

Abstract
Oxidation of methionine leads to the formation of the S and R diastereomers of methionine sulfoxide (MetO), which can be reversed by the actions of two structurally unrelated classes of methionine sulfoxide reductase (Msr), MsrA and MsrB, respectively. Although MsrAs have long been demonstrated in numerous bacteria, their physiological and biochemical functions remain largely unknown in Actinomycetes. Here, we report that a Corynebacterium glutamicum methionine sulfoxide reductase A (CgMsrA) that belongs to the 3-Cys family of MsrAs plays important roles in oxidative stress resistance. Deletion of the msrA gene in C. glutamicum resulted in decrease of cell viability, increase of ROS production, and increase of protein carbonylation levels under various stress conditions. The physiological roles of CgMsrA in resistance to oxidative stresses were corroborated by its induced expression under various stresses, regulated directly by the stress-responsive extracytoplasmic-function (ECF) sigma factor SigH. Activity assays performed with various regeneration pathways showed that CgMsrA can reduce MetO via both the thioredoxin/thioredoxin reductase (Trx/TrxR) and mycoredoxin 1/mycothione reductase/mycothiol (Mrx1/Mtr/MSH) pathways. Site-directed mutagenesis confirmed that Cys56 is the peroxidatic cysteine that is oxidized to sulfenic acid, while Cys204 and Cys213 are the resolving Cys residues that form an intramolecular disulfide bond. Mrx1 reduces the sulfenic acid intermediate via the formation of an S-mycothiolated MsrA intermediate (MsrA-SSM) which is then recycled by mycoredoxin and the second molecule of mycothiol, similarly to the glutathione/glutaredoxin/glutathione reductase (GSH/Grx/GR) system. However, Trx reduces the Cys204-Cys213 disulfide bond in CgMsrA produced during MetO reduction via the formation of a transient intermolecular disulfide bond between Trx and CgMsrA. While both the Trx/TrxR and Mrx1/Mtr/MSH pathways are operative in reducing CgMsrA under stress conditions in vivo, the Trx/TrxR pathway alone is sufficient to reduce CgMsrA under normal conditions. Based on these results, a catalytic model for the reduction of CgMsrA by Mrx1 and Trx is proposed.
INTRODUCTION
Aerobic metabolism and oxidative stress lead to the production and accumulation of reactive oxygen species (ROS). ROS induce oxidative damage to macromolecules, such as proteins, lipids, and DNA, ultimately inducing cell death and disease. In proteins, oxidation modification occurs mainly at the sulfur-containing amino acids, such as cysteine (Cys) and methionine (Met) (1). Oxidation of methionine leads to the formation of the R and S diastereomers of methionine sulfoxide (MetO) and results in either conformational change or loss of function of proteins (2). However, this oxidation can be reversed by the action of methionine sulfoxide reductase (Msr), a ubiquitous monomeric enzyme that reduces both free and protein-bound MetO by employing sulfenic acid chemistry (3). Two structurally unrelated classes of Msrs, MsrAs and MsrBs, reduce the S and R MetO diastereoisomers, respectively. MsrA has been implicated in physiological processes, such as protection of cells against oxidative damage and extension of the life span, and acts as a virulence factor in some bacterial pathogens (4–6).
MsrAs can be classified into three groups, 3-Cys, 2-Cys, and 1-Cys MsrAs, according to the number of redox-active Cys residues. Three-Cys MsrAs, such as the Escherichia coli and Bos taurus enzymes, have three Cys residues involved in catalysis, one of which functions as a catalytic residue and the other two as resolving residues (7, 8). Two-Cys MsrAs, such as the Saccharomyces cerevisiae and Streptococcus pneumoniae enzymes, possess a single resolving Cys in addition to the catalytic Cys (4, 9). The 3-Cys and 2-Cys MsrAs present similar catalytic mechanisms. First, the catalytic Cys residue attacks a sulfoxide moiety of the substrate and then forms a sulfenic acid intermediate, with a concomitant release of Met as a product. Second, the sulfenic acid of the catalytic Cys forms an intramolecular disulfide bond with a resolving Cys. For 2-Cys MsrAs, this disulfide bond is directly reduced by a reducing agent to regenerate the active form of the enzyme, such as thioredoxin (Trx), which is a natural reducing agent for Msrs. For 3-Cys MsrAs, which perform an extra thiol-disulfide exchange reaction, the first disulfide bond is further reduced by the second resolving Cys, resulting in the formation of a disulfide bond, eventually reduced by Trx, between the two resolving Cys residues. One-Cys MsrAs, such as the MsrA from Synechocystis sp. and selenoprotein MsrAs from Clostridium and Chlamydomonas, containing only the catalytic Cys, use different mechanisms for the regeneration of their activity (10–12). Some 1-Cys Msrs, such as Arabidopsis thaliana MsrB1 (AtMsrB1) and Clostridium sp. MsrA, can be reduced by a glutathione/glutaredoxin/glutathione reductase (GSH/Grx/GR) system (11, 13, 14). For AtMsrB1, the sulfenic acid is reduced by GSH, forming a glutathionylated intermediate, which is further attacked by Grx for reduction (14). Some mammalian and plant 1-Cys Msrs can also be reduced directly by Trx (15, 16).
Corynebacterium glutamicum, a Gram-positive bacterium of the family Actinomycetes widely used for the industrial production of amino acids and nucleotides, is also an ideal model organism in industrial biotechnology and systems biology (17). To survive under hostile conditions, C. glutamicum has developed a wide range of resistance systems. Instead of GSH, C. glutamicum produces mycothiol (MSH) (chemically, 1d-myo-inosityl-2-[N-acetyl-l-cysteinyl] amido-2-deoxy-γ-d-glucopyranoside) as the major low-molecular-weight thiol, which plays an important role in the defense against external stresses, including oxidative stress, alkylating agents, and antibiotics (18, 19) and can be considered equivalent to GSH. C. glutamicum also possesses a large battery of antioxidant enzymes, including Trx, mycoredoxin 1 (Mrx1), NrdH, and catalase, which act by scavenging the superoxide anion and H2O2 to prevent ROS-induced damage or by reducing disulfides or sulfenic acid of proteins under oxidative stress (20, 21). Although MsrAs have been extensively studied for decades, they have been poorly characterized in MSH-containing, high-G+C content, Gram-positive Actinobacteria, including a group of bacteria of medical, industrial, and environmental significance, such as members of the genera Corynebacterium, Mycobacterium, Rhodococcus, and Streptomyces. Therefore, this study was undertaken to determine whether C. glutamicum MsrA (CgMsrA) affects the survival of C. glutamicum under oxidative stress. At the same time, biochemical studies to investigate the capacities of C. glutamicum Trx and Mrx1 to serve as the reducing power for CgMsrA were also conducted.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. C. glutamicum and E. coli strains were cultured in Luria-Bertani (LB) broth aerobically on a rotary shaker (220 rpm) or on LB plates at 30 and 37°C, respectively. The ability to utilize MetO as the sole Met source was tested on mineral salts medium containing 10 μg/ml MetO as described previously (22–24). To construct in-frame deletion mutants, the pK18mobsacB derivatives were transformed into C. glutamicum RES167 by electroporation and screened as previously described (22). For overexpression or complementation in various C. glutamicum strains, the pXMJ19 derivatives were transformed into relevant C. glutamicum strains by electroporation, and expression in C. glutamicum was induced by the addition of 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strains or plasmids | Relevant genotype description | Reference |
|---|---|---|
| C. glutamicum | ||
| RES167 | Restriction-deficient mutant of ATCC 13032, Δ(cglIM-cglIR-cglIIR) | 57 |
| ΔmsrA | msrA deleted in RES167 | This study |
| Δmrx1 | mrx1 deleted in RES167 | This study |
| ΔmshC | mshC deleted in RES167 | 58 |
| ΔsigH | sigH deleted in RES167 | 59 |
| ΔmsrA ΔmshC Δmrx1 | msrA, mrx1, and mshC deleted in RES167 | This study |
| ΔmetE ΔmetH | metE and metH deleted in RES167 | This study |
| ΔmetE ΔmetH ΔmsrA | msrA, metE, and metH deleted in RES167 | This study |
| ΔmetE ΔmetH ΔmshC | metE, metH, and mshC deleted in RES167 | This study |
| ΔmsrA(pXMJ19-msrA) | Complementation of msrA in msrA mutant | This study |
| ΔmsrA ΔmshC Δmrx1(pXMJ19-msrA) | Complementation of msrA in ΔmsrA ΔmshC Δmrx1 mutant | This study |
| ΔmsrA ΔmshC Δmrx1(pXMJ19-msrA:C91SC204S) | Complementation of msrA(C91S,C204S) in ΔmsrA ΔmshC Δmrx1 mutant | This study |
| ΔmsrA ΔmshC Δmrx1(pXMJ19-msrA:C91SC213S) | Complementation of msrA(C91S,C213S) in ΔmsrA ΔmshC Δmrx1 mutant | This study |
| ΔmsrA ΔmshC Δmrx1(pXMJ19-msrA:C91SC204SC213S) | Complementation of msrA(C91S,C204S,C213S) in ΔmsrA ΔmshC Δmrx1 mutant | This study |
| ΔmetE ΔmetH ΔmsrA(pXMJ19-msrA) | Complementation of msrA in ΔmetE ΔmetH ΔmsrA mutant | This study |
| ΔmetE ΔmetH ΔmshC(pXMJ19-mshC) | Complementation of mshC in ΔmetE ΔmetH ΔmshC mutant | This study |
| ΔsigH(pXMJ19-sigH) | Complementation of sigH in ΔsigH mutant | This study |
| E. coli | ||
| BL21(DE3) | Host for expression vector pET28a | Novagen |
| JM109 | Host for cloning | Stratagene |
| Plasmids | ||
| pK18mobsacB | Suicide vector; Kmr | 60 |
| pK18mobsacB-ΔmsrA | Construct used for in-frame deletion of msrA | This study |
| pK18mobsacB-ΔsigH | Construct used for in-frame deletion of sigH | 59 |
| pK18mobsacB-Δmrx1 | Construct used for in-frame deletion of mrx1 | This study |
| pK18mobsacB-ΔmetE | Construct used for in-frame deletion of metE | This study |
| pK18mobsacB-ΔmetH | Construct used for in-frame deletion of metH | This study |
| pK18mobsacB-PmsrA::lacZ | PmsrA::lacZ fusion in pK18mobsacB | This study |
| pXMJ19 | Shuttle vector (Ptac lacIq pBL1 oriVCg pK18 oriVEc); Cmr | 61 |
| pXMJ19-msrA | msrA cloned into pXMJ19 for complementation and overexpression | This study |
| pXMJ19-msrA:C91SC204S | msrA(C91S,C204S) cloned into pXMJ19 for complementation | This study |
| pXMJ19-msrA:C91SC213S | msrA(C91S,C213S) cloned into pXMJ19 for complementation | This study |
| pXMJ19-msrA:C91SC204SC213S | msrA(C91S,C204S,C213S) cloned into pXMJ19 for complementation | This study |
| pXMJ19-sigH | sigH cloned into pXMJ19 for complementation | 59 |
| pET28a | Expression vector with N-terminal hexahistidine affinity tag | Novagen |
| pET28a-trx | trx in pET28a | 21 |
| pET28a-trx:C35S | trx(C35S) mutant in pET28a | This study |
| pET28a-trxR | trxR in pET28a | 21 |
| pET28a-msrA | msrA in pET28a | This study |
| pET28a-msrA:C56S | msrA(C56S) in pET28a | This study |
| pET28a-msrA:C91S | msrA(C91S) in pET28a | This study |
| pET28a-msrA:C204S | msrA(C204S) in pET28a | This study |
| pET28a-msrA:C213S | msrA(C213S) in pET28a | This study |
| pET28a-msrA:C56SC91S | msrA(C56S,C91S) in pET28a | This study |
| pET28a-msrA:C91SC204S | msrA(C91S,C204S) in pET28a | This study |
| pET28a-msrA:C91SC213S | msrA(C91S,C213S) in pET28a | This study |
| pET28a-msrA:C56SC204SC213S | msrA(C56S,C204S,C213S) in pET28a | This study |
| pET28a-msrA:C56SC91SC213S | msrA(C56S,C91S,C213S) in pET28a | This study |
| pET28a-msrA:C91SC204SC213S | msrA(C91S,C204S,C213S) in pET28a | This study |
| pET28a-mrx1 | mrx1 in pET28a | 21 |
| pET28a-mrx1:C15S | mrx1(C15S) in pET28a | This study |
| pET28a-mtr | mtr in pET28a | 21 |
| pET28a-sigH | sigH in pET28a | 59 |
Plasmid construction.
The gene encoding CgMsrA (NCgl2825) was amplified by PCR using C. glutamicum RES167 genomic DNA as the template with primers listed in Table 2. These DNA fragments were digested and afterwards subcloned into similarly digested pET28a vectors, obtaining the plasmid pET28a-msrA. Site-directed mutagenesis was carried out by overlap PCR (25) to convert the active-site Cys56 of CgMsrA into Ser [CgMsrA(C56S)]. In brief, the mutant msrA(C56S) DNA segment was amplified by two rounds of PCR. Primer pairs DmsrA-F1/msrA-C56S-R and msrA-C56S-F/DmsrA-R1 were used to amplify segments 1 and 2, respectively. The second round of PCR was carried out by using primer pair msrA-F/msrA-R with fragments 1 and 2 as templates to obtain CgMsrA(C56S), which contained a mutation at Cys56 of the msrA gene. msrA(C91S), msrA(C204S), msrA(C213S), msrA(C56S,C91S), msrA(C91S,C204S), msrA(C91S,C213S), msrA(C56S,C91S,C204S), msrA(C56S,C91S,C213S), msrA(C91S,C204S,C213S), trx(C35S), and mrx1(C15S) fragments were constructed by similar methods. The resulting mutant DNA fragments were digested and cloned into similarly digested pET28a to obtain the corresponding plasmids. The plasmids pK18mobsacB-ΔmsrA, pK18mobsacB-Δmrx1 (mycoredoxin 1; NCgl0808), pK18mobsacB-ΔmetE (5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase; NCgl1094), and pK18mobsacB-ΔmetH (5-methyltetrahydrofolate-homocysteine methyltransferase; NCgl1450) used to construct C. glutamicum deletion mutants were made by overlap PCR (25) with primers listed in Table 2. msrA, msrA(C91S), msrA(C91S,C204S), msrA(C91S,C213S), and msrA(C91S,C204S,C213S) fragments were digested and cloned into the similarly digested pXMJ19 vector to yield the corresponding pXMJ19 derivatives. The lacZ fusion reporter vector pK18mobsacB-PmsrA::lacZ was made by fusion of the msrA promoter to the lacZY reporter gene by overlap PCR with the primer pairs PmsrA-F1/PmsrA-R and lacZY-F/lacZY-R (Table 2). The resulting fusion PCR fragment was digested and inserted into the suicide vector pK18mobsacB to obtain the pK18mobsacB-PmsrA::lacZ fusion construct (25). The fidelity of all the constructs was confirmed by DNA sequencing (Sangon Biotech, Shanghai, China).
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′)a | Purpose |
|---|---|---|
| msrA-F | CGCGGATCCATGGCATGGTTTTTCGCACCCG (BamHI) | For cloning msrA wild type and mutants into pXMJ19 or pET28a |
| msrA-R | ACGCGTCGACTTAGATAGGGAGATTCTCCTC (SalI) | |
| DmsrA-F1 | CGCGGATCCGTTGAAGACTCCAAAAAGGGCATC (HindIII) | To generate pK18mobsacB-ΔmsrA |
| DmsrA-R1 | GCGAAAAACCATGCCATAACAGG | |
| DmsrA-F2 | CCTGTTATGGCATGGTTTTTCGCACAAGAATCCCGATGGCTACTG | |
| DmsrA-R2 | ACATGCATGCGGTCACCTGGTAGTTCGATGTCC (SphI) | |
| PmsrA-F1 | TCCCCCGGGCTGCCCGGCAACAGTGTG (SmaI) | To generate pK18mobsacB-PmsrA::lacZY |
| PmsrA-R | TCTAGAGAAAAACCATGCCATAACAGG (XbaI) | |
| PmsrA-F2 | GCGCCTGCAACGTAGGTTGCG | To gain 400-bp msrA promoter DNA segment |
| lacZY-F | CCTGTTATGGCATGGTTTTTCTCTAGAACTAGTATGACCATGATTACGGATTC(SpeI) | To generate lacZY fragment |
| lacZY-R | AAAACTGCAGTTAAGCGACTTCATTCACCTG (PstI) | |
| msrA-C56S-F | TGGATTGGCCTTGGTAGTTTCTGGGGTGTGG | To generate pXMJ19-msrA:C56S and pET28a-msrA:C56S |
| msrA-C56S-R | CCACACCCCAGAAACTACCAAGGCCAATCCA | |
| msrA-C91S-F | CACTTATCGCGAGGTGAGTTCTGGGCGCACCG | To generate pXMJ19-msrA:C91S and pET28a-msrA:C91S |
| msrA-C91S-R | CGGTGCGCCCAGAACTCACCTCGCGATAAGTG | |
| msrA-C204S-F | GAATCCCGATGGCTACAGCCCTCATCACTCCAC | To generate pXMJ19-msrA:C204S and pET28a-msrA:C204S |
| msrA-C204S-R | GTGGAGTGATGAGGGCTGTAGCCATCGGGATTC | |
| msrA-C213S-F | CCACGGGCATCCCGAGCGGGGTAGAAGCTT | To generate pXMJ19-msrA:C213S and pET28a-msrA:C213S |
| msrA-C213S-R | AAGCTTCTACCCCGCTCGGGATGCCCGTGG | |
| trx-F | CGCGGATCCATGAGCAATGTTGTTGCAGTAAC (BamHI) | To generate pET28a-trx and pET28a-trx:C35S |
| trx-R | CCGCTCGAGCTAGAGGTGCTTCTCTAGTTTTTC (XhoI) | |
| DmetE-F1 | GGAAGATCTATCGATGCGGAGAGCATCAG (BglII) | To generate pK18mobsacB-ΔmetE |
| DmetE-R1 | CCATTCCAGTAGCCTTCGAGCGC | |
| DmetE-F2 | GCGCTCGAAGGCTACTGGAATGGGCTAAGCAGGCTCGTGAGAAAATCG | |
| DmetE-R2 | ACGCGTCGACGTTCTGGTGGGGTGGATCAG (SalI) | |
| DmetH-F1 | CGCGGATCCGCTATTTGCCACTCCGATGTTTG (BamHI) | To generate pK18mobsacB-ΔmetH |
| DmetH-R1 | TGTTGTGGGCTGGTGAAGTAAC | |
| DmetH-F2 | GTTACTTCACCAGCCCACAACATCTACCACCCAGAGGCAAAG | |
| DmetH-R2 | ACGCGTCGACATACTCGGCTGCAATCCAGAC (SalI) | |
| Dtrx-F1 | CGCGGATCC GCTGACCTCAACCCCATCATGTTC (BamHI) | To generate pET28a-trx:C35S |
| Dtrx-R2 | CCCAAGCTT GCTGCTGGGATCATCCATGTGC (HindIII) | |
| trx-C35S-F | GAATGGTGTGGCCCCAGCAAGAAGCTCAGCC | |
| trx-C35S-R | GGCTGAGCTTCTTGCTGGGGCCACACCATTC | |
| mrx1-F | CCGGGATCCATGAGCAACGTAACCATTTACGCC (BamHI) | To generate pET28a-mrx1 and pET28a-mrx1:C15S |
| mrx1-R | CCCGTCGACTTAGGCTAATGCTTCGATTTTGG (SalI) | |
| Dmrx1-F1 | CGCGGATCCATTGTCTCGGCGTGGAATGTTTCC (BamHI) | To generate pK18mobsacB-Δmrx1 and pET28a-mrx1:C15S |
| Dmrx1-R1 | GATTACTTCCGCAGCTAGGGGATTGAGGAGGGATCGGCAGTAAG | |
| Dmrx1-F2 | ATCCCCTAGCTGCGGAAGTAATC | |
| Dmrx1-R2 | ACGCGTCGACACCCATTTGTATTCGCCCGTG (SalI) | |
| mrx1-C15S-F | GATTGGTGCCCTTACAGCCGATCCCTCCTC | To generate pET28a-mrx1:C15S |
| mrx1-C15S-R | GAGGAGGGATCGGCTGTAAGGGCACCAATC | |
| Control-F | ATGACCACCAGCAACCCC | To generate control DNA for EMSA |
| Control-R | CCCTCGGGGGAACTTCCCGG |
Underlining indicates restriction enzyme cutting sites added for cloning. Italics denote the mutation sites in overlap PCR for site-directed mutagenesis.
Overexpression and purification of recombinant proteins.
To express and purify His6-tagged proteins, recombinant pET28a plasmids were transformed into E. coli BL21(DE3). The strains were grown at 37°C in LB medium to an A600 of 0.4, shifted to 22°C, induced with 0.4 mM IPTG, and cultivated for an additional 12 h. Harvested cells were disrupted by sonication and purified with the His·Bind Ni-nitrilotriacetic acid (NTA) resin (Novagen, Madison, WI) according to the manufacturer's instructions. The purified recombinant proteins were dialyzed against phosphate-buffered saline (PBS) overnight at 4°C and stored at −80°C until use. Protein concentrations were determined using the Bradford assay according to the manufacturer's instructions (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA) as the standard.
Sensitivity assays for oxidants, alkylating agents, and heavy metals.
To measure the responses to various stress conditions, overnight-grown cultures of C. glutamicum (LB broth; 30°C) were diluted 100-fold with LB medium and exposed to various oxidants, alkylating agents, and heavy metals at 30°C with shaking for 30 min. After treatment, the cultures were serially diluted, spread on LB plates, and incubated at 30°C for 36 h. Percent survival was calculated as follows: [(CFU ml−1 with stress)/(CFU ml−1 without stress)] × 100.
Measurement of intracellular ROS levels and determination of cellular protein carbonylation.
Intracellular levels of ROS were analyzed by the 2′,7′-dichlorofluorescein diacetate (DCFH-DA)-based method as described previously by Schurig-Briccio et al. (26). Protein carbonylation assays were performed based on the method described by Si et al. (21) and Vinckx et al. (27).
Enzyme assay of CgMsrA.
The CgMsrA activity with MetO as the substrate was determined as described previously (28) by monitoring the rate of NADPH oxidation. In brief, the catalytic properties of CgMsrA were determined using a reduced-Trx-generating system (4 μM thioredoxin reductase [TrxR] and 40 μM Trx) and a Mrx1 system (500 μM MSH, 4 μM mycothione reductase [Mtr], and 40 μM Mrx1) as possible electron donors. The assays were carried out in a total volume of 500 μl containing TE buffer (50 mM Tris-HCl [pH 8.0] and 2 mM EDTA), 250 μM NADPH, 2 μM CgMsrA wild type (WT) or its variants, and an electron donor (the Trx system or Mrx1 system). The reaction was started by the addition of 100 mM MetO following a 5-min preincubation. The catalytic parameters for one substrate were obtained by varying its concentration at saturating concentrations of the other substrate (between 0 and 120 μM for Trx or Mrx1 and between 0 and 150 mM for MetO). NADPH oxidation was monitored as the A340. The activity was determined after subtracting the spontaneous reduction rate observed in the absence of CgMsrA, and the number of micromoles of NADPH oxidized per second per micromole of enzyme (i.e., the turnover number, s−1) was calculated using the molar absorption coefficient of NADPH at 340 nm (ε340) of 6,220 M−1 · cm−1. Three independent experiments were performed at each substrate concentration. The kcat and Km values of CgMsrA for Trx, Mrx1, and MetO were calculated by nonlinear regression using the program GraphPad Prism 5. CgMsrA activity was also detected by using high-performance liquid chromatography (HPLC) assays (29).
AMS analysis of the disulfide bond.
The redox states of CgMsrA WT and its variants were measured by covalent modification with the thiol-reactive probe 4-acetamido-4′-maleimidyldystilbene-2,2′-disulfonic acid (AMS) (Molecular Probes, Eugene, OR) as described previously (30) with minor modifications. In brief, CgMsrA WT and its variants (25 μM) were treated by incubation with 10 mM dithiothreitol (DTT) at room temperature for 30 min, and the excess DTT was removed by ultrafiltration. The resulting proteins were treated at room temperature with 10 mM MetO for 30 min or were directly used as negative controls. After being completely precipitated by trichloroacetic acid (TCA) (10% [wt/vol]), the proteins were redissolved in 100 mM Tris-HCl (pH 8.0) containing 1% SDS and 15 mM AMS (Invitrogen, Carlsbad, CA). After incubation for 2 h in the dark, 10 μM aliquots of the proteins were separated by nonreducing SDS-PAGE.
NBD-Cl analysis of the sulfenic acid state.
To study the formation of cysteine sulfenic acid (Cys-SOH) as a reaction intermediate, CgMsrA(C56S,C91S,C213S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C91S,C204S,C213S) proteins labeled with 4-chloro-7-nitrobenzofurazan (NBD-Cl) were assayed as described previously (31, 32) with minor modifications. The CgMsrA(C56S,C91S,C213S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C91S,C204S,C213S) proteins (25 μM) were pretreated with 10 mM DTT for 30 min and then were made anaerobic by repeated flushing with argon gas and vacuum in alternating cycles for 20 min. An anaerobic solution of NBD-Cl (25 mM in dimethyl sulfoxide [DMSO]) was prepared by bubbling argon through the solution for 10 min. Under anaerobic conditions, the CgMsrA(C56S,C91S,C213S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C91S,C204S,C213S) proteins were divided into two portions, one of which was treated with MetO (50 μM) while the other was directly used as an untreated sample (negative control). The MetO-treated and untreated proteins were incubated with NBD-Cl (5 mM) for 30 min at 25°C in the dark. Excess NBD-Cl was removed by ultrafiltration, and the protein samples were analyzed at 200 to 600 nm (DU 7500 diode array spectrophotometer; Beckman, Fullerton, CA).
Quantitative analysis of sulfhydryl groups.
Free sulfhydryl groups in CgMsrA WT and its variants were determined using 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) (33). The proteins (25 μM) were treated with 10 mM MetO and 10 mM DTT at room temperature for 30 min, followed by removing the DTT or MetO on a PD10 desalting column (GE Healthcare, Piscataway, NJ). The resulting proteins (10 μM) were added to DTNB (2 mM) in 50 mM Tris-HCl buffer (pH 8.0), and the A412 was recorded against a reference DTNB solution (2 mM). The amount of reactive sulfhydryl groups was determined using a molar absorption coefficient at 412 nm (ε412) of 13,600 M−1 cm−1 (34).
S-Mycothiolation of CgMsrA in vivo and in vitro.
Iodoacetamide (IAM)-biotin-tagged, demycothiolated proteins were obtained as described previously (35) with minor modifications. In brief, ΔmsrA(pXMJ19-His6-msrA) and ΔmshC ΔmsrA(pXMJ19-His6-msrA) strains were grown to an A600 of 0.8 aerobically. Each culture was divided into two parts. One was exposed to 55 mM H2O2 for 30 min at 30°C, while the other was grown without H2O2 (negative control). Cells were harvested and resuspended in urea–{3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} (CHAPS) alkylation buffer (100 mM Tris-HCl, pH 8.0, 1 mM EDTA, 8 M urea, 1% CHAPS, 100 mM IAM) for 30 min in the dark prior to sonication on ice. The protein extracts were obtained by centrifugation and continued to be alkylated for 30 min in the dark. His6-CgMsrA was purified with His·Bind Ni-NTA resin (Novagen, Madison, WI) according to the manufacturer's instructions. Purified His6-CgMsrA was demycothiolated using 20 μM recombinant Mrx1 in the presence of 1 mM MSH, 250 μM NADPH, and 10 μM Mtr for 30 min at room temperature. The mixture was then treated with 5 mM biotin-maleimide dissolved in dimethyl sulfoxide for 30 min. Unreacted biotin-maleimide was removed by ice-cold acetone precipitation three times for 1 h each time, followed by centrifugation at 10,000 × g for 30 min. The pellet was redissolved in Tris-HCl buffer, separated using nonreducing SDS-PAGE, and detected by Western blotting with anti-His antibody. The resulting His6-CgMsrA (10 μM) was also separated by reducing and nonreducing SDS-15% PAGE and stained with Coomassie brilliant blue. Protein bands were excised, in-gel digested with trypsin, and analyzed by ABI 4700 matrix-assisted laser desorption ionization–time of flight tandem mass spectrometry (MALDI-TOF MS-MS) (Applied Biosystems, Foster City, CA). S-Mycothiolation of CgMsrA was also performed in vitro according to the method of Chi et al. (36). MSH was purified from C. glutamicum RES167 as described previously (37). CgMsrA(C91S) (50 μM) was incubated with an excess of reduced MSH (500 μM) prior to the addition of 10 mM MetO. After 30 min incubation, the sample was loaded on a PD10 desalting column, and MSH and MetO were removed by washing (50 mM HEPES, pH 8.0, 500 mM NaCl). S-Mycothiolated CgMsrA(C91S) was eluted in the same buffer.
MALDI-TOF MS-MS analysis.
Reduced His6-Mrx1(C15S), His6-Trx(C35S), His6-CgMsrA WT, and His6-CgMsrA variants, MetO-treated His6-CgMsrA variants, and the dead-end intermediate between CgMsrA variants and Trx(C35S) variants were incubated with 10 mM IAM for 30 min at room temperature. The resulting proteins were subjected to nonreducing SDS-PAGE, and Coomassie brilliant blue-stained bands were excised, in-gel digested with trypsin, and analyzed by MALDI-TOF MS (Voyager-DE STR; Applied Biosystems).
Formation and separation of heterodimers.
Assays were performed based on the method of Rouhier et al. (38) with minor modifications. CgMsrA variants (15 μM) and Trx(C35S) or Mrx1(C15S) variants (10 μM) were mixed in the presence of TE buffer (30 mM Tris-HCl, pH 8.0, 1 mM EDTA) to a final volume of 20 μl. This reaction mixture was incubated at room temperature for 15 min before the addition of 100 μM MetO. After incubation for 30 min, the mixture was subjected to SDS-15% PAGE in the presence of an equal volume of nonreducing sample buffer (0.5 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, and bromophenol blue). CgMsrA-SSM (15 μM) was mixed with Mrx1(C15S) (10 μM) in TE buffer to a final volume of 20 μl. The rest of the procedure was the same as described above.
SDS-PAGE analysis of complexes formed between CgMsrA variants and Trx(C35S).
The complex linked with a disulfide bond was formed based on the method described by Lian et al. (39). Trx(C35S), CgMsrA(C91S,C204S,C213S), CgMsrA(C56S,C91S,C213S), and CgMsrA(C56S,C91S,C204S) were reduced by 10 mM DTT and then desalted by ultrafiltration. The DTT-treated Trx(C35S) was incubated with an excess of DTNB at 25°C to form a mixed disulfide between the active-site Cys32 of Trx and DTNB. After removing the excess DTNB, the resulting Trx(C35S) (10 μM) was incubated with 20 μM CgMsrA(C91S,C204S,C213S), CgMsrA(C56S,C91S,C213S), and CgMsrA(C56S,C91SC,204S) at 25°C for 1 h. Each sample was divided into two parts, in the presence or absence of 10 mM DTT, and subjected to SDS-PAGE to test the quantity of the complex.
Construction of chromosomal fusion reporter strains and β-galactosidase activity assay.
The lacZ fusion reporter plasmid pK18mobsacB-PmsrA::lacZ was transformed into the C. glutamicum wild-type, ΔsigH(pXMJ19), and ΔsigH(pXMJ19-sigH) strains by electroporation, and the chromosomal pK18mobsacB-PmsrA::lacZ fusion reporter strain was selected by plating on LB-kanamycin plates (21). The resulting strains were grown in LB medium to an A600 of 0.6 to 0.7 and then treated with different oxidative reagents at various concentrations at 30°C for 30 min. β-Galactosidase activity was assayed with o-nitrophenyl-β-galactoside (ONPG) as the substrate (40). The data presented are the means of triplicate analyses. Statistical analysis was carried out with Student's t test.
EMSA.
The electrophoretic mobility shift assay (EMSA) was performed as described by Hu et al. (41) with minor modifications. In brief, a 400-bp PmsrA fragment was amplified from the msrA promoter region of the corresponding pK18mobsacB-PmsrA::lacZ reporter vector by using primers PmsrA-F2 and PmsrA-R. The reaction mixture (10 μl) contained 20 mM Tris-HCl (pH 7.4), 4 mM MgCl2, 100 mM NaCl, 1 mM DTT, 10% glycerol, 20 ng PmsrA DNA fragment, and increasing concentrations of His6-SigH (0 to 4 μg). The binding reaction mixture, incubated for 30 min at room temperature, was subjected to electrophoresis on a 6% native polyacrylamide gel containing 5% glycerol in 0.5× Tris-borate-EDTA (TBE) buffer, following staining with SYBR green (Promega, Fitchburg, WI). As negative controls, 240-bp fragments from the msrA coding region amplified with primers Control-F and Control-R instead of the msrA promoter and bovine serum albumin (BSA) instead of His6-SigH were included.
Western blot analysis.
Samples resolved by SDS-PAGE were transferred onto polyvinylidene fluoride (PVDF) or nitrocellulose (NC) membranes. After blocking with 4% milk for 2 h at room temperature, the membranes were probed with the appropriate primary antibody at 4°C overnight: anti-His, 1:1,000; antidinitrophenyl, 1:500; streptavidin-horseradish peroxidase (HRP), 1:300. The blots were washed several times in PBST buffer (PBS buffer containing 0.2% Tween 20) and then incubated with a 1:5,000 dilution of horseradish peroxidase-conjugated secondary antibody (Shanghai Genomics Inc., Shanghai, China) in PBST for 1 h. The protein bands were visualized using an ECL plus kit (GE Healthcare, Piscataway, NJ) following the manufacturer's specified protocol.
qRT-PCR analysis.
Quantitative real-time PCR (qRT-PCR) analysis (7500 Fast Real-Time PCR; Applied Biosystems, Foster City, CA) was performed as described previously (42). The primers used are listed in Table 2. For standardization of results, the relative abundance of 16S rRNA was used as the internal standard.
RESULTS
Roles of CgMsrA in oxidative stress response in C. glutamicum.
To confirm whether CgMsrA has antioxidant functions in C. glutamicum, the sensitivities of the wild type, the ΔmsrA mutant, and the complementary strain to H2O2 (100 mM), cumene hydroperoxide (CHP) (11 mM), 2,4-dinitrochlorobenzene (CDNB) (70 mM), and CdCl2 (300 μM) were tested (Fig. 1A). The survival rates of the ΔmsrA mutant decreased by about 31.4 to 52.6% compared to the wild type. However, the hypersensitive phenotype of the ΔmsrA mutant to oxidative stresses was completely reversed by complementation with the msrA gene. In addition, msrA overexpression markedly improved the resistance of the wild-type strain to H2O2, CHP, CDNB, and CdCl2 treatment (Fig. 1A). Hence, CgMsrA plays an important role in oxidative stress resistance.
FIG 1.

Requirement for CgMsrA in resistance to multiple oxidative stresses. (A) Survival rates of C. glutamicum strains after being challenged with H2O2 (100 mM), CHP (11 mM), CDNB (70 mM), and CdCl2 (300 μM) for 30 min. (B) The ROS levels in the indicated C. glutamicum strains were measured by using the DCFH-DA fluorescence determination assay after exposure to oxidative reagents. A.U., arbitrary units. (C) Protein carbonyl contents were analyzed by Western blotting with anti-DNP antibody after exposure to various oxidative reagents for 30 min at 30°C. A parallel run stained with Coomassie brilliant blue is shown in the lower gel. Total proteins were extracted from C. glutamicum wild-type cells and msrA mutant cells. (D) β-Galactosidase analyses of msrA promoter activities using the transcriptional PmsrA::lacZ chromosomal fusion reporter expressed in the indicated strains under stress conditions. (E) Quantitative RT-PCR analyses of msrA expression in the indicated strains under stress conditions. (F) EMSA was carried out to analyze interactions between His6-SigH and the msrA promoter. The increasing amounts of His6-SigH used were 0, 0.5, 1.0, 1.5, 2.5, and 3.5 μg. As negative controls, a 240-bp fragment from the msrA coding open reading frame instead of the msrA promoter (lane 8) and BSA instead of His6-SigH (lane 7) were included in the binding assays.  , free DNA; , major DNA-protein complex. Mean values with standard deviations (SD) (error bars) from at least three repeats are shown. *, P ≤ 0.05; **, P ≤ 0.01.
, free DNA; , major DNA-protein complex. Mean values with standard deviations (SD) (error bars) from at least three repeats are shown. *, P ≤ 0.05; **, P ≤ 0.01.
Previous studies have shown that MsrA overexpression in PC12 cells can reduce ROS levels and prevent loss of mitochondrial membrane potential under hypoxic and hyperoxic conditions, suggesting that a major role of MsrA is detoxifying ROS under oxidative stress conditions (43). To test the function of CgMsrA in ROS reduction upon oxidative stress, ROS levels were examined in various strains using DCFH-DA, a membrane-permeable dye that passively diffuses into cells. As shown in Fig. 1B, the ΔmsrA mutant had a significantly higher level of ROS than the wild type after oxidative stress treatment. However, the ROS level in the ΔmsrA mutant was completely restored to that of the wild type by complementation (Fig. 1B).
ROS escaping from the antioxidant defense system were more apt to react with Cys thiol groups of proteins, which resulted in the formation of irreversible sulfoxidation products, inter- or intraprotein disulfides, and mixed disulfides with low-molecular-weight thiol and eventually led to protein carbonylation (44, 45). Since CgMsrA is involved in reducing ROS accumulation, it was assumed that CgMsrA might also function in protecting against protein oxidation under oxidative stress conditions. To test this assumption, total proteins were isolated from the wild type and the ΔmsrA mutant grown under oxidative stress, and the carbonyl contents of the protein samples were investigated by Western blotting with anti-DNP antibodies. The protein carbonylation of extracts from the wild type was significantly lower than that of the ΔmsrA mutant (Fig. 1C). These results demonstrate that CgMsrA plays important roles in protecting C. glutamicum against oxidative stress induced by different agents via reducing intracellular ROS accumulation and protein carbonylation.
Next, msrA expression in response to various oxidants was investigated by chromosomal PmsrA::lacZ fusion reporter analysis. As shown in Fig. 1D, the promoter activity of PmsrA in the wild type increased by 48.2, 72.5, and 59.9% upon H2O2 (40 mM), CDNB (35 mM), and CdCl2 (75 μM) treatment, respectively, compared to the untreated control. Moreover, the expression of the PmsrA::lacZ fusion exhibited a dose-dependent increase in response to the oxidative reagents tested (Fig. 1D). The induced expression of msrA by various oxidative reagents suggests that a transcriptional regulator might be involved in its regulation. As SigH, the stress-responsive extracytoplasmic-function (ECF) sigma factor, was reported to improve the resistance of C. glutamicum to multiple stresses and to regulate the expression of many oxidative stress resistance genes (42, 46), we further examined the role of SigH in msrA expression. After 30 min exposure to different oxidative stresses, a marked decrease in the level of msrA promoter activity was observed in the ΔsigH mutant compared to the wild-type strain (Fig. 1D). Indeed, the decrease of msrA expression in the ΔsigH mutant was observed even in the absence of H2O2, CDNB, and CdCl2 treatment. Moreover, msrA expression in the ΔsigH mutant was largely recovered when the regulatory protein was complemented (Fig. 1D). A similar pattern of msrA expression in response to various stresses was observed in the qRT-PCR analysis (Fig. 1E). SigH-dependent expression of msrA was further confirmed by determining the direct interaction between His6-SigH and the msrA promoter by EMSA (Fig. 1F). Collectively, these results indicate that multiple oxidative stresses induce SigH-dependent expression of msrA, which in turn directly contributes to cell tolerance for these adverse stresses.
Activities of CgMsrA by using Trx and Mrx1 as electron donors.
As reported previously, when MsrA reduces oxidized Met (free or peptide bound) to reduced Met, the formation of intramolecular bridges or sulfenic acid occurs in oxidized MsrA (29). To accomplish the catalytic cycle of the reaction in vivo, reducing systems were used to regenerate the sulfhydryl group at the active-site Cys of MsrA. Besides the Trx system (Trx/TrxR), C. glutamicum has an alternative Mrx1 reducing system (Mrx1/Mtr/MSH) that is unique to MSH-producing high-G+C Gram-positive Actinobacteria and functionally equivalent to the widely distributed Grx/GR/GSH system in most Gram-negative bacteria (20, 47). The efficiencies of both Trx and Mrx1 systems in reducing MetO by CgMsrA were investigated. As shown in Fig. 2A, the ability of the Trx/TrxR system to reduce MetO was measured following NADPH oxidation. No activity was detected when CgMsrA, MetO, or Trx was omitted from the assay system. The negative controls accounted for any nonenzymatic background due to auto-oxidation of NADPH. However, the activity of CgMsrA was significantly higher in the presence of CgMsrA, Trx, and MetO (Fig. 2A), indicating that the reaction is dependent on the presence of these factors, and CgMsrA can actually reduce MetO to Met via the Trx/TrxR reducing system. Similarly, reduction of MetO by CgMsrA via the Mrx1/Mtr/MSH system was observed (Fig. 2B). The roles of Trx and Mrx1 in reducing CgMsrA were also corroborated by monitoring the generation of Met in the HPLC assay (see Table S1 in the supplemental material).
FIG 2.

Reductase activities of CgMsrA using different reducing powers. (A and B) CgMsrA activities were measured by recording NADPH oxidation at 340 nm. The reaction mixture contained 50 mM Tris-HCl (pH 8.0), 2 mM EDTA, 250 μM NADPH, 2 μM CgMsrA, 100 mM MetO, and the Trx system (4 μM TrxR and 40 μM Trx) (A) or the Mrx1 system (500 μM MSH, 4 μM Mtr, and 40 μM Mrx1) (B). (C and D) Michaelis-Menten plots of CgMsrA activity versus MetO, Trx, and Mrx1. The reaction mixtures contained 50 mM Tris-HCl (pH 8.0), 2 mM EDTA, 250 μM NADPH, 2 μM CgMsrA, 0 to 150 mM MetO, and the Trx system (4 μM TrxR and 40 μM Trx) or the Mrx1 system (500 μM MSH, 4 μM Mtr, and 40 μM Mrx1) (C) or 100 mM MetO and the Trx system (4 μM TrxR and 0 to 120 μM Trx) or the Mrx1 system (500 μM MSH, 4 μM Mtr, and 0 to 120 μM Mrx1 (D). The data are represented as means ± SD of three independent experiments.
The catalytic constants of CgMsrA with the Trx and Mrx1 systems as the recycling reductants were determined under steady-state conditions at saturating concentrations of the reductants (40 μM) and different concentrations of MetO (0 to 150 mM). As shown in Fig. 2C, the kcat values of CgMsrA for MetO with the Trx and Mrx1 systems were 2.1 ± 0.1 s−1 and 0.8 ± 0.1 s−1, respectively, while the respective Km values were 3.0 ± 0.4 mM and 8.6 ± 1.1 mM. This corresponds to a catalytic efficiency of 7.0 × 102 M−1 s−1 and 0.9 × 102 M−1 s−1, respectively. The Km of 3.0 mM (CgMsrA for MetO with Trx) was slightly higher than the Km of 1.9 mM reported for E. coli MsrA (48).
Next, CgMsrA activity was determined at a fixed concentration of MetO (100 mM) and different concentrations of Trx and Mrx1 (0 to 120 μM). The Km values, kcat values, and catalytic efficiencies of CgMsrA for Trx were calculated to be 13.3 ± 1.4 μM, 2.1 ± 0.1 s−1, and 15.9 × 104 M−1 s−1, while the values for Mrx1 were 25.3 ± 2.7 μM, 0.8 ± 0.1 s−1, and 3.2 × 104 M−1 s−1 (Fig. 2D). These data further indicate that although CgMsrA employs both Trx and Mrx1 regeneration systems in reducing MetO, it has higher affinity for Trx than for Mrx1 in vitro. Instead of Mrx1/Mtr/MSH with the Grx/GR/GSH system, regeneration of MsrA with two different reduction systems (Trx/TrxR and Grx/GSH/GR) has been reported for poplar MsrA recently (28).
Cys56 is the peroxidatic Cys.
CgMsrA contains four Cys residues at positions 56, 91, 204, and 213 and a conserved N terminus binding motif at positions 55 to 59 consisting of Gly-Cys-Phe-Trp-Gly (see Fig. S1 in the supplemental material). Sequence alignment indicates that Cys56 might be the peroxidatic (CP) Cys residue while Cys204 and Cys213 might be the resolving (CR) Cys residues of CgMsrA. We first investigated whether Cys56 acted as the peroxidatic Cys residue of CgMsrA. During catalysis, the labile peroxidatic Cys-SOH is easily attacked by the other Cys of MsrA to form the redox-active disulfide. To stabilize and trap Cys-SOH, the C-terminal Cys of the active-site disulfide pair must be removed. Thus, the triple variants of CgMsrA, namely, CgMsrA(C91S,C213S,C204S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C56S,C91S,C213S), were constructed and treated with NBD-Cl under anaerobic conditions with and without previous exposure to MetO. As shown in Fig. 3A, MetO-treated and NBD-labeled CgMsrA(C91S,C213S,C204S) had an absorbance maximum (λmax) of 347 nm, representing the NBD-modified product Cys-S(O)-NBD (31), which clearly signified the detection and trapping of approximately stoichiometric amounts of SOH at Cys56, the only Cys in this variant. However, non-MetO-treated CgMsrA(C91S,C213S,C204S) modified with NBD-Cl produced a new covalently attached spectral species with a λmax of 420 nm, consistent with previously characterized thiol adducts with NBD-Cl (Cys-S-NBD). The difference of 0.67 thiol groups between non-MetO-treated and treated CgMsrA(C91S,C213S,C204S) was consistent with formation of the SOH following treatment with MetO (Fig. 3B). The absorbance and the thiol contents of CgMsrA(C56S,C91S,C204S) and CgMsrA(C56S,C91S,C213S) were unchanged upon incubation with NBD-Cl either before or after exposure to MetO, implying no SOH formation on Cys204 or Cys213 (Fig. 3A and B). Collectively, these results suggest that Cys56 is the peroxidatic Cys of CgMsrA oxidized to a sulfenic acid upon MetO treatment.
FIG 3.

Formation of sulfenic acid in Cys56. (A) Spectrophotometric analysis of NBD-labeled CgMsrA variants. Open symbols, non-MetO-treated CgMsrA(C91S,C204S,C213S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C56S,C91S,C213S); solid symbols, MetO-treated CgMsrA(C91S,C204S,C213S), CgMsrA(C56S,C91S,C204S), and CgMsrA(C56S,C91S,C213S). (B) Free sulfhydryl groups in the CgMsrA WT and its variants were determined using DTNB. Non-MetO-treated and MetO-treated proteins were added to DTNB (2 mM) in 50 mM Tris-HCl buffer (pH 8.0), and the absorbance at 412 nm was measured against a 2 mM DTNB solution as the reference. Mean values with standard deviations (error bars) from at least three repeats are shown.
Cys204 and Cys213 form an intramolecular disulfide bond upon MetO treatment.
To investigate whether Cys204 and Cys213 act as the resolving Cys residues of CgMsrA, the free sulfhydryl states of different variants were determined by AMS modification after preincubation with or without MetO. AMS covalently modifies free thiol groups, retarding electrophoretic mobility in proportion to the number of free thiol groups in proteins. As shown in Fig. 4A, both MetO-treated and untreated CgMsrA(C56S) were modified by AMS and have the same retarded electrophoretic mobility, and a similar phenomenon was observed for CgMsrA(C56S,C91S). These data indicate that these proteins are not oxidized by MetO and that Cys204/Cys213 still exists in the thiol state.
FIG 4.

Redox response of CgMsrA wild type and its variants. (A) Oxidation of cysteine residues of CgMsrA variants was determined as follows. Proteins (25 μM) treated with 10 mM DTT were further incubated without (−) or with (+) 10 mM MetO, and then free thiol groups were modified with AMS. The modified samples were separated by nonreducing SDS-15% PAGE. (B and C) Analysis by MALDI-TOF MS-MS of disulfide bond and sulfenic acid formation. Bands (labeled # [B] and * [C]) excised from nonreducing gels in panel A were treated with trypsin and subjected to MS analysis. Only the relevant portion of each mass spectrum is shown.
The CgMsrA(C91S) treated with MetO was also modified by AMS, and two bands are shown in Fig. 4A, far-right lane. MS analysis of the upper band (marked with an asterisk) led to the identification of a peak with a mass of 5,470.4 Da, which was 499 Da higher than predicted for the 45-to-88 peptide of CgMsrA(C91S) (calculated and observed mass, 4,971.4 Da [see Fig. S2A in the supplemental material]), consistent with the addition of one AMS molecule (500 Da) (see Fig. S3A in the supplemental material). A peak with a mass of 1,951.6 Da, which is 2.2 Da lower than predicted for the 199-to-217 peptide of CgMsrA(C91S) (calculated mass, 1,953.8 Da, and observed mass, 2,321.6 Da), was formed by adding IAM (185 Da) to Cys 204 and Cys 213 (see Fig. S2A in the supplemental material), indicating formation of a disulfide bond between Cys204 and Cys213 (Fig. 4B). The peak with a mass of 1,951.6 Da was also found in the lower band (data not shown). In addition, a peak with a mass of 4,987.8 Da, 16.4 Da higher than that of the 45-to-88 peptide of reduced CgMsrA(C91S) (calculated and observed mass, 4,971.4 Da), occurred in the lower band, consistent with the addition of one oxygen atom to the SH group (Fig. 4C). Together, these results indicate the formation of a disulfide bond between Cys204 and Cys213 in the CgMsrA(C91S) variant upon MetO treatment, and Cys56 exists in either a thiol state (the upper band) or a sulfenic acid state (the lower band). These results were further confirmed by measuring the thiol content of MetO-treated and untreated CgMsrA variants with the DTNB assay (Fig. 3B). Non-MetO-treated CgMsrA(C91S) was found to contain 2.47 ± 0.17 thiol groups per monomer, but the thiol content of MetO-treated CgMsrA(C91S) decreased to 0.56 ± 0.15. The difference of 1.91 thiol groups between the two preparations is related to the coexistence of two CgMsrA(C91S) states after MetO treatment.
MsrA(C91S,C204S) treated with MetO was not modified by AMS, because AMS- and MetO-treated CgMsrA(C91S,C204S) migrated more rapidly than the AMS-modified, non-MetO-treated form and migrated the same as the MetO-treated, non-AMS-modified form. These data indicate that CgMsrA(C91S,C204S) is fully oxidized by MetO to form a disulfide bond between Cys56 and Cys213. The formation of the Cys56-Cys213 disulfide bond was confirmed by MS analysis, with the identification of a mass of 6,907.8 Da, which corresponds to the 45-to-87 peptide (calculated and observed mass, 4,971.4 Da) and the 199-to-217 peptide (calculated and observed mass, 1,937.4 Da) of the CgMsrA(C91S,C204S) variant connected by a disulfide bridge that does not appear in non-MetO-treated CgMsrA(C91S,C204S) (see Fig. S3B in the supplemental material).
However, CgMsrA(C91S,C213S), treated with MetO and modified with AMS, migrated faster than its non-MetO-treated, AMS-modified form and slower than the MetO-treated, non-AMS-modified form (Fig. 4A, lanes 3 to 5 from left), indicating that despite modification, it was not fully oxidized. Cys56 might become the sulfenic acid while Cys204 remained in thiol form. Consistently, the difference of 0.87 thiol groups between MetO-treated and untreated CgMsrA(C91S,C213S) was observed with the DTNB assay (Fig. 3B). Collectively, these results indicate that Cys204 and Cys213 are the resolving Cys residues of CgMsrA that form an intramolecular disulfide bond upon MetO treatment.
Enzymatic properties of the CgMsrA variants.
Next, the capacities of the Mrx1 and Trx reducing systems to provide electrons to CgMsrA variants were compared (Fig. 5A). No significant effect was observed for the C91S variant using either the Mrx1/Mtr/MSH or the Trx/TrxR reducing system, thus excluding a role for Cys91 in catalysis. All variants containing a Cys56→Ser mutation were inactive in the presence of both the Trx/TrxR and Mrx1/Mtr/MSH reducing systems, consistent with the prediction that Cys56 acts as the peroxidatic Cys residue that is essential for CgMsrA activity. The variants containing Cys56 and Cys204 [CgMsrA(C91S,C213S)], or even Cys56 alone [CgMsrA(C91S,C213S,C204S)], showed higher activity than the wild type in the presence of the Mrx1/Mtr/MSH system (Fig. 5A and Table 3). These data suggest that Mrx1 can function as a reductant for various CgMsrA variants, as long as a sulfenic acid is maintained on the peroxidatic Cys residue, Cys56. Conversely, no activity was observed for the CgMsrA(C91S,C204S) variant, in which Cys56 was kept in a disulfide bond with Cys213 (Fig. 5A and Table 3).
FIG 5.

Effects of Cys mutation on CgMsrA activity. (A) The reductase activities of CgMsrA variants were determined by monitoring the rates of NADPH oxidation as described in Materials and Methods. (B) CgMsrA-mediated protection against oxidative stress is dependent on the active-site cysteines, i.e., Cys56, Cys204, and Cys213. The ΔmsrA and ΔmsrA ΔmshC Δmrx1 mutants were transformed with pXMJ19, pXMJ19-msrA, pXMJ19-msrA:C91SC204S, pXMJ19-msrA:C91SC213S, and pXMJ19-msrA:C91SC204SC213S, respectively, and tested for sensitivity to H2O2 (100 mM) by survival assay. The data are means of the values obtained from three independent assays. Mean values with standard deviations (error bars) from at least three repeats are shown. *, P ≤ 0.05.
TABLE 3.
Kinetics parameters of CgMsrA wild type and variants for reduction of MetO by the Mrx1/Mtr/MSH and Trx/TrxR pathwaysa
| Enzyme | Mrx1/Mtr/MSH |
Trx/TrxR |
||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km × 104 (M−1 s−1) | Km (μM) | kcat (s−1) | kcat/Km × 104 (M−1 s−1) | |
| WT | 26.8 ± 0.1 | 0.8 ± 0.1 | 3.0 | 14.5 ± 0.2 | 2.0 ± 0.1 | 13.8 |
| C91S | 26.2 ± 0.1 | 0.8 ± 0.2 | 3.1 | 13.7 ± 0.7 | 1.9 ± 0.2 | 13.9 |
| C91S,C56S | ND | ND | ND | ND | ND | ND |
| C91S,C204S | ND | ND | ND | 237.0 ± 3.0 | 0.5 ± 0.1 | 0.2 |
| C91S,C213S | 20.2 ± 0.8 | 1.3 ± 0.1 | 6.4 | ND | ND | ND |
| C91S,C204S,C213S | 19.0 ± 0.5 | 1.5 ± 0.1 | 7.9 | ND | ND | ND |
| C91S,C56S,C204S | ND | ND | ND | ND | ND | ND |
| C91S,C56S,C213S | ND | ND | ND | ND | ND | ND |
For determination of the Michaelis constant, activities were measured in the reaction mixtures containing 50 mM Tris-HCl (pH 8.0), 2 mM EDTA, 250 μM NADPH, 2 μM CgMsrA (WT or variants), 100 mM MetO, the Mrx1 system (4 μM Mtr, 500 μM MSH, and 0 to 120 μM Mrx1) or the Trx system (4 μM TrxR and 0 to 120 μM Trx). The data are presented as means of the values obtained from three independent assays ± standard deviations. ND, not detectable under the conditions used.
Trx was unable to regenerate CgMsrA variants with mutations in any two of the Cys56, Cys204, and Cys213 residues [i.e., the triple variants CgMsrA(C56S,C91S,C204S), CgMsrA(C56S,C91S,C213S), and CgMsrA(C91S,C213S,C204S)], as well as the double variants CgMsrA(C56S,C91S) and CgMsrA(C91S,C213S), but retained 25% of the turnover number (s−1) for CgMsrA(C91S,C204S), which contained a disulfide bond between Cys56 and Cys213 (Fig. 5A and Table 3). We concluded that Trx can efficiently reduce CgMsrA variants containing a disulfide bond between Cys204 and Cys213 under oxidative stress but Trx is not able to reduce a 1-Cys CgMsrA variant [CgMsrA(C91S,C213S,C204S)] that does not contain a disulfide bond (Fig. 5A and Table 3). Thus, it is clear that Mrx1 and Trx employ different mechanisms in reducing CgMsrA.
The Mrx1 reducing system functions as an electron donor for CgMsrA under oxidative stress conditions but not normal conditions.
The assumption that Mrx1 and Trx employ different mechanisms in reducing CgMsrA was further confirmed in vivo by comparing the abilities of CgMsrA variants to complement the oxidative-sensitive phenotype of the ΔmsrA mutant (containing both Trx/TrxR and Mrx1/Mtr/MSH regeneration pathways) and the ΔmsrA ΔmshC Δmrx1 mutant (containing the Trx/TrxR regeneration pathway only) (Fig. 5B; see Fig. S4 in the supplemental material). While wild-type msrA substantially recovered the survival rates of the ΔmsrA mutant under H2O2, CdCl2, and CDNB stress, its complementation efficiency was partially abolished (33.4 to 41.5%) in the ΔmsrA ΔmshC Δmrx1 mutant, which contains the Trx/TrxR regeneration pathway only. Although the CgMsrA(C91S,C204S,C213S), CgMsrA(C91S,C204S), and CgMsrA(C91S,C213S) variants partially (21.7 to 38.0%) recovered the survival rates of the ΔmsrA mutant compared to CgMsrA WT, only CgMsrA(C91S,C204S) maintained this ability in the ΔmsrA ΔmshC Δmrx1 mutant, consistent with its Trx/TrxR pathway-dependent but not Mrx1/Mtr/MSH pathway-dependent feature in reduction. On the other hand, the Mrx1/Mtr/MSH pathway-dependent CgMsrA(C91S,C204S,C213S) and CgMsrA(C91S,C213S) completely lost the complementation ability in the ΔmsrA ΔmshC Δmrx1 mutant lacking the Mrx1/Mtr/MSH regeneration pathway (Fig. 5B). These data indicate that both the Mrx1 and Trx pathways are operative in regeneration of CgMsrA in vivo under stress conditions.
To further clarify the roles of both the Mrx1 and Trx systems under normal conditions, we tried to delete mshC (essential for MSH synthesis), msrA, and trx in the ΔmetH ΔmetE mutant, which was reported to be auxotrophic for Met (49, 50). Unfortunately, consistent with a recent report that trx seems to be an essential gene in C. glutamicum (20), no viable colonies were obtained after multiple attempts to knock out trx. We therefore evaluated the growth of ΔmetH ΔmetE, ΔmetH ΔmetE ΔmsrA, and ΔmetH ΔmetE ΔmshC mutants under normal conditions. As shown in Fig. 6, the ΔmetH ΔmetE ΔmsrA mutant displayed a growth defect under Met-depleted conditions with MetO as the sole Met source compared to that in the presence of Met, and complementation of the triple mutant with the msrA gene completely restored growth, indicating that CgMsrA plays an important role in providing Met to the strain by reducing MetO. However, mshC gene deletion did not cause a reduction of ΔmetH ΔmetE mutant growth in the presence of MetO (Fig. 6), indicating that the Mrx1/MSH/Mtr system was not necessary for CgMsrA activity and the endogenous Trx system alone was sufficient to support this activity in normal cells. However, consistent with the finding that the Mrx1 pathway is functional in regeneration of CgMsrA in vivo under stress conditions (Fig. 5B; see Fig. S4 in the supplemental material), the growth of the ΔmetH ΔmetE ΔmshC mutant under Met-depleted conditions with MetO as the sole Met source was reduced upon H2O2 treatment, and the growth defect under H2O2 stress was restored by complementation with the mshC gene (Fig. 6). Collectively, these results demonstrate that although both the Mrx1 and Trx pathways are functional in regeneration of CgMsrA under stress conditions, the Trx system alone is sufficient to act as an electron donor for CgMsrA under normal conditions.
FIG 6.

Growth phenotype of methionine-auxotrophic C. glutamicum strains on MetO with or without H2O2 challenge. Overnight-grown ΔmetH ΔmetE(pXMJ19), ΔmetH ΔmetE ΔmshC(pXMJ19), ΔmetH ΔmetE ΔmshC(pXMJ19-mshC), ΔmetH ΔmetE ΔmsrA(pXMJ19), and ΔmetH ΔmetE ΔmsrA(pXMJ19-msrA) strains (optical density at 600 nm [OD600] = 1.6 to 1.7) were spun down, washed twice with mineral salts medium, and resuspended in mineral salts medium, and then, 5 μl of serial dilutions (from left to right) were spotted on solid mineral salts medium plates containing 0.5 mM IPTG, 10 μg/ml MetO (+MetO), or 10 μg/ml MetO and 5 mM H2O2 (+MetO+H2O2). A control assay shows all cells growing equally on minimal medium plates containing 10 μg/ml methionine (+Met). The plates were incubated at 30°C for 2 days and then photographed.
CgMsrA forms a mixed disulfide with Trx.
During MsrA regeneration, the intramolecular disulfide bond is subject to attack by Trx and thus switches to a transient intermolecular disulfide bond to link MsrA with Trx (9–11). To determine which of the Cys residues in CgMsrA is the favored Cys in the formation of the transient disulfide intermediate with Trx, the formation of the heterodimer between MetO-treated CgMsrA variants and Trx(C35S), a Trx variant that can stabilize the heterodimers between Trx and their interacting partners (38, 44, 51), was examined using SDS-PAGE. As shown in Fig. 7A, heterodimer formation was observed between Trx(C35S) and CgMsrA(C91S) or CgMsrA(C91S,C204S), indicating that Trx can reduce disulfide bonds formed in CgMsrA(C91S) and CgMsrA(C91S,C204S). However, no heterodimer formed when Trx(C35S) reacted with MetO-treated CgMsrA(C91S,C213S) or CgMsrA(C91S,C204S,C213S) that contained sulfenic acid but no intramolecular disulfide bond. The heterodimer formed between Trx(C35S) and CgMsrA(C91S) was confirmed by MS analysis, with the observation of a mass of 4,530.4 Da, which corresponds to the 199-to-217 peptide of the CgMsrA(C91S) variant (calculated mass, 1,937.4 Da, and observed mass, 2,321.6 Da, which was formed by adding IAM to Cys204 and Cys213) (see Fig. S2A in the supplemental material) and the 14-to-36 peptide of the Trx(C35S) variant (calculated mass, 2,588.2 Da, and observed mass, 2,773.2 Da, formed by adding IAM to Cys32) (see Fig. S2B in the supplemental material) connected by a disulfide bridge (Fig. 7B). These results indicate that Cys32 of Trx attacks Cys213 of the intramolecular disulfide of CgMsrA, generating an intermolecular disulfide. Consistent with this indication, the complex formed between DTNB-treated Trx(C35S) and CgMsrA(C56S,C91S,C204S), but not CgMsrA(C91S,C213S,C204S) or CgMsrA(C56S,C91S,C213S), was clearly observed (Fig. 7C, right, far-right lane). Collectively, these results provide direct evidence that the Trx/TrxR system reduces the disulfide bond of CgMsrA formed during the antioxidative process.
FIG 7.

Interaction between CgMsrA and Trx in vitro. (A) CgMsrA was able to create a stable association with Trx(C35S). Nonreducing SDS-15% PAGE shows each CgMsrA variant incubated with Trx(C35S) in the presence of MetO to generate protein complexes. Ox-MsrA, oxidized MsrA. (B) Analysis of heterodimer by MALDI-TOF MS-MS. A band (labeled with an asterisk) from the nonreducing gel in panel A was excised and treated with trypsin. Only the relevant portion of the mass spectrum is shown. The peaks with m/z 4,530.4 were from dipeptides linked by a disulfide bond between Cys32 in Trx(C35S) and Cys213 in CgMsrA(C91S,C204S). (C) SDS-PAGE analysis of the disulfide-linked protein complexes between CgMsrA variants and Trx(C35S). DTNB-treated Trx(C35S), CgMsrA(C91S,C204S,C213S), CgMsrA(C56S,C91S,C213S), CgMsrA(C56S,C91S,C204S), CgMsrA(C91S,C204S,C213S)/DTNB-treated Trx(C35S), CgMsrA(C56S,C91S,C213S)/DTNB-treated Trx(C35S), and CgMsrA(C56S,C91S,C204S)/DTNB-treated Trx(C35S) were treated or not with DTT. M, protein markers. Addition of DTT led to the disappearance of the disulfide-linked complex.
S-Mycothiolated CgMsrA was reduced by Mrx1 coupled with Mtr/MSH.
To clarify the mechanism of Mrx1-dependent CgMsrA regeneration, MetO-treated CgMsrA variants [CgMsrA(C91S), CgMsrA(C91S,C213S), CgMsrA(C91S,C204S), and CgMsrA(C91S,C204S,C213S)] were incubated with Mrx1(C15S) with a conserved exposed active-site Cys12, but the coreacting Cys15 was changed to Ser in the CXXC active site. Such an Mrx1 variant was known to make dead-end intermediates with its substrates and can be used to identify Mrx1-targeted Cys residues (39, 47). No heterodimer was detected between Mrx1(C15S) and any of these CgMsrA variants (Fig. 8A), indicating that Mrx1 directly reduces neither CgMsrA containing disulfide bonds nor CgMsrA containing sulfenic acid.
FIG 8.

CgMsrA was reduced by Mrx1 with a monothiolic mechanism. (A) Nonmycothiolated CgMsrA was unable to create any stable association with Mrx1. Nonreducing 15% SDS-PAGE shows each CgMsrA incubated with each Mrx1 in the presence of MetO. Lanes a to d, CgMsrA(C91S), CgMsrA(C91S,C204S), CgMsrA(C91S,C213S), and CgMsrA(C91S,C204S,C213S), respectively; lane e, Mrx(C15S); lanes f to i, CgMsrA(C91S), CgMsrA(C91S,C204S), CgMsrA(C91S,C213S), and CgMsrA(C91S,C204S,C213S) incubated with Mrx1(C15S) to generate mixture proteins, respectively. (B) S-Mycothiolation of CgMsrA was monitored by biotin switch assay. IAM-alkylated protein extracts of ΔmsrA(pXMJ19-msrA) and ΔmsrA ΔmshC(pXMJ19-msrA) strains were harvested before and 30 min after H2O2 stress and subjected to His·Bind Ni-NTA resin purification. The resulting His6-CgMsrA was demycothionylated using the Mrx1/MSH/MTR system, and the free protein thiol was tagged with biotin-maleimide, followed by separation on nonreducing 15% SDS-PAGE, and blotted onto nitrocellulose membranes. The membranes were reacted with HRP-conjugated streptavidin to detect S-mycothiolation of CgMsrA and incubated with the anti-His (α-His) antibody to indicate the same amount of His6-CgMsrA used for demycothionylation analysis. (C) MALDI-TOF MS-MS analysis of CgMsrA(C91S) in the presence of both MetO and MSH (top), or MSH only (bottom). An increase of 483.1 Da was observed for CgMsrA(C91S) after treatment with MetO and MSH. (D) S-Mycothiolated CgMsrA can be reactivated by Mrx1 or Mrx1(C15S) in vitro. The Mrx1 electron transfer pathway composed of Mrx1/Mrx1(C15S), MSH, Mtr, and NADPH was reconstructed with reduced or S-mycothiolated CgMsrA added as the substrate. (E) There is no formation of heterodimers between S-mycothiolated CgMsrA(C91S,C204S,C213S) and Mrx1(C15S) in vitro. S-Mycothiolated CgMsrA(C91S,C204S,C213S) was incubated with Mrx1(C15S) in the presence of MetO, followed by separation on nonreducing 15% SDS-PAGE. Lanes: 1, Mrx1(C15S); 2, S-mycothiolated CgMsrA(C91S,C204S,C213S); 3, a mixture of Mrx1(C15S) and S-mycothiolated CgMsrA(C91S,C204S,C213S).
Recently, Chi et al. (36) identified the S-mycothiolation modification of CgMsrA under hypochlorous acid stress in a thiol-redox proteomics analysis. The S-mycothiolation modification of CgMsrA was also identified with the reversible biotinylation switch assay under H2O2-induced stress. C. glutamicum ΔmsrA(pXMJ19-msrA) and ΔmshCΔmsrA(pXMJ19-msrA) strains overexpressing His6-MsrA were treated or not with H2O2 stress. Then, the purified His6-CgMsrA proteins from different cell extracts treated according to the biotin switch assay were visualized by Western blotting. As shown in Fig. 8B, His6-CgMsrA overexpressed in the ΔmsrA mutant pretreated with H2O2 exhibited a strong band of S-mycothiolation. However, no S-mycothiolation signal was observed in the non-H2O2-treated ΔmsrA(pXMJ19-msrA) strain. Similarly, no S-mycothiolation signal was observed for His6-CgMsrA overexpressed in the ΔmshC ΔmsrA double mutant, which does not produce MSH with or without H2O2 treatment (Fig. 8B). Interestingly, in vitro S-mycothiolation formed on the catalytic Cys56 was also observed by peptide mass spectroscopy after tryptic digestion of MetO/MSH-treated CgMsrA(C91S), with the identification of a mass of 5,454.9 Da, which was 483.1 Da higher than that of the Cys56-containing 45-to-88 peptide of the reduced CgMsrA (calculated and observed mass, 4,971.8 Da), consistent with the results from the addition of MSH (Fig. 8C, top). However, this observation did not occur after treatment with MSH alone (Fig. 8C, bottom). These data indicate that CgMsrA could be S-mycothiolated under oxidative stresses.
This finding prompted us to test whether Mrx1 can reduce CgMsrA by the mycothiolation/demycothiolation mechanism, similar to the glutathionylation/deglutathionylation mechanism proposed for Grx-dependent 1-Cys MsrAs. Thus, studies of the interaction between the S-mycothiolated 1-Cys variant CgMsrA(C91S,C204S,C213S) and Mrx1(C15S) and the reduction activity of S-mycothiolated CgMsrA(C91S,C204S,C213S) with the Mrx1/MSH/Mtr and Mrx1(C15S/MSH/Mtr) electron transfer pathway as a reducing substrate in vitro were performed. The enzyme assay confirmed that S-mycothiolated, but not nonmycothiolated, CgMsrA(C91S,C204S,C213S) could be reduced by the Mrx1/MSH/Mtr and Mrx1(C15S/MSH/Mtr) electron transfer pathway. As shown in Fig. 8D, only the sample with CgMsrA(C91S,C204S,C213S)-SSM coupled with the Mrx1/MSH/Mtr and Mrx1(C15S/MSH/Mtr) electron transfer pathways showed consumption of NADPH, but no NADPH consumption was observed when Mrx1 and Mrx1(C15S) were omitted from the assay or when nonmycothiolated CgMsrA(C91S,C204S,C213S) was used instead of S-mycothiolated CgMsrA(C91S,C204S,C213S). As expected, the formation of the CgMsrA(C91S,C204S,C213S)-SS-Mrx1(C15S) mixed disulfide was not observed (Fig. 8E, lane 3), suggesting that the cysteine in Mrx1(C15S) reacted with the sulfur atom of MSH in CgMsrA(C91S,C204S,C213S)-SSM, yielding reduced CgMsrA(C91S,C204S,C213S) and Mrx1(C15S)-SSM through a monothiol mechanism of reduction, in line with the result of Hugo et al. reported for Mycobacterium tuberculosis AphE (52). Thus, CgMsrA could be reduced through the Mrx1/MSH/Mtr pathway via a 1-Cys mechanism upon oxidative stress, with its Cys56, in this case, first becoming the sulfenic acid form and then being S-mycothiolated.
DISCUSSION
Crucial information arising from this study is the description of the Mrx1/MSH/Mtr pathway-dependent regeneration of CgMsrA, extending our knowledge about the reductants for MsrAs. This situation was not completely unexpected, as growing evidence has suggested that there is an alternative reductant, Grx, for certain MsrAs that is not reducible by Trx (11). Previous studies have shown that Grx could employ the monothiol mechanism for reduction of dithiol groups, in which glutathionylation/deglutathionylation on the sulfenic acid is involved (47). Here, we demonstrate that Mrx1, functionally equivalent to Grx in Actinomycetes lacking GSH, could employ a similar mechanism for C. glutamicum MsrA regeneration. Our data revealed the presence of a stable sulfenic acid intermediate in a 1-Cys CgMsrA mutant after MetO reduction (Fig. 3). Although MSH alone is not able to reduce the sulfenic derivative of CgMsrA, it attacks the sulfenic acid and forms a mixed disulfide with the enzyme, as confirmed by mass spectrometry (Fig. 8C). The mixed disulfide formed between CgMsrA and MSH is further reduced by Mrx1 (Fig. 8D), resulting in the regeneration of CgMsrA. Consistent with the monothiol mechanism of reduction, the catalytic consumption of NADPH was detected when Mrx1 was replaced by Mrx1(C15S) in a reaction mixture consisting of NADPH/Mtr/MSH/Mrx1/MsrA/MetO (Fig. 8D). Interestingly, the mycothiolation/demycothiolation regeneration mechanism mediated by Mrx1 has also been reported for M. tuberculosis AhpE regeneration recently (52). Thus, besides protecting metabolic enzymes, such as methionine synthase (MetE) and maltodextrin phosphorylase (MalP), mycothiolation appears to play a role in regeneration of antioxidative enzymes, such as MsrA and AhpE, under oxidative stresses (36, 52).
Use of two reducing pathways for regeneration has so far been reported only for MsrA in poplar, in which both Trx and Grx pathways were involved (28). Both CgMsrA and poplar MsrAs belong to the 3-Cys MsrA family, with an N-terminal catalytic Cys and two resolving Cys residues located in the C terminus. For 3-Cys MsrAs, the first disulfide bond formed between the catalytic Cys and the first resolving Cys is further reduced by the second resolving Cys, leading to the formation of a disulfide bond between the two resolving Cys residues (7, 8). Interestingly, in C. glutamicum and poplar MsrAs, the first disulfide bond is formed between the catalytic Cys and the most C-terminal Cys (28), whereas in E. coli and B. taurus 3-Cys MsrAs that use the Trx pathway only for regeneration, the most C-terminal Cys acts as the second resolving residue (7, 8). The different behaviors of these two kinds of MsrAs could be explained based on their structure difference. In E. coli and B. taurus MsrAs, a glycine-rich region between the two C-terminal resolving Cys residues was observed (see Fig. S1 in the supplemental material). In E. coli, the glycine-rich region was reported to render the C-terminal extended chain more flexible and thus could bring the intermediate resolving Cys residue (Cys198) close to the catalytic Cys51 (48, 53) very easily. However, the glycine-rich region was not observed in C. glutamicum and poplar MsrAs (see Fig. S1 in the supplemental material).
By comparing the survival rates of both ΔmsrA (containing both Trx/TrxR and Mrx1/Mtr/MSH regeneration pathways) and ΔmsrA ΔmshC Δmrx1 (containing the Trx/TrxR regeneration pathway alone) strains that were complemented with different CgMsrA variants in trans, we found that both Trx and Mrx1 reducing pathways are active in regeneration of CgMsrA with different mechanisms under oxidative stresses in vivo. As shown in Fig. 5B and Fig. S4 in the supplemental material, the Mrx1-dependent variants CgMsrA(C91S,C213S) and CgMsrA(C91S,C204S,C213S) partially restored the oxidative-tolerant phenotype in the ΔmsrA mutant compared to the WT CgMsrA but completely lost the complementary activity in the ΔmsrA ΔmshC Δmrx1 mutant lacking the Mrx1/Mtr/MSH pathway (Fig. 5B). On the other hand, the Trx-dependent variant CgMsrA(C91S,C204S) partially restored the oxidative-tolerant phenotype in both the ΔmsrA mutant and the ΔmsrA ΔmshC Δmrx1 mutant (Fig. 5B). Under normal conditions, however, the Trx/TrxR pathway alone is sufficient to facilitate CgMsrA reduction of MetO, as disruption of the Mrx1/Mtr/MSH pathway does not cause a reduction of growth of the methionine-auxotrophic ΔmetH ΔmetE mutant using MetO as the sole Met source. Accordingly, no mycothiolation of CgMsrA was observerd under normal conditions (Fig. 8B), further confirming that the Mrx1/Mtr/MSH system is not operative under normal conditions. In E. coli, the GSH/Grx/GR system cannot supply the Trx/TrxR system for the regeneration of the MsrA activity (53), as a Met-auxotrophic E. coli strain cannot grow in the presence of MetO when the genes coding for Trx1 and Trx2 are inactivated (24). However, it is technically difficult to investigate whether the Mrx1/Mtr/MSH system could supply the Trx/TrxR system, because the trx gene in C. glutamicum seems to be essential, and it cannot be deleted (20).
Simultaneous use of two recycling pathways has been reported for poplar MsrA, though this has not been verified in vivo. The reason why CgMsrA uses two recycling pathways for regeneration is possibly the different mechanisms of Mrx1 and Trx employed in reducing CgMsrA. It has been shown that in the CgMsrA/Cys91S mutant upon MetO treatment, Cys56 exists in either the thiol state or the sulfenic acid state with Cys204 and Cys213 under the disulfide state. This could be an indication that the rate of formation of the second sulfenic acid intermediate is lower than that of the first. Under normal conditions, the Trx/TrxR recycling system is sufficient to regenerate oxidized CgMsrA by reducing the disulfide bond between Cys204 and Cys213, regardless of whether Cys56 is in the sulfenic acid or thiol state. This is consistent with the finding that no mycothiolation of Cys56 in CgMsrA was observed under normal conditions (Fig. 8B). As the Trx/TrxR system is also known to be employed by many biochemical systems for regeneration, we speculate that the Trx/TrxR system would be rate limiting and thus would need another recycling process to assist its function under stress conditions. Consistent with our speculation, strong upregulation of genes involved in MSH synthesis and regeneration pathways (mshC, mca, and mtr) upon oxidative stress has been observed (36, 42), indicating that the MSH content may significantly increase when cells are treated with oxidants. Along with the highly induced expression of the msrA gene and limitation of the Trx/TrxR system in C. glutamicum under stress conditions, more sulfenic acid intermediates with Cys204 and Cys213 under the disulfide state would be accumulated in the cell. Although Trx is also able to reduce the Cys204-Cys213 disulfide bond when Cys56 is under the sulfenic acid state and then to form CgMsrA under the Cys56-SOH state with Cys213 and Cys204 free, as reported for E. coli MsrA (see reference 54 and Scheme 1 therein), the Mrx1/Mtr/MSH system seems to be more efficient in reducing the sulfenic acid intermediate (Fig. 5A and Table 3). Mrx1 reduces the sulfenic acid intermediate via the formation of an S-mycothiolated MsrA intermediate (MsrA-SSM), which is then recycled by mycoredoxin and the second molecule of mycothiol, similarly to the GSH/Grx/GR system. Interestingly, the 1-Cys CgMsrA variant [CgMsrA(C91S,C204S,C213S)], equivalent to the Cys56-SH, Cys204-Cys213 disulfide bond intermediate, showed a significantly increased turnover number for reducing MetO compared to the CgMsrA WT using the Mrx1/Mtr/MSH system (kcat, 1.5 ± 0.1 s−1 compared to 0.8 ± 0.1 s−1) while showing a turnover number similar to that of the CgMsrA WT using the Trx system, suggesting that the Mrx1/Mtr/MSH system may prefer to regenerate the sulfenic acid intermediate (Fig. 5A and Table 3). The ability to use both the Trx/TrxR and Mrx1/Mtr/MSH pathways provides 3-Cys CgMsrA an obvious advantage in resisting oxidative stresses. Indeed, compared to 2-Cys MsrAs characterized in M. tuberculosis (55) and Mycobacterium smegmatis (56), while CgMsrA plays roles in defending C. glutamicum cells against both H2O2 and cumene hydroperoxide, deletion of msrA in M. tuberculosis and M. smegmatis has no effect on the sensitivity to H2O2, and even to cumene hydroperoxide in M. tuberculosis. The physiological advantages and biochemical mechanisms of simultaneously using both regeneration pathways need to be further investigated in the future.
Based on our results, a catalytic model for reduction of CgMsrA by Mrx1 and Trx can be hypothesized (Fig. 9). The first step consists of MetO reduction, with the concomitant formation of a stable sulfenic acid intermediate on catalytic Cys56. A nucleophilic attack by Cys213 on the sulfenic acid intermediate leads to the release of 1 molecule of Met and the formation of a transient disulfide bond between Cys56 and Cys213. This disulfide bond is reduced via Cys204, leading to the formation of a new intramolecular disulfide bond between Cys204 and Cys213. Next, the peroxidatic Cys is free, and the second disulfide bond between Cys204 and Cys213 is reduced by the Trx/TrxR recycling system under both normal and stress conditions. The freed peroxidatic Cys is also able to attack another molecule of MetO, leading to the Cys56 sulfenic acid intermediate with Cys204 and Cys213 under the disulfide state. Under normal conditions, the Cys204-Cys213 disulfide bond in the sulfenic acid intermediate is reduced by the Trx/TrxR recycling system. However, under stress conditions, the sulfenic acid intermediate also reacts with MSH to form an S-mycothiolated Cys56 intermediate. S-Mycothiolated Cys56 and the disulfide bond between Cys204 and Cys213 are reduced by the Mrx1/MSH/Mtr and Txr/TxrR recycling systems, respectively. Unlike the Trx/TrxR pathway, which is operative under both normal and stress conditions, the Mrx1/MSH/Mtr pathway is operative only under stress conditions.
FIG 9.
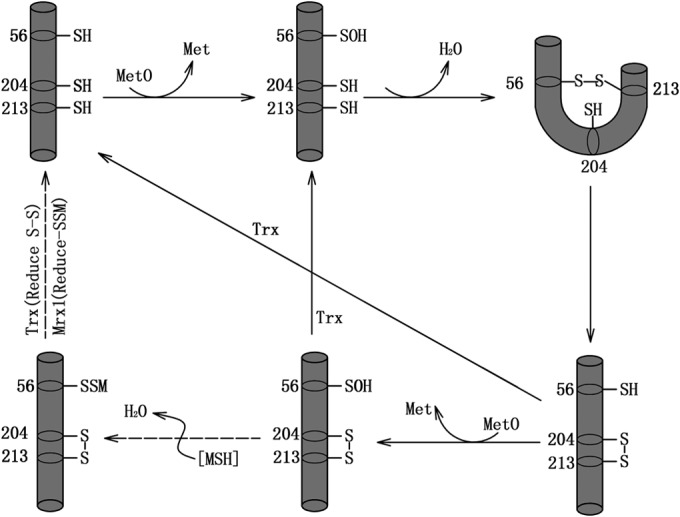
Proposed reaction mechanism for CgMsrA catalysis. The solid arrows indicate reactions occurring under both normal and stress conditions, and the dashed arrows indicate reactions occurring under stress conditions only.
According to mutation analysis, the two resolving Cys residues play different roles in CgMsrA regeneration. Mutation of Cys204 to Ser completely abrogated its use of the Mrx1 pathway but partially retained the ability to use the Trx pathway for regeneration, consistent with the detection of the disulfide bond by MS analysis (Fig. 4A and 5A; see Fig. S3 in the supplemental material). On the other hand, mutation of Cys213 to Ser completely abrogated its use of the Trx pathway but retained the ability to use the Mrx1 pathway for regeneration (Fig. 5A). Consistent with this, the CgMsrA(C91S,C213S) and CgMsrA(C91S,C204S,C213S) mutants also retained the ability to use the Mrx1 pathway for regeneration, indicating Cys56 first formed SOH and then reacted with MSH, but not in CgMsrA(C91S,C204S) (Fig. 3A, 4A, and 5A). From an evolutionary point of view, 3-Cys MsrAs that use double reducing pathways are hybrids of 1-Cys and 2-Cys MsrAs that use Grx/Mrx1 and Trx reducing pathways, respectively. The appearance of the second resolving Cys leads to a drastic change in the regeneration system, helping Trx-dependent MsrAs to gain the ability to simultaneously use the Grx/Mrx1 pathway for regeneration. Further investigations are needed to determine the precise mechanisms used by Trx and Mrx1 in simultaneously reducing CgMsrA.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National High Technology Research and Development Program of China (863 Program, grant 2013AA102802); the National Natural Science Foundation of China (31270078 and 31100001); the Key Science and Technology R&D Program of Shaanxi Province, China (2014K02-12-01); and the Opening Project of the State Key Laboratory of Microbial Resource, Institute of Microbiology, Chinese Academy of Sciences (no. SKLMR-20120601).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04221-14.
REFERENCES
- 1.Vogt W. 1995. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med 18:93–105. doi: 10.1016/0891-5849(94)00158-G. [DOI] [PubMed] [Google Scholar]
- 2.Davies MJ. 2005. The oxidative environment and protein damage. Biochim Biophys Acta 1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Kim HY, Kim JR. 2008. Thioredoxin as a reducing agent for mammalian methionine sulfoxide reductases B lacking resolving cysteine. Biochem Biophys Res Commun 371:490–494. doi: 10.1016/j.bbrc.2008.04.101. [DOI] [PubMed] [Google Scholar]
- 4.Moskovitz J, Berlett BS, Poston JM, Stadtman ER. 1997. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc Natl Acad Sci U S A 94:9585–9589. doi: 10.1073/pnas.94.18.9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hassouni ME, Chambost JP, Expert D, Van Gijsegem F, Barras F. 1999. The minimal gene set member msrA, encoding peptide methionine sulfoxide reductase, is a virulence determinant of the plant pathogen Erwinia chrysanthemi. Proc Natl Acad Sci U S A 96:887–892. doi: 10.1073/pnas.96.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM, Stadtman ER. 1998. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc Natl Acad Sci U S A 95:14071–14075. doi: 10.1073/pnas.95.24.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowther WT, Brot N, Weissbach H, Honek JF, Matthews BW. 2000. Thiol-disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc Natl Acad Sci U S A 97:6463–6468. doi: 10.1073/pnas.97.12.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tete-Favier F, Cobessi D, Boschi-Muller S, Azza S, Branlant G, Aubry A. 2000. Crystal structure of the Escherichia coli peptide methionine sulfoxide reductase at 1.9Å resolution. Structure 8:1167–1178. doi: 10.1016/S0969-2126(00)00526-8. [DOI] [PubMed] [Google Scholar]
- 9.Kim YK, Shin YJ, Lee WH, Kim HY, Hwang KY. 2009. Structural and kinetic analysis of an MsrA-MsrB fusion protein from Streptococcus pneumoniae. Mol Microbiol 72:699–709. doi: 10.1111/j.1365-2958.2009.06680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarrago L, Laugier E, Rey P. 2009. Protein-repairing methionine sulfoxide reductases in photosynthetic organisms: gene organization, reduction mechanisms, and physiological roles. Mol Plant 2:202–217. doi: 10.1093/mp/ssn067. [DOI] [PubMed] [Google Scholar]
- 11.Kim HY, Zhang Y, Lee BC, Kim JR, Gladyshev VN. 2009. The selenoproteome of Clostridium sp. OhILAs: characterization of anaerobic bacterial selenoprotein methionine sulfoxide reductase A. Proteins 74:1008–1017. doi: 10.1002/prot.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HY, Fomenko DE, Yoon YE, Gladyshev VN. 2006. Catalytic advantages provided by selenocysteine in methionine-S-sulfoxide reductases. Biochemistry 45:13697–13704. doi: 10.1021/bi0611614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarrago L, Laugier E, Zaffagnini M, Marchand C, Le Marechal P, Rouhier N, Lemaire SD, Rey P. 2009. Regeneration mechanisms of Arabidopsis thaliana methionine sulfoxide reductases B by glutaredoxins and thioredoxins. J Biol Chem 284:18963–18971. doi: 10.1074/jbc.M109.015487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vieira Dos Santos C, Laugier E, Tarrago L, Massot V, Issakidis-Bourguet E, Rouhier N, Rey P. 2007. Specificity of thioredoxins and glutaredoxins as electron donors to two distinct classes of Arabidopsis plastidial methionine sulfoxide reductases B. FEBS Lett 581:4371–4376. doi: 10.1016/j.febslet.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 15.Tarrago L, Laugier E, Zaffagnini M, Marchand CH, Le Marechal P, Lemaire SD, Rey P. 2010. Plant thioredoxin CDSP32 regenerates 1-Cys methionine sulfoxide reductase B activity through the direct reduction of sulfenic acid. J Biol Chem 285:14964–14972. doi: 10.1074/jbc.M110.108373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HY, Gladyshev VN. 2005. Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-R-sulfoxide reductases. PLoS Biol 3:e375. doi: 10.1371/journal.pbio.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen XH, Zhou NY, Liu SJ. 2012. Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium? Appl Microbiol Biotechnol 95:77–89. doi: 10.1007/s00253-012-4139-4. [DOI] [PubMed] [Google Scholar]
- 18.Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46:3348–3355. doi: 10.1128/AAC.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newton GL, Buchmeier N, Fahey RC. 2008. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev 72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ordóñez E, Van Bellem K, Roos G, De Galan S, Letek M, Gil JA, Wyns L, Mateos LM, Messens J. 2009. Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J Biol Chem 284:15107–15116. doi: 10.1074/jbc.M900877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Si MR, Zhang L, Yang ZF, Xu YX, Liu YB, Jiang CY, Wang Y, Shen XH, Liu SJ. 2014. NrdH-redoxin enhances resistance to multiple oxidative stresses by acting as a peroxidase cofactor in Corynebacterium glutamicum. Appl Environ Microbiol 80:1750–1762. doi: 10.1128/AEM.03654-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen XH, Jiang CY, Huang Y, Liu ZP, Liu SJ. 2005. Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl Environ Microbiol 71:3442–3452. doi: 10.1128/AEM.71.7.3442-3452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ezraty B, Bos J, Barras F, Aussel L. 2005. Methionine sulfoxide reduction and assimilation in Escherichia coli: new role for the biotin sulfoxide reductase BisC. J Bacteriol 187:231–237. doi: 10.1128/JB.187.1.231-237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob C, Kriznik A, Boschi-Muller S, Branlant G. 2011. Thioredoxin 2 from Escherichia coli is not involved in vivo in the recycling process of methionine sulfoxide reductase activities. FEBS Lett 585:1905–1909. doi: 10.1016/j.febslet.2011.04.070. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Wang Y, Song Y, Wang T, Xu S, Peng Z, Lin X, Zhang L, Shen XH. 2013. A type VI secretion system regulated by OmpR in Yersinia pseudotuberculosis functions to maintain intracellular pH homeostasis. Environ Microbiol 15:557–569. doi: 10.1111/1462-2920.12005. [DOI] [PubMed] [Google Scholar]
- 26.Schurig-Briccio LA, Farias RN, Rodriguez-Montelongo L, Rintoul MR, Rapisarda VA. 2009. Protection against oxidative stress in Escherichia coli stationary phase. Arch Biochem Biophys 483:106–110. doi: 10.1016/j.abb.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 27.Vinckx T, Wei Q, Matthijs S, Noben JP, Daniels R, Cornelis P. 2011. A proteome analysis of the response of a Pseudomonas aeruginosa OxyR mutant to iron limitation. Biometals 24:523–532. doi: 10.1007/s10534-010-9403-4. [DOI] [PubMed] [Google Scholar]
- 28.Couturier J, Vignols F, Jacquot JP, Rouhier N. 2012. Glutathione- and glutaredoxin-dependent reduction of methionine sulfoxide reductase A. FEBS Lett 586:3894–3899. doi: 10.1016/j.febslet.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 29.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. 2000. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem 275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 30.Ehira S, Ohmori M. 2012. The redox-sensing transcriptional regulator RexT controls expression of thioredoxin A2 in the cyanobacterium Anabaena sp. strain PCC 7120. J Biol Chem 287:40433–40440. doi: 10.1074/jbc.M112.384206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellis HR, Poole LB. 1997. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry 36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 32.Baker LM, Poole LB. 2003. Catalytic mechanism of thiol peroxidase from Escherichia coli. Sulfenic acid formation and overoxidation of essential CYS61. J Biol Chem 278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellman GL. 1959. Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 34.Gething MJH, Davidson BE. 1972. Molar absorption-coefficient of reduced Ellmans reagent: 3-carboxylato-4-nitro-thiophenolate. Eur J Biochem 30:352–353. [Google Scholar]
- 35.Jaffrey SR, Snyder SH. 2001. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 86:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 36.Chi BK, Busche T, Laer KV, Basell K, Becher D, Clermont L, Seibold GM, Persicke M, Kalinowski J, Messens J, Antelmann H. 2014. Protein S-mycothiolation functions as redox-switch and thiol protection mechanism in Corynebacterium glutamicum under hypochlorite stress. Antioxid Redox Signal 20:586–605. doi: 10.1089/ars.2013.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng J, Che Y, Milse J, Yin YJ, Liu L, Ruckert C, Shen XH, Qi SW, Kalinowski J, Liu SJ. 2006. The gene ncgl2918 encodes a novel maleylpyruvate isomerase that needs mycothiol as cofactor and links mycothiol biosynthesis and gentisate assimilation in Corynebacterium glutamicum. J Biol Chem 281:10778–10785. doi: 10.1074/jbc.M513192200. [DOI] [PubMed] [Google Scholar]
- 38.Rouhier N, Gelhaye E, Jacquot JP. 2002. Glutaredoxin-dependent peroxiredoxin from poplar. J Biol Chem 277:13609–13614. doi: 10.1074/jbc.M111489200. [DOI] [PubMed] [Google Scholar]
- 39.Lian FM, Yu J, Ma XX, Yu XJ, Chen Y, Zhou CZ. 2012. Structural snapshots of yeast alkyl hydroperoxide reductase Ahp1 peroxiredoxin reveal a novel two-cysteine mechanism of electron transfer to eliminate reactive oxygen species. J Biol Chem 287:17077–17087. doi: 10.1074/jbc.M112.357368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria, vol 1 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 41.Hu Y, Lu P, Wang Y, Ding L, Atkinson S, Chen S. 2009. OmpR positively regulates urease expression to enhance acid survival of Yersinia pseudotuberculosis. Microbiology 155:2522–2531. doi: 10.1099/mic.0.028381-0. [DOI] [PubMed] [Google Scholar]
- 42.Busche T, Silar R, Picmanova M, Patek M, Kalinowski J. 2012. Transcriptional regulation of the operon encoding stress-responsive ECF sigma factor SigH and its anti-sigma factor RshA, and control of its regulatory network in Corynebacterium glutamicum. BMC Genomics 13:445. doi: 10.1186/1471-2164-13-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yermolaieva O, Xu R, Schinstock C, Brot N, Weissbach H, Heinemann SH, Hoshi T. 2004. Methionine sulfoxide reductase A protects neuronal cells against brief hypoxia/reoxygenation. Proc Natl Acad Sci U S A 101:1159–1164. doi: 10.1073/pnas.0308215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skaar EP, Tobiason DM, Quick J, Judd RC, Weissbach H, Etienne F, Brot N, Seifert HS. 2002. The outer membrane localization of the N. gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc Natl Acad Sci U S A 99:10108–10113. doi: 10.1073/pnas.152334799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boschi-Muller S, Olry A, Antoine M, Branlant G. 2005. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim Biophys Acta 1703:231–238. doi: 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 46.Ehira S, Teramoto H, Inui M, Yukawa H. 2009. Regulation of Corynebacterium glutamicum heat shock response by the extracytoplasmic-function sigma factor SigH and transcriptional regulators HspR and HrcA. J Bacteriol 191:2964–2972. doi: 10.1128/JB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson ME. 1998. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact 111-112:1–14. [DOI] [PubMed] [Google Scholar]
- 48.Boschi-Muller S, Azza S, Branlant G. 2001. E. coli methionine sulfoxide reductase with a truncated N terminus or C terminus, or both, retains the ability to reduce methionine sulfoxide. Protein Sci 10:2272–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee HS, Hwang BJ. 2003. Methionine biosynthesis and its regulation in Corynebacterium glutamicum: parallel pathways of transsulfuration and direct sulfhydrylation. Appl Microbiol Biotechnol 62:459–467. doi: 10.1007/s00253-003-1306-7. [DOI] [PubMed] [Google Scholar]
- 50.Rückert C, Pühler A, Kalinowski J. 2003. Genome-wide analysis of the l-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J Biotechnol 104:213–228. doi: 10.1016/S0168-1656(03)00158-5. [DOI] [PubMed] [Google Scholar]
- 51.Rouhier N, Kauffmann B, Tete-Favier F, Palladino P, Gans P, Branlant G, Jacquot JP, Boschi-Muller S. 2007. Functional and structural aspects of poplar cytosolic and plastidial type a methionine sulfoxide reductases. J Biol Chem 282:3367–3378. doi: 10.1074/jbc.M605007200. [DOI] [PubMed] [Google Scholar]
- 52.Hugo M, Van Laer K, Reyes AM, Vertommen D, Messens J, Radi R, Trujillo M. 2014. Mycothiol/mycoredoxin 1-dependent reduction of the peroxiredoxin AhpE from Mycobacterium tuberculosis. J Biol Chem 289:5228–5239. doi: 10.1074/jbc.M113.510248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boschi-Muller S, Branlant G. 2014. Methionine sulfoxide reductase: chemistry, substrate binding, recycling process and oxidase activity. Bioorg Chem 57:222–230. doi: 10.1016/j.bioorg.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 54.Kriznik A, Boschi-Muller S, Branlant G. 2014. Kinetic evidence that methionine sulfoxide reductase A can reveal its oxidase activity in the presence of thioredoxin. Arch Biochem Biophys 548:54–59. doi: 10.1016/j.abb.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Lee WL, Gold B, Darby C, Brot N, Jiang X, de Carvalho LP, Wellner D, St John G, Jacobs WR Jr, Nathan C. 2009. Mycobacterium tuberculosis expresses methionine sulphoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol Microbiol 71:583–593. doi: 10.1111/j.1365-2958.2008.06548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Douglas T, Daniel DS, Parida BK, Jagannath C, Dhandayuthapani S. 2004. Methionine sulfoxide reductase A (MsrA) deficiency affects the survival of Mycobacterium smegmatis within macrophages. J Bacteriol 186:3590–3598. doi: 10.1128/JB.186.11.3590-3598.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tauch A, Kirchner O, Loffler B, Gotker S, Pühler A, Kalinowski J. 2002. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the plasmid pGA1. Curr Microbiol 45:362–367. doi: 10.1007/s00284-002-3728-3. [DOI] [PubMed] [Google Scholar]
- 58.Liu YB, Long MX, Yin YJ, Si MR, Zhang L, Lu ZQ, Wang Y, Shen XH. 2013. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch Microbiol 195:419–429. doi: 10.1007/s00203-013-0889-3. [DOI] [PubMed] [Google Scholar]
- 59.Si MR, Long MX, Chaudhry MT, Xu YX, Zhang P, Zhang L, Shen XH. 2014. Functional characterization of Corynebacterium glutamicum mycothiol S-conjugate amidase. PLoS One 9:e115075. doi: 10.1371/journal.pone.0115075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schäfer A, Tauch A, Jager W, Kalinowshi J, Thierbach G, Pühler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 61.Jakoby M, Ngouoto-Nkili CE, Burkovski A. 1999. Construction and application of new Corynebacterium glutamicum vectors. Biotechnol Techniques 13:437–441. doi: 10.1023/A:1008968419217. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.