

Background: Mutations in LEPREL1, the gene encoding prolyl 3-hydroxylase-2 (P3H2), cause severe nonsyndromic myopia.
Results: Collagens I and IV from P3h2-null mouse eye tissues were significantly reduced in 3-hydroxylation compared with wild-type littermates.
Conclusion: Loss of P3h2 causes altered collagen prolyl 3-hydroxylation from multiple tissues.
Significance: Improved understanding of molecular mechanisms of myopia could aid in early diagnosis and treatment of irreversible vision loss.
Keywords: Animal Model, Collagen, Extracellular Matrix, Mass Spectrometry (MS), Post-translational Modification (PTM), 3-Hydroxyproline, Prolyl 3-Hydroxylase, Sclera
Abstract
Myopia, the leading cause of visual impairment worldwide, results from an increase in the axial length of the eyeball. Mutations in LEPREL1, the gene encoding prolyl 3-hydroxylase-2 (P3H2), have recently been identified in individuals with recessively inherited nonsyndromic severe myopia. P3H2 is a member of a family of genes that includes three isoenzymes of prolyl 3-hydroxylase (P3H), P3H1, P3H2, and P3H3. Fundamentally, it is understood that P3H1 is responsible for converting proline to 3-hydroxyproline. This limited additional knowledge also suggests that each isoenzyme has evolved different collagen sequence-preferred substrate specificities. In this study, differences in prolyl 3-hydroxylation were screened in eye tissues from P3h2-null (P3h2n/n) and wild-type mice to seek tissue-specific effects due the lack of P3H2 activity on post-translational collagen chemistry that could explain myopia. The mice were viable and had no gross musculoskeletal phenotypes. Tissues from sclera and cornea (type I collagen) and lens capsule (type IV collagen) were dissected from mouse eyes, and multiple sites of prolyl 3-hydroxylation were identified by mass spectrometry. The level of prolyl 3-hydroxylation at multiple substrate sites from type I collagen chains was high in sclera, similar to tendon. Almost every known site of prolyl 3-hydroxylation in types I and IV collagen from P3h2n/n mouse eye tissues was significantly under-hydroxylated compared with their wild-type littermates. We conclude that altered collagen prolyl 3-hydroxylation is caused by loss of P3H2. We hypothesize that this leads to structural abnormalities in multiple eye tissues, but particularly sclera, causing progressive myopia.
Introduction
Collagen post-translational modifications are essential to animal life. Most have evolved to impart a structural stability to the collagen triple helix and subsequent higher order assemblies. Indeed, one in four amino acids in mammalian fibrillar collagen α-chains are prolyl residues, about 40% of which are 4-hydroxylated. This modification has been shown to add structural stability to the collagen triple helix through hydrogen bonding (1). Although 3-hydroxyproline (3Hyp)2 was discovered in collagens more than a half-century ago (2), no clear function is yet known for this unique modification (3). Prolyl 3-hydroxylation is a quantitatively minor yet highly conserved collagen post-translational modification found throughout the animal kingdom (4, 5). Only 1 or 2 3Hyp residues occur per α-chain in collagen types I and II of most tissues, compared with 3–6 residues per α-chain of collagen types V and XI (5) and over 10 residues per α-chain of collagen type IV in basement membranes (6, 7). In type I collagen, the one primary 3Hyp residue is formed by an enzyme complex composed of prolyl 3-hydroxylase-1 (P3H1), cartilage-associated protein, and peptidyl prolyl isomerase B. Gene mutations that disrupt expression of any protein in this complex can cause recessive osteogenesis imperfecta (8–13). The roles of cartilage-associated protein and P3H1 in collagen homeostasis and osteogenesis imperfecta pathology have been thoroughly investigated over the last decade (14). However, P3H1 is one of a family of genes that includes three isoenzymes, P3H1 (LEPRE1), P3H2 (LEPREL1), and P3H3 (LEPREL2) (15). It is believed these isoenzymes have evolved differences in their preferred collagen substrate and tissue specificities (16). Indeed, P3H2 mutations have recently been associated with autosomally recessive nonsyndromic severe myopia in humans (17, 18). The first study described individuals with a homozygous mutation of a highly conserved residue (p.Gly508Val) in LEPREL1 (18). A second study identified individuals with a homozygous loss-of-function mutation in LEPREL1 resulting in a premature termination codon after the fourth translated amino acid of P3H2 (17). These data suggest that loss of function of P3H2 in humans produces a highly selective and tissue-specific phenotype.
Myopia, a condition believed to result from an increase in the axial diameter of the eye, is the leading cause of visual impairment worldwide (19). High myopia is quantitatively defined as having an optical power of ≥ −6 diopters and an eye diameter of ≥26 mm (20). The cause of increased eye length is not fully understood but is believed to be associated with altered tissue properties in the eye. Indeed, the mechanical properties of scleral tissue were shown to be significantly altered in a myopic animal model (21). Sclera is a unique structural tissue that can regulate physiological eye distortions that occur during eye movement and intraocular pressure changes (22). In a myopic animal model, the scleral walls were observed to thin and weaken, which resulted in the inability to accommodate normal intraocular pressure fluctuations (23).
Several syndromic forms of high myopia exist, including Stickler syndrome, Marshall syndrome, and Weissenbacher-Zweymuller syndrome, which also present clinically with other connective tissue disorders and deafness. These conditions are all caused by mutations in Col2A1 and/or Col11A1. Interestingly, until quite recently very few cases of severe nonsyndromic myopias have been identified (24–27), and any pathobiological understanding behind the observed phenotypes is still unclear.
A recent report by Pokidysheva et al. (28) suggested that loss of P3h2 in mice may be associated with embryonic lethality. In this study, a new line of P3h2-null (P3h2n/n) mice was generated to address the apparent divergent observations between the Pokidysheva mouse model and the reported human cases of LEPREL1-associated high myopia (17, 18). The biochemical phenotype of the P3h2n/n mice from this study was characterized as a model to help study potential pathomechanisms leading to human high myopia. The P3h2n/n mice were viable and morphologically indistinguishable from wild-type littermates. Collagens were solubilized from the lens capsule, sclera, cornea, and vitreous and screened for prolyl 3-hydroxylation abnormalities. We hypothesize that loss of P3h2 results in altered collagen prolyl 3-hydroxylation, which cause structural abnormalities in eye tissues that drive early onset and progressive myopia.
MATERIALS AND METHODS
Production of Knock-out Mice
Embryonic stem cells were obtained from the European conditional mouse mutagenesis (EUCOMM) resource and injected in-house to generate the knock-out allele mice (P3h2+/−). Briefly, a floxed allele mouse (P3h2c/+) was generated by removing a lacZ expression cassette, using Rosa26-Flipase mice (Fig. 1A). The null allele mouse (P3h2n/+) was also generated by removing exon 3 using the CMV-Cre mouse line. Mice were maintained on a C57/BL6J background and were housed at the Baylor College of Medicine vivarium.
FIGURE 1.
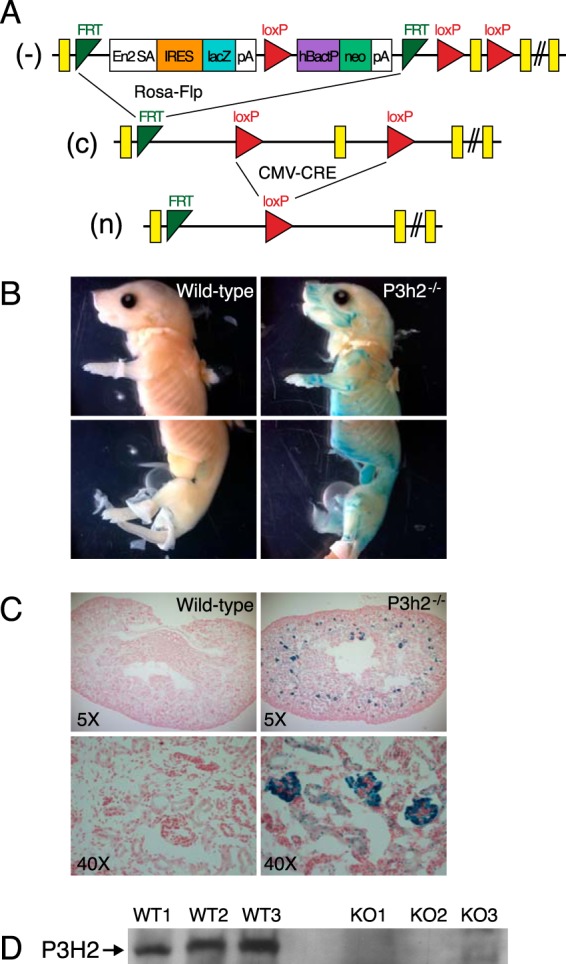
Generation and expression analysis of P3h2-null (P3h2n/n) mice. A, knock-out first allele mice (−, P3h2+/−) were generated using embryonic stem cells obtained from EUCOMM. Conditional wild-type mice (c, P3h2c/+) were generated by removing the lacZ expression cassette, using Rosa26-Flipase. Null mice (n, P3h2n/+) were generated by removing exon 3 using CMV-Cre. Mice were maintained on a C57/BL6J background. P3h2 expression was found throughout multiple tissues of the whole-mount mouse (B) but was localized to the glomeruli of kidney sections (C) as determined by β-galactosidase staining. D, immunoblot analysis using a polyclonal P3h2 antibody against total protein extracts from whole kidneys of wild-type (WT) and P3h2−/− (KO) mice supported the knock-out of protein expression in the P3h2n/n mice.
Genotyping was performed using primers flanking exon 3 for the wild-type allele (5′-CTTTTGACTCCCTCTCCCC-3′) and (5′-GCTTTACCTGCGATTGCCTC-3′). The mutant allele was identified by primers in the Bactin/neo cassette (5′-GCACAGAGCCTCGCCTTTG-3′) and (5′-TCATTCAGGGCACCGGACAG-3′) as well as primers specific for the last loxP site (5′-CGATACCACGATATCAACAAG-3′) and (5′-CCTGGGCAACTTAGTGGAG-3′).
Western Blot
Whole mouse kidneys were homogenized in RIPA buffer containing inhibitors (complete inhibitor with PMSF, Roche Applied Science). The supernatant was collected, and protein concentration was measured using a Bio-Rad Bradford assay kit. Total protein was resolved on a 4–15% gradient gel (Bio-Rad) and transblotted to a PVDF membrane. Membranes were blocked in 5% BSA, probed with Leprel1 polyclonal antibody (1:500 dilution, ProteinTech), and detected with an HRP-conjugated secondary antibody (1:5000, anti-rabbit). Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and exposed on autoradiography film.
Expression Analysis
The EUCOMM allele contains a splice acceptor with a β-galactosidase cassette (Fig. 1A), allowing for expression characterization of P3h2 by β-galactosidase staining. Homozygous mice and their wild-type littermates were collected after timed mating at P0. Samples were then fixed for 2 h in 4.0% paraformaldehyde. For whole-mount studies, the samples were then rinsed in PBS and stained in X-Gal buffer for 24 h (2 mm MgCl2, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 1 mg/ml X-Gal in 1× PBS). Samples were washed in PBS, fixed in formalin, rinsed, and then stored and imaged in 50% glycerol/PBS. For sectioned samples, fixed samples were rinsed in PBS, soaked in 30% sucrose overnight, and then embedded in OCT media. Frozen sections were collected at 12 μm thickness and stored at −80 °C. Slides were then fixed in 0.2% glutaraldehyde at room temperature for 10 min. Samples were washed three times for 5 min at room temperature in 2 mm MgCl2, 0.01% sodium deoxycholate, 0.02% Nonidet P-40, and PBS. Slides were stained overnight at 37 °C in staining solution (above) in the dark. Slides were rinsed with PBS and then with water, and counterstained with Nuclear Fast Red for 2 min. They were washed three times in water, dehydrated in ethanol, cleared in xylenes, and then mounted. Slides were imaged using a Zeiss Axioplan 2 microscope.
Collagen Extraction and Separation
Bovine tissues were purchased from Sierra for Medical Science (Whittier, CA). Bovine (18 months) and mouse (9 months) eye tissues were isolated using a dissecting microscope. Mouse eyes were prepared using a dissecting microscope. Cornea (transparent anterior region) and sclera (opaque posterior region) were easily distinguished using this approach. Vitreous (transparent inner gel) was collected upon the initial excision in the eye. The lens capsule collagen was directly isolated from the crystalline lens by pepsin digestion of the whole lens. Type I collagen was solubilized from tendon, sclera, and cornea by heat denaturation for 5 min at 100 °C in Laemmli buffer (SDS extraction), 3% acetic acid at 4 °C for 24 h, or cyanogen bromide digestion in 70% formic acid at room temperature for 24 h (5). Type II (vitreous) and type IV (lens capsule) collagen α-chains were solubilized by pepsin digestion or cyanogen bromide digestion as described previously (7). Collagen α-chains were resolved by SDS-PAGE and stained with Coomassie Blue R-250 (Sigma). Bovine sclera and cornea were isolated and collagenase-digested as described previously (29). Total collagenase digests were resolved into peptide fractions on a C8 column by reverse-phase HPLC and monitored at 220 nm.
Mass Spectrometry
Mass spectrometric analysis of 3Hyp content within collagen α-chains was performed as described previously (5, 30). Collagen α-chains were cut from SDS-polyacrylamide gels and subjected to in-gel trypsin digestion. Electrospray mass spectrometry was carried out on the tryptic peptides using an LTQ XL linear quadrupole ion-trap mass spectrometer equipped with in-line Accela 1250 liquid chromatography and automated sample injection (ThermoFisher Scientific). Proteome Discoverer software (ThermoFisher Scientific) was used for peptide identification. Tryptic peptides were also identified manually by calculating the possible MS/MS ions and matching these to the actual MS/MS spectrum using Thermo Xcalibur software. Hydroxyl differences were determined manually by averaging the full scan MS over several minutes to include all the post-translational variations of a given peptide. Protein sequences used for MS analysis were obtained from the Ensembl genome database. Mass spectral quantitation of 3Hyp residues was based on 16-Da mass increases at known sites of prolyl 3-hydroxylation at the Xaa position of Gly-Xaa-4Hyp sequences previously confirmed by Edman degradation (5, 28, 30, 31).
RESULTS
Phenotypic Characterization of P3h2n/n Mouse
Recent descriptions of two families presenting with recessively inherited nonsyndromic high myopia caused by LEPREL1 gene mutations (17, 18) prompted us to examine post-translational variations in eye tissues from the P3h2n/n mouse. The P3h2n/n mice were viable and indistinguishable from their wild-type littermates by gross physical appearance at birth (Fig. 1B). Micro-computed tomography analysis showed similar skeletal phenotypes for the P3h2n/n and control littermates (data not shown).
Expression Analysis of P3H2
The β-gal cassette in the knock-out mouse construct serves to replace the Leprel1 transcript with β-galactosidase in a targeted manner (Fig. 1A). Therefore, in the knock-out mice positive β-galactosidase staining will identify those tissues that normally express P3h2. Staining for β-galactosidase activity was performed in both whole-mount and sectioned kidney samples at P0 from knock-out mice using wild-type littermates that lack the β-galactosidase cassette as controls. In whole-mount animals, β-galactosidase staining for P3h2 was found in prehypertrophic regions of long bone throughout the whole animal (Fig. 1B). In kidney sections, P3h2 expression was localized to the glomeruli as determined by β-galactosidase staining (Fig. 1C). Immunoblot analysis using a polyclonal Leprel1 antibody against total renal protein extracts showed the absence of protein expression in the P3h2n/n mice (Fig. 1D). The expression pattern of P3h2 generated from this approach correlated with multiple previously reported expression analysis studies (15, 32, 33).
P3h2-positive β-galactosidase staining was localized to the cornea and sclera of 2-month-old mice (Fig. 2A). At higher magnification (×40), the β-galactosidase staining was observed to be specific to the outer basement membrane-lined epithelial layer of the cornea (Fig. 2B). No P3h2 expression was evident in the corneal stroma, which is primarily extracellular matrix-rich in types I and V collagens (Fig. 2B). At the posterior region of the eye, a weak staining signal appeared to be specific to the sclera, which also has primarily a type I collagen-based matrix (Fig. 2C).
FIGURE 2.
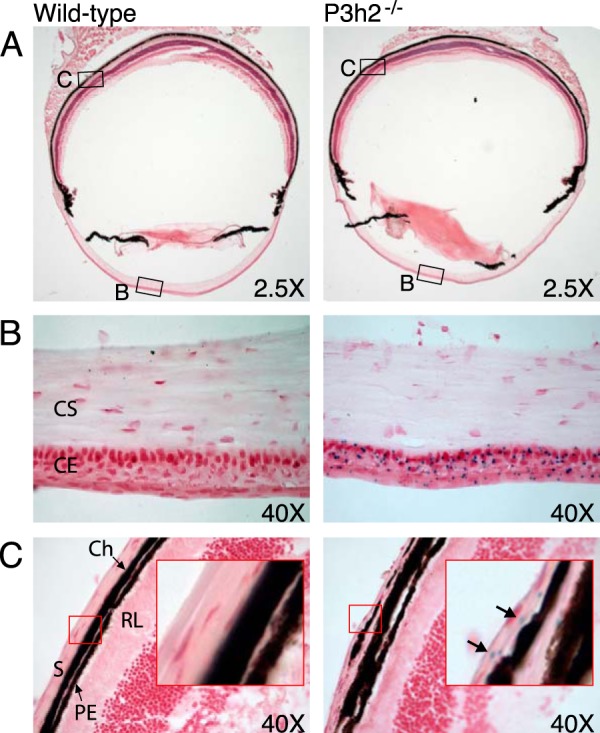
P3h2 expression in the mouse eye. A, β-galactosidase staining revealed P3h2 expression in the cornea (B) and sclera (C) of mouse eye sections. B, at the anterior of the eye, staining was localized to the corneal epithelium (CE) with no P3h2 expression observed in the corneal stroma (CS). C, at the posterior of the eye, staining was localized exclusively to the sclera (S). The red inset box displays a digitally magnified image of sclera. The black arrows indicate positive staining in sclera. The following abbreviations were used: Ch, choroid; PE, pigment epithelium; and RL, receptor layer.
Prolyl 3-Hydroxylation in Sclera
Sclera, lens capsule, and vitreous collagens were selected as collagen sources to analyze for potential P3h2 sites. Type I collagen was extracted from mouse scleral tissue by SDS extraction or pepsin extraction and digested in-gel with trypsin. Mass spectral analysis of peptides containing the wild-type mouse scleral α1(I)Pro-986 and α2(I)Pro-707 sites revealed prolyl 3-hydroxylation levels similar to those from mouse tail tendon (Table 1) (34). It has been previously shown, using genetically engineered P3h1-null mice, that both these hydroxylation sites are P3h1 substrates (34, 35). Indeed, the Pro-986 site was fully 3-hydroxylated in both wild-type and P3h2n/n mouse sclera (Fig. 3A). Hydroxylation of the α2(I)Pro-707 site dropped from 70 to 20% in the null mouse compared with wild type, suggesting that this site is a substrate for both P3h1 and P3h2 (Fig. 3B).
TABLE 1.
Summary of 3Hyp occupancy in collagens from bovine and mouse tissues
Percentage of 3Hyp at each major substrate site in type I collagen in tendon (T), sclera (S), and cornea (C); type II collagen in vitreous (V), and type IV collagen in lens capsule (LC) and kidney (K). The percentages were determined based on the ratio the m/z peaks of each post-translational variant as described previously (31). No notable differences were observed in type I collagen 3Hyp occupancy between mouse sclera and cornea (cornea not shown).

FIGURE 3.
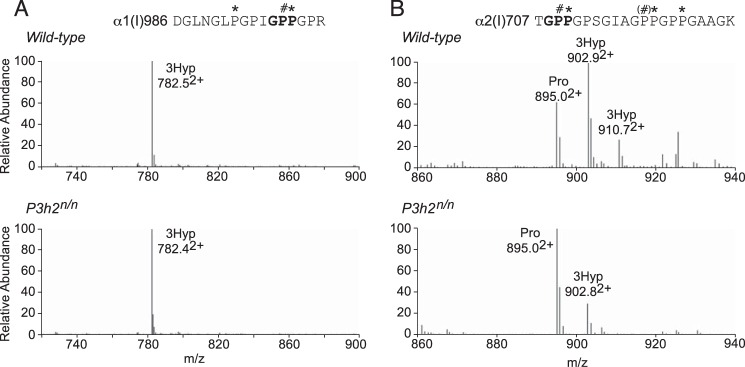
P3h1 substrate sites in type I collagen from mouse eye scleral tissue. LC-MS profiles of in-gel trypsin digests of the type I collagen α-chains from wild-type and P3h2n/n mouse sclera. A, MS profile of the α1-chain from the P3h2n/n mouse confirms no effect on Pro-986 3-hydroxylation (782.52+). B, MS spectrum from the α2-chain shows significant reduction of 3-hydroxylation at Pro-707 (895.02+) in the P3h2n/n mouse. A similar phenomenon was observed in type I collagen from cornea and tendon (data not shown). The trypsin-digested peptide is shown with P# indicating 3Hyp and P* indicating 4Hyp.
Interestingly, wild-type mouse sclera exhibited moderate levels of 3-hydroxylation at α1(I)Pro-707 (Fig. 4A), a tissue-specific prolyl 3-hydroxylation site that had previously been shown to be significantly hydroxylated only in type I collagen of tendon and bone but not skin (34, 36). Neither sclera nor tendon contained detectable levels of prolyl 3-hydroxylation at this site in P3h2n/n mice (Fig. 4B). Type I collagen also has additional tendon-specific prolyl 3-hydroxylation sites in the C-terminal (GPP)n sequence of the triple helix (30, 31). From the present findings it is clear that as in tendon, scleral type I collagen from wild-type mice exhibits this characteristic “hydroxylation ladder” (Fig. 5A). Prolyl 3-hydroxylation levels were similar in the (GPP)n site of tendon (70%) and sclera (65%) from the wild-type mouse (Fig. 5A and Table 1). The hydroxylation ladder was absent in collagens isolated from P3h2n/n mouse tissues (Fig. 5B), supporting previous cell culture knockdown assays that suggested the (GPP)n motif was a P3H2 substrate (16).
FIGURE 4.

Pro-707 site in α1(I) is a tissue-specific substrate unique to P3h2. LC-MS profiles of in-gel trypsin digests of the collagen α1(I) chain from tendon and sclera of wild-type and P3h2n/n mice. A, wild-type mouse tendon and sclera show 65 and 45% 3-hydroxylation at Pro-707 (929.02+), respectively; B, P3h2n/n mouse tendon and sclera show an almost complete loss of prolyl 3-hydroxylation at Pro-707 (922.02+). The trypsin digested peptide is shown with P# indicating 3Hyp and P* indicating 4Hyp.
FIGURE 5.

Post-translational similarities between sclera and tendon at the C-terminal (GPP)n motif from type I collagen. LC-MS profiles of in-gel trypsin digests of the collagen α1(I) chain from tendon and sclera of wild-type and P3h2n/n mice. A, MS profiles from mouse tendon and sclera reveal a similar hydroxylation ladder. B, complete loss of prolyl 3-hydroxylation is observed in the α1(I) - digested peptide is shown with P# indicating 3Hyp, P* indicating 4Hyp, and K* indicating Hyl at the C terminus where trypsin cleaves in the C-telopeptide.
Prolyl 3-Hydroxylation in Vitreous
The collagen of vitreous is a post-translational variant of type II collagen that has unusually high levels of prolyl 3-hydroxylation (5). Wild-type mouse vitreous type II collagen was found to have similarly high levels of 3Hyp at Pro-944 as shown previously in bovine vitreous (70% occupancy, data not shown). We tried but were unable to isolate sufficient type II collagen from P3h2n/n mouse vitreous for analysis. It is unclear at this point whether this was due to a lack of collagen deposition in the vitreal matrix.
Prolyl 3-Hydroxylation in Lens Capsule
Although it is well established that type IV collagen contains more prolyl 3-hydroxylation sites than any other collagen types (∼10 3Hyp per α-chain), it remains to be unequivocally determined which isoenzyme(s) are responsible. In vitro analysis with synthetic substrates using recombinant P3Hs has supported a role for P3H2 in collagen IV prolyl 3-hydroxylation (33). Collagen IV is found in many tissues in the eye, including lens capsule, epidermal and endodermal membranes of the cornea, and the inner limiting membrane and Bruch's membrane of the retina. Here, by using mass spectrometry, we identified peptides from two sites of prolyl 3-hydroxylation in type IV collagen from wild-type mouse lens capsule (Pro-602 from α1(IV) (Fig. 6, A and B) and Pro-197 from α2(IV) (mass spectral data not shown). Prolyl 3-hydroxylation was completely missing from the same peptides isolated from P3h2n/n mouse lens capsule (Fig. 6, C and D). These 3Hyp sites were initially identified in type IV collagen isolated from wild-type mouse kidney and, consistent with lens capsule, were also unhydroxylated in the α1(IV) and α2(IV) chains of P3h2n/n mouse kidney (data not shown).
FIGURE 6.

Prolyl 3-hydroxylation absent in collagen IV from lens capsule of P3h2n/n mice. LC-MS profiles of in-gel trypsin digests of the collagen α1(IV) chain from the lens capsule of wild-type and P3h2n/n mice. A, MS profile reveals a hydroxylation ladder containing partial prolyl 3-hydroxylation at Pro-602 and Pro-605. B, MS/MS fragmentation spectrum of the parent ion (1149.14+) from mouse lens capsule. The b and y ion breakages establish the added 16 Da on Pro-602. C, complete loss of prolyl 3-hydroxylation is observed at Pro-602 and Pro-605 in α1(IV) from P2h2n/n lens capsule. D, MS/MS fragmentation spectrum of the parent ion (1144.94+) from mouse lens capsule. The b and y ion breakages confirm the loss of 16 Da on Pro-602. The trypsin-digested peptide is shown with P# indicating 3Hyp, P* indicating 4Hyp, and galglc indicating glucosyl-galactosyl; M-1624+ indicates the parent ion with hexose loss after fragmentation.
Prolyl 3-Hydroxylation in Cornea
Potentially equally significant to the unique post-translational similarities between sclera and tendon type I collagens was a marked difference between type I collagens from sclera and cornea from 18-month-old steer (bovine) eyes. Bovine type I collagen from cornea and sclera differed in prolyl 3-hydroxylation of their C-terminal (GPP)n sequences (Fig. 7). Bovine scleral type I collagen gives the same characteristic C-terminal (GPP)n hydroxylation ladder as bovine tendon type I collagen (Fig. 7B) (30, 31), whereas type I collagen from bovine cornea has no C-terminal (GPP)n 3Hyp (Fig. 7A). This finding is consistent with the lack of P3h2 expression by mouse corneal stroma (Fig. 2B). In contrast, the level of wild-type mouse corneal type I collagen prolyl 3-hydroxylation was indistinguishable from that of wild-type mouse sclera (data not shown). Accordingly, these known P3h2 substrate sites completely lacked 3Hyp in P3h2n/n mouse corneal type I collagen (data not shown).
FIGURE 7.
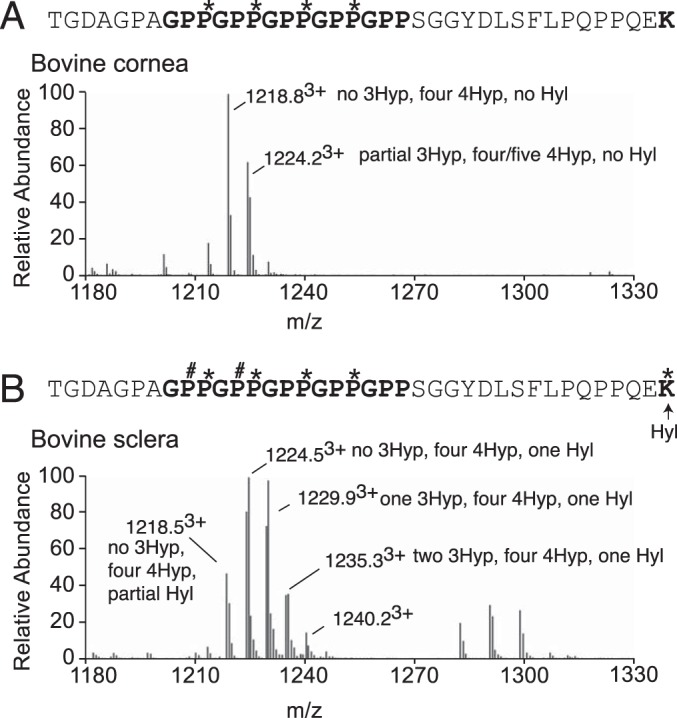
Tissue-specific hydroxylation patterns in type I collagen from bovine eye tissues. LC-MS profiles of in-gel trypsin digests of the α1(I) collagen chains from bovine sclera and cornea. A, MS profile of α1(I) from bovine cornea reveals no 3-hydroxylation at the (GPP)n. B, MS profile of the α1-chain from bovine scleral type I collagen reveals a similar hydroxylation to that observed in mouse tendon and sclera. The trypsin digested peptide is shown with P# indicating 3Hyp and P* indicating 4Hyp.
Another striking post-translational difference was observed between cornea and sclera in type I collagen cross-linking. The helical cross-linking site (Lys-87) in bovine cornea α1(I) and α2(I) was found to be completely hydroxylated and glycosylated as either glucosyl-galactosyl-hydroxylysine or galactosyl-hydroxylysine, whereas the same site from bovine sclera was found to be fully hydroxylated but not glycosylated (Fig. 8). This difference between cornea and sclera is also seen between skin and tendon, respectively. Both sclera and cornea collagens are known to be exclusively cross-linked by the lysine aldehyde pathway. Neither tissue contains detectable levels of pyridinoline cross-links (37), so it is noteworthy that in addition to a distinctive prolyl 3-hydroxylation profile, sclera and tendon also share a distinctive lack of glycosylation of the Lys-87 cross-linking site for the α1(I) C-telopeptide3 (38).
FIGURE 8.

Post-translational variances in cross-linking lysines between cornea and sclera type I collagen. A, profile of collagenase-digested whole tissue separated on C8 column (bovine sclera shown). B, LC-MS profile of C8 fraction 28 from bovine sclera reveals no glycosylation at cross-linking α1(I) Lys-87 (502.14+). C, MS/MS fragmentation spectrum of the parent ion (501.84+) from bovine sclera. The b and y ion breakages confirm no glycosylation on α1(I) Hyl87. D, LC-MS profile of C8 fraction 28 from bovine cornea reveals complete glycosylation of cross-linking α1(I) Hyl87 as glucosyl-galactosyl (galglc) (582.84+). E, MS/MS fragmentation spectrum of the parent ion (583.14+) from bovine cornea. The b and y ion breakages reveal gain of 340 Da (glucosyl-galactosyl) on α1(I) Hyl87. The trypsin-digested peptide is shown with K* indicating Hyl, K(*) indicating partial Hyl and galglc indicating glucosyl-galactosyl.
DISCUSSION
P3h2n/n mice and their wild-type littermates were indistinguishable by outward physical appearance. Furthermore, no discernible bone phenotype was observed in the null mice at birth or during later growth to 12 months by micro-computed tomography analysis. The lack of an obvious skeletal phenotype in these mice is consistent with the nonsyndromic high myopia caused by human LEPREL1 mutations (17, 18). Eye tissue collagens from P3h2n/n and wild-type mice were screened for known sites of prolyl 3-hydroxylation using tandem mass spectrometry. Previously identified novel sites of 3Hyp at Pro-707 and in the C-terminal (GPP)n of the type I collagen molecule, both peculiar to tendon type I collagen, were also found to be hydroxylated in normal scleral type I collagen. From P3h2n/n mice, these sites in α1(I) and α2(I) chains from tendon and scleral tissue had no 3Hyp. Isolated type IV collagen from lens capsule also showed missing 3Hyp in P3h2n/n compared with wild-type lens capsule collagen IV.
In a recent report, P3h2-null mice were created in which the neomycin selection cassette was retained (28). Embryos died by day 8.5, which was caused by abnormal maternal blood clotting. The mechanism proposed was clotting triggered by glycoprotein VI binding to 3Hyp-deficient type IV collagen of the embryos. The present P3h2n/n mice and an additional P3h2−/− hypomorphic strain4 showed no such pathological clotting during embryogenesis or in postnatal mice. One plausible explanation for the more severe phenotype reported by Pokidysheva et al. (28) is the differences in the two targeting constructs. Although there is no doubt it generates a loss of P3h2 as in our mice, it may also have long distance transcriptional inhibitory effects or a bystander gene effect due to the strong promoter inserted by the neomycin selection cassette. This is a well recognized phenomenon (39). Several earlier gene knock-outs have been reported to have such neighborhood gene effects from retained neomycin selection cassettes (40, 41). The reported rescue of the phenotype by crossing mouse lines to create a double P3h2/glycoprotein VI knock-out is consistent with a pathology driven by glycoprotein VI interacting with perhaps another gene product, not necessarily underhydroxylated type IV as proposed by Pokidysheva et al. (28). It is notable in the two mouse strains we studied, P3h2n/n and P3h2−/−, that 3Hyp was missing from all sites we could access in types I, II, and IV collagen with no apparent overactive clotting disorder.
A key question is how a deficiency in collagen prolyl 3-hydroxylation could lead to high myopia. The current findings suggest that a defective structure or turnover of collagen types I, II, and/or IV underlies the pathogenesis of the high myopia in the families previously reported to be caused by autosomal recessive mutations in P3H2. The mechanism for eye elongation is still unknown, but it may involve a disturbance in or remodeling of collagens within the sclera (collagen type I), vitreous body (collagen type II), or various basement membranes of the retina, cornea, and/or lens capsule (type IV collagen) (42) rather than simply a structural collagen defect affecting anatomy during development. Our P3h2n/n mouse model supports an effect on multiple basement membrane-containing tissues as we found complete loss of multiple 3Hyp sites in basement membrane type IV collagens from lens capsule, kidney, and aorta (data not shown). Unfortunately, sufficient levels of type II collagen could not be isolated from P3h2n/n mouse vitreous to confirm that 3Hyp is missing at P3h2-specific sites as appears likely. The uniquely high level of Pro-944 and C-terminal (GPP)n 3-hydroxylation of vitreous type II collagen compared with non-eye cartilage type II makes it a candidate source of an eye-specific pathology caused by a P3H2 mutation.
Mammalian myopia is associated with scleral thinning, tissue loss, and altered tissue morphology (22). Furthermore, matrix remodeling within the sclera has been demonstrated to contribute to the development of myopia in mammals and birds (43, 44). In the mammalian myopic eye, scleral thinning is greatest at the posterior pole of the eye, where the retinal photoreceptors are of the highest density (22). In this study, bovine scleral tissue was also found to have reduced collagen hydroxylation toward the posterior regions of the eye (data not shown). It is predicted that alterations in scleral matrix properties have a function in the pathobiology of myopia (21). For example, collagen and proteoglycan expression is reduced in the sclera of a myopic animal model (45), and collagen fibril ultrastructure is altered with an increased number of smaller diameter collagen fibrils (23). This loss of matrix was shown to result from reduced collagen and proteoglycan production as well as increased collagen degradation by matrix metalloproteinases (44, 45). Interestingly, only type I collagen (not type III or type V) has exhibited reduced mRNA expression levels in studies of myopic sclera (45).
No obvious function has yet emerged for the novel sites of 3Hyp occupancy that collagen types I, II, or IV from eye tissues share with various musculoskeletal tissues. As yet, the P3h2n/n mice have shown no musculoskeletal abnormalities. Likewise, in the description of the patients with high myopia caused by P3H2 recessive mutations, no skeletal or other abnormalities were noted, only severe progressive myopia (17, 18). We propose that the aberrantly modified collagens identified in this study could yield defective supramolecular structures that contribute to the pathogenic mechanism behind the high myopia in these families. It could be that the collagens are laid down normally, but the post-translational abnormality affects some aspect of subsequent turnover or tissue remodeling related to cross-linking or other higher order interaction of the fibril polymers. Abnormalities may then only appear during mature tissue remodeling in response to the mechanical stresses the mature eye experiences. Further studies are needed to test this concept.
Acknowledgment
We thank Geoffrey Traeger for help with figure preparation.
This work was supported, in whole or in part, by National Institutes of Health Grants AR37694 and AR37318 from NIAMS (to D. R. E.), HD22657 and HD070394 from NICHD (to B. H. L. and D. R. E.), AR063616 from NIAMS (to K. S. J.), P30 HD024064 from the Eunice Kennedy Shriver NICHD (to the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center), and AI036211, P30 CA125123, and RR024574 (to the Baylor College of Medicine Advanced Technology Cores). This work was also supported by the Ernest M. Burgess Endowed Chair research program of the University of Washington.
D. R. Eyre, unpublished data.
B. H. Lee and D. R. Eyre, unpublished observations.
- 3Hyp
- 3S,2S-l-hydroxyproline
- P3H
- prolyl 3-hydroxylase
- 4Hyp
- 4R,2S-l-hydroxyproline
- EUCOMM
- European conditional mouse mutagenesis.
REFERENCES
- 1. Berg R. A., Prockop D. J. (1973) The thermal transition of a non-hydroxylated form of collagen. Evidence for a role for hydroxyproline in stabilizing the triple-helix of collagen. Biochem. Biophys. Res. Commun. 52, 115–120 [DOI] [PubMed] [Google Scholar]
- 2. Ogle J. D., Arlinghaus R. B., Logan M. A. (1962) 3-Hydroxyproline, a new amino acid of collagen. J. Biol. Chem. 237, 3667–3673 [PubMed] [Google Scholar]
- 3. Hudson D. M., Eyre D. R. (2013) Collagen prolyl 3-hydroxylation: a major role for a minor post-translational modification? Connect. Tissue Res. 54, 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hudson D. M., Weis M., Eyre D. R. (2011) Insights on the evolution of prolyl 3-hydroxylation sites from comparative analysis of chicken and Xenopus fibrillar collagens. PLoS One 6, e19336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weis M. A., Hudson D. M., Kim L., Scott M., Wu J. J., Eyre D. R. (2010) Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J. Biol. Chem. 285, 2580–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dean D. C., Barr J. F., Freytag J. W., Hudson B. G. (1983) Isolation of type IV procollagen-like polypeptides from glomerular basement membrane. Characterization of pro-α1(IV). J. Biol. Chem. 258, 590–596 [PubMed] [Google Scholar]
- 7. Risteli J., Bächinger H. P., Engel J., Furthmayr H., Timpl R. (1980) 7-S collagen: characterization of an unusual basement membrane structure. Eur. J. Biochem. 108, 239–250 [DOI] [PubMed] [Google Scholar]
- 8. Morello R., Bertin T. K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F. H., Vranka J., Bächinger H. P., Pace J. M., Schwarze U., Byers P. H., Weis M., et al. (2006) CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127, 291–304 [DOI] [PubMed] [Google Scholar]
- 9. Barnes A. M., Chang W., Morello R., Cabral W. A., Weis M., Eyre D. R., Leikin S., Makareeva E., Kuznetsova N., Uveges T. E., Ashok A., Flor A. W., Mulvihill J. J., Wilson P. L., Sundaram U. T., et al. (2006) Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 355, 2757–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cabral W. A., Chang W., Barnes A. M., Weis M., Scott M. A., Leikin S., Makareeva E., Kuznetsova N. V., Rosenbaum K. N., Tifft C. J., Bulas D. I., Kozma C., Smith P. A., Eyre D. R., Marini J. C. (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 39, 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T. K., Pace J. M., Pepin M. G., Weis M., Eyre D. R., Walsh J., Lambert D., Green A., Robinson H., Michelson M., Houge G., et al. (2008) CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 29, 1435–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Dijk F. S., Nesbitt I. M., Zwikstra E. H., Nikkels P. G., Piersma S. R., Fratantoni S. A., Jimenez C. R., Huizer M., Morsman A. C., Cobben J. M., van Roij M. H., Elting M. W., Verbeke J. I., Wijnaendts L. C., Shaw N. J., et al. (2009) PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 85, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barnes A. M., Carter E. M., Cabral W. A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C. N., Eyre D. R., Raggio C. L., Marini J. C. (2010) Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 362, 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marini J. C., Cabral W. A., Barnes A. M. (2010) Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 339, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Järnum S., Kjellman C., Darabi A., Nilsson I., Edvardsen K., Aman P. (2004) LEPREL1, a novel ER and Golgi resident member of the Leprecan family. Biochem. Biophys. Res. Commun. 317, 342–351 [DOI] [PubMed] [Google Scholar]
- 16. Fernandes R. J., Farnand A. W., Traeger G. R., Weis M. A., Eyre D. R. (2011) A role for prolyl 3-hydroxylase 2 in post-translational modification of fibril-forming collagens. J. Biol. Chem. 286, 30662–30669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo H., Tong P., Peng Y., Wang T., Liu Y., Chen J., Li Y., Tian Q., Hu Y., Zheng Y., Xiao L., Xiong W., Pan Q., Hu Z., Xia K. (2014) Homozygous loss-of-function mutation of the LEPREL1 gene causes severe non-syndromic high myopia with early-onset cataract. Clin. Genet. 86, 575–579 [DOI] [PubMed] [Google Scholar]
- 18. Mordechai S., Gradstein L., Pasanen A., Ofir R., El Amour K., Levy J., Belfair N., Lifshitz T., Joshua S., Narkis G., Elbedour K., Myllyharju J., Birk O. S. (2011) High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am. J. Hum. Genet. 89, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hornbeak D. M., Young T. L. (2009) Myopia genetics: a review of current research and emerging trends. Curr. Opin. Ophthalmol. 20, 356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Young T. L., Ronan S. M., Drahozal L. A., Wildenberg S. C., Alvear A. B., Oetting W. S., Atwood L. D., Wilkin D. J., King R. A. (1998) Evidence that a locus for familial high myopia maps to chromosome 18p. Am. J. Hum. Genet. 63, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Phillips J. R., McBrien N. A. (1995) Form deprivation myopia: elastic properties of sclera. Ophthalmic Physiol. Opt. 15, 357–362 [PubMed] [Google Scholar]
- 22. McBrien N. A., Jobling A. I., Gentle A. (2009) Biomechanics of the sclera in myopia: extracellular and cellular factors. Optom. Vis. Sci. 86, E23–E30 [DOI] [PubMed] [Google Scholar]
- 23. McBrien N. A., Cornell L. M., Gentle A. (2001) Structural and ultrastructural changes to the sclera in a mammalian model of high myopia. Invest. Ophthalmol. Vis. Sci. 42, 2179–2187 [PubMed] [Google Scholar]
- 24. Guo H., Jin X., Zhu T., Wang T., Tong P., Tian L., Peng Y., Sun L., Wan A., Chen J., Liu Y., Li Y., Tian Q., Xia L., Zhang L., et al. (2014) SLC39A5 mutations interfering with the BMP/TGF-beta pathway in non-syndromic high myopia. J. Med. Genet. 51, 518–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tran-Viet K. N., Powell C., Barathi V. A., Klemm T., Maurer-Stroh S., Limviphuvadh V., Soler V., Ho C., Yanovitch T., Schneider G., Li Y. J., Nading E., Metlapally R., Saw S. M., Goh L., et al. (2013) Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am. J. Hum. Genet. 92, 820–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi Y., Li Y., Zhang D., Zhang H., Li Y., Lu F., Liu X., He F., Gong B., Cai L., Li R., Liao S., Ma S., Lin H., Cheng J., et al. (2011) Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 7, e1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aldahmesh M. A., Khan A. O., Alkuraya H., Adly N., Anazi S., Al-Saleh A. A., Mohamed J. Y., Hijazi H., Prabakaran S., Tacke M., Al-Khrashi A., Hashem M., Reinheckel T., Assiri A., Alkuraya F. S. (2013) Mutations in LRPAP1 are associated with severe myopia in humans. Am. J. Hum. Genet. 93, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pokidysheva E., Boudko S., Vranka J., Zientek K., Maddox K., Moser M., Fässler R., Ware J., Bächinger H. P. (2014) Biological role of prolyl 3-hydroxylation in type IV collagen. Proc. Natl. Acad. Sci. U.S.A. 111, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hanson D. A., Eyre D. R. (1996) Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone. J. Biol. Chem. 271, 26508–26516 [DOI] [PubMed] [Google Scholar]
- 30. Eyre D. R., Weis M., Hudson D. M., Wu J. J., Kim L. (2011) A novel 3-hydroxyproline (3Hyp)-rich motif marks the triple-helical C terminus of tendon type I collagen. J. Biol. Chem. 286, 7732–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hudson D. M., Werther R., Weis M., Wu J. J., Eyre D. R. (2014) Evolutionary origins of C-terminal (GPP)n 3-hydroxyproline formation in vertebrate tendon collagen. PLoS One 9, e93467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vranka J., Stadler H. S., Bächinger H. P. (2009) Expression of prolyl 3-hydroxylase genes in embryonic and adult mouse tissues. Cell Struct. Funct. 34, 97–104 [DOI] [PubMed] [Google Scholar]
- 33. Tiainen P., Pasanen A., Sormunen R., Myllyharju J. (2008) Characterization of recombinant human prolyl 3-hydroxylase isoenzyme 2, an enzyme modifying the basement membrane collagen IV. J. Biol. Chem. 283, 19432–19439 [DOI] [PubMed] [Google Scholar]
- 34. Pokidysheva E., Zientek K. D., Ishikawa Y., Mizuno K., Vranka J. A., Montgomery N. T., Keene D. R., Kawaguchi T., Okuyama K., Bächinger H. P. (2013) Posttranslational modifications in type I collagen from different tissues extracted from wild type and prolyl 3-hydroxylase 1 null mice. J. Biol. Chem. 288, 24742–24752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vranka J. A., Pokidysheva E., Hayashi L., Zientek K., Mizuno K., Ishikawa Y., Maddox K., Tufa S., Keene D. R., Klein R., Bächinger H. P. (2010) Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J. Biol. Chem. 285, 17253–17262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ishikawa Y., Vranka J. A., Boudko S. P., Pokidysheva E., Mizuno K., Zientek K., Keene D. R., Rashmir-Raven A. M., Nagata K., Winand N. J., Bächinger H. P. (2012) Mutation in cyclophilin B that causes hyperelastosis cutis in American Quarter Horse does not affect peptidylprolyl cis-trans isomerase activity but shows altered cyclophilin B-protein interactions and affects collagen folding. J. Biol. Chem. 287, 22253–22265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eyre D. (1987) Collagen cross-linking amino acids. Methods Enzymol. 144, 115–139 [DOI] [PubMed] [Google Scholar]
- 38. Henkel W., Rauterberg J., Stirtz T. (1976) Isolation of a crosslinked cyanogen-bromide peptide from insoluble rabbit collagen. Tissue differences in hydroxylation and glycosylation of the crosslink. Eur. J. Biochem. 69, 223–231 [DOI] [PubMed] [Google Scholar]
- 39. Olson E. N., Arnold H. H., Rigby P. W., Wold B. J. (1996) Know your neighbors: three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell 85, 1–4 [DOI] [PubMed] [Google Scholar]
- 40. Ren S. Y., Angrand P. O., Rijli F. M. (2002) Targeted insertion results in a rhombomere 2-specific Hoxa2 knockdown and ectopic activation of Hoxa1 expression. Dev. Dyn. 225, 305–315 [DOI] [PubMed] [Google Scholar]
- 41. Kaufman R. M., Lu Z. H., Behl R., Holt J. M., Ackers G. K., Ley T. J. (2001) Lack of neighborhood effects from a transcriptionally active phosphoglycerate kinase-neo cassette located between the murine β-major and β-minor globin genes. Blood 98, 65–73 [DOI] [PubMed] [Google Scholar]
- 42. Halfter W., Winzen U., Bishop P. N., Eller A. (2006) Regulation of eye size by the retinal basement membrane and vitreous body. Invest. Ophthalmol. Vis. Sci. 47, 3586–3594 [DOI] [PubMed] [Google Scholar]
- 43. McBrien N. A., Lawlor P., Gentle A. (2000) Scleral remodeling during the development of and recovery from axial myopia in the tree shrew. Invest. Ophthalmol. Vis. Sci. 41, 3713–3719 [PubMed] [Google Scholar]
- 44. Liu H. H., Gentle A., Jobling A. I., McBrien N. A. (2010) Inhibition of matrix metalloproteinase activity in the chick sclera and its effect on myopia development. Invest. Ophthalmol. Vis. Sci. 51, 2865–2871 [DOI] [PubMed] [Google Scholar]
- 45. Gentle A., Liu Y., Martin J. E., Conti G. L., McBrien N. A. (2003) Collagen gene expression and the altered accumulation of scleral collagen during the development of high myopia. J. Biol. Chem. 278, 16587–16594 [DOI] [PubMed] [Google Scholar]