

Abstract
As a result of a whole-exome sequencing study, we report three mutant alleles in SEC24D, a gene encoding a component of the COPII complex involved in protein export from the ER: the truncating mutation c.613C>T (p.Gln205∗) and the missense mutations c.3044C>T (p.Ser1015Phe, located in a cargo-binding pocket) and c.2933A>C (p.Gln978Pro, located in the gelsolin-like domain). Three individuals from two families affected by a similar skeletal phenotype were each compound heterozygous for two of these mutant alleles, with c.3044C>T being embedded in a 14 Mb founder haplotype shared by all three. The affected individuals were a 7-year-old boy with a phenotype most closely resembling Cole-Carpenter syndrome and two fetuses initially suspected to have a severe type of osteogenesis imperfecta. All three displayed a severely disturbed ossification of the skull and multiple fractures with prenatal onset. The 7-year-old boy had short stature and craniofacial malformations including macrocephaly, midface hypoplasia, micrognathia, frontal bossing, and down-slanting palpebral fissures. Electron and immunofluorescence microscopy of skin fibroblasts of this individual revealed that ER export of procollagen was inefficient and that ER tubules were dilated, faithfully reproducing the cellular phenotype of individuals with cranio-lentico-sutural dysplasia (CLSD). CLSD is caused by SEC23A mutations and displays a largely overlapping craniofacial phenotype, but it is not characterized by generalized bone fragility and presented with cataracts in the original family described. The cellular and morphological phenotypes we report are in concordance with the phenotypes described for the Sec24d-deficient fish mutants vbi (medaka) and bulldog (zebrafish).
Main Text
Osteogenesis imperfecta (OI, types I to XV [MIM 166200, 166210, 259420, 166220, 610967, 613982, 610682, 610915, 259440, 613848, 610968, 613849, 614856, 615066, and 615220]) is a heterogeneous group of disorders associated with reduced bone mass, increased bone fragility, bone deformity, and growth deficiency. Up to 90% of individuals with a classical OI phenotype have heterozygosity for mutations in the genes COL1A1 (MIM 120150) or COL1A2 (MIM 120160).1 These genes encode for the two pro-alpha chains of type I collagen, which cotranslationally translocate into the lumen of the endoplasmic reticulum (ER). Within the ER, several molecular chaperons and enzymes are required for post-translational procollagen folding and modification.2,3 The majority of the 11 genes in which mutations have been identified in autosomal-recessive types of OI encode for proteins with a role in these processes.4–6 Procollagen export from the ER occurs via membrane-bound vesicles or carriers that are generated by a set of cytoplasmic coat proteins called the COPII complex.7,8
Several rare syndromes have been described that clinically overlap with OI but display additional symptoms. Two well-known examples are Bruck syndrome (MIM 259450 and 609220), previously known as “OI with congenital joint contractures,”9 and osteoporosis-pseudoglioma syndrome (MIM 603506), first described as “ocular form of OI.”10,11 In 1986, Cole and Carpenter described two infants with bone deformities and multiple fractures reminiscent of OI who also had ocular proptosis with orbital craniosynostosis, hydrocephalus, and distinctive facial features (MIM 112240).12 A biochemical collagen analysis was performed in one of these infants and showed normal results. Subsequently, four additional individuals with Cole-Carpenter syndrome have been described in the literature.13–16 The molecular cause and mode of inheritance are unknown.
At the Tübingen University Hospital, we have treated a boy with a syndromic form of OI that we clinically classified as Cole-Carpenter syndrome, based on the history of multiple pre- and postnatal fractures and the presence of distinct craniofacial malformations. The detailed clinical history of this individual is summarized in the Supplemental Data. When he was 7 years old, we recruited the boy and his unaffected parents (family 1) for a research project on OI to identify the underlying molecular mechanism. The study was approved by the Ethics Committee of the LMU Munich, and written informed consent was obtained from the affected individuals’ parents for molecular genetics studies. At this age, the boy had moderately reduced bone mineral density (−2.0 SD in a DXA whole-body measurement, Table S1). He had been treated with a total of 23 cycles of intravenous bisphophonates. He displayed macrocephaly (head circumference > 97th percentile) with postnatal onset and short stature, and had normal mobility, intelligence, and hearing. His facial features were highly reminiscent of Cole-Carpenter syndrome (Figures 1A and 1B; see Figure S1 for comparison of the facial features with those of the individuals originally described by Cole and Carpenter). The most salient clinical symptom was a large fronto-parietal apical ossification defect of the skull (Figure 1C), which made it necessary for the child to wear a helmet during physical activities. Cranial CT scans that had been taken at the ages of 1 year and 4 years also documented dilated ventricles, multiple wormian bones (Figure 1D), and narrow coronal, lambda, and temporal sutures with a premature ossification pattern. Upon recruitment to our research project, the boy was assessed in the specialized unit for OI at the Children’s Hospital of the University of Cologne by the in-house standard protocol. The clinical findings are summarized in Table S1. Mutations in the genes COL1A1 and COL1A2 as well as in eight other genes associated with OI or increased bone fragility were excluded by Sanger sequencing of genomic DNA (see Table S2 for details). Bone mineral density of the parents measured by DXA and peripheral quantitative computertomography was in the normal age-adjusted range.
Figure 1.
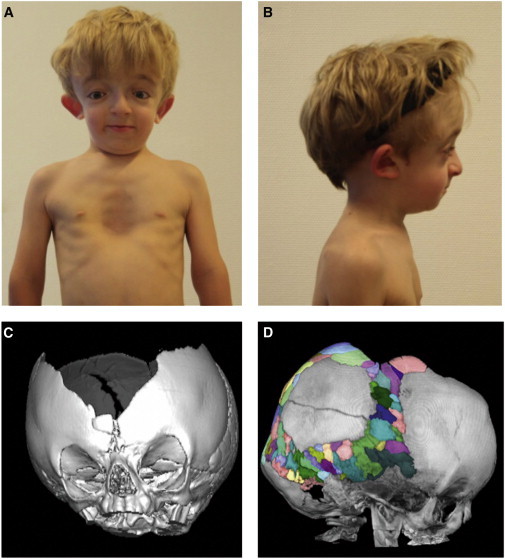
Photographs and CT Scans of the Affected Individual from Family 1
(A) Affected individual from family 1 at the age of 7 years. Note facial dysmorphism with down-slanting palpebral fissures, hypertelorism, dysplastic right ear, and pectus excavatus.
(B) Lateral view. Note mid-face hypoplasia, micrognathia, frontal bossing, and dysplastic right ear.
(C) Three-dimensional CT scan of the cranium at the age of 14 months. Note wide sagittal suture with a broad ossification defect fronto-parietal apical. Sutura metopica is displaced to the bottom, as indicated by surrounding wormian bones. Coronal sutures, lambda sutures, and temporal sutures appear narrow and with premature ossification pattern without clear synostosis, but with multiple wormian bones enclosed.
(D) Lateral view of a cranial CT scan taken at age 4 years and 4 months, with multiple wormian bones highlighted in different colors. Note an intraparietal suture on the left side (described as unilateral intraparietal suture30). The large ossification defect fronto-parietal apical persisted. The sphenoid wings appear with an increased density and with multiple erosions.
We performed whole-exome sequencing (WES) of the affected boy and his unaffected parents. Exonic and adjacent intronic regions were enriched from genomic DNA derived from peripheral blood via the 50 MB SureSelect All Exon V4 target enrichment kit from Agilent Technologies, and paired-end sequencing was performed on the Illumina platform (Genome Analyzer IIx or HiSeq2000). The mean on-target coverage was 52 for the affected individual from family 1 and for his father, and 54 for his mother. The percentage of the WES target covered by at least 10 reads was 87% for the affected individual and for his mother and 86% for his father. Alignment against the GRCh37 human reference genome was performed with a Burrows-Wheeler Aligner (BWA, v.0.6.2), PCR-duplicates marking and removal with Picard (v.1.84), indel realignment, base quality recalibration, and variant calling with the Genome Analysis Toolkit (GATK, v.2.3-4), and annotation with Annovar (v.2013_Feb21). The resulting variants were filtered to exclude the following: (1) variants with a MAF > 0.001 in dbSNP, in the Exome Variant Server, the 1000 Genomes Project, or in our in-house database and (2) variants that were not predicted to affect protein sequence or exon splicing.
Filtering the WES data for deleterious variants under an autosomal-recessive disease model provided potentially causative variants in five candidate genes (Table S3). Among these, SEC24D (MIM 607186; RefSeq accession number NM_014822.2) was a perfect candidate for several reasons. First, SEC24D is a component of the COPII complex involved in protein export from the ER. The COPII complex is responsible for ER export of procollagen, among many other secretory proteins.8,17 Second, mutations in SEC23A (MIM 610511), encoding a binding partner of SEC24D in the COPII complex, cause the autosomal-recessive disorder cranio-lenticulo-sutural dysplasia (CLSD, also known as Boyadjiev-Jabs syndrome [MIM 607812]).18–20 CLSD is characterized by facial dysmorphism, cataracts, and skeletal defects. Intriguingly, large skull ossification defects as observed in the affected individual from family 1 are one of the hallmarks in CLSD, and moreover, one of the seven individuals with CLSD described in the literature displayed mild osteopenia as an additional symptom.20 Third, the medaka mutant vbi, caused by a sec24d nonsense mutation, is characterized by short body length, OI, and craniofacial malformations—including an impaired ossification of the neurocranium—due to defective collagen secretion into the extracellular matrix.21 Thus, this animal model recapitulates in detail the phenotype of the affected individual from family 1 (of note, Sec24d-null mice are embryonic lethal prior to skeletal development22).
Sanger sequencing confirmed compound heterozygosity for a SEC24D nonsense mutation (c.613C>T [p.Gln205∗]) and for a missense mutation (c.3044C>T [p.Ser1015Phe]) in the affected individual from family 1 (Figure 2). The missense mutation affects a highly conserved amino acid (Figure S2A), is absent on ∼13,000 chromosomes from the Exome Variant Server (EVS), is not annotated in the 1000 Genomes project, is not listed in the ExAc Browser of the Exome Aggregation Consortium, and is predicted to be deleterious by the algorithms MutationTaster, PolyPhen-2, and SIFT. The parents were confirmed to be heterozygous for the nonsense mutation (father) and for the missense mutation (mother). In the non-affected sibling of the affected boy, compound heterozygosity was excluded. Finding compound-heterozygous mutations rather than a homozygous disease-causing mutation fitted the fact that the father of the affected individual was of Greek and his mother of Southern German descent.
Figure 2.
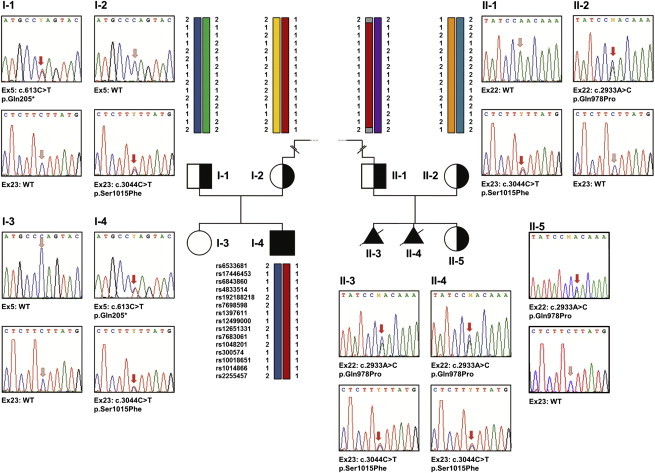
Validation of the SEC24D Mutations by Sanger Sequencing of Genomic DNA
Pedigrees of the two analyzed families and electropherograms of the respective SEC24D mutations in exons 5, 22, and 23 are shown. In family 1, each parent carries one of the two mutations and the affected son (I-4) is compound heterozygous for the SEC24D mutations c.613C>T (p.Gln205∗) and c.3044C>T (p.Ser1015Phe). The healthy sister of the affected individual did not carry either of the two mutations. Also in family 2, each parent is heterozygous carrier of one of the two SEC24D mutations and both affected fetuses (II-3 and II-4) are compound heterozygous for c.2933A>C (p.Gln978Pro) and c.3044C>T (p.Ser1015Phe). The healthy sister (II-5) is a heterozygous carrier of only one of these mutations. The red arrows in the electropherograms indicate either the mutation or the respective position in the wild-type sequence. In addition, for several family members the haplotype combination at the SEC24D locus is shown, and 15 of the 166 consecutive good-quality SNPs used for reconstructing the 14 Mb disease haplotype are depicted. Note that individuals I-2, I-4, and II-1 share the SEC24D mutation c.3044C>T and an identical haplotype between the SNPs rs6533681 and rs2255457. The dashed line between I-2 and II-2 indicates that the mutation c.3044C>T is a founder mutation inherited from a distant common ancestor.
At the time we recruited family 1, we also offered WES as part of the same OI research project to a healthy German couple who had chosen to terminate two pregnancies with female fetuses suspected to be affected by a severe type of OI. Written informed consent for molecular genetics studies was obtained from both individuals. The couple (family 2) was non-consanguineous and lived in the same region in southwestern Germany as family 1. Mutations in the genes COL1A1 and COL1A2 and in nine other genes associated with OI or increased bone fragility were excluded by Sanger sequencing of genomic DNA of one of the fetuses (see Table S2 for details). Because there was not enough high-quality fetal DNA available for WES analyses in family 2, we performed WES on genomic DNA from both parents and searched for deleterious heterozygous variants in the same gene in both parental WES datasets, assuming an autosomal-recessive mode of inheritance of the phenotype. The mean on-target coverage was 47 and 49, and the percentage of the WES target covered by at least 10 reads was 93% and 94% for the mother and for the father, respectively.
Filtering the WES datasets of family 2 (mother and father of the affected fetuses) for heterozygous deleterious mutations in the same gene resulted in a list of 17 candidate genes (Table S4). Unexpectedly, SEC24D was among these candidates, with the father being heterozygous for the same missense mutation c.3044C>T (p.Ser1015Phe) we had identified in family 1. The mother was heterozygous for the SEC24D missense mutation c.2933A>C (p.Gln978Pro). This mutation also affects a highly conserved amino acid (Figure S2B), is absent on ∼13,000 chromosomes from the Exome Variant Server (EVS), is not annotated in the 1000 Genomes project, is not listed in the ExAc Browser of the Exome Aggregation Consortium, and is predicted to be deleterious by the algorithms MutationTaster, PolyPhen-2, and SIFT. Only one other gene appeared on the candidate gene lists for both families 1 and 2: LMO7 (MIM 604362).
Sanger sequencing revealed that the affected fetuses had discordant LMO7 genotypes, excluding this gene from the list of candidates. Further analyses by Sanger sequencing confirmed heterozygosity for the SEC24D mutations in the parents and, most importantly, demonstrated that both affected fetuses were compound heterozygous for the SEC24D mutations c.3044C>T (p.Ser1015Phe) and c.2933A>C (p.Gln978Pro). The healthy 10-year-old sister of the fetuses was a heterozygous carrier of only one of the two mutations (Figure 2). Because the mother of the affected boy of family 1 and the father of the fetuses of family 2 both descended from families living in southwestern Germany (central region of Baden-Württemberg), we hypothesized that the mutation c.3044C>T (p.Ser1015Phe) might be a founder mutation. To test this hypothesis, we used SNP genotypes from the WES datasets to reconstruct the haplotypes in which the mutations were embedded. We identified a haplotype extending for 14 Mb, supported by 166 consecutive good-quality SNPs between rs6533681 and rs2255457 (chr4: 114,309,589–128,554,154), which was shared by the affected individual from family 1, by his mother, and by the father of the affected fetuses from family 2 (Figure 2). Hence, this haplotype analysis clearly demonstrated that the mutation c.3044C>T (p.Ser1015Phe) is indeed a founder mutation, strongly suggesting that it is disease causing in both families.
We collected more detailed clinical information on the affected fetuses. Both had—in addition to multiple fractures of the long bones and mildly bent extremities—a thin, poorly ossified skull upon ultrasound analysis in the 23rd and 20th week of gestation. In one of the fetuses, an autopsy was performed and X-rays of the entire body were taken (Figure S4). These analyses revealed that the extremities, the vertebrae, the base of the skull, and the facial bones were almost regularly ossified, whereas the ossification of the calvarium was strongly reduced and largely absent. The ribs were thin, and single fractures were noted, but the thorax was not severely hypoplastic, suggesting that the skeletal defects were non-lethal. Taken together, these two fetuses not only shared one of the SEC24D mutations with the affected individual from family 1, but also the highly unusual clinical finding of a severe ossification defect of the calvarium contrasted by a normal ossification of the skull base and only mildly disturbed ossification and shape of the rest of the skeleton. We concluded that all three affected individuals from these two families had the same skeletal disorder, and that the substitutions p.Gln205∗, p.Ser1015Phe (resulting from the founder mutation), and p.Gln978Pro are disease causing.
To elucidate the underlying cellular pathogenesis, we analyzed cultured skin fibroblasts from family 1 (affected individual and both parents). Functional studies in individuals with the SEC23A-associated disorder CLSD had noted inefficient ER export of type I procollagen.18,20 Thus, we expected that the SEC24D mutations block ER export of type I procollagen, resulting in accumulation of collagen in the ER in the cells of the affected individual. To test whether the ER export of procollagen is altered, we monitored the intracellular distribution of procollagen by employing immunofluorescent labeling with antibodies for a type I procollagen, COL1A1, and the ER marker PDI. The cells of the affected individual retained more procollagen in the ER than the control fibroblasts with significant statistical difference (Figure 3). These data indicate that ER export of procollagen is compromised in the fibroblasts of the affected individual. However, conventional immunoblotting did not yield conclusive results on cellular accumulation of procollagen (data not shown), suggesting that there is no gross accumulation of procollagen in the fibroblasts of the affected individual. Thus, we conclude that ER export of procollagen is mildly defective in these fibroblasts.
Figure 3.
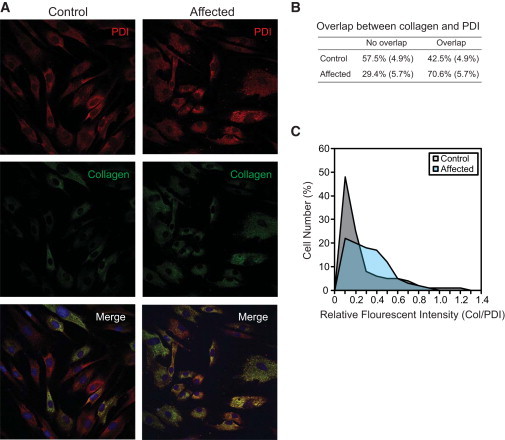
Retention of Procollagen in the ER
(A) Double immunofluorescent labeling of control and the affected individual’s fibroblasts for type I procollagen and protein disulfide isomerase (PDI), an ER marker. Images were obtained by standard confocal microscopy.
(B) The number of cells with an indicated overlap of procollagen with PDI was shown. Standard deviations from three independent experiments are shown in parentheses.
(C) ImageJ was used to normalize fluorescent intensities of procollagen with those of PDI from the images obtained via confocal microscopy. Procollagen is retained more in the fibroblasts of the affected individual (light blue) than in the control fibroblasts (gray) with statistical significance (Student’s t test, p = 0.0001, n = 467 cells for control fibroblast, n = 554 cells for the affected individual’s fibroblasts).
Depletion of COPII components causes ER retention of cargo molecules, leading to dilation of ER cisternae. To monitor an ER export defect, we measured the thickness of ER cisternae via electron micrographs in a blinded analysis setting (Figure 4). Because ER tubules range from about 100 to 150 nm in thickness in the vast majority of cells, those that are thicker than 150 nm were defined as distended (Figure 4E). Based on this criterion, about 18% of control, 22% of maternal, 13% of paternal, and 63% of the ER tubules of the affected individual were dilated. A statistical analysis confirmed that the ER tubules of the cells from the affected individual were significantly distended, compared to the control and parental cells (see legend to Figure 4). These results clearly support the notion that ER export is defective in the fibroblasts of the affected individual.
Figure 4.
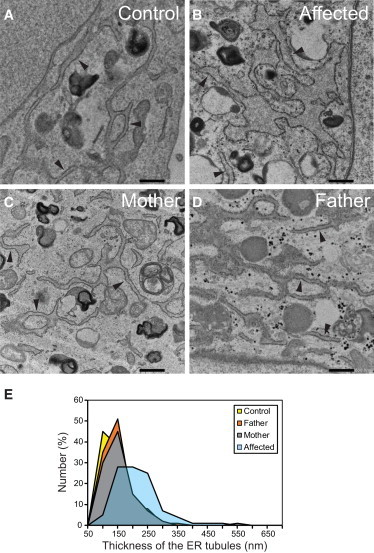
Dilation of ER in the Fibroblasts of the Affected Individual from Family 1
(A–D) Micrographs of thin-section electron microscopy for fibroblasts from control (A), affected individual (B), mother of affected individual (C), and father of affected individual (D). Tubular ER elements studded with ribosomes are indicated by arrowheads. Scale bars represent 500 nm.
(E) Summary of the thickness of ER tubules in fibroblasts. 100 nm in the x axis represents a thickness that is larger than 50 nm but equal to or less than 100 nm. The average thickness of the tubules is about 189 nm, 120 nm, 116 nm, and 125 nm for the cells from the affected individual (light blue), control (yellow), father (orange), and mother (gray), respectively. The ER tubules of the fibroblasts from the affected individual were thicker than those of other fibroblasts with statistical significance according to a one-way ANOVA with Tukey’s honest significant difference test (p < 0.01; n = 183 tubules for control, 212 for father, 171 for mother, and 181 for the affected individual).
COPII-coated vesicles, which play a critical role in exporting the majority of proteins from the ER, are composed of three proteins complexes: SAR1, SEC23/SEC24, and SEC13/SEC31.17 These COPII proteins deform the lipid bilayer into buds at ER exit sites, load cargo molecules into the nascent buds, and complete vesicle scission. There exist multiple paralogs for each COPII gene except SEC13 in vertebrates. In particular, there are four SEC24 paralogs (SEC24A, SEC24B, SEC24C, and SEC24D). SEC24 is mainly responsible for sorting cargo molecules through direct or indirect interactions during COPII vesicle assembly. Defects in SEC24, therefore, will cause inefficient loading of cargo molecules into COPII vesicles, resulting in accumulation of cargo molecules in the ER. Each SEC24 possesses multiple cargo-binding sites,23 which increases the repertoire of cargo molecules packaged into COPII vesicles.
We analyzed the structure of SEC24D to obtain further insights into the functional effects of the identified mutations (Figure 5). The SEC24D Ser1015 residue substituted in families 1 and 2 is highly conserved in SEC24s (Figure S2A). It is located near the IxM pocket of SEC24D.23 This pocket is responsible for recognizing the IxM ER export signal. We expect that the p.Ser1015Phe substitution disrupts the IxM pocket and interferes with binding to the IxM signal. Immunoblot analysis of SEC24D in family 1 showed that the levels of SEC24D were reduced in the fibroblasts of the affected individual as well as in the paternal fibroblasts (Figure 5C). Probably, these reductions are caused by haploinsufficiency of the p.Gln205∗ allele. The SEC24D Gln978 residue substituted in family 2 is located in an alpha helix of the gelsolin-like domain of SEC24D. Intriguingly, Gln978 is found in the structurally equivalent region where the CLSD-causing SEC23A substitutions p.Met702Val and p.Phe382Leu are positioned (Figure 5D). Previous studies have established that Met702 and Phe382 of SEC23A constitute a portion of the SEC31-binding groove.24,25 Although SEC23 is mainly responsible for the interaction between the SEC23-SEC24 complex and SEC31, it is possible that the region including Gln978 of SEC24D might also contribute to SEC31 binding. Alternatively, the p.Gln978Pro substitution might simply destabilize SEC24D, resulting in a reduction of the levels of SEC24D. Unfortunately, skin fibroblasts from family 2 are not available, and therefore we currently cannot test these hypotheses.
Figure 5.
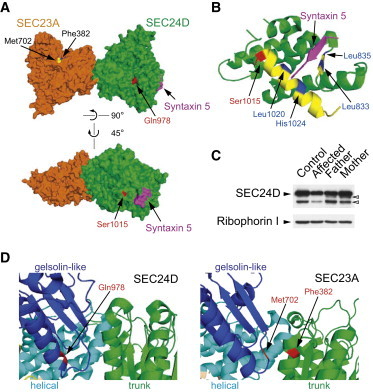
Position of the Mutant Residues in the 3D Structure of SEC24D
Note that the available human SEC24D structures were solved with the isoform 2 (O94855-2) (1,033 aa). Here we used the nomenclature of the isoform 1 (O94855-1) (1,032 aa) of SEC24D because this form has been chosen as the canonical sequence in the databases. PDB 3EFO structure was used.23
(A) Gln978 (red, upper panel) is located in the gelsolin-like domain of SEC24D. The location of Gln978 SEC24D is structurally similar to that of Met702 (yellow) and Phe382 (blue) of SEC23A. A more detailed structural comparison was shown in (D). Ser1015 (red, lower panel) is found in the IxM pocket (I, x, and M stand for Ile, any amino acid, and Met). A fragment of syntaxin 5 (magenta) contains an IxM signal. A more detailed structure is shown in (B).
(B) The helical domain of SEC24D is shown. Ser1015 (red) is located in an α-helix (yellow) that contains key residues (Leu1020 and His1024, blue) of the IxM pocket. Additional key residues (Leu833 and Leu835, blue) of this pocket are located in a β-strand (yellow).
(C) Total lysates (10 μg) of indicated fibroblasts derived from family 1 were prepared, resolved by SDS-PAGE, and processed for immunoblotting. Control foreskin fibroblasts were obtained from the American Type Culture Collection (ATCC, CRL-091). Ribophorin 1 (an ER-resident protein) was probed as loading controls. Bands (open arrowheads) below the main SEC24D band probably represent partial degradation products of SEC24D or nonspecific proteins.
(D) The gelsolin-like, the helical, and the trunk domains of SEC24D (left) and SEC23A (right) were colored blue, cyan, and green, respectively. CLSD-linked mutations were colored red. Met702 and Phe382 of SEC23A constitute a part of the SEC31-binding pocket.24,25
Summing up the results of the cellular studies, we have shown that ER export of procollagen was inefficient in the skin fibroblasts obtained from the affected individual of family 1, and ER tubules were dilated, faithfully reproducing the cellular phenotypes of individuals with CLSD resulting from SEC23A mutations.18,20,24,25 These results are also in concordance with the cellular phenotype observed in the sec24d-deficient medaka fish vbi21 and zebrafish bulldog.26
The similarities between SEC23A and SEC24D mutant fibroblasts prompted us to ask whether the diagnosis of Cole-Carpenter syndrome is the best clinical classification for the phenotype we report. As mentioned above, the affected individual from family 1 also shared many of the key symptoms described for CLSD (but has no detectable SEC23A mutation), and the facial phenotype was very similar. However, a cataract, one of the leading symptoms of CLSD, which presented within the first years of life in the original family described,19 was missing in the affected individual we report. Upon thorough ophthalmologic examination, there was also no evidence of a double-ring sign of the lens, as described in an individual with a single heterozygous SEC23A missense mutation (i.e., without a detectable second mutant allele) who had craniofacial, skeletal, and cellular features characteristic of CLSD and who in addition had osteopenia and macrocephaly.20 Interestingly, this individual with a heterozygous SEC23A mutation (and no detectable SEC24D mutation, data not shown) also had esotropia and optic nerve hypoplasia, and the affected individual from family 1 had strabism and small optic discs of normal color. Nevertheless, increased bone fragility with prenatal onset, one of the clinical hallmarks of all three affected individuals we report, has not been described for individuals with SEC23A mutations. We therefore regard the phenotype we report as a clinical entity that can be distinguished from the phenotype of the individuals with CLSD described in the literature, although the symptoms clearly overlap.
We reviewed all five published case reports of Cole-Carpenter syndrome12–16 and compared the clinical and radiological findings of the six described individuals in detail with the affected individual from family 1 (Table S5). Information on skull ossification defects—one of the most obvious symptoms in the affected individuals we describe—are incomplete or missing in some of the reports, but overall, the similarities are striking, and consequently, we still consider Cole-Carpenter syndrome to be the best classification for the phenotype. However, we were unable to obtain DNA samples from the originally reported individuals to genetically substantiate this classification, and Sanger sequencing failed to identify SEC24D mutations in the individual with Cole-Carpenter syndrome described by Amor et al.13 (Figure S1C; note that we also did not detect any SEC23A mutation in this individual). This suggests that there is further genetic heterogeneity for Cole-Carpenter syndrome. In any case, there is obviously a spectrum of clinically overlapping skeletal disorders that ranges from CLSD to Cole-Carpenter syndrome and might also include other diseases. Further genetic, clinical, and functional studies are needed to comprehensively describe the phenotypic spectrum of these syndromes and to elucidate the molecular links between them. Because generalized bone fragility, apparently due to a defect in intracellular collagen transport, was one of the leading symptoms in the individuals with SEC24D mutations described here, we consider it to be reasonable to place this type of Cole-Carpenter syndrome into the osteogenesis imperfecta and decreased bone density group, as suggested by the revised Nosology and Classification of Genetic Skeletal Disorders.27
In conclusion, we report three mutant SEC24D alleles, one of them being a founder mutation shared by all three affected individuals from two families and another one leading to a premature stop codon, in a rare autosomal-recessively inherited skeletal disorder characterized by pre- and postnatal bone fragility, skull ossification defects, craniofacial dysmorphism, and short stature. The causal role of SEC24D mutations is strongly supported by the morphological and cellular phenotype observed in medaka and zebrafish models with truncating mutations in the ortholog sec24d and in humans with mutations in SEC23A. Thus, we assign the fourth human phenotype to germline mutations in components of the COPII machinery.18,28,29
Acknowledgments
We are grateful to all family members that participated in this study and to Saskia Seland for excellent technical assistance. We would like to thank Philipp Greif and Nikola Konstandin (Department of Medicine III, University of Munich) as well as Stefan Krebs, Alexander Graf, and Helmut Blum (LaFuGA, Gene Center, University of Munich) for support in exome sequencing. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of groups contributing to ExAC can be found online. The authors would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). This work was supported in part by grants to J.K. from the National Institute of Dental and Craniofacial Research/NIH (R21 DE022419) and the National Institute of General Medical Sciences/NIH (R01 GM110373), by grants to B.W. from the German Federal Ministry of Education and Research (BMBF), grant number 01GM1211A (E-RARE network CRANIRARE-2), and grant number 01GM1109C (national rare disease network FACE), and by grants to H.-S.K. from Korea Basic Science Institute (E34700) and the Bio & Medical Technology Development Program of the National Research Foundation funded by the Ministry of Science, ICT & Future Planning (2013M3A9A9050076).
Contributor Information
Jinoh Kim, Email: jinoh.kim@ucdmc.ucdavis.edu.
Christian Netzer, Email: christian.netzer@uk-koeln.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
ExAC Browser, http://exac.broadinstitute.org/
ExAC Browser contributing groups, http://exac.broadinstitute.org/about
Human Gene Mutation Database, http://www.hgmd.org/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 2.Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 3.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byers P.H., Pyott S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012;46:475–497. doi: 10.1146/annurev-genet-110711-155608. [DOI] [PubMed] [Google Scholar]
- 5.Marini J.C., Reich A., Smith S.M. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr. Opin. Pediatr. 2014;26:500–507. doi: 10.1097/MOP.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Dijk F.S., Sillence D.O. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A. 2014;164A:1470–1481. doi: 10.1002/ajmg.a.36545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller E.A., Schekman R. COPII - a flexible vesicle formation system. Curr. Opin. Cell Biol. 2013;25:420–427. doi: 10.1016/j.ceb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venditti R., Wilson C., De Matteis M.A. Exiting the ER: what we know and what we don’t. Trends Cell Biol. 2014;24:9–18. doi: 10.1016/j.tcb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Viljoen D., Versfeld G., Beighton P. Osteogenesis imperfecta with congenital joint contractures (Bruck syndrome) Clin. Genet. 1989;36:122–126. doi: 10.1111/j.1399-0004.1989.tb03174.x. [DOI] [PubMed] [Google Scholar]
- 10.Beighton P., Winship I., Behari D. The ocular form of osteogenesis imperfecta: a new autosomal recessive syndrome. Clin. Genet. 1985;28:69–75. doi: 10.1111/j.1399-0004.1985.tb01220.x. [DOI] [PubMed] [Google Scholar]
- 11.Neuhäuser G., Kaveggia E.G., Opitz J.M. Autosomal recessive syndrome of pseudogliomantous blindness, osteoporosis and mild mental retardation. Clin. Genet. 1976;9:324–332. doi: 10.1111/j.1399-0004.1976.tb01581.x. [DOI] [PubMed] [Google Scholar]
- 12.Cole D.E., Carpenter T.O. Bone fragility, craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features: a newly recognized type of osteogenesis imperfecta. J. Pediatr. 1987;110:76–80. doi: 10.1016/s0022-3476(87)80292-5. [DOI] [PubMed] [Google Scholar]
- 13.Amor D.J., Savarirayan R., Schneider A.S., Bankier A. New case of Cole-Carpenter syndrome. Am. J. Med. Genet. 2000;92:273–277. doi: 10.1002/(sici)1096-8628(20000605)92:4<273::aid-ajmg10>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 14.MacDermot K.D., Buckley B., Van Someren V. Osteopenia, abnormal dentition, hydrops fetalis and communicating hydrocephalus. Clin. Genet. 1995;48:217–220. doi: 10.1111/j.1399-0004.1995.tb04092.x. [DOI] [PubMed] [Google Scholar]
- 15.Marwaha R.K., Sarkar B., Katariya S., Jayshree K. Cole-Carpenter’s syndrome. Indian J. Pediatr. 1993;60:305–308. doi: 10.1007/BF02822197. [DOI] [PubMed] [Google Scholar]
- 16.Stopfer J., Hurt H., Magilner A., Schneider A. A variant type of osteogenesis imperfecta: confirmation of a rare phenotype. Am. J. Hum. Genet. 1992;51:A108. [Google Scholar]
- 17.Zanetti G., Pahuja K.B., Studer S., Shim S., Schekman R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 2012;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- 18.Boyadjiev S.A., Fromme J.C., Ben J., Chong S.S., Nauta C., Hur D.J., Zhang G., Hamamoto S., Schekman R., Ravazzola M. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 2006;38:1192–1197. doi: 10.1038/ng1876. [DOI] [PubMed] [Google Scholar]
- 19.Boyadjiev S.A., Justice C.M., Eyaid W., McKusick V.A., Lachman R.S., Chowdry A.B., Jabak M., Zwaan J., Wilson A.F., Jabs E.W. A novel dysmorphic syndrome with open calvarial sutures and sutural cataracts maps to chromosome 14q13-q21. Hum. Genet. 2003;113:1–9. doi: 10.1007/s00439-003-0932-6. [DOI] [PubMed] [Google Scholar]
- 20.Boyadjiev S.A., Kim S.D., Hata A., Haldeman-Englert C., Zackai E.H., Naydenov C., Hamamoto S., Schekman R.W., Kim J. Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin. Genet. 2011;80:169–176. doi: 10.1111/j.1399-0004.2010.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohisa S., Inohaya K., Takano Y., Kudo A. sec24d encoding a component of COPII is essential for vertebra formation, revealed by the analysis of the medaka mutant, vbi. Dev. Biol. 2010;342:85–95. doi: 10.1016/j.ydbio.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Baines A.C., Adams E.J., Zhang B., Ginsburg D. Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS ONE. 2013;8:e61114. doi: 10.1371/journal.pone.0061114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancias J.D., Goldberg J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 2008;27:2918–2928. doi: 10.1038/emboj.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fromme J.C., Ravazzola M., Hamamoto S., Al-Balwi M., Eyaid W., Boyadjiev S.A., Cosson P., Schekman R., Orci L. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev. Cell. 2007;13:623–634. doi: 10.1016/j.devcel.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S.D., Pahuja K.B., Ravazzola M., Yoon J., Boyadjiev S.A., Hammamoto S., Schekman R., Orci L., Kim J. The [corrected] SEC23-SEC31 [corrected] interface plays critical role for export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 2012;287:10134–10144. doi: 10.1074/jbc.M111.283382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarmah S., Barrallo-Gimeno A., Melville D.B., Topczewski J., Solnica-Krezel L., Knapik E.W. Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS ONE. 2010;5:e10367. doi: 10.1371/journal.pone.0010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones B., Jones E.L., Bonney S.A., Patel H.N., Mensenkamp A.R., Eichenbaum-Voline S., Rudling M., Myrdal U., Annesi G., Naik S. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat. Genet. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz K., Iolascon A., Verissimo F., Trede N.S., Horsley W., Chen W., Paw B.H., Hopfner K.P., Holzmann K., Russo R. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 30.Shapiro R. Anomalous parietal sutures and the bipartite parietal bone. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1972;115:569–577. doi: 10.2214/ajr.115.3.569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.