrogates HDACi mediated cell death [173,177]. HDACi increase ROS production through downregulation of thioredoxin (Trx), a thiol reductase that acts as a scavenger of ROS, and through upregulation of thioredoxin binding protein-2 (TBP-2), a protein that binds to Trx and blocks its reducing activity [178]. Treatment with vorinostat induced TBP-2 expression followed by suppression of Trx expression [179]. Together, these multifaceted mechanisms by which HDACi act upon cancer cell survival and death are depicted in Figure 4.
Figure 4.

Multiple anti-tumor pathways activated by HDACi. Extrinsic and intrinsic refer to two apoptosis pathways, and HR and NHEJ refer to two DBS repair pathways.
11. How to Obtain Novel HDAC Inhibitors?
In addition to the four well-known structural classes of HDAC inhibitors, there exist other HDAC inhibitors with different zinc binding groups, including thioesters, epoxides, epxoyketones, thiols, dithiols, ketones, hydroxypyridinethiones and hydroxypyridone. Many other structural classes that can inhibit HDAC activity may exist as well. How do we identify novel compounds that belong to these structural classes or to entirely new structural classes that can act as an anticancer agent by inhibiting the HDAC activity? Traditional high-throughput screening of libraries of compounds to identify potential inhibitors is an important and effective tool commonly used in pharmaceutical industry. In this method, tens to hundreds of thousands of small molecules are tested against a given assay to discover various novel drugs. However, this approach can be very expensive and resource intensive. In this regard, a variety of computational techniques can help to reduce the size of chemical library by focusing on those compounds that are predicted by in silico modeling to be most likely active. They include both structure-based and ligand-based drug design methods. In the structure-based drug design method, three dimensional structural information of a drug target interacting with small molecules is used to guide drug discovery, whereas the ligand-based method uses information about known ligands of a drug target of interest.
12. Molecular Modeling Based Studies
The efforts to discover more efficient and selective HDAC inhibitors have been continually intensified ever since HDAC inhibitors were found active in various clinical trials. Computer modeling has played a critical role in understanding the enzyme-drug interactions, identifying potent inhibitors and obtaining quantitative structure activity relationships of HDAC inhibitors. To this end, both ligand and structure based drug design methods have been employed. For example, in an effort to optimize the structural analogues of cyclopeptide FR235222, an HDAC inhibitor, molecular docking studies were conducted with cis and trans isomers of 10 analogues of FR235222 and a homologous protein of HDAC1 [180]. This study provided possible bioactive conformation and revealed the contribution of hydrophobic interactions to the stability of the complex. Molecular docking is a rapid process to predict the bioactive conformation of a compound in the active site of a target protein. This method is routinely used to gain insight into the interaction between the enzyme and its inhibitors, especially when the crystal structure of the complex is not available. Towards this goal, several docking studies have been reported in the literature [181,182,183]. The structural details obtained from docking studies can be used for guiding structural modifications of the inhibitors to discover more potent and specific inhibitors for different isoforms of HDAC, or for rational drug design. The same strategy can also be applied to the HDAC inhibitors that are in clinical trials for guiding structural modification to make the drug more potent and isoform specific HDAC inhibitors with potentially reduced toxicities.
For the structural modification, computer-aided scaffold replacement method can be used wherein a portion of the molecule could be replaced, or a group might be added to achieve a particular polar or steric interaction that might enhance the binding affinity. Molecular dynamics (MD) simulation is a computer method to mimic atomic and molecular interactions and observe the structural fluctuations with respect to time. Molecular dynamics simulations of chemically diverse HDAC inhibitors (SAHA, PCI-34051 and C16) and the HDAC isoforms (8, 10 and 11) of the three different classes of zinc-dependent enzyme were also done [184]. The best binding poses from the docking studies were used as the initial structures in the 5 ns MD simulations. MD simulations provided an insight into the interactions between the HDAC and the inhibitor at the molecular level. From this study, it was found that the experimental activities are mainly determined by hydrogen bonds formed by the inhibitor particularly by the metal binding part of the inhibitors and aromatic interactions observed at the tunnel and surface of the active site. Also, the calculated non-bonded interaction energies between the inhibitor and catalytic residues revealed that the subtle difference in the amino acids at the highly conserved active sites of HDAC isoform (M274 in HDAC8, E272 in HDAC10 and L268 in HDAC11) is responsible for the selectivity observed in different HDAC inhibitors. The importance of conserved tunnel forming amino acids and their influence in maintaining the integrity of the tunnel in respective isoforms were also studied by 5 ns MD simulations of wild type HDAC8, 10, and 11, and two mutants (L268M and L268E) of HDAC11 [185]. Another MD simulation study showed continuous opening and closing of hydrophobic active site channel (HASC), affecting the affinity of valproic acid to the HASC [186]. At the same time, the affinity of valproic acid toward the HASC was consistently higher than that obtained for the catalytic site, which suggested that the HASC could be involved in the mechanism of inhibition. Similarly, several MD simulation studies have been conducted to explore the structural and dynamic characterizations of different isoforms of HDAC and specific inhibitors [187,188]. Thus MD simulations of HDAC enzyme in complex with the HDAC inhibitors, especially those made to clinical studies, can aid in understanding the mechanism of action.
Ligand and structure based virtual screening (VS) techniques are also widely used in finding inhibitors of HDACs. Virtual screening is a computer-based method to process compounds from small molecule databases and to identify compounds that are likely to inhibit the biological activity of a particular therapeutic target. Compounds selected by this method should yield higher proportion of active compounds, than a random selection of the same number of molecules. Ligand and structure based VS methods were employed to identify novel non-hydroxamate HDAC inhibitors from the NCI2000 and Maybridge databases [189]. Based on a hit molecule identified by the VS method, three series of compounds were synthesized and evaluated for both HDAC1 inhibitory activity and cytotoxicity to human breast adenocarcinoma MCF-7 cells and human umbilical vein endothelial cells (HUVEC) [189]. Virtual screening against an HDAC6 homology model using the Maybridge database had identified a new HDAC6 selective inhibitor and a carbamate derivative that acts as a prodrug in cell culture, for hydroxamate derived HDAC inhibitors [190]. Similarly, another in silico screening of a database containing 167,000 compounds identified one compound with an IC50 of 1.6 μM against HDAC8 [191]. By means of virtual screening with docking simulations, six novel HDAC inhibitors with IC50 values ranging from 1 to 100 μM were identified [192]. These inhibitors were structurally diverse and had various chelating groups for the active site zinc ion, including N-[1,3,4]thiadiazol-2-yl sulfonamide, N-thiazol-2-yl sulfonamide, and hydroxamic acid moieties. In fact, a number of studies have used VS as a supporting tool for identifying potential inhibitors of a given enzyme for other diseases as well. For example, a potential inhibitor of Schistosoma mansoni HDAC8 (smHDAC8) for the treatment of schistosomiasis, a parasitic disease caused by blood flukes of the genus Schistosoma, was identified by screening the Enamine purchasable compound library [193]. The molecules exhibited an inhibitory effect on smHDAC8, and had the capacity to induce apoptosis and mortality in schistosomes.
Synthesis of several putative structures and arriving at a clinically important therapeutic agent involves arduous and careful procedures. At the same time, high-throughput screening of chemical libraries is expensive and resource intensive. Under such conditions, virtual screening of chemical libraries provides an alternative approach to finding active chemical entities and structural scaffolds for the development of novel cancer therapeutic agents. The inexpensive virtual screening method employs either a target based or a ligand based approach. The target based approach uses molecular docking procedure. Since the crystal structures of many isoforms of HDAC are already available, the target based screening can easily be carried out. Homology models of HDAC enzymes can also be used if the crystal structure of a specific isoform is unavailable. For our advantages a number of free small molecule libraries are available for screening, which can later be purchased for testing in vitro and in vivo studies.
Ligand based virtual screening detects the most effective biologically active lead compound by searching for compounds that have structural or topological similarity or pharmacophoric similarity to a biologically active compound. In our laboratory, using shape-based screening, we have succeeded in finding a lead molecule that led to the discovery of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion [194,195]. Ligand based virtual screening method has proven to be successful in other studies as well [196,197]. Shape based screening is capable of identifying new lead with similar shape as well as electrostatic properties to a lead query molecule. Using HDAC inhibitors that are already in clinical trials as the lead query molecules, shape based screening can identify new structural scaffolds with entirely different zinc binding domain, linker, or cap groups. Indeed, using this approach we have identified several hundreds of potential candidates with various structurally distinct zinc binding domain, linker and cap groups that are currently being tested by HDAC inhibitor assays.
Docking and energy-optimized pharmacophore mapping of several known HDAC inhibitors identified structural variants that are significant for interactions against Class I HDAC enzymes [198]. Apparently inhibitors with at least one aromatic ring in their linker regions showed higher affinities towards the target enzymes, whereas inhibitors without any aromatic rings were poor binders. In this method the ligand-based pharmacophore modeling and structure based protein-ligand docking are combined to rapidly screen small molecule libraries. The energy descriptors obtained from docking are mapped on to pharmacophore feature sites, which allows the sites to be quantified and ranked on the basis of the energetic terms. In the end this method leads to a final energy minimized pharmacophore hypothesis. Thus this method offers the advantages of both structure-based and ligand-based drug design methods. The same protocol was also used in identifying structural variations that regulate the interaction of HDAC inhibitors against Class II HDAC enzymes [199]. It was shown that inhibitors possessing higher number of aromatic rings in different structural regions might function better. A docking-enabled pharmacophore model also identified HDAC8 inhibitors as anticancer agents [200]. In this study, the best docked conformations of each training set compounds were used for the pharmacophore generation and the best pharmacophore model was then used in database screening to identify novel virtual leads.
13. Quantitative Structure Activity Relationship of HDAC Inhibitors
Synthesis of chemical compounds is a costly and resource intensive process. Hence estimation of chemical compounds’ property and/or activity towards a particular enzyme before their synthesis is highly desirable. In this regard, computer-modeling based quantitative structure activity relationship (QSAR) provides a convenient method to predict activity or properties of the molecules of interest. Because of its significant contribution in the drug discovery field, application of both 2D-QSAR and 3D-QSAR modeling has become an integral part of the drug discovery process. For example, 3D QSAR relationships of a series of lactam-based HDAC inhibitor were used for further evaluation of novel lactam-based HDAC inhibitors [201]. This study suggested that HDAC inhibitors which are small in overall size but possess big surface areas with stabilized aromatic cap groups would show better HDAC inhibitory activities [201]. QSAR studies also helped in the design, synthesis and biological evaluation of γ-lactam-based HDAC inhibitors [202,203]. By introducing different cap groups, such as phenyl, naphthyl, and thiophenyl, it was observed that hydrophobic and bulky cap groups can increase the potency of HDAC inhibition because of the hydrophobic interaction between the HDAC and γ-lactam inhibitor. Similarly methoxy and trifluoromethyl substitutions at the ortho-, meta-, para- position of the cap group showed increased HDAC inhibition when the substituent (trifluoromethyl) is more lipophilic. Thus lipophilicity increases the hydrophobic interaction between the surface of HDAC active site and HDAC inhibitor, which in turn improves the HDAC inhibitor activity [203]. A recent survey of published QSAR studies of HDAC inhibitors revealed that the lipophilicity is one of the most important determinants of anticancer activity [204]. Structure activity relationship studies that included the linker region and the surface recognition group to optimize HDAC inhibition identified two lead compounds that are potent inhibitors of HDAC6 and HDAC8, but inactive against HDAC1 [205]. In SAHA-like HDAC inhibitors, a triazole ring that joins the surface recognition cap group to the linker group has shown differential inhibition against HDACs [206]. Structure activity relationships of such triazole-linked hydroxamates displayed a cap group-dependent preference for either five or six methylene spacer groups, and showed several fold greater potency than SAHA. Thus the QSAR studies greatly aid in understanding the factors that affect the biological activity, which can then be applied in rational drug design.
All the molecular modeling techniques described here, including the QSAR studies, provide excellent opportunities to identify potential HDAC inhibitors, either using the known HDAC inhibitors or from scratch, and to guide the structural modifications in the synthesis of novel HDAC inhibitors.
14. Concluding Remarks
HDAC inhibitors represent a promising class of anticancer agents, with three of them now approved for cutaneous and/or peripheral T-cell lymphoma. Many HDAC inhibitors are in different stages of clinical trials for various haematological and solid tumors. While HDAC inhibitors alone have displayed anticancer activities in various cancers, a growing number of studies have demonstrated more efficient and tumor specific anticancer activities of HDAC inhibitors when they are given in combination with other drugs. Even though vorinostat, romidepsin and belinostat are approved for cutaneous and/or peripheral T-cell lymphoma, these drugs are still being studied in clinical trials for other types of cancers, either as single agents or in combination with other drugs. This clearly underscore the potential of HDAC inhibitors in cancer treatment. Besides the promising effects on anticancer activities, the use of HDAC inhibitors in other diseases, such as intestinal fibrosis, autoimmune, inflammatory diseases, metabolic disorders and many more, is also growing.
Though there are different structural classes of HDAC inhibitors, the most common HDAC inhibitors are derivatives of four structural classes; hydroxamic acid, benzamide, short chain fatty acid or cyclic peptides. The pharmacophores of these molecules include a metal-binding moiety, a surface binding moiety and a linker connecting them. Presence of an aromatic ring in the linker region seems to enhance the affinity towards the target enzyme. Similarly, hydrophobic and bulky cap groups that bind to the surface region in the HDAC can increase the inhibitor potency. In general, lipophilicity plays an important role in determining the anticancer activity of HDAC inhibitors.
Having discovered the clinical benefits of HDAC inhibitors in various diseases, especially in cancers, there is an increasing need to develop more potent and tumor-specific HDAC inhibitors. However, disruption of multiple pathways by these inhibitors and the lack of specificity of these inhibitors to a target enzyme could contribute to the cytotoxicities that were found in many of the clinical trials. Computational modeling tools, such as docking and molecular dynamics simulations, provide an alternative ways to look into the molecular level interactions between the target enzyme and the inhibitors. Structure activity relationship determines chemical groups in a drug molecule that are responsible for evoking the biological activity of a target enzyme. A combination of docking, molecular dynamics simulations, structure activity relationships and pharamacophore models greatly assist in developing more potent and enzyme specific HDAC inhibitors. Virtual screening has also assisted in finding inhibitors for a specific HDAC enzyme. Both target based and ligand based virtual screening methods are recommended for identifying novel isoform specific HDAC inhibitors. Ligand based virtual screening that identifies new leads with similar shape and electrostatic properties to a lead query molecule, including HDAC inhibitors that are in clinical studies or found in nature, is another way to extract new structural classes of HDAC inhibitors as anticancer agents. Scaffold replacement method is another highly suitable approach by which different pharmacophore regions, such as the zinc binding domain, linker and cap region, in known HDAC inhibitors including those in clinical studies can be modified to synthesize more potent and specific HDAC inhibitors. Computer modeling has emerged as a powerful complement to the experimental approach to finding more potent and specific HDAC inhibitors. As such, clinical studies in combination with basic biological research and computer modeling should enable us to discover a greater variety of HDAC inhibitors specific for a given target, and also to develop tumor specific HDAC inhibitors. This review highlights the interplay between computer modeling based research and experimental research that is essential for the development of novel HDAC inhibitors as anticancer agents.
Acknowledgments
This work was supported by the National Institute of Minority Health and Disparities of the National Institute of Health (NIMHD-NIH) through Grant number 2G12MD007595, the National Institute of General Medical Sciences of the National Institute of Health (NIGMS-NIH) through grant number 1U54GM104940 and in part by Louisiana Cancer Research Consortium (LCRC).
Author Contributions
All authors contributed to the manuscript development. M.M. wrote the manuscript. G.W. and T.L.H. edited the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Richmond T.J., Davey C.A. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 2.Khorasanizadeh S. The nucleosome: From genomic organization to genomic regulation. Cell. 2004;116:259–272. doi: 10.1016/S0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Roth S.Y., Denu J.M., Allis C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 5.Marks P.A., Richon V.M., Rifkind R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 6.Gregoretti I.V., Lee Y.M., Goodson H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Brosch G., Loidl P., Graessle S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol. Rev. 2008;32:409–439. doi: 10.1111/j.1574-6976.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X.J., Gregoire S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischle W., Kiermer V., Dequiedt F., Verdin E. The emerging role of class II histone deacetylases. Biochem. Cell Biol. 2001;79:337–348. doi: 10.1139/o01-116. [DOI] [PubMed] [Google Scholar]
- 10.Lian Z.R., Xu Y.F., Wang X.B., Gong J.P., Liu Z.J. Suppression of histone deacetylase 11 promotes expression of il-10 in kupffer cells and induces tolerance following orthotopic liver transplantation in rats. J. Surg. Res. 2012;174:359–368. doi: 10.1016/j.jss.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 11.Villagra A., Cheng F., Wang H.W., Suarez I., Glozak M., Maurin M., Nguyen D., Wright K.L., Atadja P.W., Bhalla K., et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buglio D., Khaskhely N.M., Voo K.S., Martinez-Valdez H., Liu Y.J., Younes A. HDAC11 plays an essential role in regulating ox40 ligand expression in hodgkin lymphoma. Blood. 2011;117:2910–2917. doi: 10.1182/blood-2010-08-303701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glozak M.A., Seto E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor cdt1. J. Biol. Chem. 2009;284:11446–11453. doi: 10.1074/jbc.M809394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frye R.A. Phylogenetic classification of prokaryotic and eukaryotic sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 15.Michan S., Sinclair D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonda D.J., Lee H.G., Camins A., Pallas M., Casadesus G., Smith M.A., Zhu X. The sirtuin pathway in ageing and alzheimer disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2011;10:275–279. doi: 10.1016/S1474-4422(11)70013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houtkooper R.H., Pirinen E., Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu J., Jing H., Lin H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014;6:945–966. doi: 10.4155/fmc.14.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borradaile N.M., Pickering J.G. Nad+, sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009;15:110–117. doi: 10.2174/138161209787185742. [DOI] [PubMed] [Google Scholar]
- 20.Kozako T., Suzuki T., Yoshimitsu M., Arima N., Honda S.I., Soeda S. Anticancer agents targeted to sirtuins. Molecules. 2014;19:20295–20313. doi: 10.3390/molecules191220295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 22.Fraga M.F., Ballestar E., Villar-Garea A., Boix-Chornet M., Espada J., Schotta G., Bonaldi T., Haydon C., Ropero S., Petrie K., et al. Loss of acetylation at lys16 and trimethylation at lys20 of histone h4 is a common hallmark of human cancer. Nat. Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 23.Peng L., Seto E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011;206:39–56. doi: 10.1007/978-3-642-21631-2_3. [DOI] [PubMed] [Google Scholar]
- 24.Singh B.N., Zhang G., Hwa Y.L., Li J., Dowdy S.C., Jiang S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010;10:935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H.J., Bae S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Trans. Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 26.Mann B.S., Johnson J.R., Cohen M.H., Justice R., Pazdur R. Fda approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 27.Piekarz R.L., Frye R., Turner M., Wright J.J., Allen S.L., Kirschbaum M.H., Zain J., Prince H.M., Leonard J.P., Geskin L.J., et al. Phase ii multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whittaker S.J., Demierre M.F., Kim E.J., Rook A.H., Lerner A., Duvic M., Scarisbrick J., Reddy S., Robak T., Becker J.C., et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 29.Pohlman B., Advani R., Duvic M., Hymes K.B., Intragumtornchai T., Lekhakula A., Shpilberg O., Lerner A., Ben-Yehuda D., Hillen U. Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma; Proceedings of the 51st ASH Annual Meeting and Exposition; New Orleans, LA, USA. 5–8 December 2009; p. 920. [Google Scholar]
- 30.Ueda H., Nakajima H., Hori Y., Fujita T., Nishimura M., Goto T., Okuhara M. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. (Tokyo) 1994;47:301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 31.FDA approves Beleodaq to treat rare, aggressive form of non-Hodgkin lympoma. [(accessed on 10 September 2014)]; Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm403929.htm.
- 32.Dokmanovic M., Marks P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 33.Rajak H., Singh A., Raghuwanshi K., Kumar R., Dewangan P.K., Veerasamy R., Sharma P.C., Dixit A., Mishra P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014;21:2642–2664. doi: 10.2174/09298673113209990191. [DOI] [PubMed] [Google Scholar]
- 34.Finnin M.S., Donigian J.R., Cohen A., Richon V.M., Rifkind R.A., Marks P.A., Breslow R., Pavletich N.P. Structures of a histone deacetylase homologue bound to the tsa and saha inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 35.Johnstone R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 36.Kelly W.K., O’Connor O.A., Krug L.M., Chiao J.H., Heaney M., Curley T., MacGregore-Cortelli B., Tong W., Secrist J.P., Schwartz L., et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi W., Lawrence Y.R., Choy H., Werner-Wasik M., Andrews D.W., Evans J.J., Judy K.D., Farrell C.J., Moshel Y., Berger A.C., et al. Vorinostat as a radiosensitizer for brain metastasis: A phase i clinical trial. J. Neurooncol. 2014;118:313–319. doi: 10.1007/s11060-014-1433-2. [DOI] [PubMed] [Google Scholar]
- 38.Sarfstein R., Bruchim I., Fishman A., Werner H. The mechanism of action of the histone deacetylase inhibitor vorinostat involves interaction with the insulin-like growth factor signaling pathway. PLoS One. 2011;6:e24468. doi: 10.1371/journal.pone.0024468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bokhman J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983;15:10–17. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 40.Lax S.F. Molecular genetic pathways in various types of endometrial carcinoma: From a phenotypical to a molecular-based classification. Virchows Arch. 2004;444:213–223. doi: 10.1007/s00428-003-0947-3. [DOI] [PubMed] [Google Scholar]
- 41.Ma T., Galimberti F., Erkmen C.P., Memoli V., Chinyengetere F., Sempere L., Beumer J.H., Anyang B.N., Nugent W., Johnstone D., et al. Comparing histone deacetylase inhibitor responses in genetically engineered mouse lung cancer models and a window of opportunity trial in patients with lung cancer. Mol. Cancer Ther. 2013;12:1545–1555. doi: 10.1158/1535-7163.MCT-12-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saelen M.G., Ree A.H., Kristian A., Fleten K.G., Furre T., Hektoen H.H., Flatmark K. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat. Oncol. 2012;7 doi: 10.1186/1748-717X-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oki Y., Younes A., Copeland A., Hagemeister F., Fayad L.E., McLaughlin P., Shah J., Fowler N., Romaguera J., Kwak L.W., et al. Phase I study of vorinostat in combination with standard chop in patients with newly diagnosed peripheral T-cell lymphoma. Br. J. Haematol. 2013;162:138–141. doi: 10.1111/bjh.12326. [DOI] [PubMed] [Google Scholar]
- 44.Doi T., Hamaguchi T., Shirao K., Chin K., Hatake K., Noguchi K., Otsuki T., Mehta A., Ohtsu A. Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (gi) cancer in a phase i clinical trial. Int. J. Clin. Oncol. 2013;18:87–95. doi: 10.1007/s10147-011-0348-6. [DOI] [PubMed] [Google Scholar]
- 45.Marks P.A. Discovery and development of saha as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 46.Lane A.A., Chabner B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 47.Xu W.S., Parmigiani R.B., Marks P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 48.Coiffier B., Pro B., Prince H.M., Foss F., Sokol L., Greenwood M., Caballero D., Borchmann P., Morschhauser F., Wilhelm M., et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012;30:631–636. doi: 10.1200/JCO.2011.37.4223. [DOI] [PubMed] [Google Scholar]
- 49.Piekarz R.L., Frye R., Prince H.M., Kirschbaum M.H., Zain J., Allen S.L., Jaffe E.S., Ling A., Turner M., Peer C.J., et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117:5827–5834. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karthik S., Sankar R., Varunkumar K., Ravikumar V. Romidepsin induces cell cycle arrest, apoptosis, histone hyperacetylation and reduces matrix metalloproteinases 2 and 9 expression in bortezomib sensitized non-small cell lung cancer cells. Biomed. Pharmacother. 2014;68:327–334. doi: 10.1016/j.biopha.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Robertson F.M., Chu K., Boley K.M., Ye Z., Liu H., Wright M.C., Moraes R., Zhang X., Green T.L., Barsky S.H., et al. The class I HDAC inhibitor romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Ther. Oncol. 2013;10:219–233. [PubMed] [Google Scholar]
- 52.Jones S.F., Infante J.R., Spigel D.R., Peacock N.W., Thompson D.S., Greco F.A., McCulloch W., Burris H.A., 3rd. Phase 1 results from a study of romidepsin in combination with gemcitabine in patients with advanced solid tumors. Cancer Investig. 2012;30:481–486. doi: 10.3109/07357907.2012.675382. [DOI] [PubMed] [Google Scholar]
- 53.Amiri-Kordestani L., Luchenko V., Peer C.J., Ghafourian K., Reynolds J., Draper D., Frye R., Woo S., Venzon D., Wright J., et al. Phase i trial of a new schedule of romidepsin in patients with advanced cancers. Clin. Cancer Res. 2013;19:4499–4507. doi: 10.1158/1078-0432.CCR-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poole R.M. Belinostat: First global approval. Drugs. 2014;74:1543–1554. doi: 10.1007/s40265-014-0275-8. [DOI] [PubMed] [Google Scholar]
- 55.Ramalingam S.S., Belani C.P., Ruel C., Frankel P., Gitlitz B., Koczywas M., Espinoza-Delgado I., Gandara D. Phase ii study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. 2009;4:97–101. doi: 10.1097/JTO.0b013e318191520c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mackay H.J., Hirte H., Colgan T., Covens A., MacAlpine K., Grenci P., Wang L., Mason J., Pham P.A., Tsao M.S., et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer. 2010;46:1573–1579. doi: 10.1016/j.ejca.2010.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giaccone G., Rajan A., Berman A., Kelly R.J., Szabo E., Lopez-Chavez A., Trepel J., Lee M.J., Cao L., Espinoza-Delgado I., et al. Phase ii study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011;29:2052–2059. doi: 10.1200/JCO.2010.32.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cashen A., Juckett M., Jumonville A., Litzow M., Flynn P.J., Eckardt J., LaPlant B., Laumann K., Erlichman C., DiPersio J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS) Ann. Hematol. 2012;91:33–38. doi: 10.1007/s00277-011-1240-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dizon D.S., Blessing J.A., Penson R.T., Drake R.D., Walker J.L., Johnston C.M., Disilvestro P.A., Fader A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2012;125:367–371. doi: 10.1016/j.ygyno.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dizon D.S., Damstrup L., Finkler N.J., Lassen U., Celano P., Glasspool R., Crowley E., Lichenstein H.S., Knoblach P., Penson R.T. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int. J. Gynecol. Cancer. 2012;22:979–986. doi: 10.1097/IGC.0b013e31825736fd. [DOI] [PubMed] [Google Scholar]
- 61.Kirschbaum M.H., Foon K.A., Frankel P., Ruel C., Pulone B., Tuscano J.M., Newman E.M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A california cancer consortium study. Leuk. Lymphoma. 2014;55:2301–2304. doi: 10.3109/10428194.2013.877134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas A., Rajan A., Szabo E., Tomita Y., Carter C.A., Scepura B., Lopez-Chavez A., Lee M.J., Redon C.E., Frosch A., et al. A phase I/II trial of belinostat in combination with cisplatin, doxorubicin and cyclophosphamide in thymic epithelial tumors: A clinical and translational study. Clin. Cancer Res. 2014;20:5392–5402. doi: 10.1158/1078-0432.CCR-14-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma B.B., Sung F., Tao Q., Poon F.F., Lui V.W., Yeo W., Chan S.L., Chan A.T. The preclinical activity of the histone deacetylase inhibitor PXD101 (belinostat) in hepatocellular carcinoma cell lines. Investig. New Drugs. 2010;28:107–114. doi: 10.1007/s10637-009-9219-7. [DOI] [PubMed] [Google Scholar]
- 64.Savickiene J., Treigyte G., Valiuliene G., Stirblyte I., Navakauskiene R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anticancer Drugs. 2014;25:938–949. doi: 10.1097/CAD.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 65.O’Connor O.A., Heaney M.L., Schwartz L., Richardson S., Willim R., MacGregor-Cortelli B., Curly T., Moskowitz C., Portlock C., Horwitz S., et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 66.Kelly W.K., Richon V.M., O’Connor O., Curley T., MacGregor-Curtelli B., Tong W., Klang M., Schwartz L., Richardson S., Rosa E., et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]
- 67.Duvic M., Talpur R., Ni X., Zhang C., Hazarika P., Kelly C., Chiao J.H., Reilly J.F., Ricker J.L., Richon V.M., et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, saha) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richon V. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer. 2006;95:S2–S6. doi: 10.1038/sj.bjc.6603463. [DOI] [Google Scholar]
- 69.Sholler G.S., Currier E.A., Dutta A., Slavik M.A., Illenye S.A., Mendonca M.C.F., Dragon J., Roberts S.S., Bond J.P. PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma. J. Cancer Ther. Res. 2013;2 doi: 10.7243/2049-7962-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fouliard S., Robert R., Jacquet-Bescond A., du Rieu Q.C., Balasubramanian S., Loury D., Loriot Y., Hollebecque A., Kloos I., Soria J.C., et al. Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I. Eur. J. Cancer. 2013;49:2791–2797. doi: 10.1016/j.ejca.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 71.Lopez G., Liu J., Ren W., Wei W., Wang S., Lahat G., Zhu Q.S., Bornmann W.G., McConkey D.J., Pollock R.E., et al. Combining pci-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin. Cancer Res. 2009;15:3472–3483. doi: 10.1158/1078-0432.CCR-08-2714. [DOI] [PubMed] [Google Scholar]
- 72.Quintas-Cardama A., Kantarjian H., Estrov Z., Borthakur G., Cortes J., Verstovsek S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012;36:1124–1127. doi: 10.1016/j.leukres.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Razak A.R., Hotte S.J., Siu L.L., Chen E.X., Hirte H.W., Powers J., Walsh W., Stayner L.A., Laughlin A., Novotny-Diermayr V., et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of sb939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer. 2011;104:756–762. doi: 10.1038/bjc.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zorzi A.P., Bernstein M., Samson Y., Wall D.A., Desai S., Nicksy D., Wainman N., Eisenhauer E., Baruchel S. A phase i study of histone deacetylase inhibitor, pracinostat (sb939), in pediatric patients with refractory solid tumors: Ind203 a trial of the ncic ind program/c17 pediatric phase i consortium. Pediatr. Blood Cancer. 2013;60:1868–1874. doi: 10.1002/pbc.24694. [DOI] [PubMed] [Google Scholar]
- 75.Brunetto A.T., Ang J.E., Lal R., Olmos D., Molife L.R., Kristeleit R., Parker A., Casamayor I., Olaleye M., Mais A., et al. First-in-human, pharmacokinetic and pharmacodynamic phase i study of resminostat, an oral histone deacetylase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2013;19:5494–5504. doi: 10.1158/1078-0432.CCR-13-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mandl-Weber S., Meinel F.G., Jankowsky R., Oduncu F., Schmidmaier R., Baumann P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (mm) cells. Br. J. Haematol. 2010;149:518–528. doi: 10.1111/j.1365-2141.2010.08124.x. [DOI] [PubMed] [Google Scholar]
- 77.Walewski J., Paszkiewicz-Kozik E., Borsaru G., Moicean A., Warszewska A., Strobel K., Biggi A., Hauns B., Mais A., Henning S.W. Resminostat in Relapsed or Refractory Hodgkin Lymphoma: Initial Results of the Saphire phase II Trial with a Novel Oral Histone Deacetylase (HDAC) Inhibitor; Proceedings of the 52nd ASH Annual Meeting and Exposition; Orlando, FL, USA. 4–7 December 2010. [Google Scholar]
- 78.Walewski J., Paszkiewicz-Kozik E., Warszewska A., Borsaru G., Moicean A., Hellmann A., Mayer J., Hauns B., Mais A., Henning S.W. Final Results of the Phase II Saphire Trial of Resminostat (4sc-201) in Patients with Relapsed/Refractory Hodgkin Lymphoma; Proceedings of the 53rd ASH Annual Meeting and Exposition; San Diego, CA, USA. 10–13 December 2011. [Google Scholar]
- 79.Bitzer M., Ganten T.M., Woerns M.A., Siveke J.T., Dollinger M.M., Scheulen M.E., Wege H., Giannini E.G., Cillo U., Trevisani F. Resminostat in advanced hepatocellular carcinoma (HCC): Overall survival subgroup analysis of prognostic factors in the shelter trial. J. Clin. Oncol. 2013;31:e15088. [Google Scholar]
- 80.Rambaldi A., Dellacasa C.M., Finazzi G., Carobbio A., Ferrari M.L., Guglielmelli P., Gattoni E., Salmoiraghi S., Finazzi M.C., di Tollo S., et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with jak2v617f positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010;150:446–455. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 81.Amaru Calzada A., Pedrini O., Finazzi G., Leoni F., Mascagni P., Introna M., Rambaldi A., Golay J. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2(V617F) myeloproliferative neoplasm patients. Exp. Hematol. 2013;41:253–260. doi: 10.1016/j.exphem.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 82.Furlan A., Monzani V., Reznikov L.L., Leoni F., Fossati G., Modena D., Mascagni P., Dinarello C.A. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat) Mol. Med. 2011;17:353–362. doi: 10.2119/molmed.2011.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Finazzi G., Vannucchi A.M., Martinelli V., Ruggeri M., Nobile F., Specchia G., Pogliani E.M., Olimpieri O.M., Fioritoni G., Musolino C., et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013;161:688–694. doi: 10.1111/bjh.12332. [DOI] [PubMed] [Google Scholar]
- 84.De Marinis F., Atmaca A., Tiseo M., Giuffreda L., Rossi A., Gebbia V., D’Antonio C., dal Zotto L., Al-Batran S.E., Marsoni S., et al. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer. J. Thorac. Oncol. 2013;8:1091–1094. doi: 10.1097/JTO.0b013e318293d88c. [DOI] [PubMed] [Google Scholar]
- 85.Mascarenhas J., Lu M., Li T., Petersen B., Hochman T., Najfeld V., Goldberg J.D., Hoffman R. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF) Br. J. Haematol. 2013;161:68–75. doi: 10.1111/bjh.12220. [DOI] [PubMed] [Google Scholar]
- 86.Morita S., Oizumi S., Minami H., Kitagawa K., Komatsu Y., Fujiwara Y., Inada M., Yuki S., Kiyota N., Mitsuma A., et al. Phase I dose-escalating study of panobinostat (LBH589) administered intravenously to japanese patients with advanced solid tumors. Investig. New Drugs. 2012;30:1950–1957. doi: 10.1007/s10637-011-9751-0. [DOI] [PubMed] [Google Scholar]
- 87.Fukutomi A., Hatake K., Matsui K., Sakajiri S., Hirashima T., Tanii H., Kobayashi K., Yamamoto N. A phase I study of oral panobinostat (LBH589) in japanese patients with advanced solid tumors. Investig. New Drugs. 2012;30:1096–1106. doi: 10.1007/s10637-011-9666-9. [DOI] [PubMed] [Google Scholar]
- 88.Younes A., Sureda A., Ben-Yehuda D., Zinzani P.L., Ong T.C., Prince H.M., Harrison S.J., Kirschbaum M., Johnston P., Gallagher J., et al. Panobinostat in patients with relapsed/refractory hodgkin’s lymphoma after autologous stem-cell transplantation: Results of a phase II study. J. Clin. Oncol. 2012;30:2197–2203. doi: 10.1200/JCO.2011.38.1350. [DOI] [PubMed] [Google Scholar]
- 89.Platzbecker U., Al-Ali H.K., Gattermann N., Haase D., Janzen V., Krauter J., Gotze K., Schlenk R., Nolte F., Letsch A., et al. Phase 2 study of oral panobinostat (LBH589) with or without erythropoietin in heavily transfusion-dependent ipss low or int-1 mds patients. Leukemia. 2014;28:696–698. doi: 10.1038/leu.2013.325. [DOI] [PubMed] [Google Scholar]
- 90.Dimicoli S., Jabbour E., Borthakur G., Kadia T., Estrov Z., Yang H., Kelly M., Pierce S., Kantarjian H., Garcia-Manero G. Phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in patients with low or intermediate-1 risk myelodysplastic syndrome. Am. J. Hematol. 2012;87:127–129. doi: 10.1002/ajh.22198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lai C.J., Bao R., Tao X., Wang J., Atoyan R., Qu H., Wang D.G., Yin L., Samson M., Forrester J., et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70:3647–3656. doi: 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu T., LoRusso P.M., Papadopoulos K.P., Patnaik A., Beeram M., Smith L.S., Rasco D.W., Mays T.A., Chambers G., Ma A., et al. Phase I first-in-human study of cudc-101, a multitargeted inhibitor of HDACS, egfr, and her2 in patients with advanced solid tumors. Clin. Cancer Res. 2014;20:5032–5040. doi: 10.1158/1078-0432.CCR-14-0570. [DOI] [PubMed] [Google Scholar]
- 93.Fu S., Nemunaitis J.J., Bessudo A., Bauman J.E., Hamid O., Witta S.E., Dy G.K., Lai C., Laliberte R., Voi M. A phase Ib study of CUDC-101, A Multitargeted Inhibitor of EGFR, HER2, and HDAC, in Patients with Advanced Head and Neck, Gastric, Breast, Liver, and Non-Small Cell Lung Cancer; Proceedings of 2012 ASCO Annual Meeting; Chicago, IL, USA. 1–5 June 2012. [Google Scholar]
- 94.Voi M., Fu S., Nemunaitis J., Bauman J., Bessudo A., Hamid O., Witta S., Dy G., Lai C., Laliberte R. 590 Final results of a phase Ib study of CUDC-101, a multitargeted inhibitor of EGFR, HER2, and HDAC, in patients with advanced head and neck, gastric, breast, liver, and non-small cell lung cancer. Eur. J. Cancer. 2012;48 doi: 10.1016/S0959-8049(12)72387-0. [DOI] [Google Scholar]
- 95.ClinicalTrials.gov. [(accessed on 1 September 2014)]; Available online: https://clinicaltrials.Gov.
- 96.Fakih M.G., Groman A., McMahon J., Wilding G., Muindi J.R. A randomized phase II study of two doses of vorinostat in combination with 5-fu/lv in patients with refractory colorectal cancer. Cancer Chemother. Pharmacol. 2012;69:743–751. doi: 10.1007/s00280-011-1762-1. [DOI] [PubMed] [Google Scholar]
- 97.Fakih M.G., Fetterly G., Egorin M.J., Muindi J.R., Espinoza-Delgado I., Zwiebel J.A., Litwin A., Holleran J.L., Wang K., Diasio R.B. A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin. Cancer Res. 2010;16:3786–3794. doi: 10.1158/1078-0432.CCR-10-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mahalingam D., Mita M., Sarantopoulos J., Wood L., Amaravadi R.K., Davis L.E., Mita A.C., Curiel T.J., Espitia C.M., Nawrocki S.T., et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–1414. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Millward M., Price T., Townsend A., Sweeney C., Spencer A., Sukumaran S., Longenecker A., Lee L., Lay A., Sharma G., et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig. New Drugs. 2012;30:2303–2317. doi: 10.1007/s10637-011-9766-6. [DOI] [PubMed] [Google Scholar]
- 100.Mazumder A., Vesole D.H., Jagannath S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010;10:149–151. doi: 10.3816/CLML.2010.n.022. [DOI] [PubMed] [Google Scholar]
- 101.Wilson P.M., El-Khoueiry A., Iqbal S., Fazzone W., LaBonte M.J., Groshen S., Yang D., Danenberg K.D., Cole S., Kornacki M., et al. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-fu-based chemotherapy. Cancer Chemother. Pharmacol. 2010;65:979–988. doi: 10.1007/s00280-009-1236-x. [DOI] [PubMed] [Google Scholar]
- 102.Gressette M., Vérillaud B., Jimenez-Pailhès A.-S., Lelièvre H., Lo K.-W., Ferrand F.-R., Gattolliat C.-H., Jacquet-Bescond A., Kraus-Berthier L., Depil S. Treatment of nasopharyngeal carcinoma cells with the histone-deacetylase inhibitor abexinostat: Cooperative effects with cis-platin and radiotherapy on patient-derived xenografts. PLoS One. 2014;9:e91325. doi: 10.1371/journal.pone.0091325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fournel M., Bonfils C., Hou Y., Yan P.T., Trachy-Bourget M.C., Kalita A., Liu J., Lu A.H., Zhou N.Z., Robert M.F., et al. Mgcd0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 104.Garcia-Manero G., Assouline S., Cortes J., Estrov Z., Kantarjian H., Yang H., Newsome W.M., Miller W.H., Jr., Rousseau C., Kalita A., et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor mgcd0103 in leukemia. Blood. 2008;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang G., He J., Zhao J., Yun W., Xie C., Taub J.W., Azmi A., Mohammad R.M., Dong Y., Kong W., et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS One. 2012;7:e52095. doi: 10.1371/journal.pone.0052095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blum K.A., Advani A., Fernandez L., van der Jagt R., Brandwein J., Kambhampati S., Kassis J., Davis M., Bonfils C., Dubay M., et al. Phase ii study of the histone deacetylase inhibitor mgcd0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2009;147:507–514. doi: 10.1111/j.1365-2141.2009.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siu L.L., Pili R., Duran I., Messersmith W.A., Chen E.X., Sullivan R., MacLean M., King S., Brown S., Reid G.K., et al. Phase I study of mgcd0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J. Clin. Oncol. 2008;26:1940–1947. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Younes A., Oki Y., Bociek R.G., Kuruvilla J., Fanale M., Neelapu S., Copeland A., Buglio D., Galal A., Besterman J., et al. Mocetinostat for relapsed classical hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12:1222–1228. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pili R., Salumbides B., Zhao M., Altiok S., Qian D., Zwiebel J., Carducci M.A., Rudek M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer. 2012;106:77–84. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Witta S.E., Jotte R.M., Konduri K., Neubauer M.A., Spira A.I., Ruxer R.L., Varella-Garcia M., Bunn P.A., Jr., Hirsch F.R. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012;30:2248–2255. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yardley D.A., Ismail-Khan R.R., Melichar B., Lichinitser M., Munster P.N., Klein P.M., Cruickshank S., Miller K.D., Lee M.J., Trepel J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013;31:2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gore L., Rothenberg M.L., O’Bryant C.L., Schultz M.K., Sandler A.B., Coffin D., McCoy C., Schott A., Scholz C., Eckhardt S.G. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, ms-275, in patients with refractory solid tumors and lymphomas. Clin. Cancer Res. 2008;14:4517–4525. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Su J.M., Li X.N., Thompson P., Ou C.N., Ingle A.M., Russell H., Lau C.C., Adamson P.C., Blaney S.M. Phase 1 study of valproic acid in pediatric patients with refractory solid or cns tumors: A children’s oncology group report. Clin. Cancer Res. 2011;17:589–597. doi: 10.1158/1078-0432.CCR-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mohammed T.A., Holen K.D., Jaskula-Sztul R., Mulkerin D., Lubner S.J., Schelman W.R., Eickhoff J., Chen H., Loconte N.K. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist. 2011;16:835–843. doi: 10.1634/theoncologist.2011-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wheler J.J., Janku F., Falchook G.S., Jackson T.L., Fu S., Naing A., Tsimberidou A.M., Moulder S.L., Hong D.S., Yang H., et al. Phase I study of anti-vegf monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014;73:495–501. doi: 10.1007/s00280-014-2384-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chu B.F., Karpenko M.J., Liu Z., Aimiuwu J., Villalona-Calero M.A., Chan K.K., Grever M.R., Otterson G.A. Phase i study of 5-aza-2'-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013;71:115–121. doi: 10.1007/s00280-012-1986-8. [DOI] [PubMed] [Google Scholar]
- 117.Iwahashi S., Utsunomiya T., Imura S., Morine Y., Ikemoto T., Arakawa Y., Saito Y., Ishikawa D., Shimada M. Effects of valproic acid in combination with s-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014;34:5187–5191. [PubMed] [Google Scholar]
- 118.Bauman J., Shaheen M., Verschraegen C.F., Belinsky S.A., Houman Fekrazad M., Lee F.C., Rabinowitz I., Ravindranathan M., Jones D.V., Jr. A phase i protocol of hydralazine and valproic acid in advanced, previously treated solid cancers. Trans. Oncol. 2014;7:349–354. doi: 10.1016/j.tranon.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prakash S., Foster B.J., Meyer M., Wozniak A., Heilbrun L.K., Flaherty L., Zalupski M., Radulovic L., Valdivieso M., LoRusso P.M. Chronic oral administration of ci-994: A phase 1 study. Investig. New Drugs. 2001;19:1–11. doi: 10.1023/A:1006489328324. [DOI] [PubMed] [Google Scholar]
- 120.Yoshida M., Kijima M., Akita M., Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin a. J. Biol. Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 121.Lobera M., Madauss K.P., Pohlhaus D.T., Wright Q.G., Trocha M., Schmidt D.R., Baloglu E., Trump R.P., Head M.S., Hofmann G.A., et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013;9:319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 122.Fennell K.A., Mollmann U., Miller M.J. Syntheses and biological activity of amamistatin b and analogs. J. Org. Chem. 2008;73:1018–1024. doi: 10.1021/jo7020532. [DOI] [PubMed] [Google Scholar]
- 123.Fennell K.A., Miller M.J. Syntheses of amamistatin fragments and determination of their HDAC and antitumor activity. Org. Lett. 2007;9:1683–1685. doi: 10.1021/ol070382e. [DOI] [PubMed] [Google Scholar]
- 124.Davie J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003;133:2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 125.Duan H., Heckman C.A., Boxer L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aoyama M., Kotani J., Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating gpr-41/gpr-43 pathways. Nutrition. 2010;26:653–661. doi: 10.1016/j.nut.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 127.Petrella A., D’Acunto C.W., Rodriquez M., Festa M., Tosco A., Bruno I., Terracciano S., Taddei M., Paloma L.G., Parente L. Effects of fr235222, a novel HDAC inhibitor, in proliferation and apoptosis of human leukaemia cell lines: Role of annexin a1. Eur. J. Cancer. 2008;44:740–749. doi: 10.1016/j.ejca.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 128.De Schepper S., Bruwiere H., Verhulst T., Steller U., Andries L., Wouters W., Janicot M., Arts J., van Heusden J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J. Pharmacol. Exp. Ther. 2003;304:881–888. doi: 10.1124/jpet.102.042903. [DOI] [PubMed] [Google Scholar]
- 129.Kwon S.H., Ahn S.H., Kim Y.K., Bae G.U., Yoon J.W., Hong S., Lee H.Y., Lee Y.W., Lee H.W., Han J.W. Apicidin, a histone deacetylase inhibitor, induces apoptosis and fas/fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002;277:2073–2080. doi: 10.1074/jbc.M106699200. [DOI] [PubMed] [Google Scholar]
- 130.Nakao Y., Yoshida S., Matsunaga S., Shindoh N., Terada Y., Nagai K., Yamashita J.K., Ganesan A., van Soest R.W., Fusetani N. Azumamides a-e: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge mycale izuensis. Angew. Chem. Int. Ed. Engl. 2006;45:7553–7557. doi: 10.1002/anie.200602047. [DOI] [PubMed] [Google Scholar]
- 131.Villadsen J.S., Stephansen H.M., Maolanon A.R., Harris P., Olsen C.A. Total synthesis and full histone deacetylase inhibitory profiling of azumamides a-e as well as beta(2)- epi-azumamide e and beta(3)-epi-azumamide e. J. Med. Chem. 2013;56:6512–6520. doi: 10.1021/jm4008449. [DOI] [PubMed] [Google Scholar]
- 132.Furumai R., Komatsu Y., Nishino N., Khochbin S., Yoshida M., Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin a and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA. 2001;98:87–92. doi: 10.1073/pnas.98.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taori K., Paul V.J., Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the floridian marine cyanobacterium symploca sp. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 134.Rehman M.U., Jawaid P., Yoshihisa Y., Li P., Zhao Q.L., Narita K., Katoh T., Kondo T., Shimizu T. Spiruchostatin a and b, novel histone deacetylase inhibitors, induce apoptosis through reactive oxygen species-mitochondria pathway in human lymphoma u937 cells. Chem. Biol. Interact. 2014;221:24–34. doi: 10.1016/j.cbi.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 135.Weinlander E., Somnay Y., Harrison A.D., Wang C., Cheng Y.Q., Jaskula-Sztul R., Yu X.M., Chen H. The novel histone deacetylase inhibitor thailandepsin a inhibits anaplastic thyroid cancer growth. J. Surg. Res. 2014;190:191–197. doi: 10.1016/j.jss.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Venturelli S., Berger A., Bocker A., Busch C., Weiland T., Noor S., Leischner C., Schleicher S., Mayer M., Weiss T.S., et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS One. 2013;8:e73097. doi: 10.1371/journal.pone.0073097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nian H., Delage B., Pinto J.T., Dashwood R.H. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances sp3 binding on the p21waf1 promoter. Carcinogenesis. 2008;29:1816–1824. doi: 10.1093/carcin/bgn165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Druesne N., Pagniez A., Mayeur C., Thomas M., Cherbuy C., Duee P.H., Martel P., Chaumontet C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 139.Ho E., Clarke J.D., Dashwood R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009;139:2393–2396. doi: 10.3945/jn.109.113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Salvador L.A., Park H., Al-Awadhi F.H., Liu Y., Kim B., Zeller S.L., Chen Q.Y., Hong J., Luesch H. Modulation of activity profiles for largazole-based HDAC inhibitors through alteration of prodrug properties. ACS Med. Chem. Lett. 2014;5:905–910. doi: 10.1021/ml500170r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hassig C.A., Symons K.T., Guo X., Nguyen P.M., Annable T., Wash P.L., Payne J.E., Jenkins D.A., Bonnefous C., Trotter C., et al. Kd5170, a novel mercaptoketone-based histone deacetylase inhibitor that exhibits broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008;7:1054–1065. doi: 10.1158/1535-7163.MCT-07-2347. [DOI] [PubMed] [Google Scholar]
- 142.Payne J.E., Bonnefous C., Hassig C.A., Symons K.T., Guo X., Nguyen P.M., Annable T., Wash P.L., Hoffman T.Z., Rao T.S., et al. Identification of kd5170: A novel mercaptoketone-based histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2008;18:6093–6096. doi: 10.1016/j.bmcl.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 143.Baud M.G., Leiser T., Petrucci V., Gunaratnam M., Neidle S., Meyer-Almes F.J., Fuchter M.J. Thioester derivatives of the natural product psammaplin a as potent histone deacetylase inhibitors. Beilstein J. Org. Chem. 2013;9:81–88. doi: 10.3762/bjoc.9.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Salvador L.A., Luesch H. Discovery and mechanism of natural products as modulators of histone acetylation. Curr. Drug Targets. 2012;13:1029–1047. doi: 10.2174/138945012802008973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kwon H.J., Owa T., Hassig C.A., Shimada J., Schreiber S.L. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc. Natl. Acad. Sci. USA. 1998;95:3356–3361. doi: 10.1073/pnas.95.7.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kijima M., Yoshida M., Sugita K., Horinouchi S., Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 147.Du L., Risinger A.L., King J.B., Powell D.R., Cichewicz R.H. A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J. Nat. Prod. 2014;77:1753–1757. doi: 10.1021/np500387h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frey R.R., Wada C.K., Garland R.B., Curtin M.L., Michaelides M.R., Li J., Pease L.J., Glaser K.B., Marcotte P.A., Bouska J.J., et al. Trifluoromethyl ketones as inhibitors of histone deacetylase. Bioorg. Med. Chem. Lett. 2002;12:3443–3447. doi: 10.1016/S0960-894X(02)00754-0. [DOI] [PubMed] [Google Scholar]
- 149.Jose B., Oniki Y., Kato T., Nishino N., Sumida Y., Yoshida M. Novel histone deacetylase inhibitors: Cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg. Med. Chem. Lett. 2004;14:5343–5346. doi: 10.1016/j.bmcl.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 150.Madsen A.S., Kristensen H.M., Lanz G., Olsen C.A. The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem. 2014;9:614–626. doi: 10.1002/cmdc.201300433. [DOI] [PubMed] [Google Scholar]
- 151.Bishton M.J., Harrison S.J., Martin B.P., McLaughlin N., James C., Josefsson E.C., Henley K.J., Kile B.T., Prince H.M., Johnstone R.W. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood. 2011;117:3658–3668. doi: 10.1182/blood-2010-11-318055. [DOI] [PubMed] [Google Scholar]
- 152.Galli M., Salmoiraghi S., Golay J., Gozzini A., Crippa C., Pescosta N., Rambaldi A. A phase II multiple dose clinical trial of histone deacetylase inhibitor itf2357 in patients with relapsed or progressive multiple myeloma. Ann. Hematol. 2010;89:185–190. doi: 10.1007/s00277-009-0793-8. [DOI] [PubMed] [Google Scholar]
- 153.Miller T.A., Witter D.J., Belvedere S. Histone deacetylase inhibitors. J. Med. Chem. 2003;46:5097–5116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 154.Somoza J.R., Skene R.J., Katz B.A., Mol C., Ho J.D., Jennings A.J., Luong C., Arvai A., Buggy J.J., Chi E., et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 155.Vannini A., Volpari C., Filocamo G., Casavola E.C., Brunetti M., Renzoni D., Chakravarty P., Paolini C., de Francesco R., Gallinari P., et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lauffer B.E., Mintzer R., Fong R., Mukund S., Tam C., Zilberleyb I., Flicke B., Ritscher A., Fedorowicz G., Vallero R., et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013;288:26926–26943. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kapustin G.V., Fejer G., Gronlund J.L., McCafferty D.G., Seto E., Etzkorn F.A. Phosphorus-based saha analogues as histone deacetylase inhibitors. Org. Lett. 2003;5:3053–3056. doi: 10.1021/ol035056n. [DOI] [PubMed] [Google Scholar]
- 158.Gu W., Nusinzon I., Smith R.D., Jr., Horvath C.M., Silverman R.B. Carbonyl- and sulfur-containing analogs of suberoylanilide hydroxamic acid: Potent inhibition of histone deacetylases. Bioorg. Med. Chem. 2006;14:3320–3329. doi: 10.1016/j.bmc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 159.Saito A., Yamashita T., Mariko Y., Nosaka Y., Tsuchiya K., Ando T., Suzuki T., Tsuruo T., Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA. 1999;96:4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Phiel C.J., Zhang F., Huang E.Y., Guenther M.G., Lazar M.A., Klein P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 161.Luo Y., Liu H.M., Su M.B., Sheng L., Zhou Y.B., Li J., Lu W. Synthesis and biological evaluation of piperamide analogues as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011;21:4844–4846. doi: 10.1016/j.bmcl.2011.06.046. [DOI] [PubMed] [Google Scholar]
- 162.Taddei M., Cini E., Giannotti L., Giannini G., Battistuzzi G., Vignola D., Vesci L., Cabri W. Lactam based 7-amino suberoylamide hydroxamic acids as potent HDAC inhibitors. Bioorg. Med. Chem. Lett. 2014;24:61–64. doi: 10.1016/j.bmcl.2013.11.072. [DOI] [PubMed] [Google Scholar]
- 163.Tashima T., Murata H., Kodama H. Design and synthesis of novel and highly-active pan-histone deacetylase (pan-HDAC) inhibitors. Bioorg. Med. Chem. 2014;22:3720–3731. doi: 10.1016/j.bmc.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 164.Schrump D.S., Fischette M.R., Nguyen D.M., Zhao M., Li X., Kunst T.F., Hancox A., Hong J.A., Chen G.A., Kruchin E., et al. Clinical and molecular responses in lung cancer patients receiving romidepsin. Clin. Cancer Res. 2008;14:188–198. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 165.Johnstone R.W., Ruefli A.A., Lowe S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 166.Zhang X.D., Gillespie S.K., Borrow J.M., Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol. Cancer Ther. 2004;3:425–435. doi: 10.4161/cbt.3.5.985. [DOI] [PubMed] [Google Scholar]
- 167.Bolden J.E., Shi W., Jankowski K., Kan C.Y., Cluse L., Martin B.P., MacKenzie K.L., Smyth G.K., Johnstone R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang Z., Hao C., Wang L., Liu P., Zhao L., Zhu C., Tian X. Inhibition of leukemic cells by valproic acid, an HDAC inhibitor, in xenograft tumors. Onco. Targets Ther. 2013;6:733–740. doi: 10.2147/OTT.S46135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wilson A.J., Byun D.S., Popova N., Murray L.B., L’Italien K., Sowa Y., Arango D., Velcich A., Augenlicht L.H., Mariadason J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 170.Silva G., Cardoso B.A., Belo H., Almeida A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS One. 2013;8:e53766. doi: 10.1371/journal.pone.0053766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., Moon E.J., Kim H.S., Lee S.K., Chung H.Y., et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 172.Qian D.Z., Kachhap S.K., Collis S.J., Verheul H.M., Carducci M.A., Atadja P., Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res. 2006;66:8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 173.Lee J.H., Choy M.L., Ngo L., Foster S.S., Marks P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA. 2010;107:14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Munshi A., Kurland J.F., Nishikawa T., Tanaka T., Hobbs M.L., Tucker S.L., Ismail S., Stevens C., Meyn R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005;11:4912–4922. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 175.Chen C.S., Wang Y.C., Yang H.C., Huang P.H., Kulp S.K., Yang C.C., Lu Y.S., Matsuyama S., Chen C.Y. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67:5318–5327. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 176.Adimoolam S., Sirisawad M., Chen J., Thiemann P., Ford J.M., Buggy J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA. 2007;104:19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rosato R.R., Almenara J.A., Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63:3637–3645. [PubMed] [Google Scholar]
- 178.Marks P.A. Thioredoxin in cancer—Role of histone deacetylase inhibitors. Semin. Cancer Biol. 2006;16:436–443. doi: 10.1016/j.semcancer.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Butler L.M., Zhou X., Xu W.S., Scher H.I., Rifkind R.A., Marks P.A., Richon V.M. The histone deacetylase inhibitor saha arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA. 2002;99:11700–11705. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Di Micco S., Terracciano S., Bruno I., Rodriquez M., Riccio R., Taddei M., Bifulco G. Molecular modeling studies toward the structural optimization of new cyclopeptide-based HDAC inhibitors modeled on the natural product FR235222. Bioorg. Med. Chem. 2008;16:8635–8642. doi: 10.1016/j.bmc.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 181.Wang S., Li X., Wei Y., Xiu Z., Nishino N. Discovery of potent HDAC inhibitors based on chlamydocin with inhibitory effects on cell migration. ChemMedChem. 2014;9:627–637. doi: 10.1002/cmdc.201300372. [DOI] [PubMed] [Google Scholar]
- 182.Neelarapu R., Holzle D.L., Velaparthi S., Bai H., Brunsteiner M., Blond S.Y., Petukhov P.A. Design, synthesis, docking, and biological evaluation of novel diazide-containing isoxazole- and pyrazole-based histone deacetylase probes. J. Med. Chem. 2011;54:4350–4364. doi: 10.1021/jm2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Olson D.E., Wagner F.F., Kaya T., Gale J.P., Aidoud N., Davoine E.L., Lazzaro F., Weiwer M., Zhang Y.L., Holson E.B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013;56:4816–4820. doi: 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- 184.Thangapandian S., John S., Lee K.W. Molecular dynamics simulation study explaining inhibitor selectivity in different class of histone deacetylases. J. Biomol. Struct. Dyn. 2012;29:677–698. doi: 10.1080/07391102.2012.10507409. [DOI] [PubMed] [Google Scholar]
- 185.Thangapandian S., John S., Lee Y., Arulalapperumal V., Lee K.W. Molecular modeling study on tunnel behavior in different histone deacetylase isoforms. PLoS One. 2012;7:e49327. doi: 10.1371/journal.pone.0049327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bermudez-Lugo J.A., Perez-Gonzalez O., Rosales-Hernandez M.C., Ilizaliturri-Flores I., Trujillo-Ferrara J., Correa-Basurto J. Exploration of the valproic acid binding site on histone deacetylase 8 using docking and molecular dynamic simulations. J. Mol. Model. 2012;18:2301–2310. doi: 10.1007/s00894-011-1240-z. [DOI] [PubMed] [Google Scholar]
- 187.Estiu G., West N., Mazitschek R., Greenberg E., Bradner J.E., Wiest O. On the inhibition of histone deacetylase 8. Bioorg. Med. Chem. 2010;18:4103–4110. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kalyaanamoorthy S., Chen Y.P. Ligand release mechanisms and channels in histone deacetylases. J. Comp. Chem. 2013;34:2270–2283. doi: 10.1002/jcc.23390. [DOI] [PubMed] [Google Scholar]
- 189.Lu H., Chen Y.-D., Yang B., You Q.-D. Design, synthesis and biological evaluation of novel histone deacetylase inhibitors based on virtual screening. Acta Pharm. Sin. B. 2011;1:240–247. doi: 10.1016/j.apsb.2011.10.002. [DOI] [Google Scholar]
- 190.Schlimme S., Hauser A.T., Carafa V., Heinke R., Kannan S., Stolfa D.A., Cellamare S., Carotti A., Altucci L., Jung M., et al. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem. 2011;6:1193–1198. doi: 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- 191.Zhang L., Li M., Feng J., Fang H., Xu W. Discovery of a novel histone deacetylase 8 inhibitor by virtual screening. Med. Chem. Res. 2012;21:152–156. doi: 10.1007/s00044-010-9519-7. [DOI] [Google Scholar]
- 192.Park H., Kim S., Kim Y.E., Lim S.J. A structure-based virtual screening approach toward the discovery of histone deacetylase inhibitors: Identification of promising zinc-chelating groups. ChemMedChem. 2010;5:591–597. doi: 10.1002/cmdc.200900500. [DOI] [PubMed] [Google Scholar]
- 193.Marek M., Kannan S., Hauser A.T., Moraes Mourao M., Caby S., Cura V., Stolfa D.A., Schmidtkunz K., Lancelot J., Andrade L., et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke schistosoma mansoni. PLoS Pathog. 2013;9:e1003645. doi: 10.1371/journal.ppat.1003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zheng S., Zhong Q., Jiang Q., Mottamal M., Zhang Q., Zhu N., Burow M.E., Worthylake R.A., Wang G. Discovery of a series of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion. ACS Med. Chem. Lett. 2013;4:191–196. doi: 10.1021/ml300322n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zheng S., Zhong Q., Xi Y., Mottamal M., Zhang Q., Schroeder R.L., Sridhar J., He L., McFerrin H., Wang G. Modification and biological evaluation of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion. J. Med. Chem. 2014;57:6653–6667. doi: 10.1021/jm500724x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Canela M.D., Perez-Perez M.J., Noppen S., Saez-Calvo G., Diaz J.F., Camarasa M.J., Liekens S., Priego E.M. Novel colchicine-site binders with a cyclohexanedione scaffold identified through a ligand-based virtual screening approach. J. Med. Chem. 2014;57:3924–3938. doi: 10.1021/jm401939g. [DOI] [PubMed] [Google Scholar]
- 197.Svajger U., Brus B., Turk S., Sova M., Hodnik V., Anderluh G., Gobec S. Novel toll-like receptor 4 (TLR4) antagonists identified by structure- and ligand-based virtual screening. Eur. J. Med. Chem. 2013;70:393–399. doi: 10.1016/j.ejmech.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 198.Kalyaanamoorthy S., Chen Y.P. Energy based pharmacophore mapping of HDAC inhibitors against class I HDAC enzymes. Biochim. Biophys. Acta. 2013;1834:317–328. doi: 10.1016/j.bbapap.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 199.Ganai S.A., Shanmugam K., Mahadevan V. Energy-optimised pharmacophore approach to identify potential hotspots during inhibition of class II HDAC isoforms. J. Biomol. Struct. Dyn. 2015;33:374–387. doi: 10.1080/07391102.2013.879073. [DOI] [PubMed] [Google Scholar]
- 200.Thangapandian S., John S., Sakkiah S., Lee K.W. Docking-enabled pharmacophore model for histone deacetylase 8 inhibitors and its application in anti-cancer drug discovery. J. Mol. Graph. Model. 2010;29:382–395. doi: 10.1016/j.jmgm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 201.Yang J.S., Chun T.-G., Nam K.-Y., Kim H.M., Han G. Structure-activity relationship of novel lactam based histone deacetylase inhibitors as potential anticancer drugs. Bull. Korean Chem. Soc. 2012;33:2063–2066. doi: 10.5012/bkcs.2012.33.6.2063. [DOI] [Google Scholar]
- 202.Choi E., Lee C., Park J.E., Seo J.J., Cho M., Kang J.S., Kim H.M., Park S.K., Lee K., Han G. Structure and property based design, synthesis and biological evaluation of gamma-lactam based HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011;21:1218–1221. doi: 10.1016/j.bmcl.2010.12.079. [DOI] [PubMed] [Google Scholar]
- 203.Lee C., Choi E., Cho M., Lee B., Oh S.J., Park S.K., Lee K., Kim H.M., Han G. Structure and property based design, synthesis and biological evaluation of gamma-lactam based HDAC inhibitors: Part II. Bioorg. Med. Chem. Lett. 2012;22:4189–4192. doi: 10.1016/j.bmcl.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 204.Pontiki E., Hadjipavlou-Litina D. Histone deacetylase inhibitors (HDACIs). Structure—Activity relationships: History and new qsar perspectives. Med. Res. Rev. 2012;32:1–165. doi: 10.1002/med.20200. [DOI] [PubMed] [Google Scholar]
- 205.Sodji Q.H., Patil V., Kornacki J.R., Mrksich M., Oyelere A.K. Synthesis and structure-activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors. J. Med. Chem. 2013;56:9969–9981. doi: 10.1021/jm401225q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chen P.C., Patil V., Guerrant W., Green P., Oyelere A.K. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 2008;16:4839–4853. doi: 10.1016/j.bmc.2008.03.050. [DOI] [PubMed] [Google Scholar]