

Abstract
Freeman-Sheldon syndrome, or distal arthrogryposis type 2A (DA2A), is an autosomal-dominant condition caused by mutations in MYH3 and characterized by multiple congenital contractures of the face and limbs and normal cognitive development. We identified a subset of five individuals who had been putatively diagnosed with “DA2A with severe neurological abnormalities” and for whom congenital contractures of the limbs and face, hypotonia, and global developmental delay had resulted in early death in three cases; this is a unique condition that we now refer to as CLIFAHDD syndrome. Exome sequencing identified missense mutations in the sodium leak channel, non-selective (NALCN) in four families affected by CLIFAHDD syndrome. We used molecular-inversion probes to screen for NALCN in a cohort of 202 distal arthrogryposis (DA)-affected individuals as well as concurrent exome sequencing of six other DA-affected individuals, thus revealing NALCN mutations in ten additional families with “atypical” forms of DA. All 14 mutations were missense variants predicted to alter amino acid residues in or near the S5 and S6 pore-forming segments of NALCN, highlighting the functional importance of these segments. In vitro functional studies demonstrated that NALCN alterations nearly abolished the expression of wild-type NALCN, suggesting that alterations that cause CLIFAHDD syndrome have a dominant-negative effect. In contrast, homozygosity for mutations in other regions of NALCN has been reported in three families affected by an autosomal-recessive condition characterized mainly by hypotonia and severe intellectual disability. Accordingly, mutations in NALCN can cause either a recessive or dominant condition characterized by varied though overlapping phenotypic features, perhaps based on the type of mutation and affected protein domain(s).
Main Text
Distal arthrogryposis (DA) is a group of at least ten disorders that are characterized by non-progressive congenital contractures of two or more body areas and that typically affect the wrists, hands, ankles, and feet.1 The three most common DA syndromes are DA1 (MIM 108120),1,2 DA2A (Freeman-Sheldon syndrome [MIM 193700]),3 and DA2B1,2 (Sheldon-Hall syndrome [MIM 601680]). DA1 and DA2B can be caused by variants in any one of several genes, including TPM2 (MIM 190990), TNNT3 (MIM 600692), TNNI2 (MIM 191043), and MYH3 (MIM 160720),4 whereas DA2A is only caused by mutations in MYH3. Moreover, mutations in MYH3 are found in more than 90% of persons who meet the diagnostic criteria for DA2A.5
Among a subset of DA2A-affected persons who were referred to our research program over the past decade and in whom no pathogenic mutations were identified in MYH3, we recognized a pattern of clinical characteristics suggestive of a distinctive and previously unrecognized multiple-malformation syndrome. Specifically, we identified a subset of five families, each with an affected child born to unaffected parents, in which the proband had congenital contractures of the limbs (hands and/or feet) and face; abnormal tone, most commonly manifested as hypotonia; neonatal respiratory distress; and developmental delay. These features are collectively referred to as CLIFAHDD syndrome (Table 1, families A–E; Figure 1; Figures S1 and S2).
Table 1.
Mutations and Clinical Findings For Individuals with CLIFAHDD Syndrome
| Family A | Family B | Family C | Family D | Family E | Family F | Family G | Family H | Family I | Family J | Family K | Family L | Family M | Family N | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Original Diagnosis | CLIFAHDD | CLIFAHDD | CLIFAHDD | CLIFAHDD | CLIFAHDD | DA2A | DA2A | DA2A | DA2B | DA2B | DA2B | DA2B | DA1 | DA1 |
| Mutation Information | ||||||||||||||
| Exon (NALCN) | 6 | 9 | 15 | 39 | 31 | 8 | 13 | 26 | 9 | 13 | 14 | 31 | 13 | 26 |
| cDNA change | c.530A>C | c.938T>G | c.1768C>T | c.4338T>G | c.3493A>C | c.934C>A | c.1526T>C | c.3017T>C | c.979G>A | c.1538C>A | c.1733A>C | c.3542G>A | c.1534T>G | c.3050T>C |
| Predicted protein alteration | p.Gln177Pro | p.Val313Gly | p.Leu590Phe | p.Ile1446Met | p.Thr1165Pro | p.Leu312Ile | p.Leu509Ser | p.Val1006Ala | p.Glu327Lys | p.Thr513Asn | p.Tyr578Ser | p.Arg1181Gln | p.Phe512Val | p.Ile1017Thr |
| GERP | 4.92 | 6.16 | 5.94 | 4.73 | 5.2 | 6.16 | 5.11 | 5.03 | 6.16 | 5.11 | 5.3 | 5.2 | 5.11 | 5.03 |
| CADD 1.0 (phred-like) | 19.22 | 25.9 | 23.9 | 16.08 | 21.0 | 32.0 | 22.6 | 24.2 | 36.0 | 25.5 | 23.7 | 20.5 | 24.6 | 17.54 |
| Polyphen-2 (HumVar) | 1.0 | 1.0 | 0.996 | 0.995 | 0.969 | 1.0 | 1.0 | 0.999 | 1.0 | 1.0 | 1.0 | 0.444 | 1.0 | 1.0 |
| Clinical Features: Face | ||||||||||||||
| Downslanting palpebral fissures | – | + | ND | + | + | + | + | + | ? | + | + | + | – | + |
| Strabismus | – | + | ND | + | + | + | + | + | – | + | – | – | – | – |
| Esotropia | – | + | ND | – | + | – | – | – | – | – | + | – | – | – |
| Broad nasal bridge | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Anteverted nasal tip | + | + | + | + | + | – | + | + | + | + | – | + | + | + |
| Large nares | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Short columella | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Long philtrum | + | + | + | + | + | + | + | + | + | + | + | – | – | + |
| Micrognathia | + | + | + | + | + | + | + | + | + | + | + | + | + | – |
| Pursed lips | + | + | + | + | + | – | + | – | – | + | + | + | – | ? |
| H-shaped dimpled chin | + | + | + | + | – | + | + | – | – | + | – | + | – | – |
| Deep nasolabial folds | + | + | + | + | + | – | + | + | + | – | + | + | + | + |
| Full cheeks | + | + | + | + | + | + | – | + | + | + | + | + | + | + |
| Clinical Features: Limbs | ||||||||||||||
| Camptodactyly | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Ulnar deviation | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Adducted thumbs | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Clubfoot | + | + | + | L | metatarsus adductus | + | + | mild L varus | + | + | + | – | mild varus | + |
| Calcaneovalgus deformity | – | + | – | R | – | – | – | – | – | – | – | – | + | – |
| Hip contractures | + | + | + | BL dislocation | – | – | + | – | + | + | + | + | internally rotated | + |
| Elbow contractures | + | + | + | – | – | + | + | + | – | + | – | – | ||
| Knee contractures | + | – | + | + | – | + | + | + | + | – | + | + | – | – |
| Other Clinical Features | ||||||||||||||
| Scoliosis | + | + | – | + | – | ND | + | – | – | – | + | – | lumbar lordosis | – |
| Short neck | + | + | + | + | + | ND | + | – | + | – | + | + | + | |
| Cognitive delay | NDa | + | NDa | + | + | + | – | + | + | + | + | + | + | + |
| Speech delay | Na | + | Na | + | + | + | + | + | + | + | + | + | + | + |
| Motor delay | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Seizures | + | + | – | – | – | – | – | – | – | – | – | – | – | – |
| Questionable seizure activity or abnormal breathing, loss of motor control | ND | + | ND | abnormal discharges | periods of rigidity | + | + | ND | ND | + | + | seizure-like activity with fever | ND | irregular breathing |
| MRI findings | normal | mild cerebral and cerebellar atrophy | ND | generalized cerebral atrophy, small pituitary | normal | cerebellar atrophy | normal | ND | ND | diffuse cerebellar atrophy | ND | normal | normal | white matter edema |
| Hypotonia | no | yes | yes | yes | yes | yes | no | yes | ND | no | no | no? | yes | no |
| Respiratory insufficiency | + | + | + | + | + | apneic episodes in newborn period | – | ND | ND | ND | + | ND | – | + |
| Abnormal respiratory pattern in newborn period | + | + | + | + | + | + | – | ND | ND | ND | + | + | – | + |
| Excessive drooling | ND | ND | ND | + | ND | ND | + | + | + | ND | ND | |||
| GERD | ND | + | ND | + | + | + | ND | – | + | + | + | + | in infancy | – |
| Constipation | ND | occasional | ND | – | + | ND | ND | + | + | + | ND | ND | ||
| Hernia | – | inguinal | inguinal | inguinal / peri umbilical | inguinal | – | inguinal | – | inguinal | inguinal | inguinal | ND | umbilical | – |
| Age at time of death | 6 months | 5 years | 4 months | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na |
| Other | central apnea, uric acid renal stones | 11 pairs of ribs, abnormal pelvis and scapulae | renal calcifications | suspected fatty acid oxidation defect | cleft palate | mitral valve prolapse | small VSD | |||||||
This table provides a summary of clinical features of affected individuals from families in which mutations in NALCN were identified. Clinical characteristics listed in the table are primarily features that distinguish CLIFAHDD syndrome from DA conditions. Abbreviations are as follows: +, presence of a finding; −, absence of a finding; ND, no data were available; NA, not applicable; GERP, genomic evolutionary rate profiling; CADD, combined annotation-dependent depletion; CLIFAHDD, congenital contractures of the limbs, face, hypotonia, and developmental delay; DA2A, distal arthrogryposis type 2A; DA2B, distal arthrogryposis type 2B; DA1, distal arthrogryposis type 1; VSD, ventricular septal defect; and GERD, gastresophageal reflux disease. cDNA positions are provided as named by the HGVS MutNomen web tool relative to NM_052867.2.
Child died before an assessment could be made.
Figure 1.
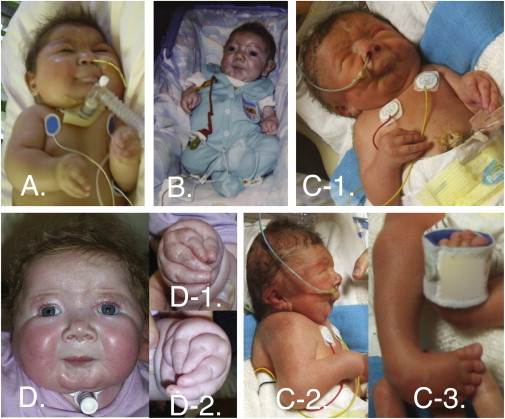
Phenotypic Characteristics of Each Individual with CLIFAHDD Syndrome
Four individuals affected by CLIFAHDD syndrome; all individuals shown have NALCN mutations. Note the short palpebral fissures, flattened nasal root and bridge, large nares, long philtrum, pursed lips, H-shaped dimpling of the chin, and deep nasolabial folds (A, B, C1, D). Camptodactyly of the digits of the hands and ulnar deviation of the wrist (A, B, C, D1, D2) or clubfoot (C3) is present in each affected person. Case identifiers for the individuals shown in this figure correspond to those in Table 1, where there is a detailed description of the phenotype of each affected individual. Figure S1 provides a pedigree of each CLIFAHDD-syndrome-affected family.
Facial characteristics shared among individuals with CLIFAHDD syndrome include downslanting palpebral fissures, a broad nasal bridge with an anteverted nasal tip and large nares, a short columella, a long philtrum, deep nasolabial folds, micrognathia, pursed lips, and chin dimpling resembling the “H-shaped” dimpling observed in persons with DA2A (Figure 1). Each person in our subset had camptodactyly with adducted thumbs, as well as positional foot deformities ranging from mild varus deformity to severe clubfoot. Additionally, contractures of the elbows, knees, and hips, as well as a short neck and scoliosis, were observed.
Neurological evaluation of each living child with CLIFAHDD syndrome revealed a global developmental delay manifesting as speech, motor, and cognitive delays that varied from mild to severe, as well as hypotonia and seizures. Magnetic resonance imaging of the brain, conducted on four of the affected persons, was reported as normal in two persons and abnormal in two. Of the latter, one person was reported to have cerebral and cerebellar atrophy and another to have cerebral atrophy and a “small pituitary.” Severe gastresophageal reflux was observed in all three individuals for whom data were available, and four of five individuals had inguinal hernias. Three of the five affected individuals died in infancy or early childhood. One of these three individuals died of hemophagocytosis complications in the newborn period, whereas the exact cause of death was unclear in the other two.
To find the mutation-harboring gene(s) in individuals with CLIFAHDD syndrome, we first screened the putatively CLIFAHDD-syndrome-diagnosed probands from each of the five families by performing Sanger sequencing for mutations in genes, other than MYH3, known to cause DA1, DA2A, and DA2B. Each proband with CLIFAHDD syndrome was also screened for copy-number variations (CNVs) by comparative genome-wide array genomic hybridization on the Illumina HumanCytoSNP-12. No pathogenic mutations or shared CNVs were identified. Next, exome sequencing was performed on four CLIFAHDD-syndrome-affected parent-child trios for whom sufficient quantities of DNA were available. All studies were approved by the institutional review boards of the University of Washington and Seattle Children’s Hospital, and informed consent was obtained from participants or their parents.
1 μg genomic DNA was subjected to a series of shotgun-library construction steps, including fragmentation through acoustic sonication (Covaris), end polishing (NEBNext End Repair Module), A-tailing (NEBNext dA-Tailing Module), and PCR amplification with ligation of 8 bp barcoded sequencing adaptors (Enzymatics Ultrapure T4 Ligase) for multiplexing. We hybridized 1 μg of barcoded shotgun library to capture probes targeting ∼36.5 Mb of coding exons (Roche Nimblegen SeqCap EZ Human Exome Library v.2.0). Library quality was determined by examination of molecular-weight distribution and sample concentration (Agilent Bioanalyzer). Pooled, barcoded libraries were sequenced via paired-end 50 bp reads with an 8 bp barcode read on Illumina HiSeq sequencers.
Demultiplexed BAM files were aligned to a human reference (UCSC Genome Browser hg19) via the Burrows-Wheeler Aligner (BWA) v.0.6.2. Read data from a flow-cell lane were treated independently for alignment and quality-control purposes in instances where the merging of data from multiple lanes was required. All aligned read data were subjected to (1) removal of duplicate reads (Picard MarkDuplicates v.1.70), (2) indel realignment (GATK IndelRealigner v.1.6-11-g3b2fab9), and (3) base-quality recalibration (GATK TableRecalibration v.1.6-11-g3b2fab9). Variant detection and genotyping were performed with GATK UnifiedGenotyper (v.1.6-11-g3b2fab9). Variant data for each sample were formatted (variant-call format) as “raw” calls that contained individual genotype data for one or multiple samples and were flagged with the filtration walker (GATK) so that lower-quality sites and potential false positives (e.g., strand bias > −0.1, quality scores (QUAL) ≤ 50, allelic imbalance (ABHet) ≥ 0.75, long homopolymer runs (HRun) > 3, and/or low quality by depth (QD) < 5).
Variants with an alternative allele frequency >0.005 in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500) or the 1000 Genomes Browser or >0.05 in an internal exome database of ∼700 individuals were excluded prior to analysis. Additionally, variants that were flagged as low quality or potential false positives (quality score ≤ 30, long homopolymer run > 5, low quality by depth < 5, within a cluster of SNPs) were also excluded from analysis. Variants that were only flagged by the strand-bias filter (strand bias > −0.10) were included in further analyses because the strand-bias flag has previously been found to be applied to valid variants. CNV calls were also generated from exome data (CoNIFER).6 Variants were annotated with the SeattleSeq137 Annotation Server, and variants for which the only functional prediction label was “intergenic,” “coding-synonymous,” “UTR,” “near-gene,” or “intron” were excluded. Individual genotypes with depth < 6 or genotype quality < 20 were treated as missing.
By analysis of variants from exome sequencing under a de novo mutation model, we identified four different de novo variants in a single gene, NALCN, encoding a sodium leak channel (NALCN [MIM 611549; Refseq accession number NM_052867.2]), in four CLIFAHDD-syndrome-affected families (Table 1 and Figure 1, Families A–D; Figures S1 and S2). Concurrent exome sequencing of six DA-affected trios in whom no pathogenic variants in genes previously associated with DA had been identified serendipitously revealed de novo NALCN variants in two families (Table 1, families G and K; Figure S3). All six variants were missense variants predicted to be deleterious and to result in amino acid substitutions of highly conserved amino acid residues (i.e., the minimum genomic evolutionary rate profile score was 4.73) in NALCN (Table 1 and Figure 2). Each variant was confirmed by Sanger sequencing to have arisen de novo, and none of the six variants were found in over 71,000 control exomes recorded in the ESP6500, 1000 Genomes phase 1 (November, 2010 release), internal databases (> 1,400 chromosomes), or the Exome Aggregation Consortium (ExAC) browser (October 20, 2014 release).
Figure 2.
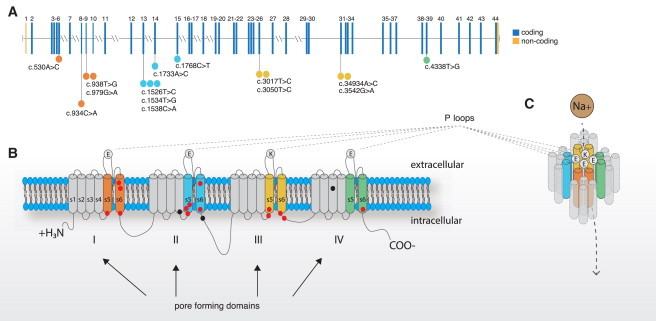
Genomic and Protein Structure of NALCN and Spectrum of Mutations that Cause CLIFAHDD Syndrome
(A) NALCN is composed of 45 exons, 43 of which are protein coding (blue) and two of which are non-coding (orange). Lines with attached dots indicate the approximate locations of 14 different de novo variants that cause CLIFAHDD syndrome. The color of each dot reflects the nearest transmembrane segment (S1–S6) depicted in (B) and (C). The two mutations between IIIS6 and IVS1 are located in the intracellular loop linking domains III and IV. This loop is known to be involved in the inactivation of voltage-gated sodium channels; however NALCN is not voltage-gated, so the specific function of this loop in NALCN is not known.
(B) The predicted protein topology of NALCN is comprised of four homologous pore-forming domains (I–IV), each composed of six transmembrane segments (S1–S6). The bold line represents the polypeptide chain linking segments and domains. The outer ring of P loop amino acid residues (EEKE) that form the ion selectivity filter is represented by filled circles. The approximate positions of variants that cause CLIFAHDD syndrome and infantile hypotonia with psychomotor retardation and characteristic facies are indicated by red-filled circles and black-filled circles, respectively.
(C) S1–S3 might have structural functions, whereas four P loops spanning from S5 to S6 form the ion selectivity filter. In other 4x6TM protein superfamily channels, such as SCN1A, the S4 segments initiate opening of the central pore (S5-P loop-S6) in response to voltage changes, however, because NALCN is not voltage-gated, the function of S4 is currently unknown.
Because the phenotype of the individuals with mutations in NALCN was variable and overlapped with other DA syndromes, we used molecular inversion probes with 5-bp molecular tags7 (smMIPs; designed with MIPGen8 v.0.9.7) to conduct targeted next-generation sequencing of NALCN in 202 additional samples from DA-affected individuals in whom no pathogenic mutation had been found. The 43 coding exons (5,214 bp total) of Ensembl transcript ENST00000251127 and the 10 bp flanking each exon (860 bp total) were targeted with smMIPs for an overall target size of 6,074 bp. Pooled and phosphorylated smMIPs were added to the capture reactions with 100 ng of genomic DNA from each individual to produce a NALCN library for each individual. The libraries were amplified during 21 cycles of PCR, during which an 8-bp sample barcode was introduced. The barcoded libraries were then pooled and purified with magnetic beads. After Picogreen quantification to determine the appropriate dilution, 10 pmol of the pool was sequenced on an Illumina MiSeq. Molecular-inversion-probe collapsing and arm trimming (MIPGenM v.1.0), alignment (BWA v.0.7.8), and multi-sample genotype calling (GATK Unified Genotyper v.3.2-2-gec30cee) were performed, and variants were annotated with SeattleSeq138. We used the same filtering strategy employed in our analysis of the exome sequences to select variants for further confirmation by Sanger sequencing.
Missense variants predicted to result in substitution of conserved amino acid residues were identified and confirmed to have arisen de novo in eight additional families: one CLIFAHDD-syndrome-affected family that had not been available for exome sequencing (Table 1), two families in which affected members were originally diagnosed with DA2A (Table 1, families F and H; Figure S1; Figure S3, family F), three families in which affected members were originally diagnosed with DA2B (Table 1, families I, J, and L; Figure S1; Figure S2, families I and J), and two families in which affected members were diagnosed with DA1 (Table 1, families M and N; Figure S1; Figure S3, family M). Altogether, we discovered unique, de novo missense NALCN mutations in fourteen simplex families in which the proband had been diagnosed with CLIFAHDD syndrome (n = 5), DA2A (n = 3), DA2B (n = 4), or DA1 (n = 2) (Table 1 and Figure 2).
The diagnosis of CLIFAHDD syndrome was considered in one additional DA2A case brought to our attention because of a pattern of congenital facial and limb contractures characteristic of DA2A in the absence of a finding of a pathogenic MYH3 mutation. However, death occurred within two hours of birth at 29 weeks gestation, and the available clinical information was therefore considered too limited. This approach proved prudent given that no mutation in NALCN was identified by exome sequencing. Instead, this child was found to be a compound heterozygote in RYR1 (MIM 180901) for a predicted nonsense variant, p. Tyr3921Ter (c.11763C>A [RefSeq NM_000540.2]), inherited from the father, and a missense variant, p.Gly341Arg (c.1021G>A [RefSeq NM_000540.2]), inherited from the mother. The missense variant has been previously reported to cause malignant hyperthermia, and persons carrying this variant had a positive “in vitro contractility test.”9 Crisponi syndrome10,11 (MIM 601378) was also considered as a possible diagnosis for this child. However, no candidate variants were identified in CRLF1 (MIM 604237), either in this child or in any of the fourteen families in which NALCN mutations were identified. In addition, we have previously found two mutations in RYR1 (i.e., c.1391A>G[p.Gln464Arg] and c.2683-1G>A) in a child diagnosed with DA2A, suggesting that mutations in RYR1 are a rare cause of DA2A.
Mutations in NALCN have recently been reported to cause an autosomal-recessive condition, infantile hypotonia with psychomotor retardation and characteristic facies (IHPRF [MIM 615419]), in one Turkish family12 and two Saudi Arabian13 families. Individuals affected by CLIFAHDD syndrome share some of the phenotypic characteristics of IHPRF (e.g., developmental delay and hypotonia). However, these are relatively non-specific findings, and IHPRF and CLIFAHDD syndrome appear to be otherwise distinct from one another. In addition, the parents of children with IHPRF are carriers of NALCN mutations but reportedly do not have any of the abnormalities found in persons with CLIFAHDD syndrome. Furthermore, each inherited NALCN variant found in the 14 individuals with CLIFAHDD syndrome was also present in ESP6500 and/or 1000 Genomes data. This finding suggests that these inherited variants are polymorphisms rather than pathogenic variants underlying CLIFAHDD syndrome. Together, these results support the hypothesis that CLIFAHDD syndrome is an autosomal-dominant condition distinct from IHPRF.
We next sought to explore the mechanism by which variants in NALCN result in CLIFAHDD syndrome. To this end, we constructed two expression plasmids carrying NALCN variants: pcDNA3-hNALCN-L509S-GFP with c.1526>T(p.Leu509Ser) found in a person with relatively mild developmental delay (Table 1, family G) and pcDNA3-hNALCN-Y578S-GFP with c.1733A>C(p.Tyr578Ser) found in a child with severe developmental delay (Table 1, family K). We then co-expressed mutant and wild-type NALCN in HEK293T cells. Immunoblot analysis demonstrated that the expression of either the p.Tyr578Ser or p.Leu509Ser (GFP-tagged) NALCN alteration nearly eliminated wild-type (HA-tagged) NALCN protein (Figure 3).
Figure 3.
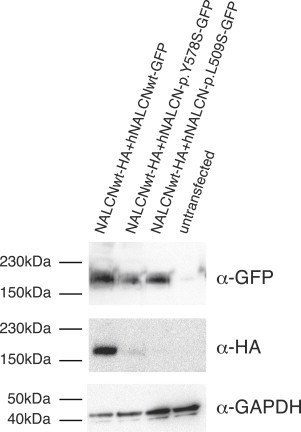
Expression of p.Tyr578Ser and p.Leu509Ser NALCN Alterations Prevents Wild-Type NALCN Detection at the Protein Level when Co-Expressed in HEK293T Cells
HA-tagged wild-type NALCN channels are not detectable when co-expressed with GFP-tagged NALCN mutant channels in HEK293T cells but are detectable when co-expressed with GFP-tagged wild-type NALCN channels. This experiment is representative of four independent experiments.
It remains to be determined whether synthesis of altered NALCN channels prevents wild-type NALCN synthesis or induces its degradation by the proteasome. The latter mechanism has been previously described14 with regard to mutations in the Cav1.2 calcium channel responsible for dominantly inherited episodic ataxia type 2 (MIM 108500). However, preliminary evidence suggests that NALCN alterations are correctly targeted to the plasma membrane (data not shown). Nevertheless, both mutations apparently induced the disappearance of wild-type NALCN to the same extent. Accordingly, an explanation for the observed phenotypic differences between the sampled cases is not yet apparent.
Collectively, these functional studies, although preliminary, are consistent with mutations in NALCN causing CLIFAHDD syndrome via a dominant-negative effect that results in haploinsufficiency. However, the finding that persons heterozygous for NALCN mutations that cause IHPRF are reportedly unaffected indicates that haploinsufficiency of NALCN alone is not adequate to cause CLIFAHDD syndrome. These observations suggest that NALCN mutations that cause CLIFAHDD syndrome might produce a mutant protein that has some residual activity (i.e., a hypomorphic allele) or that exhibits a gain of function. Moreover, mutations in NALCN could cause CLIFAHDD syndrome by more than one mechanism.
NALCN is a G-protein-coupled receptor-activated channel15 consisting of four homologous domains (domains I–IV), each of which consists of six transmembrane segments (S1–S6) separated by cytoplasmic linkers (Figure 2). Pore-forming loops (P loops) between S5 and S6 of each domain form an EEKE sodium-ion selectivity filter (Figure 2). In vertebrates and some invertebrate model organisms, such as Drosophila and C. elegans, NALCN has a sodium-selective EEKE pore and putatively functions as a sodium channel. However, it remains unclear whether NALCN is actually an ion channel rather than a sensor of sodium.16 In mammals, NALCN is most highly expressed in the central nervous system but is also found at moderate levels in the heart, lymph nodes, pancreas, and thyroid (summarized by Cochet-Bissuel et al.17). Unlike most known genes underlying DA syndromes, NALCN is not expressed in fetal skeletal muscle (Figure S4). However, homologs of NALCN are expressed in the neuromuscular junction in D. melanogaster18 as well as in motor neurons in C. elegans19 and mice.20,21 Accordingly, mutations in NALCN might cause congenital contractures by disturbing motor control of myofiber function during development, similar to the hypothesized effects of mutations in ECEL1, PIEZO2, and CHRNG (MIM 100730).
Each of the 14 de novo NALCN mutations found to cause CLIFAHDD syndrome is located in or near the predicted S5 and S6 segments, which are part of the pore-forming domain of the NALCN channel (Figure 2; Table S1). The clustering of mutations is an indication of the functional importance of these segments. In contrast, the frameshift (c.1489delT [p.Tyr497Thrfs21]) and nonsense (c.1924C>T [p.Gln642Ter]) mutations reported to cause IHPRF are predicted to result in a loss of function of NALCN, whereas the missense (c.3860G>T [p.Trp1287Leu]) mutation is predicted to alter the S3 segment of domain IV. These findings suggest that mutations in different regions of NALCN perturb different functions of the channel, result in different Mendelian conditions, and determine whether the condition is transmitted in an autosomal-recessive or an autosomal-dominant pattern. A similar relationship has recently been reported for MAB21L2 (MIM 604357) mutations that underlie a spectrum of major malformations of the eye.22
NALCN is highly conserved across vertebrates, and there is ∼98% sequence identity between human and mouse NALCN. Functional studies in model organisms have illuminated multiple physiologic roles of NALCN. In general, NALCN permits background sodium-ion leak currents that contribute to regulation of resting membrane potentials and spontaneous firing of motor neurons.23 In mice, NALCN has been shown to regulate respiration,24 intestinal pacemaking activity in the interstitial cells of Cajal,25 and serum sodium concentration.26 Mutation of NALCN homologs in D. melanogaster, C. elegans, and L. stagnalis results in abnormal locomotion,19,27–30 disturbance of circadian rhythms,29 disruption of normal respiratory rhythms,31 and altered sensitivity to ethanol and some general anesthetics30,32,33 (reviewed by Cochet-Bissuel et al.17). Homozygous Nalcn-null mice die shortly after birth and exhibit disrupted respiratory rhythm; mutant pups breath for 5 s, followed by 5 s of apnea. This disrupted respiratory rhythm results from a lack of electrical discharges from the C4 nerves that control the thoracic diaphragm.24 Interstitial cells of Cajal, which generate the electrical signals that govern gut motility, from Nalcn-null mice exhibit abnormal regulation of pacemaking activity.25 These abnormalities are reminiscent of some of the respiratory and gastrointestinal abnormalities observed in persons with CLIFAHDD syndrome. Moreover, gain-of-function mutations in exons encoding segment S6 of domain I of a NALCN homolog (nca-1) in C. elegans lead to a dominantly inherited phenotype that includes pausing and uncoordinated, exaggerated body bending19,34 and loosely recapitulates episodes of disorganized movement experienced by several individuals with CLIFAHDD syndrome. Nevertheless, these assertions are based on anecdotal clinical information and necessitate objective testing of respiratory function and gastrointestinal motility.
NALCN is part of a structurally similar superfamily (the 4x6TM family) of cation (i.e., sodium and calcium) channels with four homologous domains containing six transmembrane helices.16 Mutations in 4x6TM genes that encode voltage-gated sodium and calcium channels (e.g., SCN1A [MIM 182389], SCN9A [MIM 603415], CACNA1A [MIM 601011], and CACNA1C [MIM 114205]) can cause developmental delay, intellectual disability, seizures, myotonia, ataxia, pain hypersensitivity and insensitivity, and cardiac arrhythmias. These conditions are far more common than CLIFAHDD syndrome (hundreds of different mutations have been reported to date), and for some conditions, significant structure-function relationships have been described.35,36 Caution is warranted, however, in trying to use these observations to make inferences about structure-function relationships in NALCN because NALCN is not a paralog of these genes but instead diverged from them prior to the diversification of 4x6TM voltage-gated calcium and sodium channels in animals.37,38
In 2003, we reported that mutations in TNNI2 and TPM2 caused DA1 and hypothesized that DA syndromes are, in general, caused by mutations in genes that encode components of the contractile apparatus of fast-twitch skeletal myofibers.1 Over the past decade, we and others have identified, via a candidate-gene approach, mutations that cause DA conditions in four additional genes (i.e., TNNT3,2 MYH3,3 MYH8,39and MYBPC140 [MIM 160794]) that encode contractile proteins. However, mutations in these six genes collectively explain only ∼60% of affected families. In the past 2 years, exome sequencing has facilitated discovery of mutations in two genes, PIEZO2 (MIM 613629), which causes DA3 (Gordon syndrome [MIM 114300])41 and DA5 (MIM 108145),41,42 and ECEL1 (MIM 605896), which causes DA5D (MIM 615065).43,44 In contrast to genes that encode contractile proteins, PIEZO2 is a mechanoreceptor, and ECEL1 plays a role in the development of terminal neuronal branches at skeletal-muscle endplates. These discoveries confirmed that a subset of DA syndromes are due to a mechanism(s) other than perturbation of the contractile apparatus of skeletal myofibers. The finding that mutations in NALCN underlie some individuals’ diagnoses of atypical DA, most often DA2A with neurological impairment, suggests that NALCN should be added to the expanding list of genes that are causal for DA conditions but do not encode contractile proteins. However, in each person with a NALCN mutation, the diagnosis of DA should have been excluded because neurological abnormalities, including hypotonia and developmental delay, are explicit exclusion criteria.45 In other words, CLIFAHDD syndrome is a newly delineated Mendelian condition and one of more than 300 conditions characterized by arthrogryposis, but it is not a DA syndrome.
Descriptions of several children with clinical characteristics similar to those of CLIFAHDD syndrome have been reported previously. Specifically, in the early 1990’s four independent simplex families were described46,47 as exhibiting “distal arthrogryposis, mental retardation, whistling face, and Pierre Robin sequence,” currently designated as Illum syndrome (MIM 208155). Three of these children, including two who died at six months of age, died prematurely. In each instance, the facial characteristics, neurological findings, and natural history parallel those found in CLIFAHDD syndrome. Accordingly, we suspect that these children likely had mutations in NALCN. However, we note that Illum et al.48 reported a family with three affected siblings whose phenotypic features overlapped with but were distinctly different from those found in CLIFAHDD syndrome. Most notably, the affected members of the family reported by Illum et al. had calcium deposition in the skeletal muscle and central nervous system, whereas this is not typical of individuals with CLIFAHDD syndrome. Thus, we conclude that Illum and CLIFAHDD syndromes are different conditions.
In summary, we used exome and targeted next-generation sequencing to identify de novo mutations in NALCN as the cause of a newly delineated condition, CLIFAHDD syndrome, characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. The range of phenotypic variation in the CLIFAHDD-syndrome-affected individuals described herein is relatively narrow, but the congenital contractures were mild enough in some cases to have been overlooked, particularly in older children and young adults. Accordingly, it is possible that mutations in NALCN might also explain some cases of apparently isolated intellectual disability. Furthermore, the finding that the de novo mutations cluster in and around the pore-forming regions (i.e., S5 and S6 helices) suggests that the location and type of NALCN mutations might determine whether an individual develops IHPRF or CLIFAHDD syndrome.
Consortia
The members of the University of Washington Center for Mendelian Genomics are Michael J. Bamshad, Jay Shendure, Deborah A. Nickerson, Gonçalo R. Abecasis, Peter Anderson, Elizabeth Marchani Blue, Marcus Annable, Brian L. Browning, Kati J. Buckingham, Christina Chen, Jennifer Chin, Jessica X. Chong, Gregory M. Cooper, Colleen P. Davis, Christopher Frazar, Tanya M. Harrell, Zongxiao He, Preti Jain, Gail P. Jarvik, Guillaume Jimenez, Eric Johanson, Goo Jun, Martin Kircher, Tom Kolar, Stephanie A. Krauter, Niklas Krumm, Suzanne M. Leal, Daniel Luksic, Colby T. Marvin, Margaret J. McMillin, Sean McGee, Patrick O’Reilly, Bryan Paeper, Karynne Patterson, Marcos Perez, Sam W. Phillips, Jessica Pijoan, Christa Poel, Frederic Reinier, Peggy D. Robertson, Regie Santos-Cortez, Tristan Shaffer, Cindy Shephard, Kathryn M. Shively, Deborah L. Siegel, Joshua D. Smith, Jeffrey C. Staples, Holly K. Tabor, Monica Tackett, Jason G. Underwood, Marc Wegener, Gao Wang, Marsha M. Wheeler, and Qian Yi.
Acknowledgments
We thank the families for their participation and support; Christa Poel, Karynne Patterson, and Ian Glass for technical assistance; and J. David Spafford for helpful comments. Our work was supported in part by grants from the NIH, the National Heart, Lung, and Blood Institute (NHLBI), and the National Human Genome Research Institute (NHGRI) (1U54HG006493 to M.B., D.N., and J.S.; 1RC2HG005608 to M.B., D.N., and J.S.; 5R000HG004316 to H.K.T.), by the National Institute of Child Health and Human Development (1R01HD048895 to M.J.B. and 5K23HD057331 to A.E.B), by the Life Sciences Discovery Fund (2065508 and 0905001), and by the Washington Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHGRI, NHLBI, or NIH.
Contributor Information
Michael J. Bamshad, Email: mbamshad@uw.edu.
University of Washington Center for Mendelian Genomics:
Michael J. Bamshad, Jay Shendure, Deborah A. Nickerson, Gonçalo R. Abecasis, Peter Anderson, Elizabeth Marchani Blue, Marcus Annable, Brian L. Browning, Kati J. Buckingham, Christina Chen, Jennifer Chin, Jessica X. Chong, Gregory M. Cooper, Colleen P. Davis, Christopher Frazar, Tanya M. Harrell, Zongxiao He, Preti Jain, Gail P. Jarvik, Guillaume Jimenez, Eric Johanson, Goo Jun, Martin Kircher, Tom Kolar, Stephanie A. Krauter, Niklas Krumm, Suzanne M. Leal, Daniel Luksic, Colby T. Marvin, Margaret J. McMillin, Sean McGee, Patrick O’Reilly, Bryan Paeper, Karynne Patterson, Marcos Perez, Sam W. Phillips, Jessica Pijoan, Christa Poel, Frederic Reinier, Peggy D. Robertson, Regie Santos-Cortez, Tristan Shaffer, Cindy Shephard, Kathryn M. Shively, Deborah L. Siegel, Joshua D. Smith, Jeffrey C. Staples, Holly K. Tabor, Monica Tackett, Jason G. Underwood, Marc Wegener, Gao Wang, Marsha M. Wheeler, and Qian Yi
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ExAC Browser, http://exac.broadinstitute.org/
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
SeattleSeq Annotation 138, http://snp.gs.washington.edu/SeattleSeqAnnotation138/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Sung S.S., Brassington A.-M.E., Grannatt K., Rutherford A., Whitby F.G., Krakowiak P.A., Jorde L.B., Carey J.C., Bamshad M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genet. 2003;72:681–690. doi: 10.1086/368294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung S.S., Brassington A.-M.E., Krakowiak P.A., Carey J.C., Jorde L.B., Bamshad M. Mutations in TNNT3 cause multiple congenital contractures: a second locus for distal arthrogryposis type 2B. Am. J. Hum. Genet. 2003;73:212–214. doi: 10.1086/376418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toydemir R.M., Rutherford A., Whitby F.G., Jorde L.B., Carey J.C., Bamshad M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006;38:561–565. doi: 10.1038/ng1775. [DOI] [PubMed] [Google Scholar]
- 4.Beck A.E., McMillin M.J., Gildersleeve H.I., Kezele P.R., Shively K.M., Carey J.C., Regnier M., Bamshad M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. A. 2013;161A:550–555. doi: 10.1002/ajmg.a.35809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beck A.E., McMillin M.J., Gildersleeve H.I., Shively K.M., Tang A., Bamshad M.J. Genotype-phenotype relationships in Freeman-Sheldon syndrome. Am. J. Med. Genet. A. 2014;164A:2808–2813. doi: 10.1002/ajmg.a.36762. [DOI] [PubMed] [Google Scholar]
- 6.Krumm N., Sudmant P.H., Ko A., O’Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E., NHLBI Exome Sequencing Project Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiatt J.B., Pritchard C.C., Salipante S.J., O’Roak B.J., Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle E.A., O’Roak B.J., Martin B.K., Kumar A., Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30:2670–2672. doi: 10.1093/bioinformatics/btu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monsieurs K.G., Van Broeckhoven C., Martin J.J., Van Hoof V.O., Heytens L. Gly341Arg mutation indicating malignant hyperthermia susceptibility: specific cause of chronically elevated serum creatine kinase activity. J. Neurol. Sci. 1998;154:62–65. doi: 10.1016/s0022-510x(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 10.Crisponi L., Crisponi G., Meloni A., Toliat M.R., Nürnberg G., Usala G., Uda M., Masala M., Höhne W., Becker C. Crisponi syndrome is caused by mutations in the CRLF1 gene and is allelic to cold-induced sweating syndrome type 1. Am. J. Hum. Genet. 2007;80:971–981. doi: 10.1086/516843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dagoneau N., Bellais S., Blanchet P., Sarda P., Al-Gazali L.I., Di Rocco M., Huber C., Djouadi F., Le Goff C., Munnich A., Cormier-Daire V. Mutations in cytokine receptor-like factor 1 (CRLF1) account for both Crisponi and cold-induced sweating syndromes. Am. J. Hum. Genet. 2007;80:966–970. doi: 10.1086/513608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Köroğlu Ç., Seven M., Tolun A. Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J. Med. Genet. 2013;50:515–520. doi: 10.1136/jmedgenet-2013-101634. [DOI] [PubMed] [Google Scholar]
- 13.Al-Sayed M.D., Al-Zaidan H., Albakheet A., Hakami H., Kenana R., Al-Yafee Y., Al-Dosary M., Qari A., Al-Sheddi T., Al-Muheiza M. Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am. J. Hum. Genet. 2013;93:721–726. doi: 10.1016/j.ajhg.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mezghrani A., Monteil A., Watschinger K., Sinnegger-Brauns M.J., Barrère C., Bourinet E., Nargeot J., Striessnig J., Lory P. A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J. Neurosci. 2008;28:4501–4511. doi: 10.1523/JNEUROSCI.2844-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swayne L.A., Mezghrani A., Varrault A., Chemin J., Bertrand G., Dalle S., Bourinet E., Lory P., Miller R.J., Nargeot J., Monteil A. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. 2009;10:873–880. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senatore A., Spafford J.D. A uniquely adaptable pore is consistent with NALCN being an ion sensor. Channels (Austin) 2013;7:60–68. doi: 10.4161/chan.23981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cochet-Bissuel M., Lory P., Monteil A. The sodium leak channel, NALCN, in health and disease. Front. Cell. Neurosci. 2014;8:132. doi: 10.3389/fncel.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishikawa K., Kidokoro Y. Halothane presynaptically depresses synaptic transmission in wild-type Drosophila larvae but not in halothane-resistant (har) mutants. Anesthesiology. 1999;90:1691–1697. doi: 10.1097/00000542-199906000-00026. [DOI] [PubMed] [Google Scholar]
- 19.Yeh E., Ng S., Zhang M., Bouhours M., Wang Y., Wang M., Hung W., Aoyagi K., Melnik-Martinez K., Li M. A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C. elegans. PLoS Biol. 2008;6:e55. doi: 10.1371/journal.pbio.0060055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda M., Harris A.W., Kingham B.F., Lumpkin C.J., Opdenaker L.M., McCahan S.M., Wang W., Butchbach M.E.R. Transcriptome profiling of spinal muscular atrophy motor neurons derived from mouse embryonic stem cells. PLoS ONE. 2014;9:e106818. doi: 10.1371/journal.pone.0106818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandyopadhyay U., Cotney J., Nagy M., Oh S., Leng J., Mahajan M., Mane S., Fenton W.A., Noonan J.P., Horwich A.L. RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS ONE. 2013;8:e53575. doi: 10.1371/journal.pone.0053575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rainger J., Pehlivan D., Johansson S., Bengani H., Sanchez-Pulido L., Williamson K.A., Ture M., Barker H., Rosendahl K., Spranger J., UK10K. Baylor-Hopkins Center for Mendelian Genomics Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am. J. Hum. Genet. 2014;94:915–923. doi: 10.1016/j.ajhg.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren D. Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron. 2011;72:899–911. doi: 10.1016/j.neuron.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu B., Su Y., Das S., Liu J., Xia J., Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129:371–383. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 25.Kim B.J., Chang I.Y., Choi S., Jun J.Y., Jeon J.-H., Xu W.-X., Kwon Y.K., Ren D., So I. Involvement of Na(+)-leak channel in substance P-induced depolarization of pacemaking activity in interstitial cells of Cajal. Cell. Physiol. Biochem. 2012;29:501–510. doi: 10.1159/000338504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinke A.P., Caputo C., Tsaih S.W., Yuan R., Ren D., Deen P.M.T., Korstanje R. Genetic analysis of mouse strains with variable serum sodium concentrations identifies the Nalcn sodium channel as a novel player in osmoregulation. Physiol. Genomics. 2011;43:265–270. doi: 10.1152/physiolgenomics.00188.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jospin M., Watanabe S., Joshi D., Young S., Hamming K., Thacker C., Snutch T.P., Jorgensen E.M., Schuske K. UNC-80 and the NCA ion channels contribute to endocytosis defects in synaptojanin mutants. Curr. Biol. 2007;17:1595–1600. doi: 10.1016/j.cub.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 28.Pierce-Shimomura J.T., Chen B.L., Mun J.J., Ho R., Sarkis R., McIntire S.L. Genetic analysis of crawling and swimming locomotory patterns in C. elegans. Proc. Natl. Acad. Sci. USA. 2008;105:20982–20987. doi: 10.1073/pnas.0810359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nash H.A., Scott R.L., Lear B.C., Allada R. An unusual cation channel mediates photic control of locomotion in Drosophila. Curr. Biol. 2002;12:2152–2158. doi: 10.1016/s0960-9822(02)01358-1. [DOI] [PubMed] [Google Scholar]
- 30.Krishnan K.S., Nash H.A. A genetic study of the anesthetic response: mutants of Drosophila melanogaster altered in sensitivity to halothane. Proc. Natl. Acad. Sci. USA. 1990;87:8632–8636. doi: 10.1073/pnas.87.21.8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu T.Z., Feng Z.-P. A sodium leak current regulates pacemaker activity of adult central pattern generator neurons in Lymnaea stagnalis. PLoS ONE. 2011;6:e18745. doi: 10.1371/journal.pone.0018745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mir B., Iyer S., Ramaswami M., Krishnan K.S. A genetic and mosaic analysis of a locus involved in the anesthesia response of Drosophila melanogaster. Genetics. 1997;147:701–712. doi: 10.1093/genetics/147.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphrey J.A., Hamming K.S., Thacker C.M., Scott R.L., Sedensky M.M., Snutch T.P., Morgan P.G., Nash H.A. A putative cation channel and its novel regulator: cross-species conservation of effects on general anesthesia. Curr. Biol. 2007;17:624–629. doi: 10.1016/j.cub.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 34.Bonnett K., Zweig R., Aamodt E.J., Dwyer D.S. Food deprivation and nicotine correct akinesia and freezing in Na(+) -leak current channel (NALCN)-deficient strains of Caenorhabditis elegans. Genes Brain Behav. 2014;13:633–642. doi: 10.1111/gbb.12153. [DOI] [PubMed] [Google Scholar]
- 35.Zuberi S.M., Brunklaus A., Birch R., Reavey E., Duncan J., Forbes G.H. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011;76:594–600. doi: 10.1212/WNL.0b013e31820c309b. [DOI] [PubMed] [Google Scholar]
- 36.Meisler M.H., Kearney J.A. Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liebeskind B.J., Hillis D.M., Zakon H.H. Phylogeny unites animal sodium leak channels with fungal calcium channels in an ancient, voltage-insensitive clade. Neurology. 2012;29:3613–3616. doi: 10.1093/molbev/mss182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J.H., Cribbs L.L., Perez-Reyes E. Cloning of a novel four repeat protein related to voltage-gated sodium and calcium channels. FEBS Lett. 1999;445:231–236. doi: 10.1016/s0014-5793(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 39.Veugelers M., Bressan M., McDermott D.A., Weremowicz S., Morton C.C., Mabry C.C., Lefaivre J.-F., Zunamon A., Destree A., Chaudron J.-M., Basson C.T. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N. Engl. J. Med. 2004;351:460–469. doi: 10.1056/NEJMoa040584. [DOI] [PubMed] [Google Scholar]
- 40.Gurnett C.A., Desruisseau D.M., McCall K., Choi R., Meyer Z.I., Talerico M., Miller S.E., Ju J.-S., Pestronk A., Connolly A.M. Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010;19:1165–1173. doi: 10.1093/hmg/ddp587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMillin M.J., Beck A.E., Chong J.X., Shively K.M., Buckingham K.J., Gildersleeve H.I., Aracena M.I., Aylsworth A.S., Bitoun P., Carey J.C., University of Washington Center for Mendelian Genomics Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 2014;94:734–744. doi: 10.1016/j.ajhg.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coste B., Houge G., Murray M.F., Stitziel N., Bandell M., Giovanni M.A., Philippakis A., Hoischen A., Riemer G., Steen U. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc. Natl. Acad. Sci. USA. 2013;110:4667–4672. doi: 10.1073/pnas.1221400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMillin M.J., Below J.E., Shively K.M., Beck A.E., Gildersleeve H.I., Pinner J., Gogola G.R., Hecht J.T., Grange D.K., Harris D.J., University of Washington Center for Mendelian Genomics Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013;92:150–156. doi: 10.1016/j.ajhg.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dieterich K., Quijano-Roy S., Monnier N., Zhou J., Fauré J., Smirnow D.A., Carlier R., Laroche C., Marcorelles P., Mercier S. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum. Mol. Genet. 2013;22:1483–1492. doi: 10.1093/hmg/dds514. [DOI] [PubMed] [Google Scholar]
- 45.Bamshad M., Jorde L.B., Carey J.C. A revised and extended classification of the distal arthrogryposes. Am. J. Med. Genet. 1996;65:277–281. doi: 10.1002/(SICI)1096-8628(19961111)65:4<277::AID-AJMG6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 46.Di Rocco M., Erriu M.I., Lignana E. Distal arthrogryposis, mental retardation, whistling face, and Pierre Robin sequence: another case. Am. J. Med. Genet. 1992;44:391. doi: 10.1002/ajmg.1320440330. [DOI] [PubMed] [Google Scholar]
- 47.Schrander-Stumpel C., Fryns J.P., Beemer F.A., Rive F.A. Association of distal arthrogryposis, mental retardation, whistling face, and Pierre Robin sequence: evidence for nosologic heterogeneity. Am. J. Med. Genet. 1991;38:557–561. doi: 10.1002/ajmg.1320380412. [DOI] [PubMed] [Google Scholar]
- 48.Illum N., Reske-Nielsen E., Skovby F., Askjaer S.A., Bernsen A. Lethal autosomal recessive arthrogryposis multiplex congenita with whistling face and calcifications of the nervous system. Neuropediatrics. 1988;19:186–192. doi: 10.1055/s-2008-1052443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.