

Abstract
In this study we investigated if Wnt/β-catenin signaling in mesenchymal progenitor cells plays a role in bone fracture repair and if DKK1-Ab promotes fracture healing through activation of β-catenin signaling. Unilateral open transverse tibial fractures were created in CD1 mice and in β-cateninPrx1ER conditional knockout (KO) and Cre-negative control mice (C57BL/6 background). Bone fracture callus tissues were collected and analyzed by radiography, micro-CT (μCT), histology, biomechanical testing and gene expression analysis. The results demonstrated that treatment with DKK1-Ab promoted bone callus formation and increased mechanical strength during the fracture healing processinCD1 mice. DKK1-Ab enhanced fracture repair by activation of endochondral ossification. The normal rate of bone repair was delayed when the β-catenin gene was conditionally deleted in mesenchymal progenitor cells during the early stages of fracture healing. DKK1-Ab appeared to act through β-catenin signaling to enhance bone repair since the beneficial effect of DKK1-Ab was abrogated in β-cateninPrx1ER conditional KO mice. Further understanding of the signaling mechanism of DKK1-Ab in bone formation and bone regeneration may facilitate the clinical translation of this anabolic agent into therapeutic intervention.
Keywords: Dkk1-Ab, β-Catenin, Conditional knockout, Fracture healing, Mesenchymal progenitor cells
Introduction
Skeletal fractures may occur as a consequence of trauma as well as fragility and represent a significant public health problem. There are over 6 million adults that suffer fractures in the United States annually [1–3]. These fractures can be associated with significant morbidity, costs, and health care utilization [4], especially when people with poor outcomes suffer with fractures.
Fracture healing is a specialized postnatal repair process that recapitulates aspects of embryonic skeletal development [5–9]. Wnt/β-catenin is one of the critical signaling pathways that regulate chondrogenesis, osteogenesis, and osteoclast formation. Clinical interest in this pathway was sparked by the discovery that osteoporosis pseudoglioma (OPPG), a disease characterized by low bone mass and recurrent fractures, was caused by loss of function mutations in LRP5 [10]. Immediately after this discovery, a gain-of-function mutation in LRP5 was identified that manifests a high bone mass phenotype [11–13]. Moreover, Lrp5 KO mice (Lrp5−/−) display decreases in bone mass, mechanical properties, and mechanosensitivity [14–16].
Dickkopf-1 (DKK1) is asecreted glycoprotein, which is a potent Wnt antagonist [17] and is vital for head and limb development [17,18]. DKK1 also acts within the bone to specifically block Wnt/β-catenin signaling in osteoblasts, thereby inhibiting osteoblast development and activity [19]. DKK1 exerts this function by binding to either of the single-pass transmembrane receptor proteins Kremen 1 or Kremen 2 and also within the carboxyl-terminal propeller domains of either the LRP5 or LRP6 co-receptors [20]. It is thought that increased DKK1 levels or activity may lead to impaired osteoblast activity and bone loss [21]. In humans, unbiased global gene expression analyses have identified DKK1 as a gene associated with bone mineral density (BMD) variation in postmenopausal Caucasian women [22], and elevated serum DKK1 concentrations were inversely associated with BMD in patients with osteoporosis [23]. In mice, the physiological role of DKK1 in the bone has been examined using the heterozygous DKK1 KO mice [24] and doubleridge mice harboring a hypomorphic allele of DKK1 [25]. Though complete loss of DKK1 function led to embryonic lethality [18], diminished DKK1 levels resulting from the lack of a functional DKK1 allele resulted in alterations in bone development and patterning [25] and increased bone mass [24]. In contrast, over expression of DKK1 resulted in lower BMD due to lower rates of bone formation [26–29]. Pre-clinical studies with DKK1 neutralizing antibody (DKK1-Ab) stimulated bone formation at both cortical and trabecular sites [30], increases bone mineral density in adult ovariectomy (OVX) mice [31], and promoted fracture healing and implant fixation in rodent models [30,32]. In addition, DKK1-Ab has been shown to reverse the bone destruction pattern observed in a mouse model of rheumatoid arthritis [33].
Although DKK1 is important in fracture repair, the mechanism is still unclear. Wnt pathway components (Wnt4, Fzd2, Lrp5 and β-catenin) were up-regulated at the fracture site within 3–5 days after injury [34]. A recent report demonstrated that β-catenin was localized in the nuclei of several types of cells, including periosteal cells and osteoblasts near the fracture site, mature chondrocytes of the fracture callus and osteocytes at cortical bone 5 days post-fracture. Furthermore, activation of the Wnt pathway appears to improve bone healing in mesenchymal stem cells [35]. In the latter studies, Ad-DKK1 treatment of fractures resulted in a failure to heal and was associated with the accumulation of undifferentiated mesenchymal cells [35]. In the present studies we investigated if DKK1-Ab promotes fracture healing through activating β-catenin signaling in mesenchymal progenitor cells using β-cateninPrx1ER conditional knockout (KO) mice.
Materials and method
Experimental animals
10-week-old male CD1 mice were subjected to tibial open fracture. After surgery, mice were divided into two groups: DKK1-Ab treatment group (25 mg/kg, subcutaneous injection, twice a week for 28 days); and Vehicle (PBS) control group.
To generate Prx1-CreER;β-cateninfx/fx (β-cateninPrx1ER) mice, β-cateninfx/fx mice [36] (obtained from Jackson Laboratory) were bred with Prx1-CreER transgenic mice [37] (obtained from Dr. Malcolm Logan, National Institute for Medical Research, London, UK). 10-week-old β-cateninPrx1ER mice (Prx1-CreER;β-cateninfx/fx) and Cre-negative control mice (β-cateninfx/fx) (C57BL/6 background) were subjected to tibia fracture. Mice were treated with DKK1-Ab or Vehicle as above. Tamoxifen (TM, Sigma, St. Louis, MO) was administered immediately after fracture surgery (1 mg/10 g body weight/day, i.p. injection for 5 days). DKK1-Ab was administered after tamoxifen induction.
Mouse genotyping was determined by PCR using a DNA extraction kit (Sigma, St. Louis, MO) from tail biopsy tissues 1 month after birth. PCR primer sequences for genotyping were as follows, upper primer 5′ – AAGGTAGAGTGATGAAAGTTGTT – 3′ and lower primer 5′ – CACCATGTCCTCTGTCTATTC – 3′ (324-base-pair PCR product). All mice had free access to food and water during entire study. All procedures conducted in this study were approved by IACUC committee of University of Rochester.
Tibial fracture model
A unilateral (right side) open transverse tibial fracture with intramedullary needle fixation was used as the bone fracture model similar to that described previously [38]. This model involves a standardized surgical approach that leads to a reproducible injury that resembles repair in human tibial fractures [38]. 10-week-old mice were anesthetized with Ketamine (60 mg/Kg) by intraperitoneal injection. A 1.5 cm-long skin incision was made along the anterior-medial surface of the shaved tibia. On the medial side of the patellar ligament, a 27 gauge syringe needle was inserted into the bone marrow cavity of the tibia through the tibial plateau. The needle was removed, and a No. 11 surgical blade was used to transect the diaphysis of the tibia at the midpoint. Either a 25 or 27 gauge needle was then inserted into the tibia to stabilize the fracture. Minimal damage was made to the adjacent regions of the periosteum during the surgical procedure. A 4-0 or 5-0 silk suture was used to close the wound and buprenorphine was administered in the drinking water for pain relief for the first three days after surgery [39].
Cre-recombination efficiency
To determine if the Prx1-CreER transgene could target floxed genes specifically in mesenchymal progenitor cells at the fracture site, Prx1-CreER transgenic mice were bred with RosaLacZ (R26R) or RosamT/mG reporter mice. Tamoxifen (TM, 1 mg/10 g body weight/day, i.p. injection for 5 days) was administered immediately after fracture and mice were sacrificed 5 or 10 days later for analysis. Cre-recombination efficiency was evaluated by X-gal staining. To evaluate Cre-recombination efficiency, we counted the number of X-gal positive cells and divided by total cell number in callus tissue.
Radiographic and μCT Analyses
CD1 mice, β-cateninPrx1ER mice and Cre-negative mice were sacrificed, at days 7, 10, 14, 21 and 28 post-surgery for tissue analysis. Radiographic analysis (Faxitron X-ray, Wheeling, IL) was performed on fracture samples in both anterior–posterior and lateral orientations are performed immediately after surgery to confirm that the osteotomy was complete and pinned correctly. After mice were sacrificed, fracture healing was examined (n = 10 mice at each time point) by assessment of bridging across cortices. The extent of bridging between the fracture gap was determined qualitatively in a blinded fashion by three independent investigators using the following criteria: 1) no healing (gap present with only rudimentary evidence of repair); 2) partial healing (some gap closure with evidence of bridging); and 3) complete healing (no gap with complete bridging). Specimens were scanned at 10.5-micron isotropic resolution using a Scanco VivaCT 40 (Scanco Medical AG, Switzerland) at indicated time points. Callus total volume (TV), callus bone volume (BV), callus mineralized volume fraction (BV/TV) (%) and callus bone mineral density (BMD) were determined (n = 6 in each time point). For the CD1 mice day 28 group, some animals died or the fracture procedure failed and so radiographs, μCT and histology (below) data were not shown for this group.
Biomechanical torsion testing
Soft tissue-free full length tibia bone samples were harvested (n = 6 at days 10, 14, 21, and 28). Tissues were fixed in aluminum square tubes (0.5 cm) filled with bone cement to make sure that fracture lines were in the middle of the interval. Fracture specimens were mounted on an EnduraTec TestBench™ system with a 200 N · mm torque cell (EnduraTec TestBench™ system, Bose Corp., Minnetonka, MN) and tested in torsion at a rate of 1°/s until failure to determine the torsional stiffness and ultimate torque [39].
Quantitative gene expression analysis
The fracture callus including 1 mm of adjacent bone on either side of the fracture line was harvested and total RNA was extracted using the PureLink™ RNA Mini Kit (Invitrogen, Carlsbad, CA) (n = 3 in each time point). One microgram total RNA was used to synthesize cDNA using iScripts cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time PCR amplification was performed using gene specific primers and SYBR green real-time PCR kit (Bio-Rad, Hercules, CA). The levels of the target gene expression were normalized to that of β-actin in the cDNA sample. Real-time RT-PCR analysis was performed using murine specific primers for chondrogenesis (Sox9, Col2a1, Aggrecan, Col10, Mmp9, Mmp13), osteogenesis related genes (Runx2, osterix, osteocalcin, Dkk1, β-catenin) and osteoclastogenesis related genes (Opg and Rankl). Forward and reverse primers specific for the genes are listed in Supplemental Table 1.
Histology & histomorphometry
Bone samples were harvested for histological analysis (n=6 at days 10, 14, 21, and 28). Tissues were fixed in 10% normal buffered formaldehyde for 3 days, decalcified in 14% EDTA solution for 14 days and then embedded in paraffin. Tissue sections at the fracture site were cut longitudinally at thickness of 3-μm and prepared for Alcian blue/H&E staining and TRAP staining (days 10 and 14). Histomorphometric analysis (n = 6 in each time point) was conducted using OsteoMetrics software (Decatur, GA). The external diameter of the tibia at the fracture line was determined at baseline to define the size of the region of interest. The callus was divided into proximal and distal aspects relative to the fracture line and a length equal to 1.5× baseline length was used to define the boundary of the proximal and distal region of interest [39]. The diameter of the callus was defined as the width of the region of interest. The mineralized volume of the cortices, the area of the periosteal calluses, and the mineralized and cartilaginous volume of the calluses were measured. The structural indices were calculated as follows: 1) tibial diameter (Ti.Dm) at the fracture gap: including Ct.Wi and Ma.Dm (mm); 2) mineralized area of the cortical (Ct): Md.Ar/Ct.B.Ar (%); 3) periosteal callus area: Ps.Cl.Ar (mm2); 4) mineralized area of the periosteal callus area: Md.Ar/Ps.Cl.Ar (%); and 5) cartilage area of the periosteal callus area: Cg.Ar/Ps.Cl.Ar (%). Abbreviations: Ar, area; B, bone; Cg, cartilage; Ct, cortical; Dm, diameter; Ps, periosteal; Ma, marrow; Md, mineralized; Wi, width; and Cl, callus [39]. Three histological sections taken close to the fracture line were used for histomorphometric analysis. Due to few animals died and fracture procedure failed in the group of day 28 time point, the day28 time point was eliminated from the histological and histomorphometric analyses.
Statistical analysis
Results were presented as the mean ± standard deviation. Statistical analyses included unpaired Student’s t-tests and one-way ANOVA followed by Tukey’s test. p < 0.05 was considered as significant.
Results
Cre-recombination efficiency in Prx1CreER;RosamT/mG and Prx1-CreER; RosaLacZ transgenic mice
To evaluate the targeting efficiency and specificity, we bred Prx1-CreER mice with RosamT/mG and RosaLacZ reporter mice, respectively. Analysis of frozen sections with fluorescent microscopy and X-gal staining in 10-week-old mice showed that Cre-recombination efficiency was approximately 63% in the callus tissue when Prx1-CreER transgenic mice (n = 3) were administrated with tamoxifen immediately following fracture. The mice were sacrificed at day 5 or 10 after the fracture. X-gal positive staining included mesenchymal cells, osteoblasts, chondrocytes and bone marrow cells in the callus tissue (Supplemental Fig. 1). A recent report showed that the 2.4 kb Prx1 promoter directed Cre transgene expression in osteochondro-progenitor cells in developing limb buds and in a subpopulation of periosteal cells that is closely associated with the cortical bone [40]. Our observations were consistent with these findings.
DKK1-Ab promotes bone fracture healing in mice
The surgery and pin fixation procedure resulted in a reproducible and consistent transverse gap at the midline of the tibial diaphysis. Radiographic analyses showed that administration of DKK1-Ab enhanced bone callus formation. Compared to the Vehicle treated group, the fracture gap was repaired more quickly following DKK1-Ab treatment with evidence of accelerated bridging by days 14 and 21 compared to controls (Fig. 1 and Table 1). Quantitation of bone volume by μCT showed a significant increase in bone volume at the fracture callus in mice treated with DKK1-Ab at days 10, 14, and 21 compared to controls (Figs. 2A and B). Results of biomechanical testing showed that treatment with DKK1-Ab enhanced bone mechanical strength in CD1 mice. The maximum torque and stiffness were significantly increased at days 14, 21 and 28 in DKK1-Ab treatment group compared to controls (Figs. 2C and D).
Fig. 1.
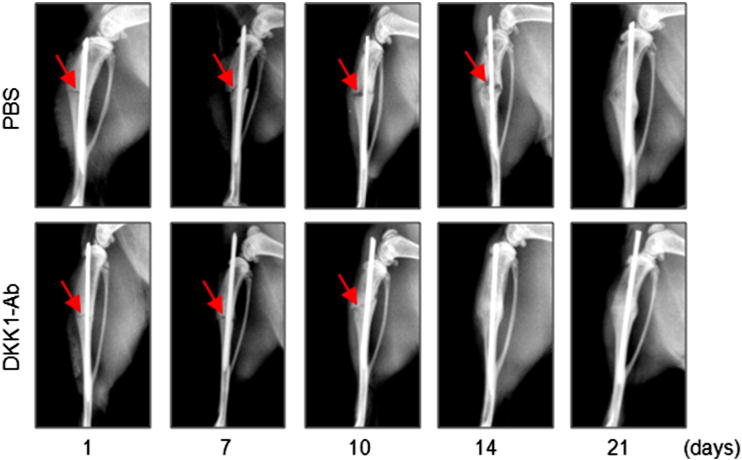
DKK1-Ab promotes bone fracture healing in CD-1 mice. Fracture procedure was conducted in 10-week-old mice treated with DKK1-Ab or Vehicle. X-ray radiographic analysis was performed in mice 1, 7, 10, 14, and 21 days after fracture. Radiographs show resolution of fracture lines at day 14 and 21 in mice receiving DKK1-Ab treatment, suggesting that fracture healing process is accelerated. Red arrows indicate the fracture lines. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Radiographic analysis of fracture gap.
| PBS
|
DKK1-Ab
|
|||||
|---|---|---|---|---|---|---|
| Healing | No | Partial | Healed | No | Partial | Healed |
| d7 | 10 | 0 | 0 | 9 | 1 | 0 |
| d10 | 7 | 3 | 0 | 5 | 5 | 0 |
| d14 | 2 | 8 | 0 | 0 | 4 | 6 |
| d21 | 0 | 6 | 4 | 0 | 0 | 10 |
N = 10 in each group.
Fig. 2.
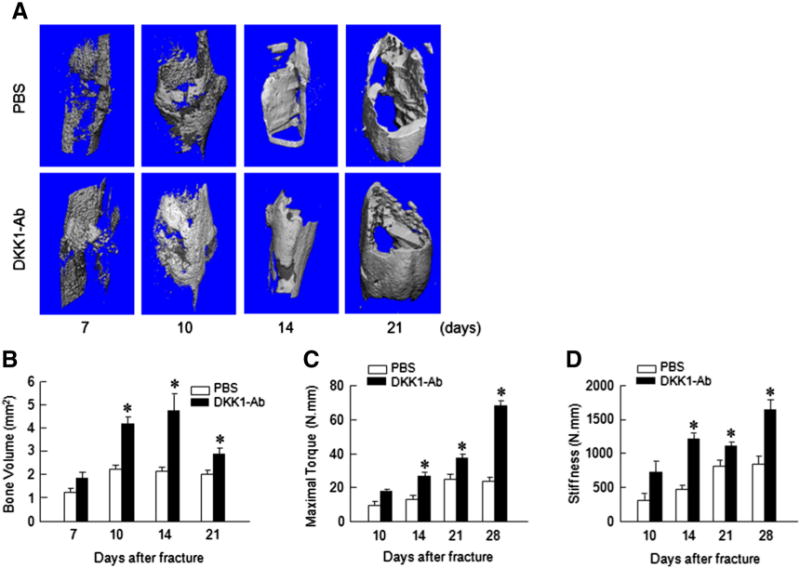
DKK1-Ab increases bone volume and mechanical strength of fracture callus. Micro-CT and mechanical testing were performed on bone samples derived from mice treated with DKK1-Ab or Vehicle at 7, 10, 14 and 21 days after fracture. Micro-CT analysis showed that bone volume and BV/TV were increased in callus tissues 10, 14 and 21 days after fracture in the mice treated with DKK1-Ab. Mechanical testing showed that maximal torque and stiffness were increased by DKK1-Ab treatment 14, 21 and 28 days after fracture. Data are presented as means ± SD. *p < 0.05, one-way ANOVA followed by Tukey’s test, n = 6.
Histologic and histomorphometric analyses also showed that the progression of fracture healing was accelerated in DKK1-Ab treated mice. Cartilage area determined by Alcian blue staining was significantly enlarged by day 7 (Figs. 3A and B) but only transiently and woven bone area was increased by days 10–21 post-operatively in DKK1-Ab treatment group (Figs. 3A and C). The increase in cartilage area is supported by gene expression data which showed that expression of the early cartilage marker gene Sox9 was increased at day 7 in the DKK1-Ab treatment group (Fig. 4A). The levels of the cartilage marker Col2a1 were also increased at days 7–14 (Fig. 4B) and this was followed by expression of the later cartilage hypertrophic marker Col10a1 which increased at days 14–28 in the DKK1-Ab treatment group (Fig. 4C). The expression of bone marker genes (Runx2 and osterix) was increased at day 14–28 in DKK1-Ab treatment group (Figs. 4D and E). The expression of osteocalcin (OC) was increased at day 21 and 28 in DKK1-Ab treatment group (Fig. 4F). These changes in bone markers are consistent with the process and timing of endochondral ossification during the fracture healing process. The expression of DKK1 was decreased at days 7 and 10 and the expression of β-catenin was up-regulated at days 7 and 10 in DKK1-Ab treatment group (Fig. 4G and H).
Fig. 3.
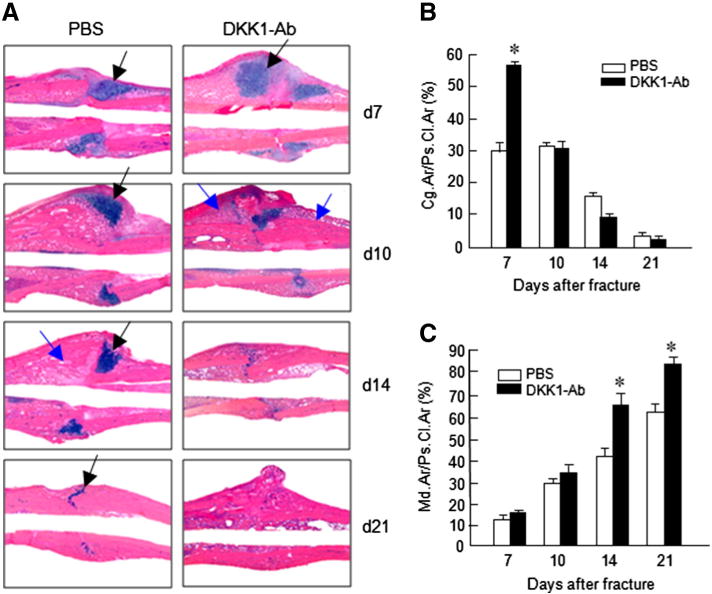
DKK1-Ab increases cartilage and bone volume of fracture callus. Histological analysis showed that fracture healing process was accelerated by DKK1-Ab treatment. On day 7, cartilage area of callus tissues (Alcian blue staining positive area) was increased after DKK1-Ab treatment (A, B). On day 10–21, new woven bone formation in callus tissues was enhanced by DKK1-Ab treated (A, C). Black arrows: cartilage area; blue arrows: woven bone area. Data are presented as means ± SD. *p < 0.05, one-way ANOVA followed by Tukey’s test, n = 6.
Fig. 4.
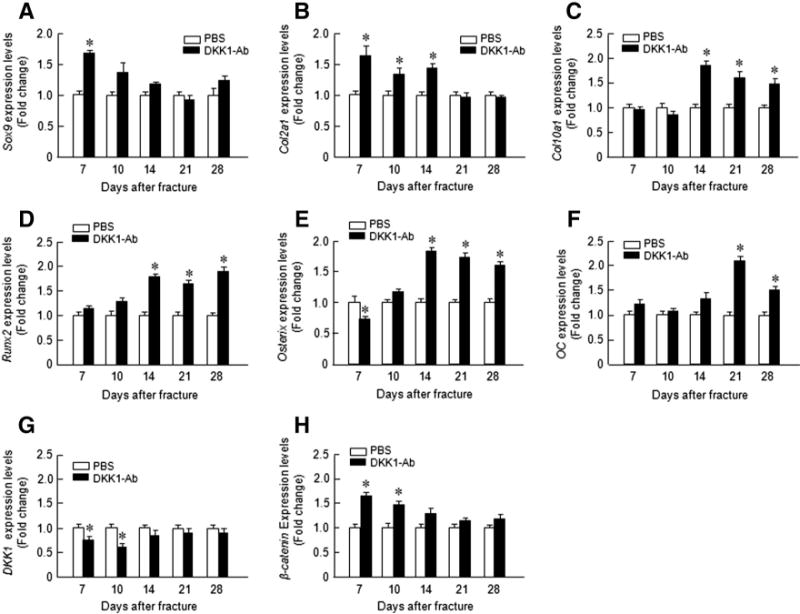
DKK1-Ab enhanced chondrocyte- and osteoblast-specific marker gene expression in callus tissues of mice. Total RNA was extracted from callus tissues 7, 10, 14, 21 and28 days after fracture in the mice treated with or without DKK1-Ab. Expression of chondrocyte-specific genes was examined. Expression of Sox9 was increased at day 7 (A); expression of Col2a1 was increased at days 7, 10, and 14 (B); and expression of Col10a1 was increased at day 14, 21, and 28 (C). Expression of Runx2 and Osterix was increased at days 14, 21, and 28 (D and E) and expression of osteocalcin(OC) was increased at days 21 and 28 (F). Expression of Dkk1 and β-catenin was examined. Expression of DKK1 was decreased at days 7 and 10 (G) and expression of β-catenin was increased at days 7 and 10 (H). Data are presented as means ± SD. *p < 0.05, one-way ANOVA followed by Tukey’s test, n = 3.
Significant bone loss in β-cateninPrx1ER conditional KO mice
To further determine the mechanism of DKK1-Ab enhanced bone fracture healing, we tested the effect of DKK1-Ab on fracture healing in β-catenin conditional knockout (KO) mice. We first crossed β-cateninflox/flox mice with Prx1-CreER mice to determine if loss of β-catenin signaling in MSC led to a change in bone mass. Tamoxifen induction was performed in 2-week-old mice and changes in bone mass were analyzed in 3-month-old mice. We found that trabecular bone volume in the femur and in tibia was significantly reduced in β-cateninPrx1ER mice by μCT and histological analyses (Figs. 5A–C). Consistent with this finding, μCT analysis also revealed that bone mineral density (BMD), trabecular numbers (Tb.N.) and connectivity density (Conn D) were significantly reduced in β-cateninPrx1ER mice (Figs. 5D, E and H) and trabecular separation (Tb.Sp.) was significantly increased in β-cateninPrx1ER mice (Fig. 5F). No significant change was found in structure model index (Fig. 5G). These results suggest that bone mass and bone mechanical properties were significantly reduced in β-cateninPrx1ER mice.
Fig.5.
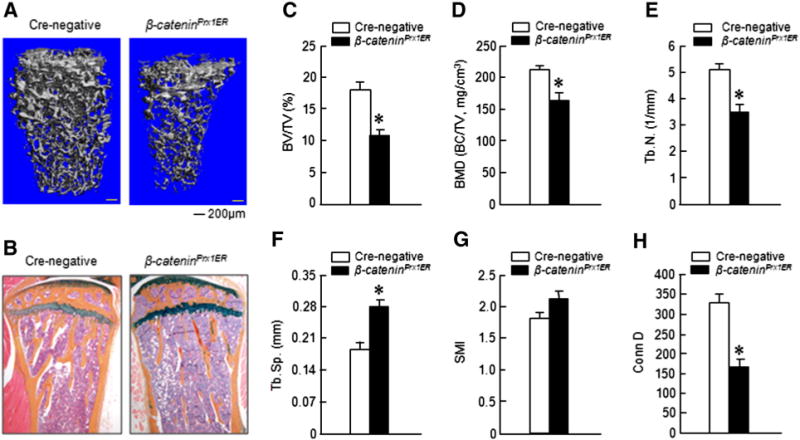
Bone mass was decreased in β-cateninPrx1ER conditional KO mice. Tamoxifen was administered to 2-week-old mice and μCT and histological analyses were performed in 3-month-old mice. Significant bone loss was observed in μCT 3D images and histological sections (A and B). Quantitative μCT analysis showed that bone volume (BV/TV), bone mineral density (BMD), trabecular numbers (Tb.N.) and connectivity density (Conn D) were significantly decreased in β-cateninPrx1ER mice (C–E and H). In contrast, trabecular separation (Tb.Sp.) was significantly increased (F). *p < 0.05, unpaired Student t-test, n = 4.
Positive effect of DKK1-Ab on fracture healing is lost in β-cateninPrx1ER conditional KO mice
Radiographic analysis showed that bone callus formation was delayed in β-cateninPrx1ER mice compared to Cre-negative control mice (Fig. 6 and Table 2). In β-cateninPrx1ER mice, the presence of a fracture gap was clearly evident in both DKK1-Ab and Vehicle treated groups at days 14, 21 and 28. These results suggest that the effects of DKK1-Ab may be mediated via β-catenin signaling. In contrast, the fracture gap showed better resolution at days 14 and 21 in Cre-negative mice with or without DKK1-Ab treatment. Moreover, there was almost no fracture gap visible at day 28 in Cre-negative with DKK1-Ab treatment, thus further confirming the beneficial role of DKK1-Ab on the normal fracture healing process (Fig. 6). μCT data showed that the bone volume of β-cateninPrx1ER mice was decreased compared to Cre-negative control group in day 21 and 28 Cre-negative groups(Figs. 7A and B). Treatment with DKK1-Ab significantly increased bone volume at days 10, 14 and 28 in Cre-negative mice, thus supporting the findings above (Fig. 7B). In contrast, however, there was no significant difference in bone volume between DKK1-Ab and Vehicle treated groups in the β-cateninPrx1ER mice (Fig. 7B). Results of biomechanical testing showed that treatment with DKK1-Ab enhanced bone strength in Cre-negative but not β-cateninPrx1ER mice (Figs. 7C and D). The maximal torque and the stiffness were significantly increased at days 10–28 with DKK1-Ab in Crenegative control mice. In contrast, the maximal torque was significantly reduced at days 21 and 28 in β-cateninPrx1ER mice compared to Cre-negative control group (Fig. 7C) and the stiffness was also significantly decreased at days 14–28 in β-cateninPrx1ER mice (Fig. 7D). DKK1-Ab had no significant effect on maximal torque and the stiffness through days 10–28 in β-cateninPrx1ER mice (Figs. 7C and D).
Fig. 6.
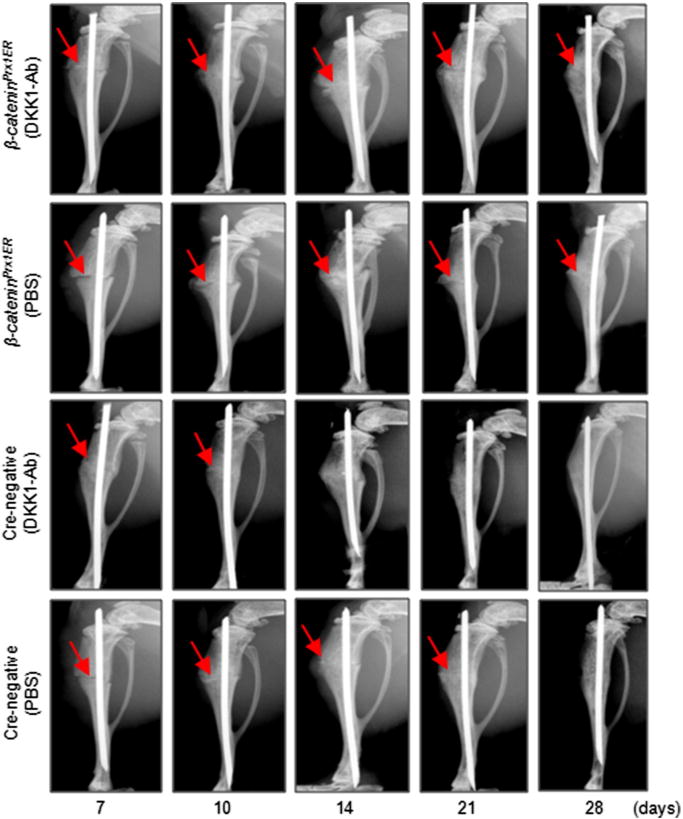
DKK1-Ab had no significant effect on fracture healing in β-cateninPrx1ER mice. Fracture procedure was performed in 10-week-old β-cateninPrx1ER and Cre-negative control mice. Tamoxifen was injected for 5 days immediately following fracture followed by treatment with DKK1-Ab. Radiographs showed that fracture healing was accelerated by DKK1-Ab treatment in Cre-negative control mice. In contrast, DKK1-Ab had no significant effect on fracture healing in β-cateninPrx1ER mice. Fracture lines are still clear at day 21 and 28 in β-cateninPrx1ER mice with or without DKK1-Ab. Red arrows: fracture lines. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Radiographic analysis of fracture gap.
| PBS (Cre−)
|
DKK1-Ab (Cre−)
|
PBS (cKO)
|
DKK1-Ab (cKO)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Healing | No | Partial | Healed | No | Partial | Healed | No | Partial | Healed | No | Partial | Healed |
| d7 | 10 | 0 | 0 | 9 | 1 | 0 | 10 | 0 | 0 | 10 | 0 | 0 |
| d10 | 7 | 3 | 0 | 5 | 5 | 0 | 8 | 2 | 0 | 8 | 2 | 0 |
| d14 | 2 | 8 | 0 | 0 | 4 | 6 | 4 | 6 | 0 | 3 | 7 | 0 |
| d21 | 0 | 6 | 4 | 0 | 0 | 10 | 1 | 7 | 2 | 1 | 8 | 1 |
| d28 | 0 | 1 | 9 | 0 | 0 | 10 | 0 | 4 | 6 | 0 | 4 | 6 |
N = 10 in each group.
Fig. 7.
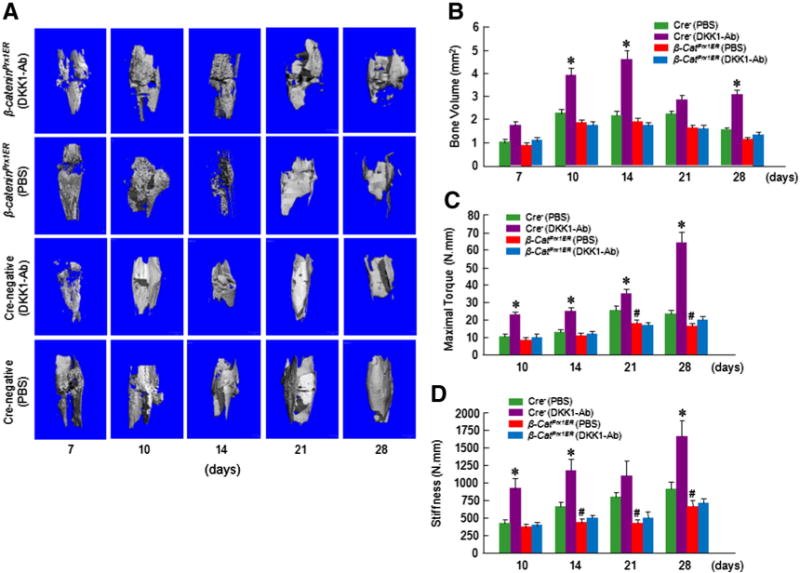
DKK1-Ab had no significant effect on bone structure and mechanical strength in β-cateninPrx1ER mice. μCT and mechanic testing were performed on bone samples collected from mice 7, 10, 14, 21 and 28 days after fracture. Results of μCT analysis and mechanical testing showed that there was a significant increase in bone volume at days 10 and 14 (A–C) and significant increases in maximal torque at day 10–28 and significant increases in the stiffness day 10, 14 and 28 in Cre-negative control mice treated with DKK1-Ab. In contrast, there were no significant changes in bone volume, maximal torque, and stiffness in β-cateninPrx1ER mice treated with DKK1-Ab (B–E). Significant reductions in maximal torque (at day 21 and 28) and the stiffness (at day 14–28) were observed in β-cateninPrx1ER mice compared to Cre-negative control mice. Data are presented as means ± SD. *p < 0.05, between DKK1-Ab and PBS groups (Cre-negative control mice); #p < 0.05, between Cre-negative and β-cateninPrx1ER groups (without DKK1-Ab); two-way ANOVA followed by Tukey’s test, n = 6.
These findings were supported by histologic and histomorphometric analyses which showed that the progression of fracture healing was accelerated by DKK1-Ab treatment only in Cre-negative control mice and not in β-cateninPrx1ER mice (Figs. 8A–C). In contrast, there was no significant difference between DKK1-Ab and Vehicle treatment groups in β-cateninPrx1ER mice (Figs. 8A–C). The cartilage area was enlarged by day 7 and the woven bone area was increased by days 14 and 21 postoperatively following DKK1-Ab treatment in Cre-negative control mice (Figs. 8B and C). There was less cartilage area at day 7 and less woven bone at day 21 in β-cateninPrx1ER mice.No significant difference between DKK1-Ab and Vehicle treatment groups was found in cartilage and woven bone areas in β-cateninPrx1ER mice (Figs. 8B and C).
Fig. 8.
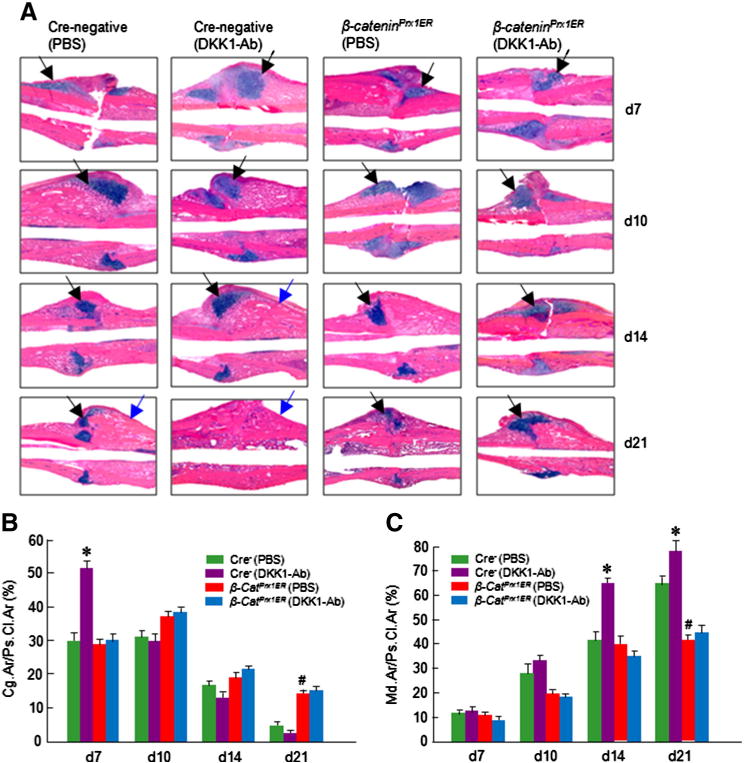
DKK1-Ab had no significant effect on cartilage and bone volume of fracture callus in β-cateninPrx1ER mice. Histological analysis was performed on day 7, 10, 14 and 21 samples and showed that cartilage volume was increased at day 7 and woven bone was increased at days 14 and 21 in DKK1-Ab treated Cre-negative control mice (A–C). In contrast, DKK1-Ab had no significant effect on cartilage and woven bone volume in fracture callus of β-cateninPrx1ER mice (A–C). Black arrows: cartilage area and blue arrows: woven bone area. Data are presented as means ± SD. *p < 0.05, between DKK1-Ab and PBS groups (Cre-negative control mice);#p < 0.05, between Cre-negative and β-cateninPrx1ER groups (without DKK1-Ab); two-way ANOVA followed by Tukey’s test, n = 6.
Gene expression data showed that the expression of cartilage marker genes (Sox9 and Col2a1) was increased at days 7 and 10 and Aggrecan (Agc1) was increased at day 14 with DKK1-Ab treatment in Cre-negative control mice (Figs. 9A–C). Col10a1 expression was increased at days 14–28 after DKK1-Ab treatment in control mice (Fig. 9D). Expression of Mmp9 and Mmp13 was increased at days 14 and 21 with DKK1-Ab treatment in control mice (Figs. 9E and F). The expression of bone marker gene (Runx2 and osterix) was increased at days 14–28 in DKK1-Ab treatment group in control mice (Figs. 9G and H). The expression of OC was increased at days 21 and 28 in DKK1-Ab treatment group in control mice (Fig. 9I). The expression of DKK1 was decreased at days 7 and 10 and the expression of β-catenin was upregulated at days 7 and 10 in DKK1-Ab treatment group in control mice (Figs. 9J and K). In β-cateninPrx1ER mice, Sox9 expression was decreased at days 7–14; Col2a1 expression was decreased at day 7; Runx2 and osterix expression was reduced at days 14 and 21 and OC expression was decreased at day 28. In addition, Dkk1 expression was increased at days 10 and 14 and β-catenin expression was decreased at days 7 and 10. Treatment with DKK1-Ab did not alter the expression of the genes mentioned above in β-cateninPrx1ER mice (Figs. 9A–K). These findings therefore support the hypothesis that the effects of DKK1-Ab in accelerating fracture healing are mediated by the β-catenin pathway.
Fig. 9.
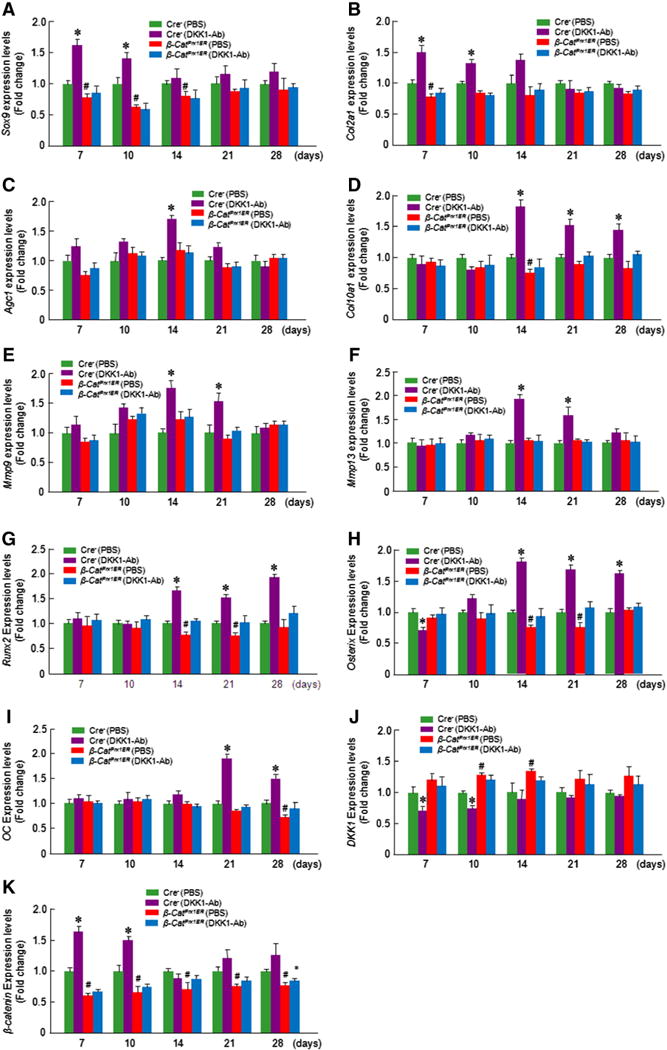
DKK1-Ab had no significant effect on gene expression of fracture callus in β-cateninPrx1ER mice. Total RNA was extracted from callus tissues 7, 10, 14, 21 and 28 days after fracture. In Cre-negative control mice treated with DKK1-Ab, the expression of Sox9 and Col2a1 was increased at day 7 and 10 (A and B), Aggrecan (Agc1) expression was increased at day 14 (C); expression of Col10a1 was increased at days 14, 21, and 28 (D); and expression of Mmp9 and Mmp13 was increased at days 14 and 21 (E and F). InCre-negative control mice treated with Dkk1-Ab, the expression of Runx2 and Osterix was increased at days 14, 21, and 28 (G and H) and expression of osteocalcin (OC) was increased at days 21 and 28 (I). Expression of DKK1 was decreased at days 7 and 10 (J); and expression of β-catenin was increased at days 7 and 10 (K) in Cre-negative control mice treated with Dkk1-Ab. There was no significantly difference between DKK1-Ab and PBS treated groups in β-catenin cKO mice (A–K). Data are presented as means ± SD. *p < 0.05, between DKK1-Ab and PBS groups (Cre-negative control mice); #p < 0.05, between Cre-negative and β-cateninPrx1ER groups (without DKK1-Ab); two-way ANOVA followed by Tukey’s test, n = 3.
We also performed TRAP staining (Fig. 10A) and examined expression of Opg and Rankl in fracture callus. Although we detected up-regulation of Opg and Rankl at days 14 and 21, no significant changes in osteoclast numbers were found after DKK1-Ab treatment (Figs. 10A–C), suggesting that DKK1-Ab has no significant effect on osteoclast formation.
Fig. 10.
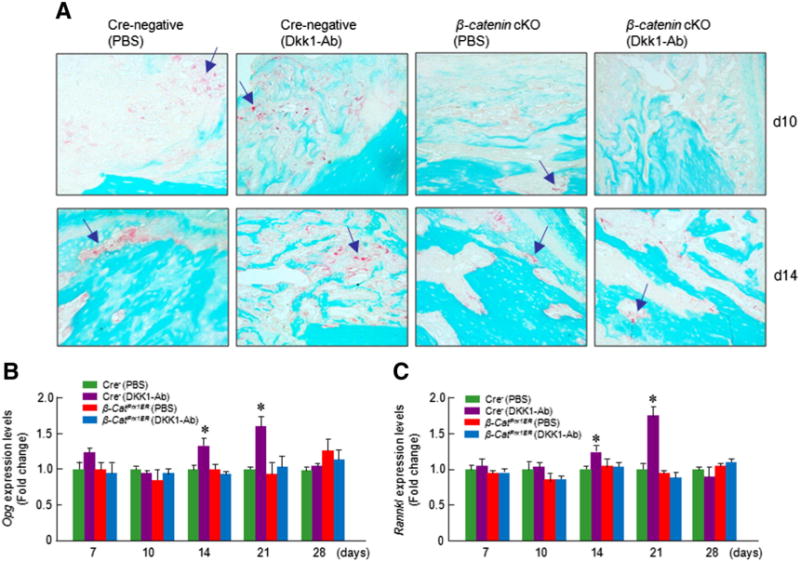
DKK1-Ab had no significant effect on osteoclast formation. TRAP staining was performed on day 10 and 14 samples and showed no significant difference in osteoclast numbers after DKK1-Ab treatment in Cre-negative control and β-cateninPrx1ER conditional KO mice (A). Expression of osteoclast-specific genes was examined. Expression of Opg and Rankl was increased at days 14 and 21 after DKK1-Ab treatment in Cre-negative control mice (B and C).
Discussion
Although fractures are a common health problem, our knowledge of the biological processes that promote and enable fracture healing is still limited. Most fractures heal following temporary immobilization and/or surgical fixation. However, a small percentage (3–10%) of fractures fail to heal ultimately resulting in non-union [41]. Agents that promote bone formation may be conducive to accelerating fracture healing and reducing fracture-associated complications (e.g., delayed union or nonunion) [42]. To date, the only anabolic drugs approved in the United States to stimulate bone formation are bone morphogenic protein (BMP) 2 and 7 and parathyroid hormone (PTH). Although effective, these biological agents have limitations, such as that some can only be given locally (e.g. BMPs) and some can be only given for a short period of time because of safety concerns (e.g. PTH). For these reasons, efforts are focused on developing safer and more easily administered agents to stimulate bone formation.
DKK1 has previously been shown to inhibit normal fracture repair. For example, increased DKK1 levels or activity in LRP5 knockout (KO) animals resulted in impaired healing response [35,43], and the study of human nonunion fractures showed elevated Dkk1 expression in stromal cells harvested from nonunion fractures [44]. In mice, the reduction of DKK1 has been reported to increase BMD due to higher rates of bone formation [45–48,24]. Furthermore, DKK1-Ab administration in rats and mice led to increased bone formation at both cortical and trabecular sites [49]. These results support the conclusion that DKK1 impedes complete healing of fractures and inhibition of DKK1 enhances bone formation, making DKK1 an attractive therapeutic target to promote bone healing. The results of the present study have identified the potential utility of antibody targeting DKK1 (DKK1-Ab) in accelerating fracture healing. In the present model, radiographic analyses represented the direct evidence that administration of DKK1-Ab enhanced bone callus formation. Bridging of the fracture gap occurred earlier at day 14 following DKK1-Ab treatment, compared to day 21 in the control group. These data are consistent with histologic and histomorphometric analyses, which demonstrated that DKK1-Ab appeared to enhance fracture healing by an early and transient increase in chondrogenesis followed by a wave of increased osteogenesis. At 1 week following the fracture, DKK1-Ab-treated mice exhibited a fracture callus with a larger cartilaginous compartment compared tocontrol mice. Moreover, the callus size was bigger in DKK1-Ab-treated mice than control mice. The increased chondrogenic response was associated with increased expression of collagen II, a marker of cartilage formation, by days 7–14. Also Sox9 used as a marker for chondrogenic differentiation [50,51], was up-regulated by DKK1-Ab at day 7. We then assessed expression levels of osteogenic marker genes, such as Runx2, osterix and osteocalcin. The up-regulation of these genes, in agreement with our histological findings, suggests an acceleration of both chondrogenic and osteogenic differentiation at the fracture sites upon the treatment with DKK1-Ab. The histomorphometric and radiographic results were confirmed by μCT analysis, which indicates higher bone volumes in DKK1-Ab treatment group than in controls. Biomechanical testing showed that the maximum torque and stiffness were significantly increased in DKK1-Ab treatment group compared to controls, further revealing that treatment with DKK1-Ab enhances the strength of the healing bones. Our findings are in accordance with the efficacy of DKK1-Ab reported previously, confirming that DKK1-Ab treatment improves bone repair [32,52].
The physiological role of Wnt/β-catenin signaling pathway in the regulation of bone mass has been well recognized [53–55]. Consistent with previous findings, suppression of the Wnt/β-catenin signaling pathway led to a dramatic increase of bone loss [56]. In this study, we report that this increase in bone loss is associated with deletion of β-catenin. Our findings further highlight the importance of Wnt/β-catenin signaling pathway in bone metabolism. Thus the above evidence suggests that Wnt/β-catenin contributes to the maintenance of bone mass and supports our hypothesis that inhibition of Wnt signaling by β-catenin deletion would attenuate bone formation.
β-Catenin signaling has been considered to be essential for bone healing [57–60]. Inhibition of Wnt signaling by Lrp5 deletion displayed impaired fracture repair [53]. Several gene coding for Wnt signaling molecules and their downstream targets are upregulated at the fracture site within 3–5 days after injury, including Wnt4, Wnt5a, Wnt5b, Frzb, β-catenin, Dvl, Tcf1, Lrp5, fibronectin, phosphatases 2A, connexin 43, as well as c-myc, suggesting that Wnt signaling molecules play an important role in the early stage fracture healing [34,60]. In the present studies, we observed that administration of DKK1-Ab increased the expression of β-catenin while decreasing the expression of DKK1, suggesting that Wnt/β-catenin signaling was activated by DKK1-Ab. This raised the intriguing possibility that DKK1-Ab facilitated fracture healing by activation of Wnt/β-catenin signaling. In order to further understand the role of Wnt/β-catenin signaling in acceleration of bone fracture healing induced by DKK1-Ab, we performed a series of tibial fracture experiments in β-cateninPrx1ER mice and wild-type littermates, as well as CD1 mice treated with DKK1 Ab. The bone fracture healing process was clearly delayed in β-cateninPrx1ER mice and the callus of β-cateninPrx1ER mice was less well developed. β-Catenin deletion influenced bone callus formation and reduced bone volume, as demonstrated by radiographic and μCT analyses. Also the maximum torque and stiffness were reduced in β-cateninPrx1ER mice. An explanation for this finding may be that loss of function of Wnt/β-catenin signaling pathway gives rise to a profound decrease of bone formation and regeneration in mice [56]. In addition, gene expression data showed that the expression of cartilage marker genes (Sox9 and Col2a1) and bone marker genes (Runx2, osterix and osteocalcin) were down-regulated in β-cateninPrx1ER mice, indicating that endochondral bone repair was inhibited. With histology, we saw a smaller callus volume, as well as reduced cartilage and woven bone areas in β-cateninPrx1ER mice. This may be due to lack of endochondral ossification resulting from inhibition of Wnt/β-catenin signaling pathway during the fracture healing process. These data suggest that β-catenin as a central component of canonical Wnt signaling is necessary to ensure normal bone repair. In contrast to Cre-negative control mice, delayed bone fracture healing in β-cateninPrx1ER mice cannot be reversed by DKK1-Ab, demonstrated by radiographic and μCT analyses, whereas bone fracture healing process in CD1 mice can be accelerated by administration of DKK1-Ab. The result was confirmed by histological and biomechanical testing, showing an increased bone content and strength of fracture callus only in Cre-negative control mice but not in β-cateninPrx1ER mice after DKK1 Ab treatment. Gene expression analysis provided genetic evidence suggesting that DKK1-Ab cannot enhance bone fracture healing in the absence of β-catenin. Taken together, our findings identify a role for Wnt/β-catenin signaling in the acceleration of bone fracture healing stimulated by DKK1-Ab and provide preclinical evidence to demonstrate that the effect of DKK1-Ab on enhancement of fracture healing is dependent on Wnt/β-catenin signaling. Although the impact of DKK1-Ab on bone fracture healing was significantly reduced compared to Cre-negative control mice, we did observe a weaker effect of DKK1-Ab on fracture healing in β-catenin conditional KO mice (Table 2). This may be due to the incomplete deletion of β-catenin in callus tissue since the recombination efficiency was estimated to be 63% in Prx1-CreER mice. Further investigation is required by choosing a different dosing regimen of tamoxifen to improve the Cre-recombination efficiency.
Fracture repair is a regenerative process that involves both intramembranous and endochondral ossification [58]. Mesenchymal stem cells directly differentiate into osteoblasts to produce a ‘hard callus’ without an intermediate cartilage template in intramembranous bone repair, whereas both chondrocytes and osteoblasts are responsible for the formation of bone via a cartilaginous model in endochondral bone repair [61,62]. Wnt/β-catenin signaling plays a key role in mesenchymal cell proliferation and differentiation and determines the commitment of cell fate towards the osteoblast or adipocyte lineage [63]. In vitro, activating the Wnt/β-catenin signaling by miR-346 positively regulates human MSC osteogenic differentiation [64]. Suppressing Wnt/β-catenin signaling by FOXOs restrains MSC from differentiating into osteoblasts while promoting adipogenesis [65]. In addition, activation of Wnt/β-catenin signaling has a dominant role in osteoblast proliferation and survival [21]. Upregulation of Lrp5 mRNA in the fracture callus and over expression of β-catenin in proliferating chondrocytes, osteoblasts and periosteal osteoprogenitor cells within the fracture callus, all indicate that Wnt/β-catenin signaling is active in endochondral and intramembranous ossification [34]. Collectively, the studies described previously provide compelling evidence that Wnt/β-catenin signaling acts on both chondrocytes and osteoblasts during fracture repair. In cartilage and bone formation phases of fracture repair, pluripotent mesenchymal stem cells (MSCs) differentiate into osteochondral progenitor cells and then further differentiate into chondrocytes and osteoblasts [58]. At this stage, precise regulation of β-catenin signaling could be important to allow differentiation of mesenchymal cells into chondrocyte and osteoblast lineage cells [58]. In the absence of β-catenin, osteochondral progenitor cells are committed to the chondrocyte lineage [58]. Active β-catenin expression in mature chondrocytes and osteoblasts was observed [58]. Once cells begin to show phenotypic features of either chondrocyte or osteoblast precursors, they exhibit β-catenin-mediated and TCF-dependent transcriptional activity, suggesting the role of β-catenin signaling in the initial phase of MSC differentiation [35]. Depletion of of β-catenin in committed osteoblasts led to a significant reduction in calcified callus formation, along with incomplete bone bridging across the fracture gap [35]. DKK1 prevents early stages of bone repair by blocking Wnt/β-catenin/TCF signaling and the differentiation of MSCs into chondrocyte or osteoblasts [66].
The findings reported here strongly suggest that, by augmenting Wnt/β-catenin signaling, DKK1-Ab contributes to the acceleration of bone fracture healing. Although not measured directly, we speculate that the most likely explanation for this finding is that DKK1-Ab activates Wnt/β-catenin signaling by blocking DKK1, thereby promoting endochondral ossification and acceleration of bone fracture healing. DKK1-Ab may be useful as a potential anabolic agent for the treatment of bone fractures and also delayed union or nonunion fractures.
Supplementary Material
Acknowledgments
This research has been supported by Contract #: 200901460 from Amgen Inc. H.J. was partially supported by grants Y211184 from Zhejiang Provincial Natural Science Foundation of China and by grants 2012R10064, 2012C37087 and 2011R50022-01 from the Science and Technology Department of Zhejiang Province. H.J. and M.Q were also supported by the Program for Zhejiang Leading Team of S&T Innovation, Key Laboratory of Zhejiang Province and Zhejiang Chinese Medical University. We would also like to acknowledge Dr. Malcolm Logan (MRC-National Institute for Medical Research, London, UK) for providing us with Prx1-CreER transgenic mice. Dr. Hua-Zhu Ke and Philip Babij are Amgen employees and they received Amgen stock or stock options.
Footnotes
Disclosures
Ke and Babij are Amgen employees and have received Amgen stock or stock options, and Chen received research contract from Amgen. All other co-authors have no conflict of interest.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bone.2014.07.039.
References
- 1.Kanis JA, McCloskey EV, Johansson H, Cooper C, Rizzoli R, Reginster JY, Scientific Advisory Board of the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO), Committee of Scientific Advisors of the International Osteoporosis of the International Osteoporosis Foundation (IOF) European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2013;24:23–57. doi: 10.1007/s00198-012-2074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanis JA, Oden A, McCloskey EV, Johansson H, Wahl DA, Cooper C, IOF Working Group on Epidemiology and Quality of Life A systematic review of hip fracture incidence and probability of fracture worldwide. Osteoporos Int. 2012;23:2239–56. doi: 10.1007/s00198-012-1964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Budhia S, Mikyas Y, Tang M, Badamgarav E. Osteoporotic fractures: a systematic review of U.S. healthcare costs and resource utilization. Pharmacoeconomics. 2012;30:147–70. doi: 10.2165/11596880-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Court-Brown CM, Caesar B. Epidemiology of adult fractures: a review. Injury. 2006;37:691–7. doi: 10.1016/j.injury.2006.04.130. [DOI] [PubMed] [Google Scholar]
- 5.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–84. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 6.Allen MR, Hock JM, Burr DB. Periosteum: biology, regulation, and response to osteoporosis therapies. Bone. 2004;35:1003–12. doi: 10.1016/j.bone.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Deschaseaux F, Sensebe L, Heymann D. Mechanisms of bone repair and regeneration. Trends Mol Med. 2009;15:417–29. doi: 10.1016/j.molmed.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson C, Alpern E, Miclau T, Helms JA. Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev. 1999;87:57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 9.Vortkamp A, Pathi S, Peretti GM, Caruso EM, Zaleske DJ, Tabin CJ. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mech Dev. 1998;71:65–76. doi: 10.1016/s0925-4773(97)00203-7. [DOI] [PubMed] [Google Scholar]
- 10.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 11.Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, et al. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18:960–74. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 12.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–21. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 14.Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, II, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–14. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A. 2005;102:17406–11. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 17.Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–62. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- 18.Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, et al. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell. 2001;1:423–34. doi: 10.1016/s1534-5807(01)00041-7. [DOI] [PubMed] [Google Scholar]
- 19.Butler JS, Queally JM, Devitt BM, Murray DW, Doran PP, O’Byrne JM. Silencing Dkk1 expression rescues dexamethasone-induced suppression of primary human osteoblast differentiation. BMC Musculoskelet Disord. 2010;11:210. doi: 10.1186/1471-2474-11-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276–87. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Drake MT. Potential role for therapies targeting DKK1, LRP5, and serotonin in the treatment of osteoporosis. Curr Osteoporos Rep. 2012;10:93–100. doi: 10.1007/s11914-011-0086-8. [DOI] [PubMed] [Google Scholar]
- 22.Reppe S, Refvem H, Gautvik VT, Olstad OK, Hovring PI, Reinholt FP, et al. Eight genes are highly associated with BMD variation in postmenopausal Caucasian women. Bone. 2010;46:604–12. doi: 10.1016/j.bone.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Butler JS, Murray DW, Hurson CJ, O’Brien J, Doran PP, O’Byrne JM. The role of Dkk1 in bone mass regulation: correlating serum Dkk1 expression with bone mineral density. J Orthop Res. 2011;29:414–8. doi: 10.1002/jor.21260. [DOI] [PubMed] [Google Scholar]
- 24.Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–45. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 25.MacDonald BT, Adamska M, Meisler MH. Hypomorphic expression of Dkk1 in the doubleridge mouse: dose dependence and compensatory interactions with Lrp6. Development. 2004;131:2543–52. doi: 10.1242/dev.01126. [DOI] [PubMed] [Google Scholar]
- 26.Guo J, Liu M, Yang D, Bouxsein ML, Saito H, Galvin RJ, et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010;11:161–71. doi: 10.1016/j.cmet.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–66. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao GQ, Wu JJ, Troiano N, Insogna K. Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab. 2011;29:141–8. doi: 10.1007/s00774-010-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Grisanti M, Fan W, Asuncion FJ, Tan HL, Dwyer D, et al. Dickkopf-1 regulates bone formation in young growing rodents and upon traumatic injury. J Bone Miner Res. 2011;26:2610–21. doi: 10.1002/jbmr.472. [DOI] [PubMed] [Google Scholar]
- 31.Glantschnig H, Scott K, Hampton R, Wei N, McCracken P, Nantermet P, et al. A ratelimiting role for Dickkopf-1 in bone formation and the remediation of bone loss in mouse and primate models of postmenopausal osteoporosis by an experimental therapeutic antibody. J Pharmacol Exp Ther. 2011;338:568–78. doi: 10.1124/jpet.111.181404. [DOI] [PubMed] [Google Scholar]
- 32.Agholme F, Isaksson H, Kuhstoss S, Aspenberg P. The effects of Dickkopf-1 antibody on metaphyseal bone and implant fixation under different loading conditions. Bone. 2011;48:988–96. doi: 10.1016/j.bone.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–63. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]
- 34.Zhong N, Gersch RP, Hadjiargyrou M. Wnt signaling activation during bone regeneration and the role of Dishevelled in chondrocyte proliferation and differentiation. Bone. 2006;39:5–16. doi: 10.1016/j.bone.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, et al. Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 37.Hasson P, Del Buono J, Logan MP. Tbx5 is dispensable for forelimb outgrowth. Development. 2007;134:85–92. doi: 10.1242/dev.02622. [DOI] [PubMed] [Google Scholar]
- 38.Schindeler A, Morse A, Harry L, Godfrey C, Mikulec K, McDonald M, et al. Models of tibial fracture healing in normal and Nf1-deficient mice. J Orthop Res. 2008;26(8):1053–60. doi: 10.1002/jor.20628. [DOI] [PubMed] [Google Scholar]
- 39.Mi M, Jin H, Wang B, Yukata K, Sheu TJ, Ke QH, et al. Chondrocyte BMP2 signaling plays an essential role in bone fracture healing. Gene. 2013;512:211–8. doi: 10.1016/j.gene.2012.09.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawanami A, Matsushita T, Chan YY, Murakami S. Mice expressing GFP and CreER in osteochondro progenitor cells in the periosteum. Biochem Biophys Res Commun. 2009;386:477–82. doi: 10.1016/j.bbrc.2009.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tzioupis C, Giannoudis PV. Prevalence of long-bone non-unions. Injury. 2007;38(Suppl. 2):S3–9. doi: 10.1016/s0020-1383(07)80003-9. [DOI] [PubMed] [Google Scholar]
- 42.Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33(5):747–83. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- 43.Kim JB, Leucht P, Lam K, Luppen C, Ten Berge D, Nusse R, et al. Bone regeneration is regulated by wnt signaling. J Bone Miner Res. 2007;22(12):1913–23. doi: 10.1359/jbmr.070802. [DOI] [PubMed] [Google Scholar]
- 44.Bajada S, Marshall MJ, Wright KT, Richardson JB, Johnson WE. Decreased osteogenesis, increased cell senescence and elevated Dickkopf-1 secretion in human fracture non union stromal cells. Bone. 2009;45:726–35. doi: 10.1016/j.bone.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 45.Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25(2):178–89. doi: 10.1359/jbmr.090730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 47.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24(10):1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 48.MacDonald BT, Joiner DM, Oyserman SM, Sharma P, Goldstein SA, He X, et al. Bone mass is inversely proportional to Dkk1 levels in mice. Bone. 2007;41(3):331–9. doi: 10.1016/j.bone.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grisanti M, Niu QT, Fan W, et al. Dkk-1 inhibition increases bone mineral density in rodents. J Bone Miner Res. 2006;21:S25. [Google Scholar]
- 50.Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–46. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bridgewater LC, Lefebvre V, de Crombrugghe B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem. 1998;273:14998–5006. doi: 10.1074/jbc.273.24.14998. [DOI] [PubMed] [Google Scholar]
- 52.Komatsu DE, Mary MN, Schroeder RJ, Robling AG, Turner CH, Warden SJ. Modulation of Wnt signaling influences fracture repair. J Orthop Res. 2010;28:928–36. doi: 10.1002/jor.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams BO, Insogna KL. Where Wnts went: the exploding field of Lrp5 and Lrp6 signaling in bone. J Bone Miner Res. 2009;24(2):171–8. doi: 10.1359/jbmr.081235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. 2009;13(4):485–96. doi: 10.1517/14728220902841961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res. 2010;25(9):1897–904. doi: 10.1002/jbmr.161. [DOI] [PubMed] [Google Scholar]
- 56.Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ. Updateon Wnt signaling in bone cell biology and bone disease. Gene. 2012;492(1):1–18. doi: 10.1016/j.gene.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi: 10.1016/j.gene.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 58.Silkstone D, Hong H, Alman BA. Beta-catenin in the race to fracture repair: in it to Wnt. Nat Clin Pract Rheumatol. 2008;4:413–9. doi: 10.1038/ncprheum0838. [DOI] [PubMed] [Google Scholar]
- 59.Sisask G, Marsell R, Sundgren-Andersson A, Larsson S, Nilsson O, Ljunggren O, et al. Rats treated with AZD2858, a GSK3 inhibitor, heal fractures rapidly without endochondral bone formation. Bone. 2013;54:126–32. doi: 10.1016/j.bone.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 60.Hadjiargyrou M, Lombardo F, Zhao S, Ahrens W, Joo J, Ahn H, et al. Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair J Biol Chem. 2002;277:30177–82. doi: 10.1074/jbc.M203171200. [DOI] [PubMed] [Google Scholar]
- 61.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 62.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 2003;335(Suppl):S7–S21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 63.Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and WNT signalling. Trends Endocrinol Metab. 2009;20(1):16–24. doi: 10.1016/j.tem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Cai J, Cai XH, Chen L. miR-346 regulates osteogenic differentiation of human bone marrow-derived mesenchymal stem cells by targeting the Wnt/β-catenin pathway. PLoS One. 2013;8(9):e72266. doi: 10.1371/journal.pone.0072266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iyer S, Ambrogini E, Bartell SM, Han L, Roberson PK, de Cabo R, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123(8):3409–19. doi: 10.1172/JCI68049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Secreto FJ, Hoeppner LH, Westendorf JJ. Wnt signaling during fracture repair. Curr Osteoporos Rep. 2009;7(2):64–9. doi: 10.1007/s11914-009-0012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.