

Abstract
Microbial taxonomy is essential in all areas of microbial science. The 16S rRNA gene sequence is one of the main phylogenetic species markers; however, it does not provide discrimination in the family Vibrionaceae, where other molecular techniques allow better interspecies resolution. Although multilocus sequence analysis (MLSA) has been used successfully in the identification of Vibrio species, the technique has several limitations. They include the fact that several locus amplifications and sequencing have to be performed, which still sometimes lead to doubtful identifications. Using an in silico approach based on genomes from 103 Vibrionaceae strains, we demonstrate here the high resolution of the fur gene in the identification of Vibrionaceae species and its usefulness as a phylogenetic marker. The fur gene showed within-species similarity higher than 95%, and the relationships inferred from its use were in agreement with those observed for 16S rRNA analysis and MLSA. Furthermore, we developed a fur PCR sequencing-based method that allowed identification of Vibrio species. The discovery of the phylogenetic power of the fur gene and the development of a PCR method that can be used in amplification and sequencing of the gene are of general interest whether for use alone or together with the previously suggested loci in an MLSA.
INTRODUCTION
In microbial science, rapid identification of isolates to the genus or species level is essential in many areas, for instance, when diagnosing an infection or unraveling the microbial diversity in different niches. Therefore, identification and classification must be reliable, reproducible, and informative and at the same time fast and user friendly. Hence, tools that allow identification should be easy and affordable for the average user (1).
Identification of microbial species was originally based on phenotypic assays, due to both the influence of other biology disciplines, such as botany, and the technological limitations at the time (2). While phenotypic identification has been an important tool, the approach was of more limited use for several microbial groups, including the family Vibrionaceae, where some species had indistinguishable phenotypes and other species could have divergent phenotypes among strains of the same species (1). More recently, genetically based methods, in particular, have been developed for species identification and phylogeny, with the “molecular clock” approach introduced by Carl Woese proving a strong tool (3). The most common genetic marker has been the 16S rRNA gene, but the sequences of a range of housekeeping genes are also being used. In the future, genome sequences (gene sequences or single-nucleotide polymorphisms [SNP]) will be used in phylogeny and identification.
The family Vibrionaceae comprises 159 species in 6 genera, of which the genus Vibrio is the largest and most extensively characterized (4, 5). For many genera, the common approach of using 16S rRNA gene similarity as the main phylogenetic species marker has failed, due to its low interspecies resolution (6).
Due to the limitations of the 16S rRNA gene phylogeny in Vibrio classification and to the development of molecular techniques, such as fluorescent amplified fragment length polymorphism (FALP) and multilocus sequence analysis (MLSA), the classification of Vibrionaceae soon evolved from using a single gene to using several gene sequences for identification and phylogeny. In particular, the introduction of an MLSA scheme using nine gene sequences (ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, topA, and the 16S rRNA gene) has provided much higher resolution for Vibrionaceae identification and taxonomy, and this has allowed the identification of new Vibrio species previously misidentified (6–11).
While the use of MLSA as described above has improved the resolution of Vibrionaceae phylogeny, it has been hampered by requiring several gene sequences. With the explosion of genome sequencing, this will become easier; however, for a great number of laboratories, the use of a single gene sequence for identification and phylogeny will be preferable for years to come. The use of the fur gene as a phylogenetic marker in bacteria has been suggested in several studies (12, 13). Also, one study (14) suggested the use of the fur gene as a discriminative phylogenetic marker between the species Alliivibrio salmonicida and Alliivibrio logei (previously Vibrio salmonicida and Vibrio logei, respectively); however, to our knowledge, no further work has addressed this possible marker. The fur gene encodes a ferric uptake regulator (Fur), which in most bacterial species is the major system for maintenance of iron homeostasis. Fur senses excess intracellular Fe2+ and binds to the promoter regions of the genes involved in iron acquisition, thereby blocking their transcription. In contrast, when iron availability is limited, derepression of the corresponding genes occurs. In addition to iron transport, Fur controls a range of other processes, such as redox stress resistance, energy metabolism, flagellar chemotaxis, and metabolic pathways (15–17).
The purpose of the present study was to determine if the fur gene could be used as a new phylogenetic marker in the identification of Vibrionaceae species. The availability of several fully genome-sequenced strains allowed us to address this in an in silico analysis. To facilitate broader use of fur as a phylogenetic marker, despite highly variable regions outside the fur gene, we developed a PCR sequencing-based method for the analysis of the fur gene in Vibrionaceae species.
MATERIALS AND METHODS
In silico analysis of fur sequences.
The whole-genome sequences (WGS) from 104 strains were used in this study: 83 Vibrio strains representing 44 species, 3 Aliivibrio strains representing 2 species, 12 Photobacterium strains representing 7 species, 2 Enterovibrio strains representing 2 species, 2 Grimontia strains representing 1 species, 1 Salinivibrio costicola strain, and 1 sequence from Shewanella xiamenensis BC01 as an unrelated Gram-negative bacillus (Table 1). The genome sequences were used in the in silico analysis of the fur gene. The genomes were all analyzed using CLC Main Workbench version 7 (CLC, Aarhus, Denmark). For the genomes annotated by NCBI, an annotation-based search was performed for the fur genes. Those not annotated were submitted to a BLAST search against the annotated fur genes and manually curated if necessary.
TABLE 1.
Vibrionaceae strains used in this study
| Species | Strain | WGS/fur GenBank accession no. |
|---|---|---|
| Strains used in the in silico studya | ||
| Aliivibrio fischeri | ZF-211 | AJYI01 |
| Aliivibrio logei | 5S-186 | AJYJ01 |
| Aliivibrio logei | ATCC 35077 | ASAH01 |
| Enterovibrio calviensis | DSM 14347T | JHZA01 |
| Enterovibrio norvegicus | FF-33 | AJYD01 |
| Grimontia hollisae | CIP 101886T | ADAQ01 |
| Grimontia sp. | AK16 | ANFM02 |
| Photobacterium aphoticum | JCM 19237 | BBMN01 |
| Photobacterium angustum | S14 | AAOJ01 |
| Photobacterium damselae subsp. damselae | CIP 102761T | ADBS01 |
| Photobacterium damselae subsp. piscicida | DI21 | AKYG01 |
| Photobacterium halotolerans | S2753 | JMIB01 |
| Photobacterium halotolerans | DSM 18316T | AULG01 |
| Photobacterium leiognathi | lrivu.4.1 | BANQ01 |
| Photobacterium leiognathi subsp. mandapamensis | svers.1.1. | BACE01 |
| Photobacterium phosphoreum | ANT220 | CCAR01 |
| Photobacterium profundum | 3TCK | AAPH01 |
| Photobacterium sp. | SKA34 | AAOU01 |
| Photobacterium sp. | AK15 | AMZO01 |
| Salinivibrio costicola subsp. costicola | ATCC 33508T = LMG 11651T | ASAI01 |
| Shewanella xiamenensis | BC01 | JAEC01 |
| Vibrio albensis = V. cholerae | VL426 | ACHV01 |
| Vibrio anguillarum | 96F | AEZA01 |
| Vibrio anguillarum | RV22 | AEZB01 |
| Vibrio azureus | NBRC 104587T | BATL01 |
| Vibrio brasiliensis | LMG 20546T | AEVS01 |
| Vibrio breoganii | ZF-55 | AJYL01 |
| Vibrio campbellii | CAIM 519T = NBRC 15631T | AMDG01 |
| Vibrio caribbeanicus | ATCC BAA-2122T | AEIU01 |
| Vibrio cholerae | MO10 | AAKF03 |
| Vibrio cholerae | CP110 | AMWF01 |
| Vibrio cholerae | TM 11079-80 | ACHW01 |
| Vibrio coralliilyticus | P1 | AEQS01 |
| Vibrio coralliilyticus | ATCC BAA-450T | ACZN01 |
| Vibrio coralliilyticus | OCN008 | AVOO01 |
| Vibrio cyclitrophicus | ZF14 | AIDH01 |
| Vibrio cyclitrophicus | FF75 | ATLT01 |
| Vibrio diazotrophicus | NBRC 103148T | BBJY01 |
| Vibrio ezurae | NBRC 102218T | BATM01 |
| Vibrio fortis | Dailan14 | JFFR01 |
| Vibrio fluvialis | PG41 | ASXS01 |
| Vibrio fluvialis | I21563 | ASXT01 |
| Vibrio halioticoli | NBRC 102217T | BAUJ01 |
| Vibrio harveyi | CAIM 1792 | AHHQ01 |
| Vibrio harveyi | NBRC 15634 = ATCC 14126T | BAOD01 |
| Vibrio harveyi | ZJ0603 | AKIH01 |
| Vibrio harveyi | AOD131 | AOMR01 |
| Vibrio harveyi | E385 | AYKI01 |
| Vibrio harveyi | VHJR4 | CAUN01 |
| Vibrio harveyi | VHJR7 | CAUO01 |
| Vibrio ichthyoenteri | ATCC 700023T | AFWF01 |
| Vibrio jasicida | 090810c | BAOC01 |
| Vibrio kanaloae | 5S-149 | AJYX01 |
| Vibrio litoralis | DSM 17657T | AUFZ01 |
| Vibrio maritimus | JCM 19240 | BBMT01 |
| Vibrio maritimus | JCM 19235 | BBMR01 |
| Vibrio metschnikovii | CIP 69.14T | ACZO01 |
| Vibrio mimicus | VM223 | ADAJ01 |
| Vibrio mimicus | MB451 | ADAF01 |
| Vibrio mimicus | CAIM 602T | AOMO01 |
| Vibrio mimicus | SX-4 | ADOO01 |
| Vibrio natriegens | NBRC 15636T = ATCC 14048T = DSM 759T | ATFJ01 |
| Vibrio nigripulchritudo | ATCC 27043T | AFWJ01 |
| Vibrio nigripulchritudo | FTn2 | CANW01 |
| Vibrio nigripulchritudo | SOn1 | CAOF01 |
| Vibrio ordalii | FS-238 | AJYS01 |
| Vibrio ordalii | 12B09 | AJYV01 |
| Vibrio ordalii | FF-93 | AJYT01 |
| Vibrio ordalii | FS-144 | AJYU01 |
| Vibrio orientalis | CIP 102891T = ATCC 33934T | ACZV01 |
| Vibrio owensii | ATCC 25919 | BANZ01 |
| Vibrio owensii | CAIM 1854T | BAOH01 |
| Vibrio pacinii | DSM 19139T | JONH01 |
| Vibrio parahaemolyticus | NIHCB0603 | AVOM01 |
| Vibrio parahaemolyticus | IDH02189 | JAHD01 |
| Vibrio proteolyticus | NBRC 13287T | BATJ01 |
| Vibrio rhizosphaerae | DSM 18581T | JONG01 |
| Vibrio rotiferianus | DAT722 | AFAJ01 |
| Vibrio rumoiensis | 1S-45 | AJYK01 |
| Vibrio sagamiensis | NBRC 104589T | BAOJ01 |
| Vibrio scophthalmi | LMG 19158T | AFWE01 |
| Vibrio shilonii = V. mediterranei | AK1T | ABCH01 |
| Vibrio sinaloensis | DSM 21326 | AEVT01 |
| Vibrio sp. | PPCK-2014 | JJMN01 |
| Vibrio splendidus | ZS-139 | AJZE01 |
| Vibrio splendidus | FF-6 | AJZI01 |
| Vibrio splendidus | ATCC 33789 | AFWG01 |
| Vibrio splendidus | 12B01 | AAMR01 |
| Vibrio splendidus | 12E03 | AJZD01 |
| Vibrio splendidus | ZF-90 | AJZF01 |
| Vibrio splendidus | 5S-101 | AJZG01 |
| Vibrio splendidus | FF-500 | AJZH01 |
| Vibrio splendidus | 1F-157 | AJZJ01 |
| Vibrio splendidus | 1S-124 | AJZL01 |
| Vibrio tasmaniensis | 1F-187 | AJZM01 |
| Vibrio tasmaniensis | 1F-155 | AJZN01 |
| Vibrio tasmaniensis | 5F-79 | AJZP01 |
| Vibrio tasmaniensis | ZS-17 | AJZQ01 |
| Vibrio tubiashii | ATCC 19109T | AFWI01 |
| Vibrio tubiashii | NCIMB 1337 = ATCC 19106 | AHHF01 |
| Vibrio variabilis | JCM 19239 | BBMS01 |
| Vibrio vulnificus | BAA87 | JDSE01 |
| Vibrio vulnificus | NBRC 15645T = ATCC 27562T | AMQV01 |
| Culture collection strains used in PCR sequencing method development | ||
| Aliivibrio fischeri | DSM 2168 | KP721366 |
| Enterovibrio calviensis | DSM 14347T | KP721381 |
| Grimontia hollisae | DSM 15132T | KP721382 |
| Photobacterium damselae | ATCC 33539T | KP721367 |
| Photobacterium halotolerans | LMG 22194T | KP721368 |
| Photobacterium angustum | S14 | KP721369 |
| Photobacterium rosenbergii | LMG 22223T | KP721370 |
| Vibrio coralliilyticus | ATCC BAA-450T = DSM 19607T | KP721371 |
| Vibrio fluvialis | NCTC 11327T | KP721372 |
| Vibrio harveyi | DSM 19623T = ATCC 14126T | KP721373 |
| Vibrio nigripulchritudo | ATCC 27043T | KP721374 |
| Vibrio owensii | DY05T | KP721375 |
| Vibrio parahaemolyticus | ATCC 17802T | KP721376 |
| Vibrio splendidus | ATCC 33125T | KP721377 |
| Vibrio vulnificus | ATCC 27562T = DSM 10143T | KP721378 |
| Vibrio anguillarum | DSM 21597T | KP721379 |
| Vibrio ponticus | DSM 16217T | KP721383 |
| Vibrio chagasii | DSM 17138T | KP721384 |
| Vibrio brasiliensis | DSM 17184T | KP721385 |
| Vibrio porteresiae | DSM 19223T | KP721386 |
| Vibrio pectenicida | DSM 19585T | KP721387 |
| Salinivibrio costicola subsp. costicola | DSM 11403T | KP721380 |
| Isolates used in the testing of the PCR sequencing method | ||
| Photobacterium halotolerans | S2753 | KP721398 |
| Vibrio anguillarum | 775 | KP721388 |
| Vibrio coralliilyticus | S2043 | KP721394 |
| Vibrio coralliilyticus | S2052 | KP721395 |
| Vibrio nigripulchritudo | S2604 | KP721397 |
| Vibrio neptunius | S2394 | KP721396 |
| Vibrio parahaemolyticus | V2 | KP721401 |
| Vibrio sp. | S188 | KP721389 |
| Vibrio sp. | S203 | KP721390 |
| Vibrio sp. | S344 | KP721391 |
| Vibrio sp. | S787 | KP721392 |
| Vibrio sp. | S1110 | KP721393 |
| Vibrio sp. | S2757 | KP721399 |
| Vibrio sp. | S4497 | KP721400 |
| Vibrio sp. | VibAn | KP721402 |
Genomes from GenBank.
Phylogenetic-data analysis.
The fur sequences isolated in silico or PCR amplified and sequenced in this study were aligned using the alignment tools in CLC Main Workbench version 7 (CLC, Aarhus, Denmark). The Gap cost settings were as follows: gap open cost of 10 points and gap extension cost of 1 point, and end gaps were treated like any other gap. The alignments obtained were used to perform a pairwise comparison of the number of differences and the percent identity using CLC Main Workbench version 7 (CLC, Aarhus, Denmark). Furthermore, maximum-likelihood phylogeny trees were also generated using the CLC Main Workbench version 7 (CLC, Aarhus, Denmark) tools. Neighbor-joining was the tree construction method used, with the Jukes-Cantor nucleotide distance measure. The design of the trees was finalized using MEGA 6 (18).
Bacterial strains and genomic-DNA extraction.
The bacterial strains used for development of the PCR method (Table 1) were grown in Marine Broth (Difco; catalog no. 279110) overnight at 25°C, and genomic DNA was isolated using the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany). The quality of the genomic DNA was checked by 1% agarose gel electrophoresis and quantified by absorbance using DeNovix DS-11.
Primer design.
The degenerate primers fur_AP _fw (5′-CCWCCATAYTGDGWMCGRTTNGCATTCCWCCATAYTGDGWMCGRTTNGCATT-3′) and fur_AP_rv (5′-ACWGTHGGYYTWCGTGATACWTGGG-3′) were designed according to the alignments done using the fur regions of several Vibrio, Aliivibrio, and Photobacterium strains. Also, an extra set of primers were designed for amplification in other Vibrio species where the AP primers did not work: fur_V_fw (5′-TAACCYYTTGAASTTGAASTTCG-3′), fur_TS_rv (5′-CGWAYDGGHTAYTTCTGTGYDGAT-3′), and fur_OM_rv (5′-GTGGCRGATAAYGTKMGHAAAGG-3′). These primers were then used to amplify the whole fur gene. Due to the different sizes of the fragments, internal primers were also designed to confirm the presence of the fur gene in the amplified fragment: fur_Sp_internal_fw (5′-CACCAYTTYGAAGGCGGYAAGTC-3′) and fur_Sp_internal_rv (5′-ATYTCTTTYTGKCGYTCTTCRAT-3′).
PCR amplification and sequencing.
Amplification reaction mixtures contained 1× PfuX7 buffer [20 mM Tris-HCl, pH 8.8, 10 mM KCl, 6 mM (NH4)2SO4, 2 mM MgSO4, 0.1 mg/ml bovine serum albumin (BSA), 0.1% Triton X-100], a 200 μM deoxynucleoside triphosphate (dNTP) mixture, 0.4 μM each primer, 1 μl of a 10-fold dilution of genomic DNA as the template, and 1 μl of PfuX7 polymerase (19) in a final reaction volume of 25 μl. The PCR amplification was carried out in a thermal cycler (Veriti 96-well thermal cycler; Applied Biosystems) as follows: a 2-min initial denaturation step at 98°C, followed by 30 cycles of 98°C for 20 s, 52°C for 20 s, and 72°C for 25 s, with a final extension step of 2 min at 72°C. The amplified products were visualized by agarose gel electrophoresis (1 or 1.5%) and ethidium bromide staining. The PCR products were enzymatically purified by treatment with exonuclease I (ExoI) (Thermo Scientific) and FastAP thermosensitive alkaline phosphatase (Thermo Scientific) before sequencing at GATC Biotech (Cologne, Germany) or Macrogen (Amsterdam, The Netherlands). The sequences were analyzed using CLC Main Workbench version 7 (CLC, Aarhus, Denmark).
Nucleotide sequence accession numbers.
The sequence data generated in this study were deposited in GenBank under the accession numbers provided in Table 1.
RESULTS AND DISCUSSION
16S rRNA gene limitations.
16S rRNA gene sequences have long been used for distinguishing and classifying new strains at the genus and species levels. This approach has been very successful in several bacterial groups, but not in the Vibrionaceae, due to low interspecies resolution achieved using the gene (6). This is in part caused by the many alleles of the 16S rRNA gene that, when cloned and sequenced individually, can identify a strain as belonging to several different species (20). To confirm this observation, we used three closed genomes as examples and used the different 16S rRNA alleles in a BLAST search in order to identify the species (Fig. 1). Not only did the number of alleles seem to be variable among vibrios, but the identification drawn from each allele pointed to a different species, and thus, the identifications made using this approach in vibrios are very questionable. Furthermore, when using next-generation sequencing techniques, such as Illumina, the length of the reads obtained does not allow differentiation of the different alleles, resulting in genome assemblies with only one 16S rRNA allele.
FIG 1.
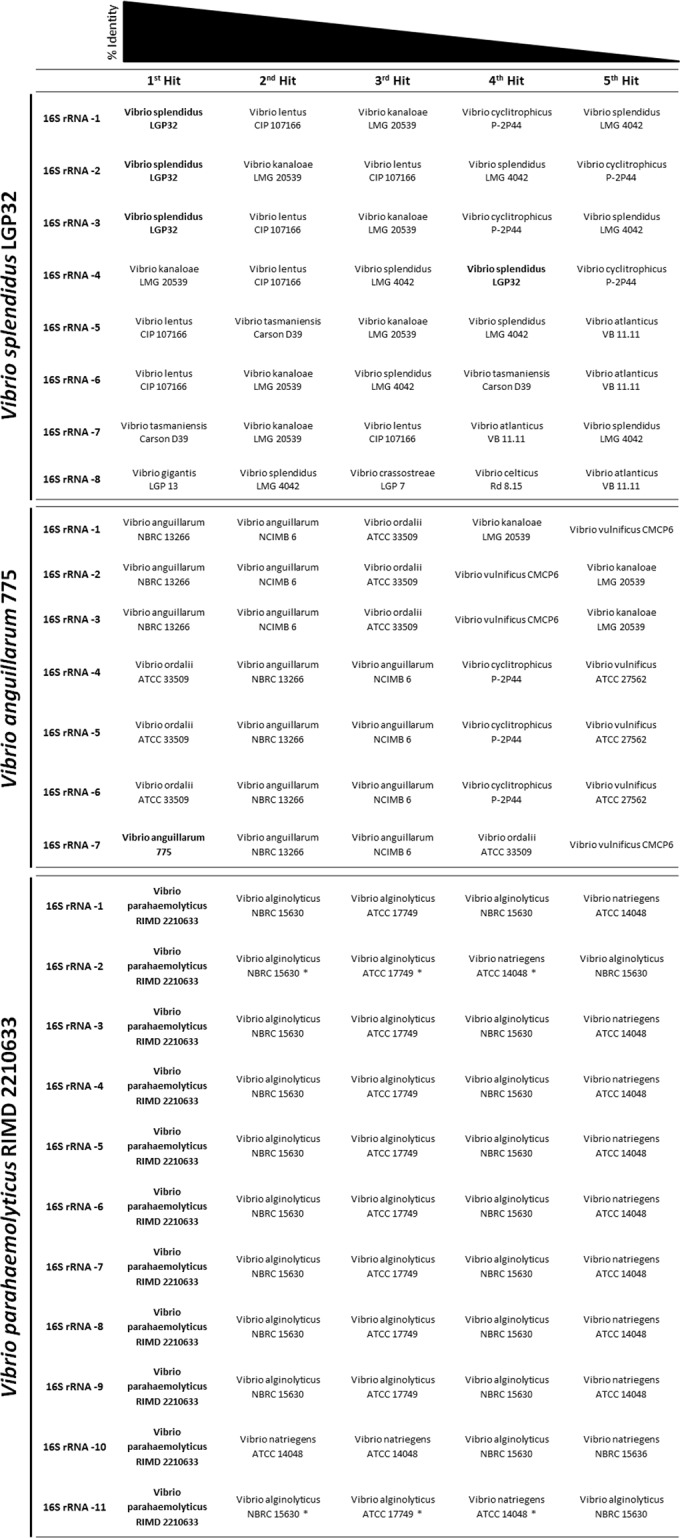
BLAST analyses and closest hits to the different 16S rRNA alleles in three different Vibrionaceae strains. Hits from the same strain are in boldface.
In silico analysis of fur sequences.
For all 103 strains analyzed, only one copy of the fur gene was identified in the whole genome. The fur sequences varied in size between 441 and 456 bp, with the exception of Shewanella xiamenensis BC01, where the fur gene was only 432 bp. The fur genes with 441 bp were from Vibrio halioticoli and Vibrio ezurae, and only Vibrio nigripulchritudo SOn1 had a 456-bp fur sequence, making the variance of the gene size mostly between 444 and 453 bp (see Fig. S1 in the supplemental material).
The maximum-likelihood phylogeny tree constructed with the alignment of the fur genes (Fig. 2) confirmed the clustering of the different species in a manner similar to what has been observed for both 16S rRNA gene- and MLSA-based trees (4, 6, 8, 21–24). The clades recently emended by Sawabe et al. (4) could also be identified (Fig. 2), although a few differences arose. Within the Vibrio species, the major differences observed were the clustering of Vibrio rotiferianus DAT722 within the Splendidus clade rather than within the Harveyi clade, as previously described (4). Other species, such as Vibrio metschnikovii and Vibrio fluvialis, did not cluster in the Cholerae clade but were clearly closely related. A similar observation was made for the species Vibrio orientalis and Photobacterium phosphoreum.
FIG 2.
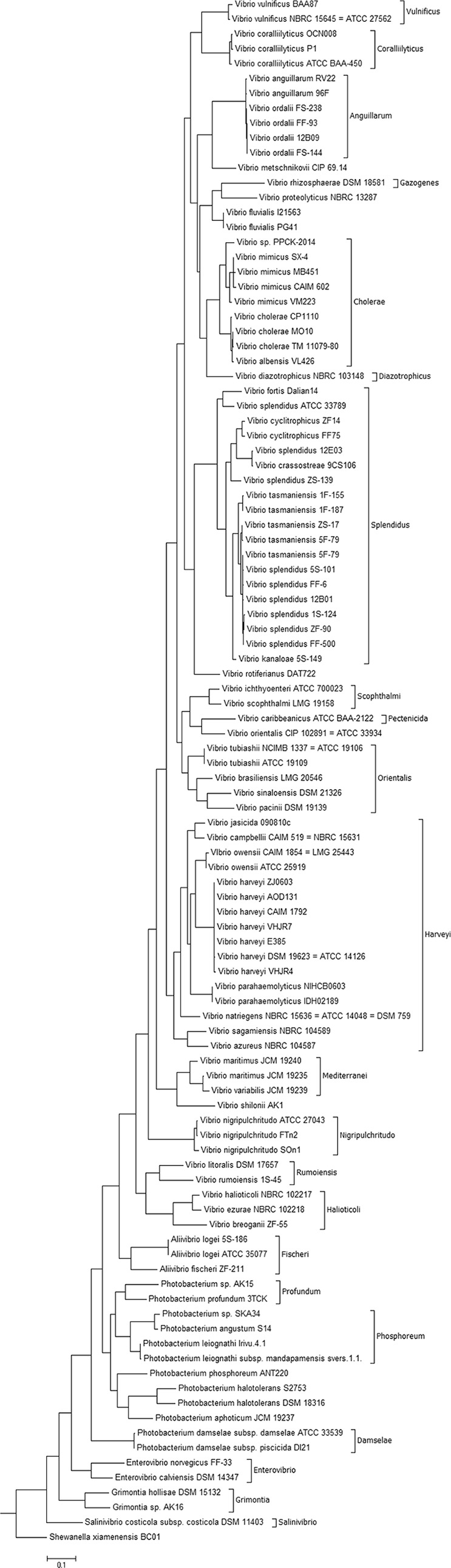
Phylogenetic tree of 103 Vibrionaceae strains. The tree is based on in silico analyses using the complete fur gene sequences and was constructed by the neighbor-joining method. S. xiamenensis BC01 was used as the outlier.
At the species level, some of the strains previously identified as Vibrio splendidus, namely, V. splendidus 12E03, V. splendidus ZS-139, and V. splendidus ATCC 33789, clustered separately from the other V. splendidus strains. In fact, V. splendidus 12E03, V. splendidus ZS-139, and V. splendidus ATCC 33789 clustered in the Vibrio cyclitrophicus and the Vibrio fortis branches of the phylogenetic tree (Fig. 2). This could be explained by the previously demonstrated genetic diversity and polyphyletic nature of V. splendidus (7, 8, 25), or it could be that these strains were misidentified, which is a recurrent problem within the genus Vibrio (22, 26, 27). The second hypothesis seems to be consistent with the genomic data index (ANI) at EzGenome (http://www.ezbiocloud.net/ezgenome/hierarchy?n=Vibrionales&d=2#), where ANI-based trees locate these strains closer to other named Vibrio species.
A comparison of the 103 sequences revealed a percent similarity in the same species of more than 97%, with the exception of two species pairs, V. splendidus and Vibrio tasmaniensis, as well as Vibrio anguillarum and Vibrio ordalii, where the above-mentioned species threshold did not apply (see Fig. S2 in the supplemental material). This is similar to what has been shown by others using the genes rpoA, recA, and pyrH, where 98%, 94%, and 94% similarities within the same species were reported (8). Among these genes, pyrH has been described as the most discriminatory (28); however, this study was performed in a small selection of strains, focusing mostly on the Harveyi, Campbellii, and Rotiferianus groups, making it difficult to do meaningful comparison between the discriminatory powers of this gene and the fur gene described here. Another gene reported as a possible good phylogenetic marker in the family Vibrionaceae is atpA (22). Compared with this gene, fur presents itself as a better phylogeny discriminator. The atpA gene phylogeny showed very high homology between species within the main identified groups, which could vary between 94 and 99%, while in the fur gene phylogeny, only the two previously described pairs (V. splendidus-V. tasmaniensis and V. anguillarum-V. ordalii) show that limitation.
Development of a PCR sequencing-based method.
The above-described in silico analysis confirmed the value of the fur sequence as a phylogenetic marker in the family Vibrionaceae, allowing the distinction of most of the strains at species level. Therefore, a PCR-based method that could be widely used by researchers working with Vibrionaceae genera would be extremely valuable as a simpler tool in the classification of Vibrio isolates or as an extra differentiation marker added to an MLSA.
To develop and validate the PCR method and the designed primers, 22 type strains obtained from several culture collections (Fig. 3) were used in the establishment of a general protocol for the amplification of fur. They included 1 Aliivibrio type strain, 4 Photobacterium type strains, 1 Enterovibrio type strain, 1 Salinivibrio type strain, 1 Grimontia type strain, and 14 Vibrio type strains. The degenerate fur_AP primers (see Materials and Methods) amplified the fur gene in most Vibrio species and in all the Photobacterium, Aliivibrio, Enterovibrio, Grimontia, and Salinivibrio species tested. The main challenge was the design of a reverse primer suitable for all the Vibrio strains, since the variation in sequence downstream of the fur gene is significant (Fig. 4). Therefore, we designed an extra set of primers, including one forward and two distinct reverse primers, allowing us to amplify the fur genes from the Vibrio strains on which the above-mentioned set of primers (AP) did not work. The primers developed here did allow amplification of all the tested strains of Vibrio species and the other genera of the family Vibrionaceae. The differences between the flanking regions of the fur gene have been described previously (13), and the conserved genetic organization of the region upstream from the fur gene was not reflected in the downstream region, where there is higher variability between species (13). We also noted in our amplification process that there was a species-dependent fragment size of the amplicon. The relationship between the fragment size amplified and the species needs further investigation, although it could possibly expedite the attribution of a provisional clade or even species at an earlier stage in the classification process.
FIG 3.
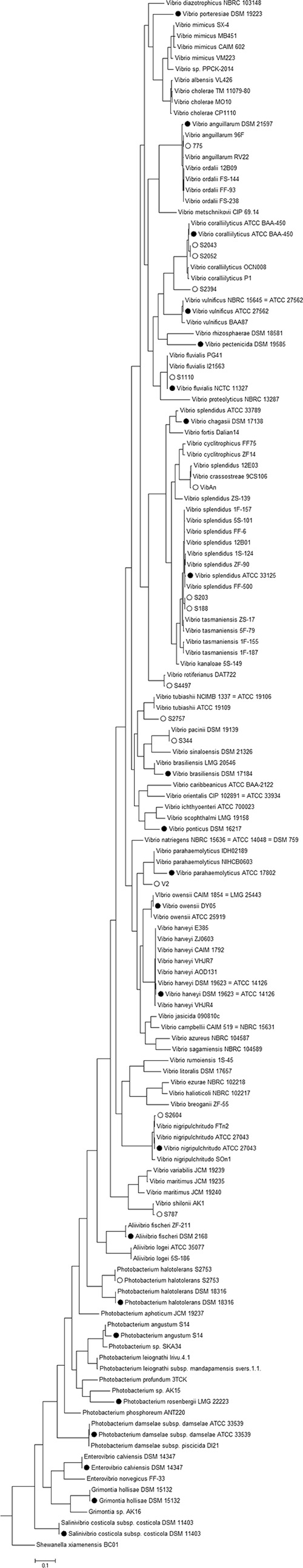
Phylogenetic tree of 140 Vibrionaceae isolates (including strains shown in Fig. 1). The tree is based on the PCR-amplified and sequenced complete fur gene sequences of representatives of each species and the fur sequences of the type strains and of the isolates used in the development and testing of the PCR sequencing-based method and was constructed using the neighbor-joining method. The circles indicate fur genes sequenced in the development and testing of the PCR sequencing-based method: the solid circles are the type strains used in the development of the method, and the open circles are the environmental strains used to test the method. S. xiamenensis BC01 was used as the outlier.
FIG 4.

Analysis of the variability downstream of the fur gene by alignment of the fur regions from six whole-genome-sequenced Vibrio species.
The limited number of genomes available from Grimontia, Salinivibrio, Enterovibrio, Photobacterium, and Aliivibrio species hampers the design of more universal primers, and once more WGS from strains belonging to these genera are available, a more in-depth analysis of the fur gene flanking regions might provide enough information for the design of primers suitable for all the strains belonging to these species. The so-called “primer problems” have been previously reported in several MLSA analyses, both for strains from the genus Vibrio and for strains of the genus Photobacterium (4, 6). This likely reflects the large genomic variability between members of the family Vibrionaceae.
After amplification and sequencing of the amplicons, the sequences were used in two approaches: (i) the fur sequences were extracted from the consensus sequence and subjected to BLAST searches against the NCBI database or (ii) the whole consensus sequence was used in the BLAST analysis. Both approaches showed a high level of identification, since the first BLAST hits in both cases corresponded to strains of the same species as the tested strains. Nevertheless, this approach using the NCBI database directly presented some limitations. Because few fur sequences are available for the Vibrio species, the hits that were obtained corresponded to whole-genome-sequenced strains, limiting the analysis to such strains. In fact, the results were more expressive and clearer when the extracted fur sequences were subjected to BLAST searches against a database created with the sequences used in the in silico analysis. This shows the importance of creating a database with more fur sequences in order to obtain more accurate identifications and to bring this method to its full potential.
Identification of Vibrio strains using the fur gene sequence.
To test the method developed, a collection of previously isolated Vibrio species was used. The strains have been categorized in our laboratory by conventional 16S rRNA gene sequence analyses (29), and some also by sequencing of recA, toxR, and rpoA genes (30). PCR, as has been described, worked well for all the tested strains (Fig. 4), so the amplicons were purified and sequenced. The fur genes were identified in all the sequenced amplicons.
The isolated fur sequences were incorporated in the phylogenetic tree, giving an overview of the distribution of the isolates between the different species and clades (Fig. 3). Of the tested isolates, strains S2757 and S2394 could not be identified with high certainty, although it is obvious that strain S2757 clustered closely with Vibrio tubiashii (Fig. 3). This inconclusive identification could indicate that strain S2757 belongs to a new species; further attempts using MLSA were also inconclusive (data not shown). With respect to strain S2394, the fact that no WGS or fur sequences from Vibrio neptunius are available led to a close association of the strain with Vibrio coralliilyticus, which makes sense, given its close phylogenetic proximity (4) (Fig. 3).
It is evident that whole-genome sequencing and bioinformatics will drive identification and taxonomy in the future. Nevertheless, it may be a while until the average laboratory can afford the whole-genome sequencing of their strains and can master the bioinformatics needed to correctly identify them. Therefore, we believe that the discovery of the phylogenetic power of the fur gene and the development of a PCR method that can be used in amplification and sequencing of the gene is of general interest, whether for use alone or together with the previously suggested loci in an MLSA.
Supplementary Material
ACKNOWLEDGMENTS
H.M. was supported by a Ph.D. grant from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7-People-2012-ITN, under grant agreement no. 317058, “BACTORY.”
We thank Paul D'Alvise for helpful discussions and for providing genome sequences and isolates of newly isolated Vibrio species.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00058-15.
REFERENCES
- 1.Amaral GRS, Dias GM, Wellington-Oguri M, Chimetto L, Campeão ME, Thompson FL, Thompson CC. 2014. Genotype to phenotype: identification of diagnostic vibrio phenotypes using whole genome sequences. Int J Syst Evol Microbiol 64:357–365. doi: 10.1099/ijs.0.057927-0. [DOI] [PubMed] [Google Scholar]
- 2.Drews G. 2000. The roots of microbiology and the influence of Ferdinand Cohn on microbiology of the 19th century. FEMS Microbiol Rev 24:225–249. doi: 10.1111/j.1574-6976.2000.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 3.Wheelis M, Kandler O, Woese C. 1992. On the nature of global classification. Proc Natl Acad Sci U S A 89:2930–2934. doi: 10.1073/pnas.89.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawabe T, Ogura Y, Matsumura Y, Feng G, Amin AR, Mino S, Nakagawa S, Sawabe T, Kumar R, Fukui Y, Satomi M, Matsushima R, Thompson FL, Gomez-Gil B, Christen R, Maruyama F, Kurokawa K, Hayashi T. 2013. Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front Microbiol 4:414. doi: 10.3389/fmicb.2013.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishiguchi MK. 2003. Evolution of symbiosis in the Vibrionaceae: a combined approach using molecules and physiology. Int J Syst Evol Microbiol 53:2019–2026. doi: 10.1099/ijs.0.02792-0. [DOI] [PubMed] [Google Scholar]
- 6.Sawabe T, Kita-Tsukamoto K, Thompson FL. 2007. Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J Bacteriol 189:7932–7936. doi: 10.1128/JB.00693-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson F, Hoste B. 2001. Genomic diversity amongst Vibrio isolates from different sources determined by fluorescent amplified fragment length polymorphism. Syst Appl Microbiol 24:520–538. doi: 10.1078/0723-2020-00067. [DOI] [PubMed] [Google Scholar]
- 8.Thompson FL, Gevers D, Thompson CC, Dawyndt P, Naser S, Hoste B, Munn CB, Swings J. 2005. Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Appl Environ Microbiol 71:5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Roux F, Gay M, Lambert C, Waechter M, Poubalanne S, Chollet B, Nicolas J, Berthe F. 2002. Comparative analysis of Vibrio splendidus-related strains isolated during Crassostrea gigas mortality events. Aquat Living Resour 15:251–258. doi: 10.1016/S0990-7440(02)01176-2. [DOI] [Google Scholar]
- 10.Gevers D, Cohan F, Lawrence J. 2005. Re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 11.Gabriel MW, Matsui GY, Friedman R, Lovell CR. 2014. Optimization of multilocus sequence analysis for identification of species in the genus Vibrio. Appl Environ Microbiol 80:5359–5365. doi: 10.1128/AEM.01206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jogler C, Lin W, Meyerdierks A, Kube M, Katzmann E, Flies C, Pan Y, Amann R, Reinhardt R, Schüler D. 2009. Toward cloning of the magnetotactic metagenome: identification of magnetosome island gene clusters in uncultivated magnetotactic bacteria from different aquatic sediments. Appl Environ Microbiol 75:3972–3979. doi: 10.1128/AEM.02701-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Achenbach LA, Yang W. 1997. The fur gene from Klebsiella pneumoniae: characterization, genomic organization and phylogenetic analysis. Gene 185:201–207. doi: 10.1016/S0378-1119(96)00642-7. [DOI] [PubMed] [Google Scholar]
- 14.Colquhoun DJ, Sørum H. 2002. Cloning, characterisation and phylogenetic analysis of the fur gene in Vibrio salmonicida and Vibrio logei. Gene 296:213–220. doi: 10.1016/S0378-1119(02)00863-6. [DOI] [PubMed] [Google Scholar]
- 15.Escolar L, Pérez-Martín J, De Lorenzo V. 1999. Opening the iron box: transcriptional metalloregulation by the Fur protein. J Bacteriol 181:6223–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McHugh JP, Rodríguez-Quinoñes F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC. 2003. Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J Biol Chem 278:29478–29486. doi: 10.1074/jbc.M303381200. [DOI] [PubMed] [Google Scholar]
- 17.Vasileva D, Janssen H, Hönicke D, Ehrenreich A, Bahl H. 2012. Effect of iron limitation and fur gene inactivation on the transcriptional profile of the strict anaerobe Clostridium acetobutylicum. Microbiology 158:1918–1929. doi: 10.1099/mic.0.056978-0. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nørholm MHH. 2010. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol 10:21. doi: 10.1186/1472-6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen S, Frost P, Torsvik VL. 2009. The nonrandom microheterogeneity of 16S rRNA genes in Vibrio splendidus may reflect adaptation to versatile lifestyles. FEMS Microbiol Lett 294:207–215. doi: 10.1111/j.1574-6968.2009.01567.x. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann M, Monday SR, Fischer M, Brown EW. 2012. Genetic and phylogenetic evidence for misidentification of Vibrio species within the Harveyi clade. Lett Appl Microbiol 54:160–165. doi: 10.1111/j.1472-765X.2011.03183.x. [DOI] [PubMed] [Google Scholar]
- 22.Thompson CC, Thompson FL, Vicente ACP, Swings J. 2007. Phylogenetic analysis of vibrios and related species by means of atpA gene sequences. Int J Syst Evol Microbiol 57:2480–2484. doi: 10.1099/ijs.0.65223-0. [DOI] [PubMed] [Google Scholar]
- 23.Urbanczyk H, Ogura Y, Hayashi T. 2013. Taxonomic revision of Harveyi clade bacteria (family Vibrionaceae) based on analysis of whole genome sequences. Int J Syst Evol Microbiol 63:2742–2751. doi: 10.1099/ijs.0.051110-0. [DOI] [PubMed] [Google Scholar]
- 24.Thompson F, Iida T, Swings J. 2004. Biodiversity of vibrios. Microbiol Mol Biol Rev 68:403–431. doi: 10.1128/MMBR.68.3.403-431.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pascual J, Macián MC, Arahal DR, Garay E, Pujalte MJ. 2010. Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int J Syst Evol Microbiol 60:154–165. doi: 10.1099/ijs.0.010702-0. [DOI] [PubMed] [Google Scholar]
- 26.Lin B, Wang Z, Malanoski AP, O'Grady EA, Wimpee CF, Vuddhakul V, Alves N Jr, Thompson FL, Gomez-Gil B, Vora GJ. 2010. Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ Microbiol Rep 2:81–89. doi: 10.1111/j.1758-2229.2009.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez-Gil B. 2004. Vibrio hispanicus sp. nov., isolated from Artemia sp. and sea water in Spain. Int J Syst Evol Microbiol 54:261–265. doi: 10.1099/ijs.0.02775-0. [DOI] [PubMed] [Google Scholar]
- 28.Chimetto LA, Brocchi M, Gondo M, Thompson CC, Gomez-Gil B, Thompson FL. 2009. Genomic diversity of vibrios associated with the Brazilian coral Mussismilia hispida and its sympatric zoanthids (Palythoa caribaeorum, Palythoa variabilis and Zoanthus solanderi). J Appl Microbiol 106:1818–1826. doi: 10.1111/j.1365-2672.2009.04149.x. [DOI] [PubMed] [Google Scholar]
- 29.Gram L, Melchiorsen J, Bruhn JB. 2010. Antibacterial activity of marine culturable bacteria collected from a global sampling of ocean surface waters and surface swabs of marine organisms. Mar Biotechnol 12:439–451. doi: 10.1007/s10126-009-9233-y. [DOI] [PubMed] [Google Scholar]
- 30.Wietz M, Mansson M, Gotfredsen CH, Larsen TO, Gram L. 2010. Antibacterial compounds from marine Vibrionaceae isolated on a global expedition. Mar Drugs 8:2946–2960. doi: 10.3390/md8122946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.