

Abstract
New-onset diabetes after transplantation (NODAT) is associated with increased risk of allograft failure, cardiovascular disease and mortality, and therefore, jeopardizes the success of renal transplantation. Increased awareness of NODAT and the prediabetic states (impaired fasting glucose and impaired glucose tolerance, IGT) has fostered previous and present recommendations, based on the management of type 2 diabetes mellitus (T2DM). Unfortunately, the idea that NODAT merely resembles T2DM is potentially misleading, because the opportunity to initiate adequate anti-hyperglycaemic treatment early after transplantation might be given away for ‘tailored’ immunosuppression in patients who have developed NODAT or carry personal risk factors. Risk factor-independent mechanisms, however, seem to render postoperative hyperglycaemia with subsequent development of overt or ‘full-blown’ NODAT, the unavoidable consequence of the transplant and immunosuppressive process itself, at least in many cases. A proof of the concept that timely preventive intervention with exogenous insulin against post-transplant hyperglycaemia may decrease NODAT was recently provided by a small clinical trial, which is awaiting confirmation from a multicentre study. However, because early insulin therapy aimed at beta-cell protection seems to contrast the currently recommended, stepwise approach of ‘watchful waiting’ prior to pancreatic decompensation, we here aim at reviewing recent concepts regarding the development, prevention and treatment of NODAT, some of which seem to challenge the traditional view on T2DM and NODAT. In summary, we suggest a novel, risk factor-independent management approach to NODAT, which includes glycaemic monitoring and anti-hyperglycaemic treatment in virtually everybody after transplantation. This approach has widespread implications for future research and is intended to tackle NODAT and also ultimately cardiovascular disease.
INTRODUCTION
Renal transplantation has become a great success story overall [1], mainly because kidney transplant recipients (KTRs) benefit from increased survival rates [2–7] and higher quality of life compared with dialysis patients [8–11]. To ensure that post-transplant outcomes may continue to improve in aging end-stage renal disease populations, the transplant community is undertaking considerable efforts. Specifically, multiple diagnostic procedures along with subsequent interventions are performed prior to wait listing [12]. Organizations responsible for organ allocation optimize immunological donor–recipient matching [13, 14]. KTRs are screened and treated for infection, rejection and cardiovascular disease. Furthermore, the search for optimal immunosuppression is undergoing constant review [15–18].
Among the leading obstacles to long-term allograft and recipient survival is new-onset diabetes after transplantation (NODAT) [19]. Data from the US renal data system (USRDS) indicate that 40% of KTRs will have developed NODAT by their third year post-transplantation [20]. This number is alarming, because NODAT is a major risk factor for cardiovascular disease [21] and mortality [22–25], and is also associated with reduced kidney graft survival [26, 27], infections [28–30] and increased health care costs [31]. Impaired glucose tolerance (IGT), which naturally precedes the onset of diabetes, has likewise been linked to mortality, indicating that an even greater number of KTRs may be at risk [22].
NODAT has commonly been viewed as resembling type 2 diabetes mellitus (T2DM) [32, 33]. Hyperglycaemia after transplantation, however, appears rapidly and the transition to full-blown diabetes is clearly much faster than in T2DM [23, 31], due to a variety of transplant-specific mechanisms. Evidence suggests that beta-cell dysfunction rather than insulin resistance is the main contributing factor for NODAT development [34–36] and early insulin-based correction of postoperative hyperglycaemia has proven beneficial, most probably through beta-cell protection [37]. However, this intervention seems to contrast sharply with the 2003 international consensus guidelines recommending a stepwise approach to NODAT treatment, based on T2DM [33].
Postoperative hyperglycaemia, despite high blood glucose levels ≥200 mg/dL, can formally not be equalized to ‘full-blown’, or ‘overt’ NODAT, which has previously also been defined as the need for antidiabetic treatment >30 days [33]. Even for overt NODAT, however, a stepwise approach might be inadequate, because beta-cell function in KTRs may suffer from iatrogenic stress (e.g. immunosuppressants) and can likely be rescued more efficiently by intensive interventions, possibly also at later time points. The aim of the present article is, therefore, to review novel concepts regarding the development, prevention and treatment of NODAT, some of which challenge traditional views on T2DM and NODAT, as well as their relation. As a consequence, we suggest a revised monitoring and management approach with the goal of protecting or rescuing beta-cell function, early after transplantation and beyond.
NODAT DEVELOPMENT
Risk factors
Review articles [32, 38, 39] and the 2003 international consensus guidelines [33] focus on the risk factors for NODAT and the potential to modify them for the sake of reducing diabetes development [33]. Traditional, ‘non-transplant-specific’ characteristics such as older age, family history, various ethnic backgrounds, obesity or hepatitis C are well known to occur at a higher rate in the non-transplanted population with T2DM compared with nondiabetics [40–42] and a large body of evidence has also shown an association between these characteristics and the risk of NODAT in KTRs (reviewed in [32, 33, 38, 39]). Linked to obesity, triglyceride levels as well as the metabolic syndrome are also well-established risk factors for NODAT [43, 44]. Perhaps less well recognized, Ghisdal et al. genotyped KTRs without diabetes at transplantation for 11 polymorphisms that associate with T2DM and found that NODAT was significantly associated with the TCF7L2 polymorphism [45], expressed in pancreatic beta cells and involved in the control of insulin secretion.
Among ‘transplant-specific’ risk factors, the contribution of immunosuppressive agents to NODAT development is predominant [32, 33, 38, 39]. Polycystic kidney disease [46–48], cytomegalovirus infection [21, 49, 50], hypomagnesemia [51] and HLA-mismatch (reviewed in [52]) have also been proposed to play a role. Importantly, episodes of acute rejection are usually treated with high-dose corticosteroids. In the multivariate analysis by Ghisdal et al. [45], genetic factors were compared with clinical aspects and NODAT was more strongly associated with the occurrence of a corticosteroid-treated acute rejection episode than with tacrolimus use [45]. A recent study moreover showed that the mean number of acute rejection episodes per KTR was significantly higher among NODAT patients and that in KTRs who experienced both overt NODAT and an acute rejection, the rejection episode almost always occurred first [53]. Rejections and subsequent corticosteroid administration may, therefore, be classified into the group of transplant-specific risk factors leading to the development of overt NODAT.
The focus on diabetes risk factors inevitably leads to the recommendation that patients with the highest risk for NODAT development should be identified and then receive individualized or ‘tailored’ immunosuppression as well as counselling on weight control and physical activity in order to prevent the disease [32, 33, 39]. However, early and severe hyperglycaemia from the first post-transplant days is the rule rather than the exception [37, 54], and may thus be viewed as a predictable consequence of mechanistic changes controlling glucose metabolism, induced by the process of renal transplantation and its encompassing medications. Post-transplant hyperglycaemia is consecutively linked to overt NODAT [55], potentially even in hyperglycaemic KTRs who do not have a pre-existing elevated risk.
Prevalence of early post-transplant hyperglycaemia
At the Mayo Clinic, 87% of KTRs without pre-transplant diabetes showed evidence of post-transplant hyperglycaemia (bedside glucose >200 mg/dL or physician-instituted insulin therapy) under tacrolimus-based immunosuppression [54]. At the Medical University of Vienna, in a recently completed prospective trial of basal insulin against NODAT (the ‘TIP-study’, Trial of Basal Insulin in Post-transplant Hyperglycaemia [37]), conventional treatment (control) patients experienced hyperglycaemia (blood glucose ≥200 mg/dL) at a similarly high rate (23/25 = 92%). Moreover, all patients in the insulin intervention group (25/25 = 100%) had blood glucose ≥140 mg/dL by the third day post-transplantation, although glucose was always measured ≥2 h after caloric intake, if not fasting. The latter finding was also observed in the control group, where all patients (25/25 = 100%) had blood glucose ≥140 mg/dL by the third day post-transplantation. Blood glucose ≥140 mg/dL in an oral glucose tolerance test (OGTT) defines impaired glucose metabolism [56].
A prototypic glucose profile early after renal transplantation from a TIP-study participant is shown in Figure 1. As illustrated by the graphs, fasting glucose was low between Days 3 and 16 after kidney transplantation in this patient, while the maximal glucose values were consistently observed during the evening hours. This effect is a well-known consequence of corticosteroids, which when administered in the morning exert their maximal hyperglycaemic effect during the course of the day, but suppress endogenous steroid secretion on the following morning [57–59].
FIGURE 1:
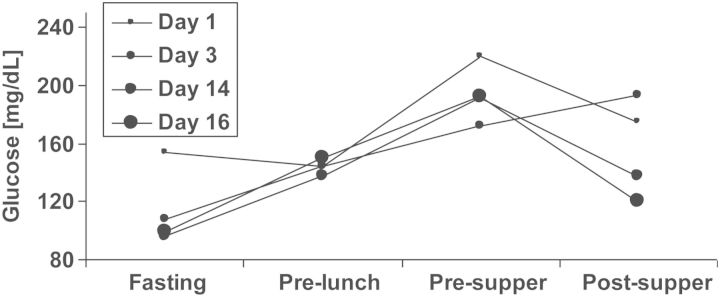
A prototypic blood glucose profile after transplantation. A 67-year-old female with body mass index 27 kg/m2, without diabetes, without family history of diabetes and without hepatitis C infection, had been on haemodialysis for 18 months (lithium-induced nephropathy) before undergoing transplantation with a deceased donor kidney. The patient agreed to participate in the treat-to-target TIP-study [37] and was randomized to the conventional treatment (control) group. HbA1c was 5.0% at baseline. The very early post-transplant glucose profile and the glucose profile in post-transplant Week 3 are displayed. In post-transplant Week 2, the patient received short acting insulin on two consecutive days, but no more insulin corrections were administered and the patient was discharged on post-transplant Day 20. Non-fasting blood glucose ≥200 mg/dL had occurred only during three independent days before discharge. At the first TIP-study control visit on post-transplant Day 87, HbA1c had increased to 7.3%, 2 h glucose value during OGTT was 238 mg/dL, OGTT-derived beta-cell function was poor [insulinogenic index (IGI) = 0.016 nmol insulin/mmol glucose], while insulin sensitivity was not clearly impaired [oral glucose insulin sensitivity (OGIS) index = 300 mL/min/m2]. This patient's decompensation of glucose metabolism had not previously been noticed by measurements of fasting glucose during any of the patient's visits in the outpatient clinic. The patient received an oral antidiabetic agent (sulphonylurea) throughout the end of the study's follow-up as well as 2 years thereafter, when she was additionally contacted. Two consecutive high-dose corticosteroid treatments for acute transplant rejection had been given during post-transplant Weeks 3 and 5, which might have contributed to her rapid impairment of glucose metabolism. As discussed in the text, self-measurements of evening glucose along with early insulin therapy might have uncovered and simultaneously prevented the rapid decline in beta-cell function. Corticosteroid-treated rejections have been shown to associate with NODAT development [45, 53] and may therefore be considered transplant-specific risk factors rather than confounders, as discussed in the text. Fasting: ∼7:30 am, pre-lunch: ∼12:00 am, pre-supper: ∼5:30 pm, post-supper: ∼9:00 pm.
As described in detail in the legend to Figure 1, this patient's history of NODAT development was drastic (for example HbA1c increased from 5.0% at transplantation to 7.3% at 87 days post-transplantation). However, such a clinical course may not be uncommon, as indicated by the finding of a high diabetes prevalence at 3 months post-transplantation in the control group of our trial (13/25 = 52%) [37]. While age and high-dose corticosteroid treatment in the reported case were clear risk factors, both ‘non-transplant specific’ as well as ‘transplant-specific’, it seems unlikely that this drastic form of disease progression could have been prevented by risk factor-dependent counselling on diet and physical activity immediately after kidney transplantation, or by tailored immunosuppression alone.
Novel terminology and collective mechanisms
In 2007, Crutchlow and Bloom proposed the term transplant-associated hyperglycaemia (TAH) [60], specifying also in a subsequent editorial, that this term encompasses the full range of new-onset, post-transplant glycaemic abnormalities, including the prediabetic states of impaired fasting glucose and IGT, as well as NODAT [61]. Although the authors did not focus on immediate postoperative TAH, their definition was a major step forward, not only by naming hyperglycaemia as the root of subsequent disease in context with the disease itself, but also by expanding the risk population to KTRs who have ‘only’ IGT. Compellingly, the authors also focussed on general mechanisms in TAH pathogenesis that must be collectively considered, independent of individual risk factors. We attempted to complete their list as follows and as illustrated in the upper part of Figure 2. From here on as well as in the figure, we will discriminate between impaired insulin secretion and increased insulin resistance (e.g. impaired insulin sensitivity), both of which may contribute to NODAT development [62], although there may be disagreement on the relative importance of either component [34–36, 63], as further discussed below.
FIGURE 2:

General mechanisms of NODAT development, (A) early postoperative prevention and (B) late postoperative treatment. Beta cell mass inside the pancreas leading to insulin secretion is depicted by the clouds. The idea that beta cells can become stressed is symbolized by the warning flashes. Insulin resistance is depicted by the circle, and contributors to insulin resistance are also symbolized by flashes. When describing early post-transplant status (upper part of the figure = development), the figure explicitly contains only general mechanistic details, but no patient-specific risk factors of NODAT development, such as age, obesity, family history, ethnic background and viral infection, which may further influence insulin secretion and insulin resistance in a patient-specific manner. Asterisk denotes that these factors are listed in the text as contributors to hyperglycaemia (a, b and c). (A) Early basal insulin and lifestyle intervention (diet and exercise) act against evening hyperglycaemia and are helping beta cells to overcome the burden imposed after renal transplantation, as symbolized by green flashes. Insulin secretion may consequently increase. (B) Long-term anti-hyperglycaemic treatment may become necessary in stable KTRs if NODAT prevention has not been established or has failed. Note that the circle symbolizing insulin resistance is neither augmented nor diminished after renal transplantation, because the progression of insulin resistance is currently still a matter of debate. However, as indicated in the text, convincing evidence indicates that insulin resistance may even increase.
‘Risk factor-independent’ mechanisms contributingto NODAT development:
Calcineurin inhibitors (CNIs), tacrolimus and cyclosporin A, are the mainstay of immunosuppressive therapy [64], but are also well known to substantially impair insulin secretion in vitro [65, 66] and in vivo [66–69], because calcineurin/NFAT (nuclear factor of activated T-cells) signalling regulates pancreatic beta-cell growth and function [70]. CNIs thereby indispensably cause hyperglycaemia (reviewed in [60]), and overt NODAT, with tacrolimus having a higher diabetogenic potential than cyclosporin A [31, 71–74].
Glucocorticoids increase dose-related [75] hepatic glucose production (via stimulation of gluconeogenesis), augment insulin resistance [76–79] and suppress insulin secretion, in addition to inducing islet cell apoptosis at higher doses [80–86].
- Hyperglycaemia by itself is a recognized stressor for beta cells, suppressing insulin secretion and/or leading to beta-cell apoptosis in vitro [87–89] via oxidative stress [90]. Beta-cell failure appears to play a major role in T2DM development [91, 92] and is thus very likely to play a key role in NODAT development as well. Additional general mechanisms contribute to hyperglycaemia in the post-transplant setting:
- Common but unhealthy dietary habits, for example excessive consumption of pure sugar, rapidly absorbed carbohydrates and saturated fatty acids (‘fast food’), in conjunction with a deficiency of aerobic exercise, are well known to increase the risk of developing T2DM [98–103] and are, therefore, very likely to also increase the risk of post-transplant hyperglycaemia and subsequent NODAT among KTRs. In addition, dietary advice intended to benefit dialysis patients, such as low protein (low phosphate), high caloric intake, may be insufficiently revised post-transplantation.
- Insulin demand increases after renal transplantation.
- As mentioned by Crutchlow and Bloom [60], healthy kidneys degrade insulin, shown by an arteriovenous decrease in insulin concentration [106, 107] and decreased insulin requirements in patients with T2DM during kidney disease progression [108]. Through sudden restoration of kidney function after renal transplantation, insulin demand increases (normalizes) in KTRs without previous T2DM.
- Insulin demand increases through hyperglycaemia alone. Consequently, beta-cell stress can in turn cause impaired insulin secretion, which may drive subsequent hyperglycaemia with even higher insulin requirements. This vicious cycle of hyperglycaemia and hypoinsulinaemia has been reported to be associated with poor hospital outcomes [109], and most likely occurs in an accelerated form during the course of NODAT development.
Because insulin resistance is prevalent in patients with chronic kidney disease [110] and the majority of stable KTRs are in chronic kidney disease Stages II and III [111], chronic allograft dysfunction and uraemia before [112], but also after renal transplantation can plausibly exert a relevant influence on insulin resistance and subsequent NODAT development.
Decreased insulin secretion versus increased insulin resistance
Although plasma triglyceride concentration and the ratio of triglyceride to high-density lipoprotein cholesterol concentrations have been shown to be useful metabolic markers for insulin resistance in the general population [113], and in turn are associated with NODAT development [43], previous reports are controversial regarding the relative importance of either increased insulin resistance and/or decreased insulin secretion in NODAT development.
Using hyperinsulinaemic euglycaemic as well as hyperglycaemic clamps combined with indirect calorimetry and infusion of tritiated glucose, Ekstrand et al. have shown that both insulin resistance and insulin deficiency are necessary for NODAT development [62]. Midtvedt et al. subsequently analysed OGTTs, and classified KTRs as having normal glucose tolerance (NGT), IGT and NODAT. The authors also performed hyperinsulinaemic euglycaemic clamps and found a significant difference in insulin sensitivity between patients with NODAT versus those with NGT, and between patients with IGT versus those with NGT. Midtvedt et al. concluded in their abstract as well as in their title that insulin resistance is a common denominator of KTRs with NODAT and IGT [63]. Although the same study simultaneously identified a significant difference in insulin secretion between KTRs with NODAT versus those with NGT, the authors did not further comment on insulin secretion. The transplant community may subsequently have attached greater importance to insulin resistance than to insulin secretion.
In the same year as Midtvedt et al., Shimizu et al. also performed hyperinsulinaemic euglycaemic clamps, and published that in comparison to haemodialysis patients, KTRs had ‘higher’ insulin sensitivity [36]. The authors concluded that renal transplantation ‘restores’ (decreases) insulin resistance found in renal failure patients, but does not ameliorate insulin secretion.
Hagen et al. analysed KTRs by OGTTs at 10 weeks post-transplantation, as well as 6 years later, and found that OGTT-derived insulin secretion was significantly reduced, whereas insulin sensitivity significantly ‘increased’ [34] at 6 years in comparison to 10 weeks post-transplantation, presumably a consequence of corticosteroid tapering.
In an adequately powered study, Nam et al. analysed OGTTs at 1 week before and 9–12 months after living-related renal transplantation [35]. The authors found significantly lower OGTT-derived insulin secretion (area under the curve) in the NODAT group after transplantation, in comparison to before. Importantly, insulin sensitivity was ‘higher’ in all groups (NGT, IGT, NODAT) after transplantation in comparison to before. Although some caution must be applied when comparing results from Asian (here: Korean) populations with those from Caucasians, it seems that the results linking NODAT and beta-cell failure are more convincing than those emphasizing the importance of insulin resistance.
In the non-transplanted general population, a previously ongoing debate has been labelled as the ‘eternal chicken and egg question’ [114] whether disease progression from NGT to T2DM is mainly caused by insulin resistance, as historically perceived [115–117], or beta-cell dysfunction [118], which in turn would necessitate treatment options that preserve beta-cell function [119]. Data from the UK prospective diabetes study have, however, shown that for patients with newly diagnosed T2DM, glycaemic deterioration is associated with progressive loss of beta-cell function [120, 121]. Moreover, although insulin resistance is a major pathogenic factor underlying progression from NGT to IGT to diabetes, deterioration in glycaemic control does not occur unless beta cells fail to compensate for insulin resistance [122]. Eventually, beta-cell failure is responsible for IGT progression to T2DM [123, 124]; among IGT subjects, a low plasma insulin concentration predicts IGT progression to T2DM in many ethnic groups [125, 126].
It is worth noting that even in NGT, beta-cell function is the best predictor for the progression of NGT to IGT and subsequently to T2DM [127]. Because insulin secretion is tightly regulated by insulin sensitivity [128], the matter of query is also about the type of relationship between the two entities. While it appears clear that insulin secretion (or better, beta-cell function) and insulin sensitivity are inversely correlated [129], a controversy exists as to whether the relationship is linear or non-linear and if, in the latter case, it is a regular hyperbola or a power function [130]. These distinctions imply different physiological interpretations, although it may not be trivial to make inferences about treatment for T2DM, as a consequence of further untangling this puzzling relationship [131].
The above-mentioned relationship between insulin secretion and insulin sensitivity in the general population may not genuinely be transferred to KTRs, where multiple processes occur simultaneously after transplantation as described above and possibly with great individual variability. Our most recent results from comparing a large cohort of stable KTRs with the general population indicate that glucose metabolism as a whole—including HbA1c, fasting glucose and OGTT-derived 2h glucose—differs substantially between KTRs and non-transplanted control subjects. Because in our analyses, an impairment in OGTT-derived insulin secretion appeared to be the predominant pathophysiological feature after renal transplantation, supportive of the results obtained by Shimizu et al. [36], Hagen et al. [34] and Nam et al. [35], we concluded that therapeutic regimens that preserve beta-cell function are potentially beneficial in this population [132].
In summary on NODAT development, risk factor-independent mechanisms occur routinely in patients undergoing renal transplantation today. These general mechanisms are very likely to explain recent clinical observations of postoperative hyperglycaemia [37, 54, 55]. The respective importance of insulin secretion versus insulin resistance has been controversially perceived [34–36, 63], possibly in analogy to the general population where a similar argument has been ongoing. Because impaired insulin secretion seems to be the predominant component of NODAT development, we suggest that beta-cell protection is a reasonable goal for NODAT prevention.
NODAT PREVENTION
Previous NODAT prevention concepts
In the 2003 international consensus guidelines, NODAT prevention is mentioned in the context of monitoring fasting glucose and OGTT-derived 2 h glucose levels [33]. Later NODAT reviews [32, 38, 39, 60, 133] emphasized the importance of preventing NODAT by modifying patient-specific risk factors: obesity [134], hyperlipidaemia [135], viral infections [136, 137] and an activated renin–angiotensin system [138]. ‘Tailoring’ of the immunosuppression has received the greatest attention among NODAT prevention strategies in the 2003 international consensus guidelines and subsequent reviews. Specifically, pre-transplant individualization of immunosuppression based on cardiovascular and diabetes risk factors [32, 33, 38, 39, 60], and post-transplant withdrawal of corticosteroids or conversion from tacrolimus to cyclosporin A have been advocated [33] and described [139–143]. During the late course of transplantation, Luan et al. showed a lack of difference in NODAT risk between cyclosporin A- and tacrolimus-treated, nondiabetic KTRs [144], indicating perhaps that conversion might be of even greater benefit during the early postoperative period. Greater efficacy of early post-transplant conversion might mechanistically be plausible, according to previous data on insulin release in tacrolimus- and cyclosporin A-treated patients, respectively [67].
Regarding corticosteroid withdrawal, although most available literature suggest reduced NODAT risk with early corticosteroid withdrawal [27, 145–150], an overall beneficial effect of corticosteroid sparing strategies has not been uniformly demonstrated [148]. Systematic reviews and meta-analyses recently undertaken suggest that corticosteroid withdrawal between 3 and 6 months after transplantation had no meaningful effect on NODAT incidence [145, 149]. Early corticosteroid avoidance after only some days of treatment did show decreased NODAT incidence, but only significant when the CNI used was cyclosporine A. Interestingly, this positive effect disappeared when the immunosuppression was based on the more diabetogenic tacrolimus [145, 150]. Moreover, a mild increase in the incidence of acute rejection with corticosteroid sparing strategies might counterbalance the metabolic beneficial effect [32, 145, 149–151].
A novel NODAT prevention concept
Based on the available evidence, we posit NODAT cannot be efficiently prevented by tailored immunosuppression alone without compromising kidney graft survival. In Figure 3, we show the development of a patient who participated in the treatment group of the TIP-study and injected relatively high daily doses of basal insulin early after transplantation [37] (for the general concept, see also Panel A of Figure 2). This patient's very favourable metabolic outcomes were not an exception, as shown by 73% lower odds of NODAT and significantly lower HbA1c in the basal insulin treatment group compared with the standard-of-care control group throughout 1 year of follow-up. In addition, significantly improved beta-cell function in this exemplary case and in the entire treatment group, in comparison to the control group [37], suggests that early insulin therapy may genuinely protect beta cells against the deleterious NODAT-causing factors listed here above.
FIGURE 3:

A case of NODAT prevention after renal transplantation. A 47-year-old male with body mass index 21 kg/m2, without diabetes, with family history of diabetes and without hepatitis C infection, underwent transplantation with a deceased donor kidney. Manifestation of end-stage renal disease (glomerulonephritis) had been 25 years ago (one previous graft, functioning for 18 years). The patient agreed to participate in the Treat-to-target TIP-study [37] and was randomized to the long-acting insulin treatment group. Although not required for the study protocol, this patient measured glucose at least twice daily throughout the course of one whole year, and his mean daily glucose values are displayed in the figure. In contrast to all other treatment patients, and not explicitly foreseen for the study protocol, this patient went back to injecting long-acting insulin three times after having been weaned off (which was not initially revealed to the study investigators), because he noted a rise in glucose levels upon high-dose corticosteroid treatment for rejection. As shown in the figure, HbA1c increased from baseline to the 3-month follow-up OGTT, despite insulin therapy and continuously decreased thereafter. The 2 h glucose level during the OGTT was excellent at 12 months, but higher before (data in the figure). OGTT-derived beta-cell function (IGI, displayed in the figure) was excellent at 12 months only. Insulin sensitivity (OGIS index, displayed in the figure) increased slightly towards the 12-month follow-up visit, but not nearly as strongly as insulin secretion: Specifically, from 6 to 12 months, OGIS increased by 7%, while IGI increased by 600%. At 2 years after the end of study follow-up, when that patient was additionally contacted, he reported excellent glycaemic control revealed during sporadic self-measurements of glucose, his most recent HbA1c had been 5.3%. Asterisk denotes that the y-axis units are in mg/dL for glucose, mg for prednisone, IU for insulin. Double asterisks denote during 3 days, the patient received 1000 mg of methylprednisolone, equivalent to 1250 mg of prednisone, outside the range of the axis.
Risk of hypoglycaemia
The insulin-treated patient whose glucose profiles are shown in Figure 3 did not report clinical evidence of hypoglycaemia, although 2 of his 1064 measurements throughout the course of 1 year (0.2%) showed values <60 mg/dL (44 mg/dL and 46 mg/dL, respectively). In the entire TIP-study, we determined the number of hypoglycaemic episodes during the patients' postoperative hospitalization and arrived at a similarly low rate. Specifically, capillary blood glucose between 41 and 60 mg/dL was measured once in the control group and five times in the treatment group [37], corresponding to 0.05 versus 0.2% of all measurements and to 0.15 ± 0.8 versus 1.0 ± 2.2% days per patient (P = 0.105). These hypoglycaemic episodes were not noticed by the patients, and there were no hospitalizations due to hypoglycaemic events throughout the follow-up.
Important limitations of the TIP-study, as previously described [37], include its small sample size and nearly significant group differences at baseline (for which, however, the principal study findings were adjusted). Another limitation is its open label design, although major diabetes studies [152, 153] were similarly not blinded and had to consider insulin-unrelated benefits. Patients allocated to early insulin therapy might be very motivated to optimize diet and exercise in order to be able to wean their insulin treatment. Such an effect is currently speculative and if present would not be undesired. When exogenous insulin is combined with improved health behaviour, the relative contribution of either component on metabolic outcomes should be resolved scientifically in future work.
When considering these limitations, it becomes clear that the supportive data for a novel NODAT prevention concept are presently not broad, even with pathophysiological evidence emphasizing the crucial role of insulin hyposecretion, as well as the demonstration of a significant increase in OGTT-derived beta-cell function in the TIP-study [37]. Still in favour of such a concept, it may be stated that beta-cell protection through exogenous insulin administration is only novel for the transplant community. In the general population, the benefit of insulin therapy for long-term glycaemic control among newly diagnosed type 2 diabetic patients has been reported from 1997 onwards [154–158]. A large Chinese multicentre study reached perhaps the highest impact, after showing 51% remission from newly diagnosed T2DM as late as 1 year after a 2-week normoglycaemic treatment period using continuous subcutaneous insulin infusions [159].
Risk factor-independent recommendations
A large randomized controlled clinical trial, Insulin Therapy for the Prevention of NODAT (ITP-NODAT), NCT01683331), is currently evaluating whether the promising findings of the TIP-study can be reproduced by five additional international renal transplant centres in well over 300 patients. Based on the previous findings, one may expect that (i) essentially all KTRs display early postoperative evening glucose levels ≥140 mg/dL, (ii) KTRs may be successfully treated with basal insulin and (iii) the risk of hypoglycaemia is reasonably low. Until the results of the ITP-NODAT trial become available, it seems nevertheless rational to advocate the following, general, risk factor-independent NODAT prevention strategies:
As a prerequisite for subsequent interventions, self-measurements of blood glucose should be reinforced in all KTRs immediately post-transplantation, as has also been recommended previously [38, 133]. Evening measurements of blood glucose will uncover more cases of hyperglycaemia and are, therefore, more important than fasting measurements.
If the patient can be sufficiently supervised and thus reasonably protected against hypoglycaemia in the hospital or outpatient clinical setting, early insulin therapy could be recommended as the most rational preventive strategy. Exogenous insulin seems more reasonable in this setting than administration of first-line oral antidiabetics for patients with renal insufficiency, at least agents such as the insulin secretagogues, since they are not known to restore beta-cell function [160] and might, therefore, even worsen the insulin secretion problem over time. The right timing and the threshold for given hyperglycaemia values when insulin therapy should be started may be debateable. These queries also await more data from the currently ongoing clinical studies (ITP-NODAT, NCT01683331; SAPT-NODAT, NCT01680185). However, a relatively high glucose threshold of 200 mg/dL—evening or fasting—cannot plausibly be questioned on clinical or scientific grounds.
Active lifestyle modification including dietitian referral, exercise programme and weight loss advice benefits KTRs with IGT [134] and should be incorporated in any preventive measure. However, lifestyle modifications may be more difficult to reinforce than pharmacological interventions, particularly early after transplantation when NODAT risk is greatest.
Regarding oral anti-hyperglycaemics, their specific advantages and risks for KTRs are discussed further below. We are currently not aware of published data on the use of oral anti-hyperglycaemics against early post-transplant hyperglycaemia in order to prevent overt NODAT at later time points, but the use of oral agents that are not known to worsen beta-cell function may potentially be beneficial. An interesting safety/efficacy study that initiates sitagliptin in KTRs without a history of T2DM at 2 weeks post-transplantation in order to prevent NODAT has been started at the University of Nebraska in 2009 (NCT00936663).
In summary on NODAT prevention, previous concepts were often based on monitoring fasting glucose, although evening glucose is better suited for the detection of hyperglycaemia in patients who receive glucocorticoids. Tailored immunosuppression may only be of limited benefit. As risk factor-independent prevention strategies, we recommend early insulin therapy against severe postoperative hyperglycaemia, along with lifestyle modification rather than altering immunosuppression. In the future, more experience will be gained from randomized controlled trials, but also by further clinical application of this practice. Areas of interest include the efficacy and safety of various long-acting insulin regimens, the glucose threshold for insulin initiation, ideal glycaemic targets, the benefit of individual insulin therapy strategies (e.g. basal/basal-bolus therapy, continuous subcutaneous insulin infusion, etc.) and finally also the potential improvement of hard outcomes through control of post-transplant hyperglycaemia, which is by itself associated with mortality [22]. Finally, whether oral anti-hyperglycaemics, when used against postoperative hyperglycaemia, may prevent overt NODAT at later time points also remains to be determined in this setting.
TREATMENT OF NODAT IN THE ABSENCE OR AFTER FAILURE OF PREVENTION
As a simplification, we here define ‘stable’ KTRs as having lived ≥3 months post-transplantation without requiring haemodialysis at the time of evaluation. When such patients have not received adequate, preventive management during their early postoperative period, they may present with signs of NODAT at the outpatient transplant clinic: elevated HbA1c and/or elevated random glucose. Depending on centre practice, stable KTRs may still be undergoing or may have completed glucocorticoid tapering or withdrawal schemes and their degree of renal function may be variable. Although aggressive lifestyle modification has been shown to improve glycaemic control [134], its long-term sustainability is doubtful and pharmacological intervention will likely be necessary. Anti-hyperglycaemic treatment options for such patients will, therefore, be discussed here below.
Anti-hyperglycaemic agents
The available anti-hyperglycaemics have been previously reviewed [32, 38, 39, 133] and are summarized in Table 1. As with any medical intervention, attaining the balance between maximizing efficacy and minimizing harm is of crucial importance. Briefly, renal function must be taken into account before starting a sulphonylurea, biguanide (metformin), meglitinide (glinide), glucagon-like peptide (GLP)-1 agonist or a dipeptidase (DPP)-4 inhibitor (gliptin), while cardiovascular risk and heart failure must be considered for the thiazolidinediones (TZDs, e.g. pioglitazone). Gastrointestinal side effects may, at least for alpha-glucosidase inhibitors (acarbose), not be a concern clinically, but they limit the acceptance of treatment, especially in the context of concomitant mycophenolate mofetil immunosuppression. Regarding exogenous insulin therapy, the complex association between insulin and cancer risk identified in observational studies [161] may be less problematic with short duration of insulin administration. Hypoglycaemia, therefore, remains the predominant problem in insulin therapy, although this risk is—in attenuated form—also evident for the insulin secretagogues, especially sulphonylureas.
Table 1.
Currently available anti-hyperglycaemic agents
| Agent | Mechanism of action | Advantages | Disadvantages | Adjustment in renal allograft dysfunctiona |
|---|---|---|---|---|
| Biguanides (metformin) | Insulin sensitizing | Efficacy (micro and macrovascular end points), no hypoglycaemia, no weight gain, drug cost | Gastrointestinal side effects, risk of lactic acidosis in renal impairment | eGFR 30–45 mL/min (caution)b |
| eGFR < 30 mL/min (avoid)b | ||||
| Sulphonylureas (glipizide, gliclazide, etc.) | Stimulation of insulin secretion | Efficacy (microvascular end points), drug cost | Hypoglycaemia, weight gain, accumulates in renal failure | ⇓ dose |
| Thiazolidinediones (rosiglitzazone, pioglitazone) | Insulin sensitizing | More sustained glucosecontrol | Weight gain, oedema, drug cost, adverse cardiovascular effects, fracture risk, risk of bladder cancer | Nonspecific |
| Meglitinides (repaglinide1, nateglinide2) | Stimulation of insulin secretion | Reduces postprandial hyperglycaemia, safe with advancing renal failure1 | Hypoglycaemia, weight gain, drug cost, dose adjustment in renal failure2 | Nonspecific |
| Alpha glucosidase inhibitors (acarbose) | Decreases gastrointestinal carbohydrate absorption | No hypoglycaemia, weight neutral | Gastrointestinal side effects | eGFR < 25 mL/min (avoid) |
| GLP-1 agonists (exenatide3, liraglutide4) | Stimulates insulin secretion, decreases glucagon production, stimulates satiety | No weight gain (possible reduction), low risk of hypoglycaemia, lowers blood pressure | Gastrointestinal side effects, risk of pancreatitis altered drug absorption, drug cost, renal impairment, antibody production3 | eGFR3 30–50 mL/min (use with caution) |
| eGFR3 < 30 mL/min (avoid) | ||||
| eGFR4 < 60 mL/min (avoid) | ||||
| DPP-4 inhibitors (sitagliptin5, vildagliptin6, linagliptin6, saxagliptin7) | Decreases inactivation of incretins (GLP-1) | No weight gain | Drug cost, risk of pancreatitis, putative link to certain cancers | 5eGFR < 50 mL/min (avoid) |
| 6No dose adjustment required | ||||
| 7⇓ dose | ||||
| Insulin | Exogenous administration of primary glycaemia countering hormone | Efficacy (micro and macrovascular end points), no ceiling of treatment, range of insulin types for individualization | Weight gain, subcutaneous administration, hypoglycaemia, putative link to certain cancers | Often ⇓ requirement |
GLP-1 = glucagon-like peptide 1, DPP-4 = dipeptidase-4, CNI = calcineurin inhibitor, eGFR = estimated glomerular filtration rate.
aAdapted from British National Formulary (www.bnf.org).
bThese cut-off values may not apply outside the UK.
Available evidence for anti-hyperglycaemic treatmentin KTRs
A review of the literature highlights the lack of randomized controlled trials, and the paucity of observational and/or retrospective studies, even of short-term duration. Turk et al. compared the use of repaglinide, a short-acting meglitinide, with rosiglitazone (insulin-sensitizing TZD) in stable KTRs with NODAT (diagnosed median of 4 months post-transplantation) [162]. In this observational analysis, a similar efficacy and tolerable side effect profile was observed between the repaglinide (n = 23) and rosiglitazone groups (n = 21). Lane et al. reported on safety and efficacy of sitagliptin, a DPP-4 inhibitor that improves insulin secretion via an incretin effect, in 15 KTRs diagnosed with NODAT over a 3-month period [163].
Pietruck et al. published their experience with rosiglitazone in 22 KTRs with NODAT, diagnosed 3 months post-transplantation [164]. Over a mean follow-up of 10 months, 16 patients were deemed to have had successful treatment compared with six deemed unsuccessful. Pioglitazone has been shown to be safe in KTRs with T2DM (not NODAT) and no influences on tacrolimus metabolism have been observed [165]. Although metformin is the most established insulin sensitizer, its experience is limited to a single retrospective report of 24 KTRs [166]. This retrospective study compared metformin with TZDs in stable KTRs with NODAT or pre-existing diabetes mellitus over a mean duration of 16 months post-transplantation and showed equivalent efficacy and no safety concerns.
The results from randomized controlled trials exploring the use of anti-hyperglycaemic agents in kidney transplantation are not yet publicly available, but this gap in the literature is now being rectified. Werzowa et al. recently showed that pioglitazone (n = 16) and vildagliptin (n = 16) led to reductions in HbA1c and glucose tolerance in KTRs with IGT compared with placebo (n = 16) [167]. Another study comparing vildagliptin and placebo in KTRs with overt NODAT is still ongoing [168].
Previous and novel recommendations
The 2003 international consensus guidelines recommended a stepwise approach to the treatment of NODAT, namely non-pharmacological therapy, followed by oral agent monotherapy, oral agent combination therapy, then insulin ± oral agents and ultimately insulin monotherapy [33]. This recommendation, specifically the stepwise approach, has not been repeated by recent reviews [32, 38, 39, 133], but has formally also not yet been questioned, thereby remaining the only expert consensus at the disposal of the transplant community today. With respect to many of the aforementioned arguments, however, we disagree with the stepwise approach even for NODAT treatment in stable KTRs (not just for NODAT prevention), for the following reasons:
During the crucial period up to 6 months post-transplantation when hyperglycaemia is prominent and subsequent incidence of overt (or full-blown) NODAT is highest [23, 31, 169], the disease does not begin insidiously, as T2DM, but has a much faster onset, even if hyperglycaemia may have been overlooked early after transplantation. Thus, the treatment should be more aggressive, and not solely focussed on lifestyle interventions, in order to restore normal glucose metabolism.
In light of the reported evidence indicating that NODAT is predominantly an insulin secretion problem, oral agent monotherapy—especially with sulphonylureas—may even aggravate beta-cell decline via islet cell exhaustion.
Even in T2DM, beta-cell preservation is becoming a major focus [118, 160, 170]. In hyperglycaemic KTRs as well as stable KTRs with full-blown NODAT, insulin can be more easily administered than in type 2 diabetics because glucose levels are likely higher early on, patients are used to complex medications and have frequent control visits.
Thus, in contrast to the 2003 international consensus guidelines [33], novel recommendations could argue for intermittent insulin therapy first, potentially even for treating overt NODAT in stable KTRs. If patients have relatively high daily glucose levels (≥200 mg/dL) and can be kept under surveillance, insulin initiation may be safe, with respect to hypoglycaemia. Owing to the nature of the incidence curve of overt NODAT, bending horizontally to an almost stable incidence rate and thereby paralleling T2DM in the general population after the steep increase within the first 1–6 months post-transplantation [23, 31, 169], NODAT development at late time points could, however, be linked to the traditional risk factors more than to risk factor-independent mechanisms discussed here above.
In the absence of larger trials, we still suggest to avoid sulphonylureas, due to the risk of beta-cell exhaustion. DPP-4 inhibitors (gliptins) and biguanides (metformin) may be more reasonable alternatives in light of their specific advantages and adverse effects. DPP-4 inhibitors have been shown to preserve pancreatic beta-cell function in diabetic animal models [171, 172], which may render their application in NODAT patients potentially of interest. Alpha-glucosidase inhibitors and TZDs are not irrational choices, but these agents seem harder to endorse, due to the public debate concerning TZD safety [173] and knowing that many patients will not accept acarbose because of frequent diarrhoea and flatulence.
The metformin debate and future agents–timeto collaborate
Theoretically, metformin has many advantages in the context of transplantation that should promote it as the oral anti-hyperglycaemic agent of choice for the majority of KTRs [174]. However, there is an ongoing debate regarding the safety of metformin in the complicated transplantation milieu and polypharmacy of KTRs [175]. The heightened concerns regarding metformin use in KTRs are understandable but unsubstantiated, and run the risk of overlooking one of the most efficacious oral anti-hyperglycaemic agents available, which has virtually no drug interactions and therefore, actually minimizes the risk of polypharmacy. Because such discussions can only be attenuated in the context of a randomized controlled trial assessing safety and efficacy, it is now time to collaborate [176]. Emerging anti-hyperglycaemic agents that are currently under evaluation in various stages of development and trials are highlighted in Table 2. These drugs should also be thoroughly evaluated for use in the context of kidney transplantation.
Table 2.
Anti-hyperglycaemic agents in development
| Agent | Mechanism of action | Advantages | Disadvantages |
|---|---|---|---|
| Sodium-dependent glucose transporters 2 inhibitors | Block renal glucose reabsorption in the proximal tubule | Possible natriuretic effect, action independent of insulin, little risk of hypoglycaemia | Glycosuria may increase the risk of genitourinary infections and exacerbate pro-fibrotic pathways, risk of dehydration |
| Glucokinase inhibitors | Activate glucokinase ‘glucose-sensors’ in both pancreatic and hepatic cells | Dual action on both liver and pancreas, weight neutral (possible reduction) | Safety (glucokinase expressed in neuronal cells), effect on kidney unknown |
| Glucagon antagonists | Blocks the antagonistic action of glucagon versus insulin | Glucagon integral to whole body glucose homeostasis | Awaiting further investigation |
| Bile acid sequestrants (cholestyramine, colestimide and colesevelam) | Unknown (possible pleiotropic effect of lipid lowering) | Beneficial effects on abnormal lipid profiles, safe in renal impairment | Gastrointestinal side effects very common, disruption of fat-soluble vitamin absorption |
| Amylin analogues | Synthetic analogue of beta-cell hormone amylin —delays gastric emptying, increases satiety and inhibits glucagon production | Weight neutral (possible reduction), safe in mild-to-moderate renal impairment | Subcutaneous administration, risk of hypoglycaemia, gastrointestinal side effects, not available outside USA |
In summary on NODAT treatment, the 2003 international consensus guideline-based, stepwise approach to the management [33] does not appear reasonable, even for stable KTRs. Owing to the probable deficiency in insulin secretion, intermittent insulin therapy aimed at preserving and/or restoring beta-cell function may be the preferable option, as has even been argued for type 2 diabetics [159, 160]. This approach, however, is limited when glucose levels are relatively low. Among oral antidiabetics, the biguanide metformin could become our anti-hyperglycaemic agent of choice after transplantation [174], if safety can be shown prospectively.
PREVENTION VERSUS TREATMENT
The difference between overt or full-blown NODAT requiring long-term anti-hyperglycaemic treatment [31] and post-transplant hyperglycaemia [37] is depicted in Figure 4. This figure moreover presents a hypothesis on the consequences of early preventive intervention, as well as showing (in the right lower corner) those patients who will potentially require long-term anti-hyperglycaemic treatment. Our hypothesis that a marked reduction in NODAT incidence after renal transplantation could result in matching the diabetes incidence rate in the general population may seem provocative at first sight, but is in fact based on the results of our previous study [37], where no patient at 1-year post-transplantation required anti-hyperglycaemic treatment, but will of course undergo subsequent re-evaluation.
FIGURE 4:

Hyperglycaemia and NODAT incidence after transplantation and possible consequences of early intervention. Plus denotes that the cumulative incidence of hyperglycaemia post-transplantation was adopted from original results reported in the TIP-study [37], e.g. 21 of 25 patients (84%) had blood glucose ≥200 mg/dL by postoperative Day 10 and 23 of 25 patients (92%) by postoperative Day 18. Double plus denotes that the cumulative incidence of overt NODAT post-transplantation was adopted from original results reported by Woodward et al. in tacrolimus-treated KTRs [31], using the USRDS's February 2001 data release. As discussed by the authors, by using the second International Coding of Diseases-9 coding for a diagnosis of diabetes after transplantation to define NODAT, they obtained ‘conservative estimates’ of the disease incidence. Because NODAT coding in this data set obtained prior to 2001 was very likely based on anti-hyperglycaemic treatment (not on hyperglycaemia itself), we applied the term overt NODAT in the figure legend. Today's incidence of NODAT is much higher, namely 40% by the third year post-transplantation, as reported by the USRDS [20]. Although the figure legend describes a 'hypothesized’ effect of early intervention, this is not fully hypothetical, as no patient in the TIP-study required anti-hyperglycaemic treatment, clinically, at the 1-year post-transplant follow-up visit [37]. NODAT , new-onset diabetes after transplantation.
CONCLUSION
Reaching a thorough understanding of new-onset diabetes development after renal transplantation is more than a challenging, intellectual goal; it is a necessity for making proper treatment decisions. The remarkably high incidence of early post-transplant hyperglycaemia, when measured in the evenings rather than fasting, suggests an opportunity for intervention with immediate insulin therapy to ‘protect islet cells’ and potentially prevent subsequent overt NODAT. This new concept is derived from evidence at early stages of T2DM and from a limited trial in transplant recipients. Until a currently ongoing, major clinical trial yields further insights, it may be reasonable to apply this concept under close supervision of patients with post-transplant hyperglycaemia.
CONFLICT OF INTEREST STATEMENT
None declared.
ACKNOWLEDGEMENTS
We thank Petra Reinke, Alexander Rosenkranz and Andrea Tura for critically revising the manuscript.
REFERENCES
- 1.Morris PJ. Transplantation—a medical miracle of the 20th century. N Engl J Med. 2004;351:2678–2680. doi: 10.1056/NEJMp048256. [DOI] [PubMed] [Google Scholar]
- 2.Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341:1725–1730. doi: 10.1056/NEJM199912023412303. [DOI] [PubMed] [Google Scholar]
- 3.Rabbat CG, Thorpe KE, Russell JD, et al. Comparison of mortality risk for dialysis patients and cadaveric first renal transplant recipients in Ontario, Canada. J Am Soc Nephrol. 2000;11:917–922. doi: 10.1681/ASN.V115917. [DOI] [PubMed] [Google Scholar]
- 4.Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol. 2001;12:589–597. doi: 10.1681/ASN.V123589. [DOI] [PubMed] [Google Scholar]
- 5.Meier-Kriesche HU, Port FK, Ojo AO, et al. Effect of waiting time on renal transplant outcome. Kidney Int. 2000;58:1311–1317. doi: 10.1046/j.1523-1755.2000.00287.x. [DOI] [PubMed] [Google Scholar]
- 6.Brunkhorst R, Lufft V, Dannenberg B, et al. Improved survival in patients with type 1 diabetes mellitus after renal transplantation compared with hemodialysis: a case–control study. Transplantation. 2003;76:115–119. doi: 10.1097/01.TP.0000070225.38757.81. [DOI] [PubMed] [Google Scholar]
- 7.Merion RM, Ashby VB, Wolfe RA, et al. Deceased-donor characteristics and the survival benefit of kidney transplantation. JAMA. 2005;294:2726–2733. doi: 10.1001/jama.294.21.2726. [DOI] [PubMed] [Google Scholar]
- 8.Franke GH, Reimer J, Philipp T, et al. Aspects of quality of life through end-stage renal disease. Qual Life Res. 2003;12:103–115. doi: 10.1023/a:1022238707028. [DOI] [PubMed] [Google Scholar]
- 9.Jofre R, Lopez-Gomez JM, Moreno F, et al. Changes in quality of life after renal transplantation. Am J Kidney Dis. 1998;32:93–100. doi: 10.1053/ajkd.1998.v32.pm9669429. [DOI] [PubMed] [Google Scholar]
- 10.Overbeck I, Bartels M, Decker O, et al. Changes in quality of life after renal transplantation. Transplant Proc. 2005;37:1618–1621. doi: 10.1016/j.transproceed.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Balaska A, Moustafellos P, Gourgiotis S, et al. Changes in health-related quality of life in Greek adult patients 1 year after successful renal transplantation. Exp Clin Transplant. 2006;4:521–524. [PubMed] [Google Scholar]
- 12.Gallon LG, Leventhal JR, Kaufman DB. Pretransplant evaluation of renal transplant candidates. Semin Nephrol. 2002;22:515–525. doi: 10.1053/snep.2002.35966. [DOI] [PubMed] [Google Scholar]
- 13.Neylan JF, Sayegh MH, Coffman TM, et al. The allocation of cadaver kidneys for transplantation in the United States: consensus and controversy. ASN Transplant Advisory Group. American Society of Nephrology. J Am Soc Nephrol. 1999;10:2237–2243. doi: 10.1681/ASN.V10102237. [DOI] [PubMed] [Google Scholar]
- 14.Mayer G, Persijn GG. Eurotransplant kidney allocation system (ETKAS): rationale and implementation. Nephrol Dial Transplant. 2006;21:2–3. doi: 10.1093/ndt/gfi269. [DOI] [PubMed] [Google Scholar]
- 15.Denton MD, Magee CC, Sayegh MH. Immunosuppressive strategies in transplantation. Lancet. 1999;353:1083–1091. doi: 10.1016/S0140-6736(98)07493-5. [DOI] [PubMed] [Google Scholar]
- 16.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 17.Wong W, Venetz JP, Tolkoff-Rubin N, et al. 2005 immunosuppressive strategies in kidney transplantation: which role for the calcineurin inhibitors? Transplantation. 2005;80:289–296. doi: 10.1097/01.tp.0000168436.76784.45. [DOI] [PubMed] [Google Scholar]
- 18.Meier-Kriesche HU, Li S, Gruessner RW, et al. Immunosuppression: evolution in practice and trends, 1994–2004. Am J Transplant. 2006;6:1111–1131. doi: 10.1111/j.1600-6143.2006.01270.x. [DOI] [PubMed] [Google Scholar]
- 19.Krentz AJ, Wheeler DC. New-onset diabetes after transplantation: a threat to graft and patient survival. Lancet. 2005;365:640–642. doi: 10.1016/S0140-6736(05)17962-8. [DOI] [PubMed] [Google Scholar]
- 20.National Institutes of Health NIoDaDaKD. US Renal Data System USRDS 2011 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. http://www.usrds.org/2011/view/v2_07.asp .
- 21.Hjelmesaeth J, Hartmann A, Leivestad T, et al. The impact of early-diagnosed new-onset post-transplantation diabetes mellitus on survival and major cardiac events. Kidney Int. 2006;69:588–595. doi: 10.1038/sj.ki.5000116. [DOI] [PubMed] [Google Scholar]
- 22.Valderhaug TG, Hjelmesaeth J, Hartmann A, et al. The association of early post-transplant glucose levels with long-term mortality. Diabetologia. 2011;54:1341–1349. doi: 10.1007/s00125-011-2105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasiske BL, Snyder JJ, Gilbertson D, et al. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. 2003;3:178–185. doi: 10.1034/j.1600-6143.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- 24.Cosio FG, Pesavento TE, Kim S, et al. Patient survival after renal transplantation: IV. Impact of post-transplant diabetes. Kidney Int. 2002;62:1440–1446. doi: 10.1111/j.1523-1755.2002.kid582.x. [DOI] [PubMed] [Google Scholar]
- 25.Cole EH, Johnston O, Rose CL, et al. Impact of acute rejection and new-onset diabetes on long-term transplant graft and patient survival. Clin J Am Soc Nephrol. 2008;3:814–821. doi: 10.2215/CJN.04681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valderhaug TG, Hjelmesaeth J, Jenssen T, et al. Early posttransplantation hyperglycemia in kidney transplant recipients is associated with overall long-term graft losses. Transplantation. 2012;94:714–720. doi: 10.1097/TP.0b013e31825f4434. [DOI] [PubMed] [Google Scholar]
- 27.Matas AJ, Gillingham KJ, Humar A, et al. Posttransplant diabetes mellitus and acute rejection: impact on kidney transplant outcome. Transplantation. 2008;85:338–343. doi: 10.1097/TP.0b013e318160ee42. [DOI] [PubMed] [Google Scholar]
- 28.Pietrzak-Nowacka M, Safranow K, Dziewanowski K, et al. Impact of posttransplant diabetes mellitus on graft function in autosomal dominant polycystic kidney disease patients after kidney transplantation. Ann Acad Med Stetin. 2008;54:41–48. [PubMed] [Google Scholar]
- 29.Siraj ES, Abacan C, Chinnappa P, et al. Risk factors and outcomes associated with posttransplant diabetes mellitus in kidney transplant recipients. Transplant Proc. 2010;42:1685–1689. doi: 10.1016/j.transproceed.2009.12.062. [DOI] [PubMed] [Google Scholar]
- 30.von Kiparski A, Frei D, Uhlschmid G, et al. Post-transplant diabetes mellitus in renal allograft recipients: a matched-pair control study. Nephrol Dial Transplant. 1990;5:220–225. doi: 10.1093/ndt/5.3.220. [DOI] [PubMed] [Google Scholar]
- 31.Woodward RS, Schnitzler MA, Baty J, et al. Incidence and cost of new onset diabetes mellitus among U.S. wait-listed and transplanted renal allograft recipients. Am J Transplant. 2003;3:590–598. doi: 10.1034/j.1600-6143.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- 32.Sarno G, Muscogiuri G, De Rosa P. New-onset diabetes after kidney transplantation: prevalence, risk factors, and management. Transplantation. 2012 doi: 10.1097/TP.0b013e31824db97d. Apr 3 Epub ahead of print], PMID:22475764. [DOI] [PubMed] [Google Scholar]
- 33.Davidson J, Wilkinson A, Dantal J, et al. New-onset diabetes after transplantation: 2003 International consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation. 2003;75:SS3–S24. doi: 10.1097/01.TP.0000069952.49242.3E. [DOI] [PubMed] [Google Scholar]
- 34.Hagen M, Hjelmesaeth J, Jenssen T, et al. A 6-year prospective study on new onset diabetes mellitus, insulin release and insulin sensitivity in renal transplant recipients. Nephrol Dial Transplant. 2003;18:2154–2159. doi: 10.1093/ndt/gfg338. [DOI] [PubMed] [Google Scholar]
- 35.Nam JH, Mun JI, Kim SI, et al. beta-Cell dysfunction rather than insulin resistance is the main contributing factor for the development of postrenal transplantation diabetes mellitus. Transplantation. 2001;71:1417–1423. doi: 10.1097/00007890-200105270-00011. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu M, Iino Y, Terashi A. Improvement of insulin sensitivity after renal transplantation measured by a glucose clamp technique—abstract published in English. Nihon Ika Daigaku Zasshi. 1998;65:50–54. doi: 10.1272/jnms1923.65.50. [DOI] [PubMed] [Google Scholar]
- 37.Hecking M, Haidinger M, Doller D, et al. Early basal insulin therapy decreases new-onset diabetes after renal transplantation. J Am Soc Nephrol. 2012;23:739–749. doi: 10.1681/ASN.2011080835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yates CJ, Fourlanos S, Hjelmesaeth J, et al. New-onset diabetes after kidney transplantation-changes and challenges. Am J Transplant. 2012;12:820–828. doi: 10.1111/j.1600-6143.2011.03855.x. [DOI] [PubMed] [Google Scholar]
- 39.Pham PT, Pham PM, Pham SV, et al. New onset diabetes after transplantation (NODAT): an overview. Diabetes Metab Syndr Obes. 2011;4:175–186. doi: 10.2147/DMSO.S19027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 41.Naing C, Mak JW, Ahmed SI, et al. Relationship between hepatitis C virus infection and type 2 diabetes mellitus: meta-analysis. World J Gastroenterol. 2012;18:1642–1651. doi: 10.3748/wjg.v18.i14.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmet P. Type 2 (non-insulin-dependent) diabetes—an epidemiological overview. Diabetologia. 1982;22:399–411. doi: 10.1007/BF00282581. [DOI] [PubMed] [Google Scholar]
- 43.Porrini E, Delgado P, Alvarez A, et al. The combined effect of pre-transplant triglyceride levels and the type of calcineurin inhibitor in predicting the risk of new onset diabetes after renal transplantation. Nephrol Dial Transplant. 2008;23:1436–1441. doi: 10.1093/ndt/gfm762. [DOI] [PubMed] [Google Scholar]
- 44.Bayer ND, Cochetti PT, Anil Kumar MS, et al. Association of metabolic syndrome with development of new-onset diabetes after transplantation. Transplantation. 2010;90:861–866. doi: 10.1097/TP.0b013e3181f1543c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghisdal L, Baron C, Le Meur Y, et al. TCF7L2 polymorphism associates with new-onset diabetes after transplantation. J Am Soc Nephrol. 2009;20:2459–2467. doi: 10.1681/ASN.2008121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Mattos AM, Olyaei AJ, Prather JC, et al. Autosomal-dominant polycystic kidney disease as a risk factor for diabetes mellitus following renal transplantation. Kidney Int. 2005;67:714–720. doi: 10.1111/j.1523-1755.2005.67132.x. [DOI] [PubMed] [Google Scholar]
- 47.Ducloux D, Motte G, Vautrin P, et al. Polycystic kidney disease as a risk factor for post-transplant diabetes mellitus. Nephrol Dial Transplant. 1999;14:1244–1246. doi: 10.1093/ndt/14.5.1244. [DOI] [PubMed] [Google Scholar]
- 48.Hamer RA, Chow CL, Ong AC, et al. Polycystic kidney disease is a risk factor for new-onset diabetes after transplantation. Transplantation. 2007;83:36–40. doi: 10.1097/01.tp.0000248759.37146.3d. [DOI] [PubMed] [Google Scholar]
- 49.Hjelmesaeth J, Muller F, Jenssen T, et al. Is there a link between cytomegalovirus infection and new-onset posttransplantation diabetes mellitus? Potential mechanisms of virus induced beta-cell damage. Nephrol Dial Transplant. 2005;20:2311–2315. doi: 10.1093/ndt/gfi033. [DOI] [PubMed] [Google Scholar]
- 50.Hjelmesaeth J, Sagedal S, Hartmann A, et al. Asymptomatic cytomegalovirus infection is associated with increased risk of new-onset diabetes mellitus and impaired insulin release after renal transplantation. Diabetologia. 2004;47:1550–1556. doi: 10.1007/s00125-004-1499-z. [DOI] [PubMed] [Google Scholar]
- 51.Van Laecke S, Van Biesen W, Verbeke F, et al. Posttransplantation hypomagnesemia and its relation with immunosuppression as predictors of new-onset diabetes after transplantation. Am J Transplant. 2009;9:2140–2149. doi: 10.1111/j.1600-6143.2009.02752.x. [DOI] [PubMed] [Google Scholar]
- 52.Sharif A, Baboolal K. Complications associated with new-onset diabetes after kidney transplantation. Nat Rev Nephrol. 2012;8:34–42. doi: 10.1038/nrneph.2011.174. [DOI] [PubMed] [Google Scholar]
- 53.Nagaraja P, Ravindran V, Morris-Stiff G, et al. Role of insulin resistance indices in predicting new-onset diabetes after kidney transplantation. Transpl Int. 2012 doi: 10.1111/tri.12026. doi:10.1111/tri.12026. [Epub ahead of print] PMID:23230898. [DOI] [PubMed] [Google Scholar]
- 54.Chakkera HA, Weil EJ, Castro J, et al. Hyperglycemia during the immediate period after kidney transplantation. Clin J Am Soc Nephrol. 2009;4:853–859. doi: 10.2215/CJN.05471008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chakkera HA, Knowler WC, Devarapalli Y, et al. Relationship between inpatient hyperglycemia and insulin treatment after kidney transplantation and future new onset diabetes mellitus. Clin J Am Soc Nephrol. 2010;5:1669–1675. doi: 10.2215/CJN.09481209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35:S64–S71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beard JC, Halter JB, Best JD, et al. Dexamethasone-induced insulin resistance enhances B cell responsiveness to glucose level in normal men. Am J Physiol. 1984;247:E592–E596. doi: 10.1152/ajpendo.1984.247.5.E592. [DOI] [PubMed] [Google Scholar]
- 58.Hirsch IB, Paauw DS. Diabetes management in special situations. Endocrinol Metab Clin North Am. 1997;26:631–645. doi: 10.1016/s0889-8529(05)70271-1. [DOI] [PubMed] [Google Scholar]
- 59.Levetan CS, Magee MF. Hospital management of diabetes. Endocrinol Metab Clin North Am. 2000;29:745–770. doi: 10.1016/s0889-8529(05)70162-6. [DOI] [PubMed] [Google Scholar]
- 60.Crutchlow MF, Bloom RD. Transplant-associated hyperglycemia: a new look at an old problem. Clin J Am Soc Nephrol. 2007;2:343–355. doi: 10.2215/CJN.03671106. [DOI] [PubMed] [Google Scholar]
- 61.Reese PP, Bloom RD. Transplant-associated hyperglycemia: shedding light on the mechanisms. Clin J Am Soc Nephrol. 2010;5:560–562. doi: 10.2215/CJN.01430210. [DOI] [PubMed] [Google Scholar]
- 62.Ekstrand AV, Eriksson JG, Gronhagen-Riska C, et al. Insulin resistance and insulin deficiency in the pathogenesis of posttransplantation diabetes in man. Transplantation. 1992;53:563–569. doi: 10.1097/00007890-199203000-00014. [DOI] [PubMed] [Google Scholar]
- 63.Midtvedt K, Hartmann A, Hjelmesaeth J, et al. Insulin resistance is a common denominator of post-transplant diabetes mellitus and impaired glucose tolerance in renal transplant recipients. Nephrol Dial Transplant. 1998;13:427–431. doi: 10.1093/oxfordjournals.ndt.a027841. [DOI] [PubMed] [Google Scholar]
- 64.Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR) 2010. Annual Data Report http://www.srtr.org/annual_reports/2010/pdf/2010_SRTR_ADR.pdf .
- 65.Redmon JB, Olson LK, Armstrong MB, et al. Effects of tacrolimus (FK506) on human insulin gene expression, insulin mRNA levels, and insulin secretion in HIT-T15 cells. J Clin Invest. 1996;98:2786–2793. doi: 10.1172/JCI119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herold KC, Nagamatsu S, Buse JB, et al. Inhibition of glucose-stimulated insulin release from beta TC3 cells and rodent islets by an analog of FK506. Transplantation. 1993;55:186–192. doi: 10.1097/00007890-199301000-00035. [DOI] [PubMed] [Google Scholar]
- 67.van Hooff JP, Christiaans MH, van Duijnhoven EM. Evaluating mechanisms of post-transplant diabetes mellitus. Nephrol Dial Transplant. 2004;19:vi8–vi12. doi: 10.1093/ndt/gfh1063. [DOI] [PubMed] [Google Scholar]
- 68.Duijnhoven EM, Boots JM, Christiaans MH, et al. Influence of tacrolimus on glucose metabolism before and after renal transplantation: a prospective study. J Am Soc Nephrol. 2001;12:583–588. doi: 10.1681/ASN.V123583. [DOI] [PubMed] [Google Scholar]
- 69.Menegazzo LA, Ursich MJ, Fukui RT, et al. Mechanism of the diabetogenic action of cyclosporin A. Horm Metab Res. 1998;30:663–667. doi: 10.1055/s-2007-978954. [DOI] [PubMed] [Google Scholar]
- 70.Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 71.Vincenti F, Friman S, Scheuermann E, et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant. 2007;7:1506–1514. doi: 10.1111/j.1600-6143.2007.01749.x. [DOI] [PubMed] [Google Scholar]
- 72.European FK506 Multicentre Liver Study Group. Randomised trial comparing tacrolimus (FK506) and cyclosporin in prevention of liver allograft rejection. Lancet. 1994;344:423–428. [PubMed] [Google Scholar]
- 73.Pirsch JD, Miller J, Deierhoi MH, et al. A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation. FK506 Kidney Transplant Study Group. Transplantation. 1997;63:977–983. doi: 10.1097/00007890-199704150-00013. [DOI] [PubMed] [Google Scholar]
- 74.Mayer AD, Dmitrewski J, Squifflet JP, et al. Multicenter randomized trial comparing tacrolimus (FK506) and cyclosporine in the prevention of renal allograft rejection: a report of the European Tacrolimus Multicenter Renal Study Group. Transplantation. 1997;64:436–443. doi: 10.1097/00007890-199708150-00012. [DOI] [PubMed] [Google Scholar]
- 75.Huscher D, Thiele K, Gromnica-Ihle E, et al. Dose-related patterns of glucocorticoid-induced side effects. Ann Rheum Dis. 2009;68:1119–1124. doi: 10.1136/ard.2008.092163. [DOI] [PubMed] [Google Scholar]
- 76.Olefsky JM, Johnson J, Liu F, et al. The effects of acute and chronic dexamethasone administration on insulin binding to isolated rat hepatocytes and adipocytes. Metabolism. 1975;24:517–527. doi: 10.1016/0026-0495(75)90076-1. [DOI] [PubMed] [Google Scholar]
- 77.Olefsky JM. Effect of dexamethasone on insulin binding, glucose transport, and glucose oxidation of isolated rat adipocytes. J Clin Invest. 1975;56:1499–1508. doi: 10.1172/JCI108231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Magnuson MA, Quinn PG, Granner DK. Multihormonal regulation of phosphoenolpyruvate carboxykinase-chloramphenicol acetyltransferase fusion genes. Insulin's effects oppose those of cAMP and dexamethasone. J Biol Chem. 1987;262:14917–14920. [PubMed] [Google Scholar]
- 79.Qi D, Rodrigues B. Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am J Physiol Endocrinol Metab. 2007;292:E654–E667. doi: 10.1152/ajpendo.00453.2006. [DOI] [PubMed] [Google Scholar]
- 80.Ullrich S, Berchtold S, Ranta F, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) mediates glucocorticoid-induced inhibition of insulin secretion. Diabetes. 2005;54:1090–1099. doi: 10.2337/diabetes.54.4.1090. [DOI] [PubMed] [Google Scholar]
- 81.Billaudel B, Mathias PC, Sutter BC, et al. Inhibition by corticosterone of calcium inflow and insulin release in rat pancreatic islets. J Endocrinol. 1984;100:227–233. doi: 10.1677/joe.0.1000227. [DOI] [PubMed] [Google Scholar]
- 82.Khan A, Ostenson CG, Berggren PO, et al. Glucocorticoid increases glucose cycling and inhibits insulin release in pancreatic islets of ob/ob mice. Am J Physiol. 1992;263(4 Pt 1):E663–E666. doi: 10.1152/ajpendo.1992.263.4.E663. [DOI] [PubMed] [Google Scholar]
- 83.Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99:414–423. doi: 10.1172/JCI119175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koizumi M, Yada T. Sub-chronic stimulation of glucocorticoid receptor impairs and mineralocorticoid receptor protects cytosolic Ca2+ responses to glucose in pancreatic beta-cells. J Endocrinol. 2008;197:221–229. doi: 10.1677/JOE-07-0462. [DOI] [PubMed] [Google Scholar]
- 85.Delaunay F, Khan A, Cintra A, et al. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest. 1997;100:2094–2098. doi: 10.1172/JCI119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ranta F, Avram D, Berchtold S, et al. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55:1380–1390. doi: 10.2337/db05-1220. [DOI] [PubMed] [Google Scholar]
- 87.Ihara Y, Toyokuni S, Uchida K, et al. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48:927–932. doi: 10.2337/diabetes.48.4.927. [DOI] [PubMed] [Google Scholar]
- 88.Maedler K, Spinas GA, Lehmann R, et al. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes. 2001;50:1683–1690. doi: 10.2337/diabetes.50.8.1683. [DOI] [PubMed] [Google Scholar]
- 89.Federici M, Hribal M, Perego L, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50:1290–1301. doi: 10.2337/diabetes.50.6.1290. [DOI] [PubMed] [Google Scholar]
- 90.Ortega-Camarillo C, Guzman-Grenfell AM, Garcia-Macedo R, et al. Hyperglycemia induces apoptosis and p53 mobilization to mitochondria in RINm5F cells. Mol Cell Biochem. 2006;281:163–171. doi: 10.1007/s11010-006-0829-5. [DOI] [PubMed] [Google Scholar]
- 91.Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes—a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 92.Donath MY, Ehses JA, Maedler K, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54:S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 93.Madsen SN, Engguist A, Badawi I, et al. Cyclic AMP, glucose and cortisol in plasma during surgery. Horm Metab Res. 1976;8:483–485. doi: 10.1055/s-0028-1093601. [DOI] [PubMed] [Google Scholar]
- 94.Ewaldsson CA, Hahn RG. Beta 2-adrenergic responsiveness in vivo during abdominal surgery. Br J Anaesth. 1998;81:343–347. doi: 10.1093/bja/81.3.343. [DOI] [PubMed] [Google Scholar]
- 95.Hotamisligil GS, Murray DL, Choy LN, et al. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. 1994;91:4854–4858. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology. 1992;130:43–52. doi: 10.1210/endo.130.1.1727716. [DOI] [PubMed] [Google Scholar]
- 97.Smiley DD, Umpierrez GE. Perioperative glucose control in the diabetic or nondiabetic patient. South Med J. 2006;99:580–589. doi: 10.1097/01.smj.0000209366.91803.99. quiz 590–1. [DOI] [PubMed] [Google Scholar]
- 98.Hu EA, Pan A, Malik V, et al. White rice consumption and risk of type 2 diabetes: meta-analysis and systematic review. BMJ. 2012;344:e1454. doi: 10.1136/bmj.e1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Riserus U, Willett WC, Hu FB. Dietary fats and prevention of type 2 diabetes. Prog Lipid Res. 2009;48:44–51. doi: 10.1016/j.plipres.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malik VS, Popkin BM, Bray GA, et al. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Malik VS, Popkin BM, Bray GA, et al. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care. 2010;33:2477–2483. doi: 10.2337/dc10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Odegaard AO, Koh WP, Yuan JM, et al. Western-style fast food intake and cardiometabolic risk in an eastern country. Circulation. 2012;126:182–188. doi: 10.1161/CIRCULATIONAHA.111.084004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pereira MA, Kartashov AI, Ebbeling CB, et al. Fast-food habits, weight gain, and insulin resistance (the CARDIA study): 15-year prospective analysis. Lancet. 2005;365:36–42. doi: 10.1016/S0140-6736(04)17663-0. [DOI] [PubMed] [Google Scholar]
- 104.Lee IM, Shiroma EJ, Lobelo F, et al. Effect of physical inactivity on major non-communicable diseases worldwide: an analysis of burden of disease and life expectancy. Lancet. 2012;380:219–229. doi: 10.1016/S0140-6736(12)61031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krishnan S, Rosenberg L, Palmer JR. Physical activity and television watching in relation to risk of type 2 diabetes: the Black Women's Health Study. Am J Epidemiol. 2009;169:428–434. doi: 10.1093/aje/kwn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rubenstein AH, Mako ME, Horwitz DL. Insulin and the kidney. Nephron. 1975;15:306–326. doi: 10.1159/000180518. [DOI] [PubMed] [Google Scholar]
- 107.Rabkin R, Ryan MP, Duckworth WC. The renal metabolism of insulin. Diabetologia. 1984;27:351–357. doi: 10.1007/BF00304849. [DOI] [PubMed] [Google Scholar]
- 108.Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial. 2004;17:365–370. doi: 10.1111/j.0894-0959.2004.17346.x. [DOI] [PubMed] [Google Scholar]
- 109.Clement S, Braithwaite SS, Magee MF, et al. Management of diabetes and hyperglycemia in hospitals. Diabetes Care. 2004;27:553–591. doi: 10.2337/diacare.27.2.553. [DOI] [PubMed] [Google Scholar]
- 110.Becker B, Kronenberg F, Kielstein JT, et al. Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. J Am Soc Nephrol. 2005;16:1091–1098. doi: 10.1681/ASN.2004090742. [DOI] [PubMed] [Google Scholar]
- 111.Karthikeyan V, Karpinski J, Nair RC, et al. The burden of chronic kidney disease in renal transplant recipients. Am J Transplant. 2004;4:262–269. doi: 10.1046/j.1600-6143.2003.00315.x. [DOI] [PubMed] [Google Scholar]
- 112.Bergrem HA, Valderhaug TG, Hartmann A, et al. Glucose tolerance before and after renal transplantation. Nephrol Dial Transplant. 2010;25:985–992. doi: 10.1093/ndt/gfp566. [DOI] [PubMed] [Google Scholar]
- 113.McLaughlin T, Abbasi F, Cheal K, et al. Use of metabolic markers to identify overweight individuals who are insulin resistant. Ann Intern Med. 2003;139:802–809. doi: 10.7326/0003-4819-139-10-200311180-00007. [DOI] [PubMed] [Google Scholar]
- 114.Cerasi E. β-Cell dysfunction vs insulin resistance in type 2 diabetes: the eternal ‘chicken and egg’ question. Medicographia N° 106. 2011;33:35–41. [Google Scholar]
- 115.Himsworth HP. High Carbohydrate Diets and Insulin Efficiency. Br Med J. 1934;2:57–60. doi: 10.1136/bmj.2.3836.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Himsworth HP. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Diabet Med. 2011;28:1440–1444. doi: 10.1111/j.1464-5491.2011.3508.x. [DOI] [PubMed] [Google Scholar]
- 117.Himsworth HP. The syndrome of diabetes mellitus and its causes. Lancet. 1949;1:465–473. doi: 10.1016/s0140-6736(49)90797-7. [DOI] [PubMed] [Google Scholar]
- 118.Cerasi E, Boitard C, Efendic S, et al. The islet in type 2 diabetes: back to center stage. Diabetes. 2001;50(Suppl 1):S1–S3. doi: 10.2337/diabetes.50.2007.s1. [DOI] [PubMed] [Google Scholar]
- 119.Leahy JL, Hirsch IB, Peterson KA, et al. Targeting beta-cell function early in the course of therapy for type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:4206–4216. doi: 10.1210/jc.2010-0668. [DOI] [PubMed] [Google Scholar]
- 120.UK prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. UK Prospective Diabetes Study Group. Diabetes. 1995;44:1249–1258. [PubMed] [Google Scholar]
- 121.Kahn SE. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. doi: 10.1210/jcem.86.9.7713. [DOI] [PubMed] [Google Scholar]
- 122.DeFronzo RA, Abdul-Ghani M. Type 2 diabetes can be prevented with early pharmacological intervention. Diabetes Care. 2011;34:S202–S209. doi: 10.2337/dc11-s221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139. doi: 10.2337/diacare.2951130. [DOI] [PubMed] [Google Scholar]
- 124.Polonsky KS, Sturis J, Bell GI. Seminars in medicine of the Beth Israel hospital, Boston. Non-insulin-dependent diabetes mellitus - a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med. 1996;334:777–783. doi: 10.1056/NEJM199603213341207. [DOI] [PubMed] [Google Scholar]
- 125.Buchanan TA, Xiang AH, Peters RK, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk hispanic women. Diabetes. 2002;51:2796–2803. doi: 10.2337/diabetes.51.9.2796. [DOI] [PubMed] [Google Scholar]
- 126.Qian L, Xu L, Wang X, et al. Early insulin secretion failure leads to diabetes in Chinese subjects with impaired glucose regulation. Diabetes Metab Res Rev. 2009;25:144–149. doi: 10.1002/dmrr.922. [DOI] [PubMed] [Google Scholar]
- 127.Saad MF, Knowler WC, Pettitt DJ, et al. A two-step model for development of non-insulin-dependent diabetes. Am J Med. 1991;90:229–235. [PubMed] [Google Scholar]
- 128.Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes. 1993;42:1663–1672. doi: 10.2337/diab.42.11.1663. [DOI] [PubMed] [Google Scholar]
- 129.Ferrannini E, Mari A. Beta cell function and its relation to insulin action in humans: a critical appraisal. Diabetologia. 2004;47:943–956. doi: 10.1007/s00125-004-1381-z. [DOI] [PubMed] [Google Scholar]
- 130.Pacini G. The hyperbolic equilibrium between insulin sensitivity and secretion. Nutr Metab Cardiovasc Dis. 2006;16(Suppl 1):S22–S27. doi: 10.1016/j.numecd.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 131.Weyer C, Bogardus C, Mott DM, et al. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hecking M, Kainz A, Werzowa J, et al. Impaired glucose metabolism despite decreased insulin resistance after renal transplantation. Kidney Res Clin Pract. 2012;31:A35. manuscript submitted; parts of the results accepted for oral presentation at the first world renal nutrition week [June 26–30, 2012, Honolulu, Hawai'i], at the American Society of Nephrology [October 30-November 4, San Diego, CA], and published in the form of an abstract. [Google Scholar]
- 133.Bloom RD, Crutchlow MF. New-onset diabetes mellitus in the kidney recipient: diagnosis and management strategies. Clin J Am Soc Nephrol. 2008;3:S38–S48. doi: 10.2215/CJN.02650707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sharif A, Moore R, Baboolal K. Influence of lifestyle modification in renal transplant recipients with postprandial hyperglycemia. Transplantation. 2008;85:353–358. doi: 10.1097/TP.0b013e3181605ebf. [DOI] [PubMed] [Google Scholar]
- 135.Prasad GV, Kim SJ, Huang M, et al. Reduced incidence of new-onset diabetes mellitus after renal transplantation with 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors (statins) Am J Transplant. 2004;4:1897–1903. doi: 10.1046/j.1600-6143.2004.00598.x. [DOI] [PubMed] [Google Scholar]
- 136.Gursoy M, Koksal R, Karavelioglu D, et al. Pretransplantation alpha-interferon therapy and the effect of hepatitis C virus infection on kidney allograft recipients. Transplant Proc. 2000;32:580–582. doi: 10.1016/s0041-1345(00)00898-8. [DOI] [PubMed] [Google Scholar]
- 137.Kamar N, Toupance O, Buchler M, et al. Evidence that clearance of hepatitis C virus RNA after alpha-interferon therapy in dialysis patients is sustained after renal transplantation. J Am Soc Nephrol. 2003;14:2092–2098. doi: 10.1097/01.asn.0000079613.81511.3c. [DOI] [PubMed] [Google Scholar]
- 138.Scheen AJ. Renin–angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab. 2004;30:487–496. doi: 10.1016/s1262-3636(07)70146-5. [DOI] [PubMed] [Google Scholar]
- 139.Abouljoud MS, Kumar MS, Brayman KL, et al. Neoral rescue therapy in transplant patients with intolerance to tacrolimus. Clin Transplant. 2002;16:168–172. doi: 10.1034/j.1399-0012.2002.01054.x. [DOI] [PubMed] [Google Scholar]
- 140.Emre S, Genyk Y, Schluger LK, et al. Treatment of tacrolimus-related adverse effects by conversion to cyclosporine in liver transplant recipients. Transpl Int. 2000;13:73–78. doi: 10.1007/s001470050012. [DOI] [PubMed] [Google Scholar]
- 141.Ghisdal L, Bouchta NB, Broeders N, et al. Conversion from tacrolimus to cyclosporine A for new-onset diabetes after transplantation: a single-centre experience in renal transplanted patients and review of the literature. Transpl Int. 2008;21:146–151. doi: 10.1111/j.1432-2277.2007.00589.x. [DOI] [PubMed] [Google Scholar]
- 142.Oberholzer J, Thielke J, Hatipoglu B, et al. Immediate conversion from tacrolimus to cyclosporine in the treatment of posttransplantation diabetes mellitus. Transplant Proc. 2005;37:999–1000. doi: 10.1016/j.transproceed.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 143.Wyzgal J, Oldakowska-Jedynak U, Paczek L, et al. Posttransplantation diabetes mellitus under calcineurin inhibitor. Transplant Proc. 2003;35:2216–2218. doi: 10.1016/s0041-1345(03)00819-4. [DOI] [PubMed] [Google Scholar]
- 144.Luan FL, Zhang H, Schaubel DE, et al. Comparative risk of impaired glucose metabolism associated with cyclosporine versus tacrolimus in the late posttransplant period. Am J Transplant. 2008;8:1871–1877. doi: 10.1111/j.1600-6143.2008.02328.x. [DOI] [PubMed] [Google Scholar]
- 145.Pascual J, Zamora J, Galeano C, et al. Steroid avoidance or withdrawal for kidney transplant recipients. Cochrane Database Syst Rev. 2009:CD005632. doi:10.1002/14651858.CD005632.pub2. Review. PMID:19160257.. doi: 10.1002/14651858.CD005632.pub2. [DOI] [PubMed] [Google Scholar]
- 146.Luan FL, Steffick DE, Ojo AO. New-onset diabetes mellitus in kidney transplant recipients discharged on steroid-free immunosuppression. Transplantation. 2011;91:334–341. doi: 10.1097/TP.0b013e318203c25f. [DOI] [PubMed] [Google Scholar]
- 147.Luan FL, Steffick DE, Gadegbeku C, et al. Graft and patient survival in kidney transplant recipients selected for de novo steroid-free maintenance immunosuppression. Am J Transplant. 2009;9:160–168. doi: 10.1111/j.1600-6143.2008.02442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Woodle ES, First MR, Pirsch J, et al. A prospective, randomized, double-blind, placebo-controlled multicenter trial comparing early (7 day) corticosteroid cessation versus long-term, low-dose corticosteroid therapy. Ann Surg. 2008;248:564–577. doi: 10.1097/SLA.0b013e318187d1da. [DOI] [PubMed] [Google Scholar]
- 149.Pascual J, Royuela A, Galeano C, et al. Very early steroid withdrawal or complete avoidance for kidney transplant recipients: a systematic review. Nephrol Dial Transplant. 2012;27:825–832. doi: 10.1093/ndt/gfr374. [DOI] [PubMed] [Google Scholar]
- 150.Pascual J, Galeano C, Royuela A, et al. A systematic review on steroid withdrawal between 3 and 6 months after kidney transplantation. Transplantation. 2010;90:343–349. doi: 10.1097/TP.0b013e3181e58912. [DOI] [PubMed] [Google Scholar]
- 151.Augustine JJ, Hricik DE. Steroid sparing in kidney transplantation: changing paradigms, improving outcomes, and remaining questions. Clin J Am Soc Nephrol. 2006;1:1080–1089. doi: 10.2215/CJN.01800506. [DOI] [PubMed] [Google Scholar]
- 152.The Diabetes Control and Complications Trial Research Group . The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 153.Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ilkova H, Glaser B, Tunckale A, et al. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Care. 1997;20:1353–1356. doi: 10.2337/diacare.20.9.1353. [DOI] [PubMed] [Google Scholar]
- 155.Wajchenberg BL. Beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- 156.McFarlane SI, Chaiken RL, Hirsch S, et al. Near-normoglycaemic remission in African-Americans with Type 2 diabetes mellitus is associated with recovery of beta cell function. Diabet Med. 2001;18:10–16. doi: 10.1046/j.1464-5491.2001.00395.x. [DOI] [PubMed] [Google Scholar]
- 157.Ryan EA, Imes S, Wallace C. Short-term intensive insulin therapy in newly diagnosed type 2 diabetes. Diabetes Care. 2004;27:1028–1032. doi: 10.2337/diacare.27.5.1028. [DOI] [PubMed] [Google Scholar]
- 158.Li Y, Xu W, Liao Z, et al. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients is associated with improvement of beta-cell function. Diabetes Care. 2004;27:2597–2602. doi: 10.2337/diacare.27.11.2597. [DOI] [PubMed] [Google Scholar]
- 159.Weng J, Li Y, Xu W, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet. 2008;371:1753–1760. doi: 10.1016/S0140-6736(08)60762-X. [DOI] [PubMed] [Google Scholar]
- 160.Palumbo PJ. The case for insulin treatment early in type 2 diabetes. Cleve Clin J Med. 2004;71:385–386. doi: 10.3949/ccjm.71.5.385. 391–2, 394 passim. [DOI] [PubMed] [Google Scholar]
- 161.Johnson JA, Gale EA. Diabetes, insulin use, and cancer risk: are observational studies part of the solution-or part of the problem? Diabetes. 2010;59:1129–1131. doi: 10.2337/db10-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Turk T, Pietruck F, Dolff S, et al. Repaglinide in the management of new-onset diabetes mellitus after renal transplantation. Am J Transplant. 2006;6:842–846. doi: 10.1111/j.1600-6143.2006.01250.x. [DOI] [PubMed] [Google Scholar]
- 163.Lane JT, Odegaard DE, Haire CE, et al. Sitagliptin therapy in kidney transplant recipients with new-onset diabetes after transplantation. Transplantation. 2011;92:e56–e57. doi: 10.1097/TP.0b013e3182347ea4. [DOI] [PubMed] [Google Scholar]
- 164.Pietruck F, Kribben A, Van TN, et al. Rosiglitazone is a safe and effective treatment option of new-onset diabetes mellitus after renal transplantation. Transpl Int. 2005;18:483–486. doi: 10.1111/j.1432-2277.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 165.Luther P, Baldwin D., Jr Pioglitazone in the management of diabetes mellitus after transplantation. Am J Transplant. 2004;4:2135–2138. doi: 10.1111/j.1600-6143.2004.00613.x. [DOI] [PubMed] [Google Scholar]
- 166.Kurian B, Joshi R, Helmuth A. Effectiveness and long-term safety of thiazolidinediones and metformin in renal transplant recipients. Endocr Pract. 2008;14:979–984. doi: 10.4158/EP.14.8.979. [DOI] [PubMed] [Google Scholar]
- 167.Werzowa J, Hecking M, Haidinger M, et al. Vildagliptin and pioglitazone in patients with impaired glucose tolerance after kidney transplantation. Transplantation. 2012 doi: 10.1097/TP.0b013e318276a20e. in press. [DOI] [PubMed] [Google Scholar]
- 168.Haidinger M, Werzowa J, Voigt HC, et al. A randomized, placebo-controlled, double-blind, prospective trial to evaluate the effect of vildagliptin in new-onset diabetes mellitus after kidney transplantation. Trials. 2010;11:91. doi: 10.1186/1745-6215-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cosio FG, Pesavento TE, Osei K, et al. Post-transplant diabetes mellitus: increasing incidence in renal allograft recipients transplanted in recent years. Kidney Int. 2001;59:732–737. doi: 10.1046/j.1523-1755.2001.059002732.x. [DOI] [PubMed] [Google Scholar]
- 170.Marchetti P, Lupi R, Del Guerra S, et al. Goals of treatment for type 2 diabetes: beta-cell preservation for glycemic control. Diabetes Care. 2009;32:S178–S183. doi: 10.2337/dc09-S306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes. 2006;55:1695–1704. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- 172.Furuta Y, Horiguchi M, Sugaru E, et al. Chronic administration of DSP-7238, a novel, potent, specific and substrate-selective DPP IV inhibitor, improves glycaemic control and beta-cell damage in diabetic mice. Diabetes Obes Metab. 2010;12:421–430. doi: 10.1111/j.1463-1326.2009.01180.x. [DOI] [PubMed] [Google Scholar]
- 173.Ensuring drug safety: lessons from the thiazolidinediones. Lancet. 2007;370:1101. doi: 10.1016/S0140-6736(07)61485-8. [DOI] [PubMed] [Google Scholar]
- 174.Sharif A. Should metformin be our antiglycemic agent of choice post-transplantation? Am J Transplant. 2011;11:1376–1381. doi: 10.1111/j.1600-6143.2011.03550.x. [DOI] [PubMed] [Google Scholar]
- 175.Sharif A. Metformin safety post-transplantation—trials and tribulations. Am J Transplant. 2012;12:796. [Google Scholar]
- 176.Sharif A, de Vries AP, Porrini E, et al. Clinical trials for treatment of NODAT: time to collaborate. Transplantation. 2012;94:e23–e24. doi: 10.1097/TP.0b013e318260b1e9. [DOI] [PubMed] [Google Scholar]