

Abstract
Helicobacter pylori from different individuals exhibits substantial genetic diversity. However, the kinetics of bacterial diversification after infection with a single strain is poorly understood. We investigated evolution of H. pylori following long-term infection in the primate stomach; Rhesus macaques were infected with H. pylori strain USU101 and then followed for 10 years. H. pylori was regularly cultured from biopsies, and single colony isolates were analyzed. At 1-year, DNA fingerprinting showed that all output isolates were identical to the input strain; however, at 5-years, different H. pylori fingerprints were observed. Microarray-based comparative genomic hybridization revealed that long term persistence of USU101 in the macaque stomach was associated with specific whole gene changes. Further detailed investigation showed that levels of the BabA protein were dramatically reduced within weeks of infection. The molecular mechanisms behind this reduction were shown to include phase variation and gene loss via intragenomic rearrangement, suggesting strong selective pressure against BabA expression in the macaque model. Notably, although there is apparently strong selective pressure against babA, babA is required for establishment of infection in this model as stains in which babA was deleted were unable to colonize experimentally infected macaques.
Keywords: evolution, babA, Helicobacter pylori, colonization, outer membrane protein
Introduction
The human pathogen, Helicobacter pylori, has colonized the stomach of the majority of humans for millennia (Falush et al., 2003). While colonization by this bacterium is often asymptomatic, H. pylori infection has the potential to lead to severe disease outcomes such as peptic ulcer disease and gastric cancer. The fact that only a small percentage of infected patients develop H. pylori related disease is believed to result from a combination of bacterial, host, and environmental factors (Bridge and Merrell, 2013). Given the vast degree of genetic diversity found among H. pylori clinical isolates, it is not difficult to imagine how variations in strain background may contribute to differential disease outcomes. The degree of diversity that exists among H. pylori strains was first shown when the second genome sequence became available for this organism; strains 26695 (Tomb et al., 1997) and J99 (Alm et al., 1999) have a 6% difference in their genomic content. A more recent study expanded the comparison to over 50 strains and found that the H. pylori core genome is comprised of approximately 1,150 core genes (Gressmann et al., 2005). This translates into an average of 27% of the genome being variably present in different isolates (Gressmann et al., 2005).
Several characteristics lend to the highly dynamic genome structure of H. pylori. These include a high frequency of spontaneous mutation, gene conversion, transposition, and natural competence (Fu et al., 2014). H. pylori is missing several of the genes that are important for mismatch repair, which may result in the higher rate of spontaneous mutation (Garcia-Ortiz et al., 2011; Tomb et al., 1997). Furthermore, a large proportion of differences between strains are due to insertions or deletion of genes (Alm et al., 1999; Gressmann et al., 2005). Three common mechanisms by which deletions and/or insertions may arise in H. pylori include horizontal gene transfer, transposition and gene conversion. As a naturally competent organism, H. pylori can acquire new DNA fragments via horizontal gene transfer, and these new DNA fragments can then integrate into the chromosome through recombination. Coexistence of genetically diverse strains within a given host is a well-documented phenomenon and provides an environment where H. pylori may undergo recombination among distinct isolates (Kersulyte et al., 1999; Suerbaum et al., 1998; Taylor et al., 1995).
In addition to inter-strain rearrangements, H. pylori has several direct and indirect repeat sequences that can result in intragenomic rearrangements (Fu et al., 2014; Kennemann et al., 2011). These gene conversion events have been documented in at least two outer membrane protein (OMP) groups, the bab genes (Solnick et al., 2004) and the sab genes (Talarico et al., 2012). To further add to the genetic diversity amongst H. pylori strains, numerous genes are predicted to undergo slip-strand mismatch as a result of homopolymeric and dinucleotide repeat regions. Based on a comparative analysis of two H. pylori genomes, there are 46 candidate phase-variable genes, and of those 46 genes, there is supportive evidence for the phase-variability of 30 (Salaun et al., 2004). Interestingly, several of the predicted phase variable genes are virulence factors: many OMPs, flagellar genes, and genes involved in LPS biosynthesis (Kennemann et al., 2012; Matteo et al., 2010; Salaun et al., 2004; Solnick et al., 2004; Talarico et al., 2012). As a persistent pathogen that elicits little to no protective immunity, and in many cases results in an asymptomatic infection, one might envision a model of infection in which H. pylori constantly varies surface components as a means to evade the host immune system. In addition, it is not difficult to see how genetic variations in factors such as cagA or babA, which are known to contribute to disease and colonization, respectively, may lead to differences in virulence between H. pylori isolates.
Despite the abundance of evidence regarding the highly dynamic population structure of H. pylori (Salaun et al., 2004; Suerbaum et al., 1998), very few studies have followed the dynamics of long term colonization. Initial studies examined isolates over short time intervals post infection and found that the majority of isolates from a given patient appeared to be identical (Langenberg W, 1986; Salama et al., 2007). In addition, other work that sought to characterize H. pylori strains through familial studies found that isolates from family members and persons living within the same household were often closely related and in some cases indistinguishable (Suerbaum et al., 1998). In contrast, studies following strains during persistent colonization found remarkable genetic differences (Israel et al., 2001; Kersulyte et al., 1999; Kuipers et al., 2000). For example, one study examined the sequenced strain J99 as compared to fresh H. pylori isolates cultured from the source patient six years after J99’s original culture. Initial characterization of these new isolates showed slight differences in the random amplified polymorphic DNA (RAPD) pattern, as well as antibiotic resistance patterns as compared to the initial J99 isolate that was taken six year prior (Israel et al., 2001). Further investigation of these strains using comparative microarray hybridization revealed gene loss among the new isolates as well as the presence of genes not found within the archival J99 strain. Thus, direct interpretation of the evolution of gene content across the strains is complicated by the fact that the infection may have been caused by a mixture of strains. Additionally, it’s clear that the sensitivity of the various techniques used in the studies affects the interpretation of the degree of H. pylori evolution after infection. Furthermore, the length of time between initial colonization and sampling or between individual samplings may also affect the diversity detected. Although time between sampling can be controlled in some instances, the ability to determine the length of time from the initial infection to when the sample is taken is harder to control. For example, the J99 H. pylori strain had been residing in the stomach of the patient for an unknown number of years when it was originally isolated in 1994 (Israel et al., 2001); therefore, the available sequence of J99 provides a “snapshot” of the one isolated colony at that one point in time. Furthermore, as mentioned above, it is difficult to ascertain that all strains colonizing a given stomach at the start of the study have been isolated and characterized. If a particular strain is only a minor portion of the total population of bacterial variants present, it may not represent the colonizing strain as a whole.
In another example, an elegant study previously characterized changes in gene content seen in H. pylori J166 isolates recovered after short-term (17 week) infection of three rhesus macaques (Solnick et al., 2004). The investigators found that the output strains they examined had either undergone replacement of babA with babB or had undergone changes in the number of CT repeats at the 5′ end of the babA sequence that resulted in a frame shift mutation that truncated BabA. Thus, that early work suggested that babA undergoes selective pressure early in infection and changes in babA might represent a crucial adaptation to the host stomach. In addition, a recent publication by Linz, et al. showed that changes within the babA promoter can happen as early as 1 week post-infection in a macaque model, with larger genomic rearrangements within the bab genes occurring later (Linz et al., 2014).
In order to fully understand the panmictic population structure of H. pylori that results from frequent recombination and/or horizontal gene transfer within the host, it would be beneficial to know that an infection was seeded from a single strain and to carefully control for the length of time between initial infection and subsequent sampling. Herein, we describe such a study in which H. pylori strain USU101 (Liu et al., 2009) was experimentally inoculated into Rhesus macaques (Macaca mulatta). Evolution of USU101 within the animals was monitored across a 10–year timeframe via periodic isolation and characterization of output strains. We found that strain variation was detected within one week of infection and that the majority of output strains showed changes in the OMP, BabA. This variation was characterized in detail and shown to involve intragenic recombination as well as phase variation within babA.
Materials and Methods
Bacterial Strain Isolation and Culture Conditions
As previously reported (Liu et al., 2009), the virulent Helicobacter pylori strain, USU101 was isolated from a patient with gastric adenocarcinoma and was characterized as cagA+ (EPIYA motif ABC), vacA+ (s1, i1, m1), and babA+ using established techniques (Argent et al., 2005; Aspholm-Hurtig et al., 2004; Rhead et al., 2007). In addition, USU101 was shown to induce high levels of IL-8 upon co-culture of the strain with AGS cells (Peek, Merrell and DuBois, unpublished). USU101 and output strains were grown on commercially available blood agar plates (Campy Chocolate with TVAP, REMEL) and incubated for 3 days under microaerobic conditions (90% N2, 5% O2, 5% CO2). All strains and biopsy isolates were maintained as frozen stocks in F12 media supplemented with 30% glycerol at −80°C.
Animals
All experiments were approved by the Armed Forces Radiobiology Research Institute Institutional Animal Care and Use Committee; the protocol was monitored and reapproved yearly. Twelve domestic rhesus macaques (Macaca mulatta) (2 to 5 years old, 2 to 5kg) that had not been involved in previous research protocols and were free of H. pylori by culture and histology were utilized. Before purchase, all animals had been bred, reared, and socially housed either in indoor gang cages, in outdoor corrals, or in a large free-ranging colony (Caribbean Primate Research Center, Sabana Seca, PR). Upon arrival at our facility, the macaques were quarantined for 90 days in individual stainless steel cages in conventional holding rooms of an animal facility approved by the American Association for Accreditation of Laboratory Animal Care and were provided with tap water ad libitum, commercial primate chow, and fruits. They were negative by three intradermal tuberculin injections at 2-week intervals. After release from quarantine, animals were maintained in equivalent individual housing units. Due to the length of this study, there was some attrition of the animals as a result of death due to H. pylori related diseases; several of the monkeys given both H. pylori and the nitrosating carcinogen, ethyl-nitro-nitrosoguanidine (ENNG), succumbed to stomach hemorrhage, as did one animal given H. pylori alone.
H. pylori Infection and Periodic Isolation
Two days before inoculation of monkeys, USU101 was transferred to flasks containing 25 mLs of brain-heart infusion broth with 4% fetal calf serum and incubated with shaking for 2 days in an atmosphere of 90% N2, 5% O2, and 5% CO as previously described (Dubois et al., 1999). The twelve macaques were treated with famotidine (Pepcid; Merck, Inc.; 2mg/kg intramuscular 14h and then again 1h before inoculation) to suppress acid output. After an overnight fast, the animals were endoscoped as previously described (Liu et al., 2009), phenol red was sprayed onto the gastric body to estimate the pH of the mucosa (generally a pH between 2 and 7), and 6ml of 0.25M NaHCO3 was introduced into the antrum to neutralize gastric acid. A 1ml suspension of 108 to 109 CFU of H. pylori strain USU101 was then sprayed onto the gastric antrum of the 12 macaques, which were then divided into two test groups. Six of the animals were maintained on a normal diet after H. pylori infection (H group) and 6 were given oral ENNG (EH group) as previously described (Liu et al., 2009). The macaques were re-endoscoped and biopsied five to seven days, one month and two months after inoculation, and then generally at 3 month intervals for the following 10 years. A comprehensive study schematic showing the infections, subsequent sampling and downstream analyses is provided in Fig. 1.
FIG 1.
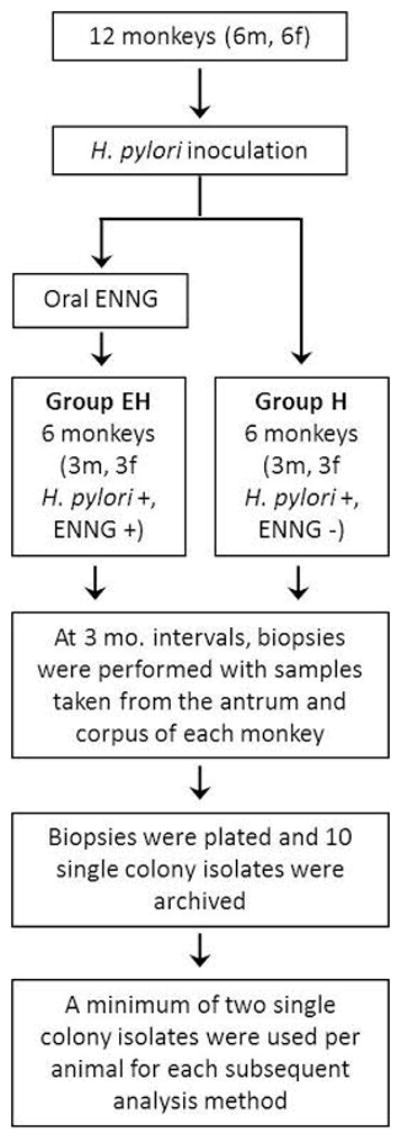
Experimental design, time course and subsequent analyses.
Macaques underwent gastroduodenal endoscopic examination under general anesthesia (atropine sulfate, 0.02mg/kg intramuscularly followed by ketamine HCl, 10mg/kg intramuscularly), using a CV160 Olympus endoscopy system. After each endoscopy, the equipment was rinsed with water and then disinfected by soaking for 20min in an activated dialdehyde solution of a 2% glutaraldehyde solution, followed by 10 min of exposure to CIDEX® (Johnson & Johnson Medical, Inc.); the instruments were then rinsed sequentially with sterile water and 70% alcohol and air dried. Six mucosal pinch biopsies of the gastric corpus and antrum were taken from every animal. Biopsies were performed at regular intervals as detailed above.
Biopsy Sample Processing
Histological examination of biopsy samples was conducted as previously described (Dubois et al., 1996). Briefly, two biopsies each from the corpus and antrum were fixed in neutral 10% buffered formalin and embedded in paraffin. Sections of 5μm thickness were stained with hematoxylin and eosin (H&E) and viewed at 200X or 400X magnifications. Antral gastritis was scored as previously described (Dubois et al., 1996). Slides also were scored for the presence of H. pylori infection following Genta staining.
Microbiological analysis of biopsy sampling was performed as previously described (Dubois et al., 1996). In brief, two biopsies each from the corpus and antrum were immediately placed in 0.26ml of sterile F-12 media on ice, coded, and homogenized with a sterile plastic cone-shaped pestle fitting a tapered 1.5ml Eppendorf tube. An aliquot (0.1ml) was spread on Campylobacter chocolate agar plates supplemented with trimethoprim, vancomycin, amphotericin B, and polymyxin B (Remel) and incubated at 37°C under microaerobic conditions. H. pylori isolates were identified as pinhead-sized, transparent colonies that grew within 3 to 7 days, as well as by microscopy as Gram-negative and curved or “gull-wing” rods. Biopsies from subsequent endoscopies done at 3-month intervals were cultured similarly and up to ten single colony isolates were isolated, expanded, and frozen for subsequent analysis.
Genomic DNA Isolation
Genomic DNA was extracted from H. pylori isolates as previously described (Akopyanz et al., 1992; Dubois et al., 1996). Single colony specimens were grown on blood agar plates for 3 days as detailed above. If required, the strains were passaged on plates to increase bacterial number. Bacterial cultures were subsequently harvested in phosphate buffered saline (1xPBS) and stored at 4°C until genomic DNA extraction was performed. gDNA was isolated using the QIAmp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions and was eluted in 200μl dH2O. DNA quality and quantity were evaluated by spectrophotometry.
DNA Fingerprinting
To examine differences among H. pylori strains, the random amplified polymorphic DNA (RAPD) fingerprinting method was used as previously described (Akopyanz et al., 1992; Berg et al., 1997; Dubois et al., 1996). Briefly, gDNA purified from cultures of individual single colony isolates was amplified with five different arbitrary primers: 1247, 1254, 1281, 1283, and 1290, where each reaction contained one primer, which acted as both the forward and reverse primer (Berg et al., 1997) (Table 1). After PCR, products were electrophoresed on 2% agarose gels containing 0.5μg/mL of ethidium bromide in Tris-borate running buffer, and bands were visualized under UV light. The pGEM DNA Marker (Promega) was used as a size marker.
TABLE 1.
Primers and Probes used in this study
| Primer Name | Primer sequence (5′ - 3′) | Reference |
|---|---|---|
| RAPD Primers | ||
| 1247 | AAG AGC CCG T | (Akopyanz et al., 1992) |
| 1254 | CCG CAG CCA A | (Akopyanz et al., 1992) |
| 1281 | AAC GCG CAA C | (Akopyanz et al., 1992) |
| 1283 | GCG ATC CCC A | (Akopyanz et al., 1992) |
| 1290 | GTG GAT GCG A | (Akopyanz et al., 1992) |
| Microarray Confirmation Primers | ||
| babA-F | TCT TCA ACC ACC ATC TTC AAC AA | This study |
| babA-R | TCG TTA GGG TTT GCT CCA CAT | This study |
| babB-F | TTT GCT TGA TGT CAA AAC CAA TT | This study |
| babB-R | CCT TGG ATC GCG CTG ATT | This study |
| babA-M1R | GGC CAA AGA ATT GCT TAT AG | This study |
| babA-M1F | ATT CAA GAG CTT TCC GAT C | This study |
| babA-M2F | TAT AAT CCT GGA GTC CAA G | This study |
| babA-M3F | TCA ATT CTT GTT AGC GCA AG | This study |
| babA Sequencing Primers | ||
| LAF | ATA GCG CAG TTT TCA ATT CCT G | This study |
| LAR | AGA AAG GTT GCA TGA CAC TAT TG | This study |
| LAF1 | TGA GCT TGG CGA GTT TGA AC | This study |
| LAR1 | ACA CAC CGC TAC CGC TTA G | This study |
| 46766 | AAA GGG AGT TTC ACA AAG | This study |
| JHP0834-R | CAA TTG ACA TTG ATG CTA AC | This study |
| HypD-F | TTT TGA GCC GGT GGA TAT ATT AG | (Colbeck et al., 2006) |
| S18-F | CTT TAA TCC CCT ACA TTG TGG A | (Colbeck et al., 2006) |
| babAR1 | TTT GCC GTC TAT GGT TTG G | (Colbeck et al., 2006) |
| 1F | GCA AGG AAA TGC GCA CAC G | This study |
| 1R | CTC GTT GGG CTC ATA ACC C | This study |
| 2F | GGC ACT AAA GGC ATG CTA GG | This study |
| 2R | CCC CTA CAT TGT GGA CAG G | This study |
| 3F | GAG CCA AGC CGT TAG ACA C | This study |
| 3R | GAG CAA ACG GCT TGA CAC TC | This study |
| T7 | GGG TTT TCC CAG TCA CGA | |
| SP6 | GCA CCC CAG GCT TTA CAC | |
| J99 RT-PCR primers | ||
| 16s-RTf | TCG GAA TCA CTG GGC GTA A | This study |
| 16s-RTr | TTC TAT GGT TAA GCC ATA GGA TTT CAC | This study |
| 16s-RT Probe | TGA CTG ACT ATC CCG CCT ACG CGC T | This study |
Microarray Based Comparative Genomic Hybridization
The microarray design and hybridization conditions were performed as previously described (Salama et al., 2000). Briefly, each strain was examined via a two-color competitive hybridization with a reference sample. To determine which genes were present in USU101, initial microarrays compared the USU101 strain to the reference preparation of an equimolar mixture of sequenced strains 26695 (Tomb et al., 1997) and J99 (Alm et al., 1999) (Salama et al., 2000). Next, to identify differences in the gene content between input and output strains, output strains were cohybridized with USU101. Thus, for the post inoculation biopsy isolates (output strains), the reference sample was the USU101 input strain used to infect the animals. A complete list of genes present in USU101 and the genes lost in the output strains can be found in Tables S1 and S2, respectively. Two isolates per animal were analyzed by microarray, which generated four potential data points for each gene. Data points were excluded due to low signal, slide abnormalities, and a regression correlation of pixel intensities in each channel of <0.6. Only genes for which at least two (and up to ten) measurements were obtained were analyzed. Data were normalized using the default-computed normalization of the Stanford Microarray Database (Gollub et al., 2003), and the mean of the log2 (red channel normalized net intensity/green channel net intensity) (log2RAT2N) was computed. Data were also excluded if the standard deviation of the log2RAT2N was greater than 1.0. For analysis of the input USU101 and output macaque isolates, a constant cutoff for absence of a gene was defined as a log2RAT2N value of −1.0 based on test hybridizations (Israel et al., 2001). Data were simplified into a binary score, analyzed with CLUSTER (http://bonsai.ims.u-tokyo.ac.jp/~mdehoon/software/cluster/) (de Hoon et al., 2004), and displayed with TREEVIEW (http://rana.lbl.gov/EisenSoftware.htm) (Eisen et al., 1998). The raw data are available in the NCBI Gene Expression Omnibus (accession number GSE60474), and a list of genes that differed in at least one sample is available in Table S2 in the supplemental material.
PCR Confirmation of Microarray Analysis Results
To confirm the presence or absence of the babA gene as determined by microarray hybridization, PCR analysis was performed on gDNA of two isolates from each of the 12 macaques. 50ng of gDNA from each sample was used as template for the PCRs in conjunction with the babA microarray probe region primers (Table 1, microarray confirmation primers) as well as babA specific primers (Table 1) that were designed based on the USU101 babA sequence that we determined by sequencing. The PCR reaction was performed using GoTaq Green Master Mix (Promega) with an initial denaturation at 95°C for 5min, followed by 35 cycles of 95°C for 15s, 58°C for 15s and 72°C for 1min, and a final 10min extension at 72°C. PCR products were visualized on agarose gels as detailed above.
babA Promoter and Genomic Localization
To identify the genomic location of babA in USU101, PCR amplifications were performed using the same primers as Colbeck, et al. (Colbeck et al., 2006) with FideliTaq PCR Master Mix (Affymetrix). Two of these primers (HypD-F and S18-F; Table 1) were designed such that the forward primer was located in the hypD or s18 genes that lie upstream of the babA and babB loci in J99, respectively. A third primer, BabAR1, which lies approximately 700bp into the babA gene, was used as the reverse primer in combination with either HypD-F or S18-F to determine the genomic location of babA. The resulting product was approximately 3000bps and spanned the babA promoter region and part of the babA gene. An additional set of primers was designed to confirm the presence/location of the bab genes within the chromosome. In this case, three sets of primers were designed such that the forward primer lies up stream and the reverse primer lies down stream of the three bab genes (babA, babB, and babC). These primers were designated 1F and 1R, 2F and 2R, 3F and 3R, and correspond to the babA, babB and babC loci from strain 26695, respectively (Table 1). PCR products were electrophoresed on 1.0 % agarose minigels with a 1kb DNA ladder size standard (GibcoBRL). DNA bands were visualized by ethidium bromide staining. The PCR products of correct sizes were purified using QIAquick PCR Purification Kit (Qiagen) and subsequently ligated into the pGEM-T Easy Vector (Promega). The resulting plasmids were transformed into Eschericia coli Top10 cells. Trasformants were selected for on LB plates containing ampicillin (100μg/mL), IPTG (1μM) and X-gal (40μg/mL). Single colony isolates from each transformation were inoculated into LB media supplemented with 25μg/mL ampicillin, and following overnight growth, plasmid DNA was extracted using the QIAprep Spin Miniprep Kit (Qiagen) per the manufacturer’s instructions. Proper insertion of the PCR products into the vector was verified by digestion with EcoRI, and plasmids containing an insertion were sequenced using the universal T7 and SP6 primers (Table 1) and the ABI BigDye Terminator 3.1 cycle sequencing kit. Sequencing results from experimental samples were then subjected to GeneBank BLAST analysis, and those with significant homology to the babA, babB, and/or babC genes of 26695 and J99 were subjected to multiple and pair wise sequence alignments using the 26695 sequenced strain as the reference (PRABI the French network of bioinformatic platforms; http://www.prabi.fr/) for further analysis.
To sequence the babA promoter region, the babA locus primers, 1R and 1F, were used to amplify the babA locus and promoter region from gDNA obtained from biopsy isolates. Following purification with the QIAquick PCR Purification Kit (Qiagen), PCR products were sequenced using the 1R primer in a manner analagous to that detailed above. Sequencing results were aligned using Seaview (http://pbil.univ-lyon1.fr/software/seaview.html) to compare the promoter regions.
Leb Adherence Radioimmunoassay (RIA)
Leb binding of bacterial populations obtained as sweeps of cells grown from antral and corpus biopsies from the animals was assessed using an adherence RIA. Adherence RIA was conducted by combining 1 ml of a 0.1 ODU bacterial suspension (8 ×108 bacteria) with 2.5 × 10−5 mCi of radiolabelled Leb conjugate (125I-HSA-Leb). The mixture was incubated for 2h at room temperature and then centrifuged. The percentage of binding was determined by comparing the relative radioactivity of the pellet and supernatant. Strain CCUG17875 (Ilver et al., 1998) and the babA1::kan babA2::cat (ΔbabA) CCUG17875 H. pylori strain were used as positive and negative controls, respectively. Strain CCUG17875 was chosen because it binds Leb well (Ilver et al., 1998).
Western Blot Analysis
BabA positive and negative control lysates were prepared from wild type H. pylori USU101 and a J99 babA knockout strain (Boren et al., 1993), respectively. H. pylori total protein was extracted using the Total Protein Extraction Kit (G-Biosciences) per the manufacturer’s protocol. Lysates were stored at −80°C. Protein concentrations were calculated using the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories) and a bovine serum albumin standard curve. Protein sample (50μg), Bio-Rad LDS buffer (Bio-Rad; 5μl) and 1x NuPAGE reducing agent (Invitrogen; 2μl) were adjusted to a final volume of 15μl with dH2Oand then heated at 70°C for 10min. These denatured samples were then loaded on 4–12% NuPAGE Novex Bis-Tris pre-cast gels (Invitrogen) with NuPAGE MOPS SDS running buffer (Invitrogen) containing 0.25% NuPAGE Antioxidant (Invitrogen). The gel was electrophoresed at 125V until the Full-range Rainbow MW Marker (Fisher Scientific) was close to the bottom of the gel. The proteins were then transferred to a nitrocellulose membrane with an iBlot® Dry Blotting System (Invitrogen) for 7min. Following transfer, the nitrocellulose membranes were incubated with blocking buffer (5% fat free dry milk in 1xPBS with 0.1% Tween 20) for 1hr at room temperature and then incubated at 4°C overnight in a 1:6000 dilution of polyclonal rabbit anti-BabA or 1:3000 dilution of polyclonal rabbit anti-BabB antibody in blocking buffer. Following incubation with the primary antibody, membranes were washed 5 times for 5min each with 1xPBS containing 0.1% Tween 20, followed by incubation with a 1:10,000 dilution of horse anti-rabbit HRP conjugated IgG (Vector Laboratories) for 1hr at room temperature in the dark. The membrane was subsequently washed and immersed in the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) with constant agitation for 5min and then visualized with FluorChem E (Cell Biosciences). Image files were analyzed using ImageJ Software (version 1.45, NIH).
Animal Infection with J99
Five H. pylori negative rhesus macaques were infected intragastricaly with 108 CFU of J99 babA::Cam; construction of J99babA::Cam was described previously in Mahdavi et al.(Mahdavi et al., 2002). Gastroscopies were performed at 1month, 4 months, and 6 months following infection to assess the macroscopic appearance of the gastric mucosa and harvest biopsies from the antrum and the corpus of the stomach. Antrum and corpus biopsies were cultured as previously described (Dubois et al., 1996). In addition, Real time (RT) PCR for H. pylori 16s rRNA and FISH were conducted to assess H. pylori colonization status. These same macaques, none of which were colonized with J99 babA::Cam, were subsequently infected with J99-GFP. J99-GFP was constructed by transforming strain J99 with EGFP-N1 (Clontech). GFP positive J99 colonies were selected based on fluorescence and morphology. Post infection with J99-GFP, endoscopies and gastric biopsies were conducted at 1, 4 and 6 months post infection. Biopsies were subsequently cultured and examined by RT-PCR for presence of H. pylori using the primers and probe listed in Table 1. In addition, fluorescence microscopy was used to visualize the H. pylori.
Results
RAPD Fingerprinting
At 1-year post inoculation, output isolates from all animals exhibited identical RAPD patterns to the input strain (Supplemental Figure 1). This was true even when multiple colonies were examined from each animal biopsy as a means to assess any variability present within the population; the RAPD pattern seen across all isolates from all animals was identical (data not shown). In contrast, at 5 years post-inoculation, output isolates from 8 of the animals demonstrated different RAPD patterns (Fig. 2 and data not shown). While no differences were seen across the isolates when using the 1254 primer (Fig. 2 bottom panel), the 1290 RAPD primer revealed banding patterns that varied among isolates (Fig. 2 top panel). Multiple isolates tested form the same animal showed similar changes (Fig. 2 and data not shown).
FIG 2. RAPD patterns of the input strain as compared to the output strains 5 years post-inoculation.

Samples from biopsies taken at 5 years post-infection were examined by RAPD finger printing in the same manner as the samples taken at 1 year post-inoculation (See Supplementary Figure 1). The two gel images show the RAPD patterns for primer 1290 (top) and 1254 (bottom). Two RAPD reactions were performed for two isolates from each monkey. Compared to USU101, several of the output strains show differences in the banding pattern; these include changes in band sizes and the loss of bands in the areas indicated by the A and B arrows.
Microarray Based Comparative Genomic Hybridization (CGH)
To further investigate the genomic changes in the output strains, CGH was performed using DNA obtained from the 5 year post inoculation isolates. Of the initial 12 animals infected, 4 EH and 5 H animals survived to this time point; two isolates from each animal were selected for analysis. Cumulatively, the CGH profile identified 52 genes that were absent in at least one of the 18 output strains tested (Table S2). Among the 52 genes, 18 had a recognized name and/or function. These included a putative type II DNA modification enzyme (methyltransferase), babA, babC, topA_3 (topoisomerase I homolog), a putative lipopolysaccharide biosynthesis protein, and a Zinc-dependent alcohol dehydrogenase, which were absent in multiple isolates. Of note, two transcriptional regulators, hspR and nikR, were also found to be absent in monkeys H4 and EH1, respectively. Additionally, 3 outer membrane proteins (OMPs), hopZ, babA (hopS), and babC (hopU) were identified as missing from multiple animals. The average number of genes not detected by microarray in any particular isolate ranged from 0 to 26 (Table 2). To determine if there was a statistically significant difference between the numbers of genes missing in H isolates as compared to EH isolates, a two-tailed Mann-Whitney test was performed. No statistical difference was found (p-value 0.4191), suggesting that the addition of ENNG to the animal diet did not induce additional changes in H. pylori gene content. Although, administration of ENNG does not appear to alter H. pylori gene loss, a more rigorous sequence-based analysis would be required to determine if the mutation rate of H. pylori was influenced by the carcinogen.
TABLE 2.
Number of Genes Missing at 5 years as Determined by CGH
| EH Group* | H Group | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Monkey | EH1 | EH3 | EH4 | EH5 | H1 | H2 | H4 | H5 | H6 | |||||||||
| Biopsy ID (isolate designation)# | 4641(3) | 4694(2) | 4689(5) | 4690(1) | 4633(1) | 4633(2) | 4637(3) | 4637(5) | 4661(3) | 4662(1) | 4723(1) | 4723(2) | 4726(1) | 4726(2) | 4721(1) | 4722(3) | 4667(2) | 4668(1) |
| Dysplasia∞ | − | − | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − | − |
| no. of missing genes | 16 | 15 | 0 | 9 | 0 | 2 | 1 | 20 | 26 | 24 | 1 | 0 | 1 | 5 | 3 | 2 | 23 | 22 |
Only 4 animals were included in this group; one animal died before CGH studies were initiated.
Two isolates per animal were analyzed by microarray
Previously published data from this study showed that disease only develops in this model when H. pylori and the oral carcinogen ENNG are administered together (Liu et al., 2009).
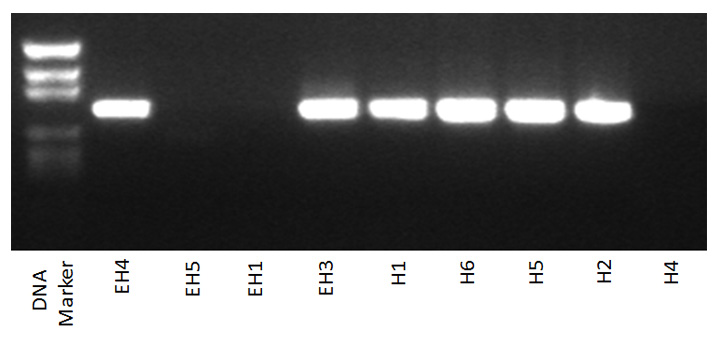
Since loss of babA has been demonstrated in previous macaque studies (Solnick et al., 2004; Styer et al., 2010), we decided to focus our efforts on this gene. First, PCR was used to confirm the microarray results. The forward primer, babAF, which is specific to the babA sequence from the USU101 strain, was paired with the reverse primer, babAR, which was used for the probe on the microarray (Table 1). We found a 100% agreement between the PCR results and the presence or absence of babA as determined by microarray hybridization. For the output strains that were determined to be babA positive by microarray (those from animals EH3, EH4, H1, H2, H5, and H6), there was no detectable change in the babA gene upon PCR analysis (Supplemental Figure 2). Conversely, when CGH showed that babA was absent (all tested isolates from animals EH1, EH5, and H4 at the 5-year post infection time point), this result was confirmed by PCR (Supplemental Figure 2). In addition, when we utilized three additional forward primers (babA-M1F, babA-M2F, babA-M3F), which were designed to target different locations within the babA gene, in combination with the babA-M1R reverse primer (Table 1), no bands were ever obtained (data not shown). These results suggest that the entire babA gene may be missing in the output strains from these three monkeys.
Changes in babA Gene Presence over Time
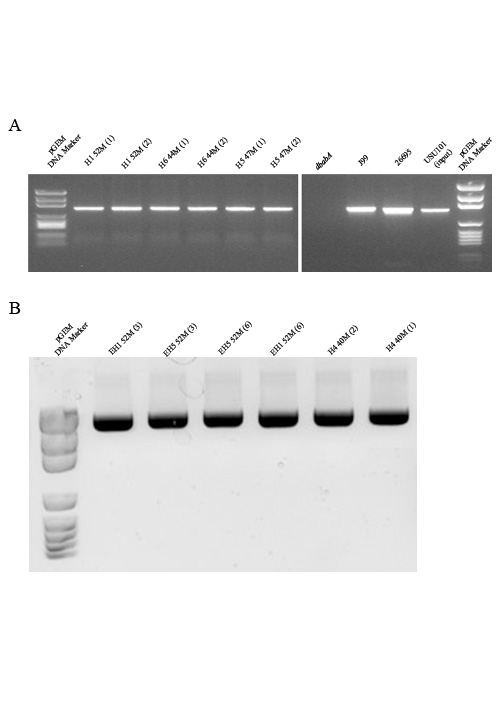
In order to determine the temporal nature of when the babA deletion occurred within the isolates that showed a loss of babA at the 5-year time point, we next analyzed isolates obtained from the same monkeys at earlier times. PCR indicated that babA was present in output strains taken from 1 week to 18 months post inoculation; bands were identical to the input strain (Fig. 3 and Supplemental Figure 3). However, 24 months after infection isolates began to yield different patterns when babA was amplified. PCR results fell into two categories: an identical pattern to what was seen prior to 24 months (isolates from EH3, EH4, H1, H2, H5, H6) or the appearance of two bands (isolates from EH1, EH5, and H4) (Fig. 3 and data not shown). We expanded the analysis for several isolates from each animal to also look at the 5 year time point. For isolates from animals in which there was no change at 24 months, the PCR pattern remained consistent at 5 years; however, in isolates obtained from animals EH1, EH5, and H4 that had 2 bands at 24 months, all of the isolates had completely lost babA by 5 years post infection (Supplemental Figure 2).
FIG 3. Assessment of the babA gene at early time points.
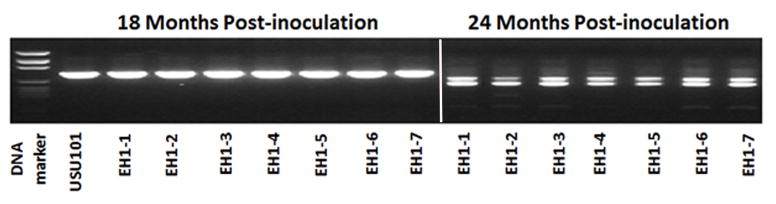
For strains that lacked the babA gene at 5 years post inoculation as determine by CGH, PCR amplification of babA was performed using primers babA-M3F and babA-M1R on 18 and 24 month post inoculation isolates. Individual isolates (numbered 1–7) from EH1 are shown here as a representative example. Seven individual isolates from 18 months post-infection and seven individual isolates from 24 months post-infection were examined. Individual strains are indicated by a “- “ followed by the isolate number. At 18 months, EH1 output strains are similar to the USU101 input strain; however, at 24 months, a doublet appears.
To specifically determine the nature of the change in babA seen in the 24 month EH1 output strains (Fig. 3), the DNA from the two bands was isolated, cloned and subsequently sequenced. The sequencing results showed that the two bands demonstrated homology to babA, babB, and babC sequences from 26695 when BLASTed against Genebank. Additionally, the sequences of the two bands were almost identical with only minor variation seen in the middle part of the sequence (data not shown) (Lasergene software, ver. 10). ClustalV analysis, sequence pair distance, and a Lasergene produced phylogenetic tree demonstrated that the larger MW band was most similar to babB and the smaller MW band was most similar to babC (data not shown). Since data indicated that all isolates from EH1, EH5, and H4 had completely lost babA at the 5-year time point, we also looked at isolates taken from 3–4 years post-infection. PCR analysis from these time points showed that the two bands were gradually lost due to the replacement of babA by babB (Supplemental Figure 4 and data not shown).
Analysis of BabA Protein Expression
Given the changes in babA that were observed by PCR and microarray, we next directly examined BabA protein expression. Western blot analysis was used to measure BabA expression in the USU101 input strain and output isolates obtained as early as 1 week post infection and as late as 107 months post infection. As a positive control, lysates from J99 were used to confirm that the anti-BabA antibody was able to detect the 75kD BabA protein (Boren et al., 1993). Additionally, lysates from a J99 babA deletion strain were used as a negative control. BabA expression was detected in J99 as well as in USU101 (Fig. 4). BabA expression was completely lost in the babA mutant and was dramatically reduced or lost in the majority of output strains. We did observe very low levels of BabA expression periodically, but the reduction in babA expression was evident within weeks of infection for some animals, as seen in the week 1 isolate from monkey H2 (Fig. 4 and data not shown). Furthermore, the reduction in BabA expression was maintained throughout the 10 year time point (Fig. 4).
FIG 4. Detection of BabA by Western blot.
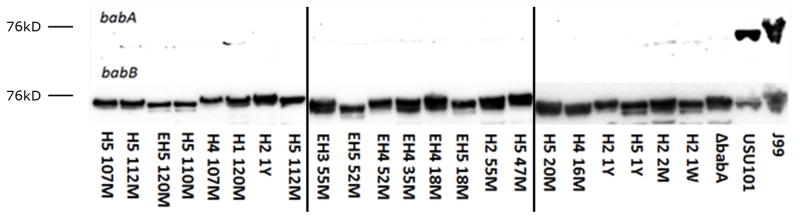
Protein was purified from single colony isolates from H and EH monkeys as early as 1 week to 120 months (10 years) post inoculation. Equal amounts of protein from lysates were run on a denaturing gel, and BabA expression was detected using poly-clonal rabbit antibodies. J99 and USU101 were included as positive controls, and a ΔbabA J99 strain was included as a negative control. Relative sizes as determined using a protein ladder are indicated; J99 BabA is known to run at approximately 75kD. BabB expression was also analyzed as a relative loading control. Lanes are labeled with the monkey designation followed by the time point. W, M, and Y stand for weeks, months, and years post inoculation, respectively.
babA Sequence Comparison Between Input and Output Strains
Since the most probable reason for the decreased expression of BabA was the presence of a promoter or gene mutation, we next sequenced babA from USU101 as well as from output strains and compared the obtained sequences to identify any changes. We utilized output strains obtained from each of the macaques at 5 years and 10 years post inoculation. Furthermore, we also utilized longitudinal samples taken from a single animal, H2, as a means to monitor the temporal nature of any observed sequence variation. Sequence analysis revealed that for all of the output strains, the core promoter elements themselves, as well as the region between the −10 promoter element and the ATG translational start codon, were unchanged as compared to the wild type USU101 strain (Fig. 5 and data not shown). Strikingly, the majority of the output strains showed variations within a poly-A tract that lies between the −35 and −10 promoter elements, as well as variation within a CT dinucleotide tract that lies downstream of the translational start codon (Fig. 5, Table 3 and data not shown). Of note, the CT repeat region has previously been shown to undergo phase variation whereby the loss or gain of CT dinucleotides within this tract results in truncation of BabA due to a frame shift mutation that results in a premature stop codon (Solnick et al., 2004). At 5 years post-inoculation, analysis of the babA gene from isolates from the H group (one isolate per animal H1, H2, H4 and H5) and isolates from the EH group (one isolate per animal EH1, EH3, EH4, EH5) showed that five of the eight isolates tested contained CT repeats that resulted in premature truncation of BabA (Table 3). The remaining three isolates (from animals H4, EH1 and EH5) no longer carried babA, but instead now carried babB at the babA locus; replacement of babA by babB was confirmed by PCR amplification and sequencing (Supplemental Figure 5 and data not shown). Analysis of the length of the poly-A tract in the 5 year isolates revealed that, of the 5 strains that still carried babA, three isolates carried a poly-A tract similar to the USU101 input strain, but two isolates carried an additional A which made the poly-A tract 14 nucleotides in length (Fig. 5 and Table 3). Interestingly, analysis of the longitudinal isolates obtained from H2 showed that changes in the number of CT repeats (increasing from 8 to 10 dinucleotides) could be observed as early as 2 months post inoculation and then remained at 9 CT repeats for all subsequent time points (Fig. 5). Similarly, the number of adenines found within the poly-A tract fluctuated at the earliest time point and varied from 13–15 across the following years (Fig. 5). Finally, analysis of isolates collected from the antrum and the corpus of the stomach showed similar genotypes with regard to the number of CT repeats and the replacement of babA with babB (data not shown).
FIG 5. Sequencing of the babA promoter region.

To further assess the loss of BabA expression, the babA promoter region was sequenced for several isolates using primer IR (Table 1). The −10 core promoter element (TATAAT), the +1 transcriptional start site (A), and the ATG translational start codon are boxed. The poly-A tract upstream of the −10 region and the CT repeat region downstream of the translational start site are underlined. In comparison to USU101 (13 A’s, 8 CT’s) the output strains differ in the number of repeats in either one or both of these regions. The lengths of poly-A tracts range from 13 to 15 and the CT repeat lengths range from 8 to 10. The addition of CT repeats, seen in all of the isolates, occurs within the coding region of babA and results in the pre-mature truncation of the protein.
TABLE 3.
Sequence Comparison of babA from Input and Output Strains
| Strain | Time post inoculation | PCR for babA | Western Blot for BabA | No. CT repeats* | No. As in PolyA tract# |
|---|---|---|---|---|---|
| USU101 | 0 mo. | + | + | 8 | 13 |
| H2 | 2 mo. | + | − | 10 | 14 |
| H2 | 16 mo. | + | − | 9 | 14 |
| H2 | 18 mo. | + | − | 9 | 15 |
| H2 | 24 mo. | + | − | 9 | 14 |
| H2 | 55 mo. | + | − | 9 | 13 |
| H1 | 52 mo. | + | − | 10 | 14 |
| H5 | 47mo. | + | − | 9 | 13 |
| H4 | 47mo. | − | − | babB | N/A |
| EH1 | 55 mo. | − | − | babB | N/A |
| EH3 | 55 mo. | + | − | 9 | 14 |
| EH4 | 52 mo. | + | − | 9 | 13 |
| EH5 | 55 mo. | − | − | babB | N/A |
babB indicates that no CT repeats were present because the gene was replaced by babB.
N/A indicates that this is not applicable because the poly- A tract was lost due to the replacement of babA by babB.
At ten years post infection only 5 animals survived (EH5, H1, H2, H4 and H5). Sequencing of babA from isolates from the ten-year time point revealed that two of the remaining animals had isolates that were babA positive but carried 9 CT repeats; therefore, babA was out of frame. The other three animals had isolates in which babA had been replaced with babB (supplemental figure 6 and data not shown). Of note, isolates from H5 that were taken at earlier time points did not contain a babB replacement; therefore, this replacement took place between 5 and 10 years. Thus, babA expression was absent in isolates from all five remaining animals; for three out of five of the 10-year isolates, the babA gene was replaced by babB and in the other two a frame shift resulted in a premature truncation.
Population based analysis of Leb binding
An obvious limitation of looking at only subsets of colonies obtained from the infected animals is that it does not allow for global analysis of the presence or absence of babA across the population of strains colonizing an animal. Therefore, to look at BabA at the functional level across the population, sweeps of colonies taken at various time points were obtained from eight monkeys and were analyzed for BabA functional interaction with Leb using adherence RIA; BabA is known to mediate binding of H. pylori to Leb (Aspholm-Hurtig et al., 2004). RIA assesses affinity of BabA for radiolabeled Leb. The percent adherence value is determined by comparing the ratio of radiolabeled Leb in the pellet (bound by the bacteria) to the supernatant (unbound Leb). Therefore the higher the affinity of the bacteria for the Leb, the higher the percentage of Leb will be in the pellet. In addition to our output isolates, we also determined Leb binding for a positive control strain, CCUG17875, which has previously been shown to be a high binding strain (Iver et al, 1998), and the input strain USU101. We also included a CCUG17875ΔbabA strain, to control for any nonspecific binding. At least one sample was tested from each animal, and in some cases, sweeps collected from both the corpus and the antrum were tested. While there was typically no apparent difference between binding of samples obtained from the antrum and corpus; one antral sweep from animal H6 (51 months) appeared different from the corresponding corpus population (Table 4). However, at a later time point (63 months) the binding appeared similar to the corpus sample. Some level of Leb binding was observed for strains from five of the nine monkeys and ranged from 1.1%–40.7% (Table 4). The isolates from the remaining 3 animals only exhibited binding affinities from 0.2%–0.4%, which are similar to the values of the negative control (Table 4). En masse, these data indicate that even though analysis of individual isolates revealed that expression of BabA was lost as early as 1 week post infection and in every animal and isolate we looked at (Figure 4), in 5 of the animals, a fraction of the bacterial population retains the ability to express BabA (as evidenced by the positive signal in the RIA assay). Conversely, BabA expression is completely lost in 3 of the animals.
TABLE 4.
RIA Analysis for Leb Binding
| Monkey | Time point* | RIA % hot Leb bound | |
|---|---|---|---|
| Antrum | Corpus | ||
| EH5 | 51 mo | N/D$ | 0.4 |
| EH5 | 60 mo | 0.4 | N/D |
| EH1 | 51 mo | N/D | 0.4 |
| EH1 | 55 mo | 0.5 | N/D |
| EH1 | 60 mo | 0.4 | 0.4 |
| H1 | 51 mo | N/D | 2.7 |
| H6 | 51 mo | 40.7 | 2.2 |
| H6 | 63 mo | 3.0 | 4.1 |
| EH3 | 55 mo | 2.8 | 3.3 |
| H5 | 51 mo | N/D | 2.8 |
| H5 | 60 mo | 1.1 | 1.7 |
| H2 | 63 mo | 3.7 | 4.3 |
| EH4 | 52 mo | N/D | 0.2 |
| CCUG17875 | Positive Control | 78.2 | |
| CCUG17875 ΔbabA | Negative Control | 0.3 | |
| USU101 | Input Strain | 36.0 | |
Time point is months post infection with H. pylori
N/D indicates “Not Determined”
babA is Required for Establishment of Infection
The dynamic nature of changes in expression and gene loss of babA that were observed in all animals in our study strongly suggests that there is a strong selective pressure against BabA within this model. Therefore, we sought to determine whether BabA was actually required for H. pylori colonization of macaques. To this end, we experimentally challenged five separate macaques with a babA mutant of strain J99 in which babA had been replaced with a chloramphenicol cassette. Post infection, animals were followed for 6 months and colonization was assessed at 1, 4 and 6 months by biopsy and culture. To detect low levels of colonization that might be missed by culture, we also assessed the presence of H. pylori 16s rRNA by RT-PCR and FISH. Post challenge with the babA mutant strain, biopsy culture showed that all 5 animals remained culture negative at each of the time points tested throughout the 6 months. Low levels of H. pylori 16s rRNA were detected in two animals at the 1 month time point, but not in any of the animals at later time points. Thus, BabA appears to be required to colonize macaques. To ensure that these animals were not resistant to infection with strain J99, these same 5 animals were next challenged with a J99 strain engineered to express GFP. Once again, colonization was assessed at 1, 4 and 6 months by biopsy and culture as well as by RT-PCR and FISH for H. pylori 16s rRNA. At 1 month post-infection all animals were H. pylori culture negative; however 2/5 monkeys showed low levels of 16s rRNA and GFP expressing bacteria were visible by microscopy. By 4 months post-infection 3/5 monkeys were cultures positive and all monkeys were positive by RT-PCR and fluorescent microscopy. At the final 6 month time point, all five monkeys were culture positive for H. pylori. Thus, these animals were susceptible to H. pylori infection. Taken together, these data indicate that BabA is required to establish infection in the macaque model.
Discussion
H. pylori has long been characterized as panmictic (Salaun et al., 1998), and as with other bacterial species, the ability of H. pylori to alter its genetic make-up likely allows the bacterium to adapt to the host environment and establish persistent infection. Indeed, the host appears to impose a selective pressure that drives variation within the bacterium (Thompson et al., 2004). Over the course of an infection, these genetic changes may alter the host’s immune response to the bacterium and therefore, may contribute to different clinical outcomes (Cover et al., 2003). Thus, a greater understanding of the process of genetic variation seen within persistently colonizing strains has the potential to elucidate factors that are key to disease progression as well as information concerning the immune response to H. pylori. As such, numerous studies have sought to understand genetic variation during colonization (Israel et al., 2001; Kersulyte et al., 1999; Kuipers et al., 2000; Langenberg W, 1986; Linz et al., 2014; Salama et al., 2007; Solnick et al., 2004).
Whereas previous studies looked only at relatively early time points post infection, we sought to further our understanding of the long term, chronic effects of colonization on the H. pylori genome by following the H. pylori infected rhesus macaques for over ten years. Of note, macaques represent the most “human-like” animal model available to study H. pylori, and to our knowledge, this study timeline far exceeds any other experimental colonization studies carried out to date.
Isolates recovered at 1 year post-infection showed a similar RAPD pattern to USU101, and it was not until 5 years post infection that RAPD differences were apparent (Fig. 2 and data not shown). Isolates recovered at 5 years post infection were further analyzed by CGH, and a total of 52 genes did not hybridize in at least one of the 18 output isolates that were tested (Tables S1 and S2). Interestingly, the number of genes not detected by microarray in any particular isolate ranged from 0 to 26 (Table 2) and was similar among the H and EH group of macaques (SEM of H group = 11.0+3.6 and EH group = 7.9+2.9; p-value 0.56). Given that we did not see a significant difference between H and EH group monkey strains, we concluded that while long term colonization of the gastric niche can lead to specific H. pylori genetic changes, these changes occur independent of the dietary carcinogen. Instead, the changes are likely due to selective pressure imposed by the host immune response. Also of note, at this resolution no single gene was uniformly lost in all animals. Thus, evolution of the H. pylori genome within a single animal is subject to the specific microenvironmental pressures present within that particular host.
While analysis of the 5-year time point provided a snapshot of the genetic variation that occurred in the H. pylori strains after long term colonization, we wished to understand the temporal nature of adaptation to the host environment since this has not been evaluated over such an extensive infection period. To this end, we focused on babA, which was among the genes identified as absent in 3 animals at the 5 year time point by microarray (Table S1). For these three animals, PCR analysis using babA specific primers identified a window between 18 and 24 months where dramatic changes in babA appeared to occur (Fig 3). Interestingly, in the three isolates that were negative for babA, these changes were reflected by PCR not necessarily by RAPD (Fig. 2). This suggests that changes in the genome occurred at focused regions and were not the result of drastic chromosomal rearrangement or deletion. By 24 months post infection, PCR of the babA gene from EH1 showed two smaller MW bands suggesting that the babA sequence was substantially modified in these isolates (Fig. 3). Sequencing of these PCR products showed that the fragments were very similar to the babA gene sequence as well as to the 26695 babB and babC gene sequences (data not shown). Indeed, ClustalV analysis, sequence pair distance, and phylogenetic tree analyses demonstrated that the larger MW band had the most similarity to babB while the smaller MW band had the most similarity to babC. As seen in previous studies, these findings suggest that the babA gene undergoes recombination and/or replacement events due to its significant homology to babB and babC (Linz et al., 2014; Solnick et al., 2004).
In terms of the temporal nature of babA changes, Western blot analysis revealed that even though genomic replacement of babA was not universal, all isolates obtained within a few weeks to months post inoculation showed little to no BabA expression (Fig. 4 and data not shown). This was true even among isolates that still appeared to carry an intact babA gene. Indeed, in contrast to the genetic rearrangements leading to babA replacement, which took approximately 2 years, BabA expression was lost much earlier and in all tested isolates. Analysis of the babA promoter from strains that maintained babA but had lost protein expression showed that the babA gene contains two variable regions of repetitive DNA: a poly-A tract in the promoter region between the −10 and −35 regions as well as a dinucleotide CT repeat within the coding region approximately 20bps from the start codon. While the input babA expressing strain, USU101, contains a 13 bp poly-A tract and eight CT repeats, output strains that carried babA but no longer expressed the protein contained a poly- A tract that ranged from 13–15 bps and an additional 1 or 2 CT dinucleotide repeats; the CT insertions result in a frame shift within the coding sequence of the babA gene leading to a premature stop codon (Solnick et al., 2004). Conversely, the significance of changes in the length of the poly-A tract within the promoter is unclear; however, it is possible that increasing the length between the −10 and −35 regions may affect ribosomal and/or regulatory protein binding to the babA promoter. If this were the case, this could represent a novel mechanism by which modulation of babA expression is controlled. In fact, two recent publications regarding the regulation of another OMP, sabA, suggest that changes in the length of a poly-A tract leads to changes in the DNA structure and subsequently effect the binding of the RNA polymerase (Aberg et al., 2014; Harvey et al., 2014).
Similar to the previous studies that looked at babA (Linz et al., 2014; Solnick et al., 2004), the CT changes were identified at very early time points post inoculation. Although this would suggest that babA is dispensable for infection, we found that babA null mutants were not able to colonize the macaque. In the context of infection, these two pieces of evidence suggest that babA expression is important for establishment of infection, but is detrimental to the persistence of H. pylori within the macaque model. It is also plausible that BabA may also play a role in transmission of H. pylori between hosts. It has been shown that BabA can bind salivary mucins (Linden et al., 2008), which could aid in the process of oral-oral transmission. Thus, even when BabA expression appears to be totally lost within an animal, it is tempting to speculate that a small percentage of Leb binders are maintained within the population as a means to promote more efficient transmission of H. pylori.
In terms of establishment of colonization, binding of Lewis antigens on the inner layers of the gastric mucosa by BabA likely helps to establish H. pylori infection (Linden et al., 2008); however, if a strong immune response is mounted, H. pylori may silence expression of certain adhesins to distance itself from host defenses such as reactive oxygen species and responding immune cells. Since we found that at later time points the babA gene was lost through recombination with the other bab loci, simply turning off babA expression appears insufficient for immune evasion. Perhaps if the gene is still present, low levels of BabA are still able to be expressed through normal rates of phase variation at this locus. Alternatively, recombination of babA with the other bab loci may result in increased expression of BabB or BabC. In turn, this increased expression may provide some additional unknown benefit for H. pylori. In order to survive within the host, H. pylori must maintain a delicate balance in terms of interaction with host cells. The bacteria must remain close enough to host cells to access nutrients, but if the bacteria attach to the host cells too tightly, they risk mechanical clearance through the sloughing of the mucosa. Furthermore, overly intimate interaction with the host cells could prompt immune stimulation that could clear infection. Regulation of outer membrane proteins could help to modulate the level of bacterial attachment and help to facilitate persistent H. pylori infection, and perhaps BabB and BabC play crucial roles in this process.
Interestingly, while loss of BabA in the macaques occurs in 100% of animals either through replacement or a frame shift, studies looking at the presence of babA in human infections have found that expression of babA is highly variable. (Aspholm-Hurtig et al., 2004). A recent study that compared sequential isolates from human patients found changes in Leb binding in only 5/23 individuals (Nell et al., 2014). Furthermore, in that patient population changes in Leb binding were due predominantly to changes in the amino acid sequence of babA rather than a frame shift or replacement as seen in the animal studies (Nell et al., 2014). While the reason for differences in BabA loss between macaques and humans is unclear at present, it may reflect different host-specific microenvironments and immune responses that exist between the macaques and humans. Conversely, it may reflect differences induced during experimental infection versus natural infection.
Taken en masse, our data suggest that while BabA may be important in the earliest stages of infection in Rhesus macaques, clearly there is some negative selection that drives for babA loss. Given its surface exposure, we hypothesize that BabA may be lost as a result of selective pressure from the immune system. Furthermore, since H. pylori has a wide repertoire of outer membrane proteins, we propose that coordinated regulation of these proteins allows for H. pylori to evade and/or alter the host immune system while maintaining a certain level of attachment. Overall, temporal colonization studies are starting to shed light on how H. pylori adapts to the host environment over the course of long term colonization.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. R. Peek for providing strain J99. Research in the laboratories of D. Scott Merrell and Andre Dubois is/was made possible by grant CA082312 from the NIH. Research conducted in the laboratory of Thomas Boren was supported by grants from Vetenskapsrådet/VR and Cancerfonden, and the J.C. Kempe and Seth M. Kempe Memorial Foundation (TB). During the conduction of these studies, Dr. Andre Dubois passed away unexpectedly. Though ill for a length of time, Dr. Dubois did not want others to be concerned and was silent about this fact. Throughout his illness, he worked diligently and passionately on his research. His wisdom, generosity and expertise are sorely missed by his colleagues and the research community. The opinions and assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the NIH, the Department of Defense, or the Uniformed Services University of the Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberg A, Gideonsson P, Vallstrom A, Olofsson A, Ohman C, Rakhimova L, Boren T, Engstrand L, Brannstrom K, Arnqvist A. A repetitive DNA element regulates expression of the Helicobacter pylori sialic acid binding adhesin by a rheostat-like mechanism. PLoS pathogens. 2014;10:e1004234. doi: 10.1371/journal.ppat.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopyanz N, Bukanov NO, Westblom TU, Kresovich S, Berg DE. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acids Res. 1992;20:5137–5142. doi: 10.1093/nar/20.19.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Argent RH, Zhang Y, Atherton JC. Simple method for determination of the number of Helicobacter pylori CagA variable-region EPIYA tyrosine phosphorylation motifs by PCR. J Clin Microbiol. 2005;43:791–795. doi: 10.1128/JCM.43.2.791-795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, Lundberg C, Arnqvist A, Mahdavi J, Nilsson UJ, Velapatino B, Gilman RH, Gerhard M, Alarcon T, Lopez-Brea M, Nakazawa T, Fox JG, Correa P, Dominguez-Bello MG, Perez-Perez GI, Blaser MJ, Normark S, Carlstedt I, Oscarson S, Teneberg S, Berg DE, Boren T. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- Berg DE, Lelwala-Guruge J, Incecik ET, Srivastava K, Akopyants NS. H. pylori DNA Fingerprinting Using the Arbitrarily Primed PCR (AP-PCR) or Random Amplified Polymorphic DNA (RAPD) Method. Methods Mol Med. 1997;8:117–132. doi: 10.1385/0-89603-381-3:117. [DOI] [PubMed] [Google Scholar]
- Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- Bridge DR, Merrell DS. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut microbes. 2013;4:101–117. doi: 10.4161/gmic.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbeck JC, Hansen LM, Fong JM, Solnick JV. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect Immun. 2006;74:4375–4378. doi: 10.1128/IAI.00485-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover TL, Krishna US, Israel DA, Peek RM., Jr Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–957. [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Dubois A, Berg DE, Incecik ET, Fiala N, Heman-Ackah LM, Del Valle J, Yang M, Wirth HP, Perez-Perez GI, Blaser MJ. Host specificity of Helicobacter pylori strains and host responses in experimentally challenged nonhuman primates. Gastroenterology. 1999;116:90–96. doi: 10.1016/s0016-5085(99)70232-5. [DOI] [PubMed] [Google Scholar]
- Dubois A, Berg DE, Incecik ET, Fiala N, Heman-Ackah LM, Perez-Perez GI, Blaser MJ. Transient and persistent experimental infection of nonhuman primates with Helicobacter pylori: implications for human disease. Infect Immun. 1996;64:2885–2891. doi: 10.1128/iai.64.8.2885-2891.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang X, Achtman M, Suerbaum S. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- Fu Y, Zepeda-Gurrola RC, Aguilar-Gutierrez GR, Lara-Ramirez EE, De Luna-Santillana EJ, Rodriguez-Luna IC, Sanchez-Varela A, Carreno-Lopez R, Moreno-Medina VR, Rodriguez-Perez MA, Lopez-Vidal Y, Guo X. The detection of inherent homologous recombination between repeat sequences in H. pylori 26695 by the PCR-based method. Curr Microbiol. 2014;68:211–219. doi: 10.1007/s00284-013-0466-7. [DOI] [PubMed] [Google Scholar]
- Garcia-Ortiz MV, Marsin S, Arana ME, Gasparutto D, Guerois R, Kunkel TA, Radicella JP. Unexpected role for Helicobacter pylori DNA polymerase I as a source of genetic variability. PLoS Genet. 2011;7:e1002152. doi: 10.1371/journal.pgen.1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollub J, Ball CA, Binkley G, Demeter J, Finkelstein DB, Hebert JM, Hernandez-Boussard T, Jin H, Kaloper M, Matese JC, Schroeder M, Brown PO, Botstein D, Sherlock G. The Stanford Microarray Database: data access and quality assessment tools. Nucleic Acids Res. 2003;31:94–96. doi: 10.1093/nar/gkg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, Kraft C, Suerbaum S, Meyer TF, Achtman M. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 2005;1:e43. doi: 10.1371/journal.pgen.0010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey VC, Acio CR, Bredehoft AK, Zhu L, Hallinger DR, Quinlivan-Repasi V, Harvey SE, Forsyth MH. Repetitive sequence variations in the promoter region of the adhesin-encoding gene sabA of Helicobacter pylori affect transcription. Journal of bacteriology. 2014;196:3421–3429. doi: 10.1128/JB.01956-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM., Jr Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci U S A. 2001;98:14625–14630. doi: 10.1073/pnas.251551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennemann L, Brenneke B, Andres S, Engstrand L, Meyer TF, Aebischer T, Josenhans C, Suerbaum S. In vivo sequence variation in HopZ, a phase-variable outer membrane protein of Helicobacter pylori. Infect Immun. 2012;80:4364–4373. doi: 10.1128/IAI.00977-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, Reinhardt R, Correa P, Meyer TF, Josenhans C, Falush D, Suerbaum S. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci U S A. 2011;108:5033–5038. doi: 10.1073/pnas.1018444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersulyte D, Chalkauskas H, Berg DE. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol Microbiol. 1999;31:31–43. doi: 10.1046/j.1365-2958.1999.01140.x. [DOI] [PubMed] [Google Scholar]
- Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van Der Ende A, van Der Hulst RW, Wirth HP, Hook-Nikanne J, Thompson SA, Blaser MJ. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis. 2000;181:273–282. doi: 10.1086/315173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberg WRE, WidjojoKusumo A, Tytgat GN, Zanen HC. Identification of Campylobacter pyloridis isolates by restriction endonuclease DNA analysis. J Clin Microbiol. 1986;24:414–417. doi: 10.1128/jcm.24.3.414-417.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden SK, Wickstrom C, Lindell G, Gilshenan K, Carlstedt I. Four modes of adhesion are used during Helicobacter pylori binding to human mucins in the oral and gastric niches. Helicobacter. 2008;13:81–93. doi: 10.1111/j.1523-5378.2008.00587.x. [DOI] [PubMed] [Google Scholar]
- Linz B, Windsor HM, McGraw JJ, Hansen LM, Gajewski JP, Tomsho LP, Hake CM, Solnick JV, Schuster SC, Marshall BJ. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nature communications. 2014;5:4165. doi: 10.1038/ncomms5165. [DOI] [PubMed] [Google Scholar]
- Liu H, Merrell DS, Semino-Mora C, Goldman M, Rahman A, Mog S, Dubois A. Diet synergistically affects Helicobacter pylori-induced gastric carcinogenesis in nonhuman primates. Gastroenterology. 2009;137:1367–1379. e1361–1366. doi: 10.1053/j.gastro.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteo MJ, Armitano RI, Granados G, Wonaga AD, Sanches C, Olmos M, Catalano M. Helicobacter pylori oipA, vacA and dupA genetic diversity in individual hosts. J Med Microbiol. 2010;59:89–95. doi: 10.1099/jmm.0.011684-0. [DOI] [PubMed] [Google Scholar]
- Nell S, Kennemann L, Schwarz S, Josenhans C, Suerbaum S. Dynamics of Lewis b Binding and Sequence Variation of the babA Adhesin Gene during Chronic Helicobacter pylori Infection in Humans. mBio. 2014;5 doi: 10.1128/mBio.02281-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133:926–936. doi: 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci U S A. 2000;97:14668–14673. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Gonzalez-Valencia G, Deatherage B, Aviles-Jimenez F, Atherton JC, Graham DY, Torres J. Genetic analysis of Helicobacter pylori strain populations colonizing the stomach at different times postinfection. J Bacteriol. 2007;189:3834–3845. doi: 10.1128/JB.01696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun L, Audibert C, Le Lay G, Burucoa C, Fauchere JL, Picard B. Panmictic structure of Helicobacter pylori demonstrated by the comparative study of six genetic markers. FEMS Microbiol Lett. 1998;161:231–239. doi: 10.1111/j.1574-6968.1998.tb12953.x. [DOI] [PubMed] [Google Scholar]
- Salaun L, Linz B, Suerbaum S, Saunders NJ. The diversity within an expanded and redefined repertoire of phase-variable genes in Helicobacter pylori. Microbiology. 2004;150:817–830. doi: 10.1099/mic.0.26993-0. [DOI] [PubMed] [Google Scholar]
- Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A. 2004;101:2106–2111. doi: 10.1073/pnas.0308573100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, Benghezal M, Marshall BJ, Peek RM, Jr, Boren T, Solnick JV. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect Immun. 2010;78:1593–1600. doi: 10.1128/IAI.01297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talarico S, Whitefield SE, Fero J, Haas R, Salama NR. Regulation of Helicobacter pylori adherence by gene conversion. Mol Microbiol. 2012;84:1050–1061. doi: 10.1111/j.1365-2958.2012.08073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NS, Fox JG, Akopyants NS, Berg DE, Thompson N, Shames B, Yan L, Fontham E, Janney F, Hunter FM, et al. Long-term colonization with single and multiple strains of Helicobacter pylori assessed by DNA fingerprinting. J Clin Microbiol. 1995;33:918–923. doi: 10.1128/jcm.33.4.918-923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LJ, Danon SJ, Wilson JE, O’Rourke JL, Salama NR, Falkow S, Mitchell H, Lee A. Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect Immun. 2004;72:4668–4679. doi: 10.1128/IAI.72.8.4668-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.