

Abstract
Objective
The roles of microRNAs (miRNAs) in coronary heart disease (CHD) have not been well characterized. This study sought to systematically characterize the complex genomic architecture of CHD by integrating whole blood miRNA and mRNA expression with genetic variation in 186 CHD cases and 186 controls.
Approach and Results
At FDR<0.2, 15 miRNAs were differentially expressed between CHD cases and controls. To explore regulatory mechanisms, we integrated miRNA and mRNA expression with genotype data genome-wide to investigate miRNA and mRNA associations and relations of genetic variation to miRNAs. We identified a large number of correlated miRNA-mRNA pairs and genetic loci that appear to regulate miRNA levels. Subsequently, we explored the relations of these complex molecular associations to CHD status. We identified a large difference in miRNA-mRNA associations between CHD cases and controls, as demonstrated by a significantly higher proportion of inversely correlated miRNA-mRNA pairs in cases vs. controls (80% vs. 30%; p<1e-16), suggesting a genome-wide shift in the regulatory structure of the transcriptome in CHD. The differentially co-expressed miRNA-mRNA pairs showed enrichment for CHD risk genetic variants affecting both miRNA and mRNA expression levels, implicating a putatively causal role in CHD. Furthermore, three miRNAs (miR-1275, miR-365a-3p, and miR-150-5p) were associated with an mRNA co-expression module that was causally linked to CHD and reflected dysregulation of B-cell centered immune function.
Conclusions
Our results provide novel evidence that miRNAs are important regulators of biological processes involved in CHD via genetic control and via their tight co-expression with mRNAs.
Keywords: microRNA expression, coronary heart disease, systems biology, genetics
Introduction
Coronary heart disease (CHD) is a multifactorial disease involving the complex interaction of genetic and environmental factors. In recent years, genetic and genomic approaches have been applied, revealing a variety of genes associated with CHD phenotypes. At the genetic level, genome-wide association studies (GWAS) and candidate gene studies have identified scores of loci associated with CHD1, 2. At the transcriptomic level, associations of CHD with mRNA expression levels of protein-coding genes have been reported in several independent studies3–6. Most of these investigations identified relatively few genes that were significantly differentially expressed between CHD cases and controls, and there is little overlap among the genes identified. The failure to identify replicable CHD gene signatures may reflect the small effect sizes and complexity of differentially expressed genes in CHD. By using an integrative network analysis strategy, we recently identified a co-expression network module that we suspect of being causally associated with CHD7.
MicroRNAs (miRNAs), a class of small noncoding RNAs, represent an important mediator of post-transcriptional regulation of mRNA expression or mRNA translation into protein8–10. The importance of miRNAs in cardiovascular disorders is becoming increasingly recognized. Highly specific miRNA expression patterns have been correlated with different cardiovascular disorders11, such as heart failure12–14, myocardial infarction15 and vascular remodeling11.
Despite several early successes identifying molecular underpinnings of CHD at the levels of genetic sequence and of mRNA and miRNA expression, the relations between these three molecular elements and their multidimensional interactions in relation to CHD susceptibility remain largely unknown. Recent studies have revealed that mRNA and miRNA transcript levels behave as heritable quantitative traits and display significant association with genetic variation16–20. The association of single nucleotide polymorphisms (SNPs) with the expression of mRNAs or miRNAs has been proposed as a mechanism that can affect disease risk and drug treatment response because mRNA (or miRNA) expression-associated SNPs (eSNPs) affect gene expression or miRNA targeting affinity21. Given the complexity of the molecular underpinnings of complex diseases like CHD, which is likely to be due in part to disrupted heterotypic SNP–miRNA–mRNA associations, integrative genomic analyses that incorporate all three dimensions of molecular data are necessary to investigate and characterize the complex changes in the regulatory machinery and their effects on biological functions and disease risks.
To this end, we assayed miRNA expression in 186 CHD cases and 186 age, sex, and statin treatment matched controls from the Framingham Heart Study (FHS), and integrated miRNA profiling with SNP and mRNA expression data from the same individuals. Using an integrative analysis framework that utilized these comprehensive genome-wide resources (Figure 1), we examined the complex multidimensional SNP-miRNA-mRNA relations in the context of CHD. At first, we sought to establish links between genome-wide associations of genetic variants with miRNA and mRNA expression by identifying miRNA eSNPs, mRNA eSNPs, and miRNA-mRNA co-expression pairs (Figure 1A). Second, we sought to find molecular associations between SNPs, miRNAs, and mRNAs in relation to CHD status (Figure 1B). We identified differentially expressed miRNAs (miRNA signatures) associated with CHD, differential miRNA-mRNA co-expression pairs in relation to CHD, and miRNAs associated with mRNA differential co-expression modules in the context of CHD. These CHD related miRNAs and mRNAs were, in turn, integrated with results from CHD GWAS1 and with eSNPs, to determine inferentially, which transcriptomic differences were putatively causal for CHD and which were downstream responses to CHD. In this way, we dissected the role of miRNAs in regulating gene networks associated with CHD.
Figure 1. Integrative Genomics Analysis Framework Depicting the Genome-wide Evaluation of the Relations between SNPs, miRNAs, and mRNAs, and the Identification of CHD-related Signals.
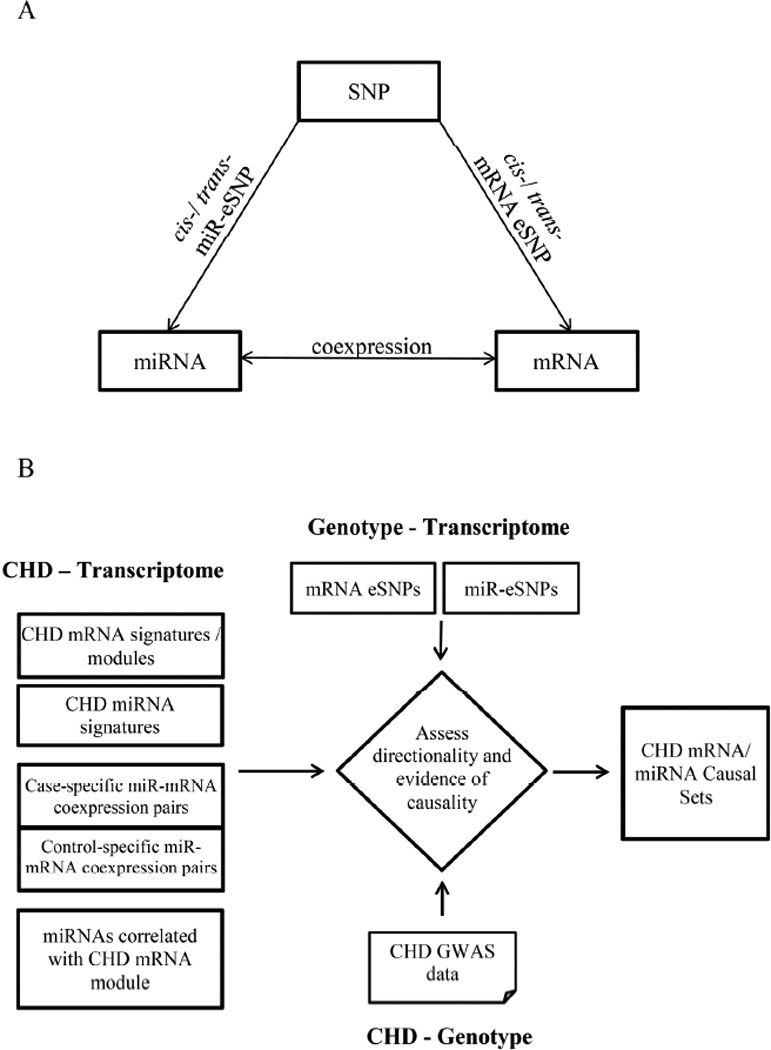
A) Genome-wide evaluation of SNP-miRNA-mRNA associations. The genome-wide evaluation includes the identification of mRNA/miRNA eSNPs, and miRNA-mRNA expression pairs. B) Identification of CHD-related molecular relationships in CHD case-control samples. The overall analysis includes five steps. First, identify CHD differential mRNA signatures and mRNA co-expression module (this step and results were reported in our previous paper7). Second, identify differentially expressed miRNA signatures in CHD vs. controls. Third, identify miRNA-mRNA co-expression patterns in CHD cases and controls, and then identify differential miRNA-mRNA co-expression pairs between the two groups. Fourth, identify miRNAs correlated with CHD differential mRNA co-expression module. Fifth, integrate the CHD miRNA and mRNA sets identified in Step 1 to 4 with CHD GWAS as well as miRNA and mRNA eSNPs by the SNP set enrichment method35, in order to assess the directionality of causality.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Demographics of the study population
We designed a CHD case control study consisting of prevalent CHD cases and age, sex, and statin-use matched controls in the FHS. In two recent reports 3, 7, we explored differential mRNA expression and derived gene networks associated with CHD in 188 case-control pairs. Among the 188 pairs, 186 pairs had successful miRNA profiling and were used in subsequent analyses. The clinical characteristics of the 186 CHD case-control pairs are described in Supplementary Table I.
Influence of blood cell types on CHD associated miRNA expression differences
As miRNA expression levels might be influenced by differences in the proportions of different cell types in whole blood, we assessed the correlations between miRNAs and cell type proportions imputed from whole blood mRNA expression (see the Methods). At false discovery rate (FDR) <0.05, 20 miRNAs showed significant correlations with one or more of the seven cell types (Supplementary Table II). This analysis suggested that cell type proportions affect only a small fraction of miRNAs. The remaining majority of miRNAs may reflect signals relatively independent of cell types. However, we cannot exclude the possibility of small cell type effects. Therefore, we carried out two sets of analysis: one using cell count-adjusted miRNA data and the other using unadjusted data. Adjusting for cell counts did not have a major influence on our results. Therefore, in the subsequent sections, we focus on results from the unadjusted analysis, but also report differences if any were observed.
Identification of differentially expressed miRNAs between CHD cases and controls
Among the 752 miRNAs assayed, 271 miRNAs were expressed in more than 50 individuals and were used for differential expression analysis. None of the miRNAs was found to be differentially expressed between cases and controls at FDR <0.05; 15 miRNAs were differentially expressed at FDR<0.2 (Table 1). A volcano plot (Supplementary Figure I) revealed that these 15 miRNAs all had increased expression in CHD cases compared to controls (fold change ranged from 1.20 to 1.55 in average miRNA expression levels). Among the 15 “signature” miRNAs, miR-423-5p was previously identified as a circulating biomarker for heart failure in plasma13; miR-422a was reported to be expressed in both monocytes and atherosclerotic plaque22. Analysis of miRNA data from three previously published studies revealed associations with cardiovascular disease for miR-19113, miR-58913, miR-423-5p13, 23, miR-339-3p23, miR-422a23, 24, miR-42523, miR-50023 and miR-55024 at p<0.05 in plasma13, 23 or platelets24. To our knowledge, the other miRNA signatures that we identified have not been reported previously in relation to CHD.
Table 1.
Differentially Expressed miRNAs in Coronary Heart Disease with FDR<0.2
| miRNA | FC* | p-Value | FDR† | Peak miR-eSNP | miR-eSNP p-Value |
|---|---|---|---|---|---|
| miR-550 | 1.36 | 4e-4 | 0.11 | ||
| miR-339-3p | 1.25 | 0.003 | 0.20 | rs13242526‡ | 8.2e-7 |
| miR-425-3p | 1.26 | 0.004 | 0.20 | rs2239675 | 3.4e-9 |
| miR-502-3p | 1.25 | 0.004 | 0.20 | rs17376328 | 7.4e-7 |
| miR-342-3p | 1.36 | 0.005 | 0.20 | rs12978278 | 5.1e-7 |
| miR-324-5p | 1.28 | 0.007 | 0.20 | rs709713 | 7.2e-7 |
| miR-423-5p | 1.30 | 0.007 | 0.20 | ||
| miR-191-5p | 1.55 | 0.008 | 0.20 | rs7118671 | 3.6e-7 |
| miR-422a | 1.21 | 0.009 | 0.20 | ||
| miR-616-3p | 1.31 | 0.011 | 0.20 | ||
| miR-1275 | 1.23 | 0.009 | 0.20 | rs12977021 | 7.0e-7 |
| miR-505-5p | 1.23 | 0.010 | 0.20 | rs11858860 | 6.4e-7 |
| miR-500a-5p | 1.26 | 0.010 | 0.20 | ||
| miR-589-3p | 1.20 | 0.011 | 0.20 | ||
| miR-339-5p | 1.20 | 0.011 | 0.20 | rs6951245‡ | 1.8e-10 |
FC indicated fold change of miRNA average expression values adjusted for age, sex, mean difference and diabetes status. The positive FC values indicated the miRNA upregulation in CHD cases.
FDR was estimated by Benjamini–Hochberg method.
miR-eSNPs marked with ‡ were cis-miR-eSNPs and the others were trans-miR-eSNPs.
Identification of miRNA regulatory SNPs (miR-eSNPs)
To gain insight into genetic regulation of miRNAs, we analyzed the associations between genome-wide SNP genotypes and miRNA expression by pooling CHD cases and controls together (N=361). At FDR<0.05, we identified 123 cis-miR-eSNPs for 13 miRNAs (the full list is provided in Supplementary Table III; the top cis-miR-eSNPs are reported in Table 2). Among the top signals in Table 2, miR-100 and miR-125 had two cis-miR-eSNPs (rs10750218 and rs11218891) that overlapped with those identified by Borel et al. for the same miRNAs20, supporting the reliability of the cis signals identified in our study. This finding also supports the notion that miRNAs have their own cis-regulatory elements. rs7941030 a cis-miR-eSNP for both miR-100 (p=3.7e-6) and miR-125 (p=1.9e-5), was previously found to be associated with high-density lipoprotein cholesterol and total cholesterol in genome-wide association testing25. Among these cis-miR-eSNPs, 72% are located in the 5’-upstream region, suggesting that these miRNA-eSNPs may regulate miRNA expression by perturbing the regulatory regions of miRNAs. We did not find any cis-miR-eSNPs in the seed regions of the miRNAs.
Table 2.
Top cis-miR-eSNPs
| miRNA | Chr. (miRNA) |
Peak miR-eSNP |
Chr. (SNP) |
Allele | SNP Location | miR-eSNP R2 |
miR-eSNP p-Value |
miR-eSNP FDR |
|---|---|---|---|---|---|---|---|---|
| miR-100-5p | 11 | rs10892855 | 11 | A/C | Intergenic | 0.23 | <1e-16 | <1e-16 |
| miR-125b-5p | 11 | rs11218646 | 11 | C/T | Intergenic | 0.18 | <1e-16 | <1e-16 |
| miR-133a | 18, 20 | rs6062278 | 20 | A/G | Intronic (C20orf200) | 0.13 | 1.1e-12 | 9.8e-8 |
| miR-339-5p | 7 | rs6951245 | 7 | G/A | Intronic (C7orf50) | 0.10 | 1.8e-10 | 8.0e-6 |
| miR-127-3p | 14 | rs9671394 | 14 | C/T | Intergenic | 0.09 | 7.1e-9 | 8.0e-5 |
| miR-30a-3p | 6 | rs9364167 | 6 | G/A | Intronic (C6orf155) | 0.18 | 2.9e-9 | 1.6e-4 |
| miR-654-5p | 14 | rs12882815 | 14 | C/G | Intergenic | 0.09 | 3.6e-8 | 6.5e-4 |
| miR-218-5p | 4, 5 | rs7726430 | 5 | A/T | Intronic (SLIT3) | 0.07 | 1.9e-7 | 2.6e-3 |
| miR-339-3p | 7 | rs13242526 | 7 | C/G | Intronic (COX19) | 0.06 | 8.2e-7 | 8.6e-3 |
| miR-370 | 14 | rs7141210 | 14 | T/C | Intergenic | 0.07 | 1.2e-6 | 0.01 |
| miR-204-5p | 9 | rs567490 | 9 | T/C | Intronic (TRPM3) | 0.13 | 3.4e-6 | 0.03 |
| miR-941 | 20 | rs2427565 | 20 | C/A | Intronic (DNAJC5) | 0.07 | 3.1e-6 | 0.03 |
| miR-543 | 14 | rs17099614 | 14 | C/G | Intergenic | 0.06 | 7.2e-6 | 0.04 |
As miR-eSNPs that affect miRNA expression may in turn affect their target mRNAs, we compared the cis-miR-eSNPs identified in this study with the mRNA-eSNPs identified in the same individuals in our previous study. In doing so, we found little overlap (overlapping eSNPs are listed in Supplementary Table IV). Considering that cis-miR-eSNPs are likely trans-mRNA-eSNPs whereas trans-eSNPs are difficult to detect, we speculate that the lack of overlap between the mRNA and miRNA eSNPs might result from the fact that a large proportion of SNP-miRNA associations were not detectable given the moderate sample size of our study.
Identification of CHD-related miRNA-mRNA co-expression pairs
To further explore the regulatory mechanisms involving miRNAs in CHD, we performed association analysis between miRNAs and mRNAs. At FDR<0.05, we identified 5539 miRNA-mRNA co-expression pairs in the CHD case group, 6558 pairs in the control group, and 26,115 pairs in the pooled sample (Supplementary Table V and Supplementary Figure II). These results support the existence of extensive associations between miRNAs and mRNAs.
There were 691 miRNA-mRNA co-expression pairs common to the CHD case and control groups (Figure 2A) and all of these pairs showed consistent directions in their correlation coefficients (Figure 2B). The genes in this consistent set of miRNA-mRNA pairs were found to be enriched for intracellular transport (Bonferroni correction [BF] p=3.6e-10) and cellular protein localization (BF p=1.2e-6) (Table 3). This set most likely reflects the common regulatory relations between miRNA and mRNA under different physiological or pathological conditions.
Figure 2. Distinct Patterns of miRNA-mRNA Correlation in CHD Cases and Controls.
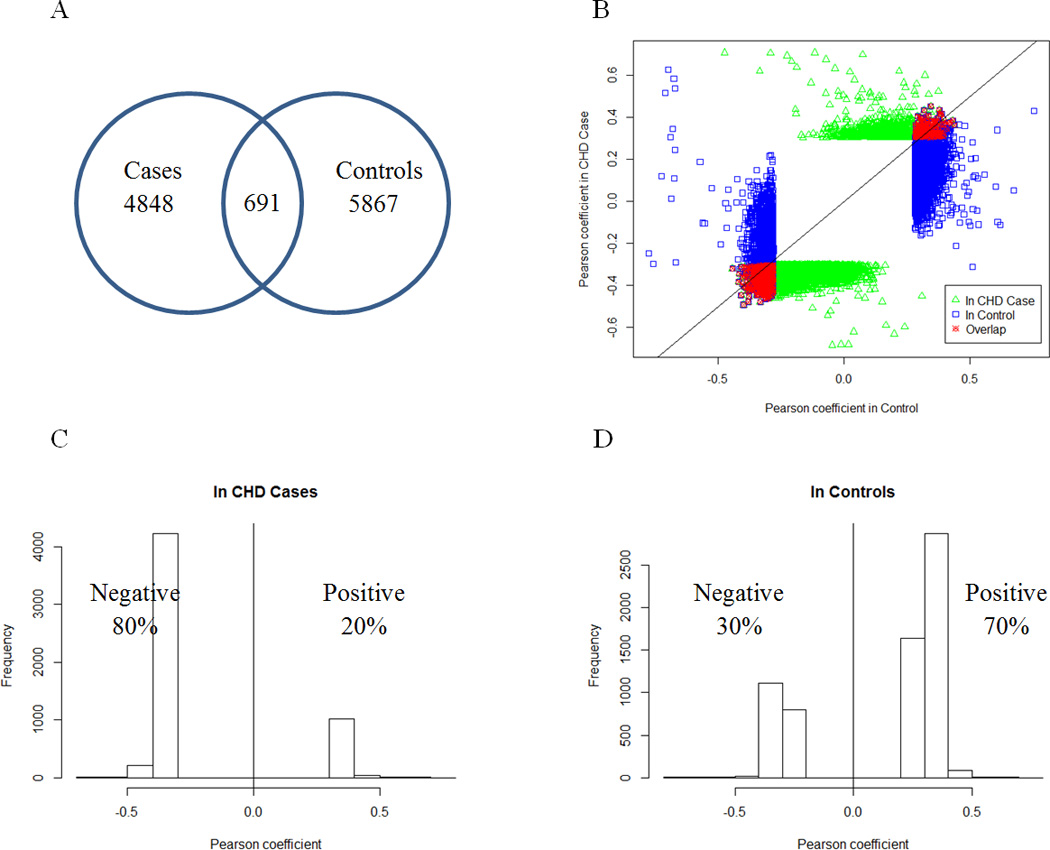
A) Venn diagram of the overlap in the correlated miRNA-mRNA pairs between cases and controls; B) Comparison of the correlation coefficients of correlated miRNA-mRNA pairs between cases (Y-axis) and controls (X-axis). Each dot indicates a miRNA-mRNA pair. The blue and green dots indicate the miRNA-mRNA correlation pairs at FDR<5% in CHD cases and controls respectively, and the red dots indicate the miRNA-mRNA correlation pairs at FDR<5% in both CHD cases and controls. C) Distribution of the Pearson correlation coefficients in CHD cases shows a dominance of negative correlations. D) Distribution of the Pearson correlation coefficients in controls shows a dominance of positive correlations.
Table 3.
Gene Ontology Enrichment of miRNA-mRNA co-expression Sets
| Gene Ontology | Overlap | Fold Enrichment | p-Value* |
|---|---|---|---|
| miRNA-mRNA co-expression pairs in both CHD cases and controls | |||
| Intracellular transport | 51 | 3.2 | 4.3e-13 |
| Cellular protein localization | 34 | 3.4 | 1.5e-9 |
| Nucleocytoplasmic transport | 20 | 5.3 | 7.3e-9 |
| Mitotic cell cycle | 26 | 2.9 | 3.9e-6 |
| Chromosome organization | 30 | 2.5 | 7.2e-6 |
| case-specific miRNA-mRNA co-expression pairs | |||
| Intracellular transport | 61 | 2.0 | 3.2e-7 |
| Gene silencing by RNA | 10 | 6.5 | 1.3e-5 |
| Intracellular protein transport | 37 | 2.1 | 2.6e-5 |
| Chromatin modification | 30 | 2.4 | 2.8e-5 |
| Negative regulation of macromolecule metabolic process | 59 | 1.7 | 4.2e-5 |
| control-specific miRNA-mRNA co-expression pairs | |||
| M phase of mitotic cell cycle | 25 | 3.1 | 1.9e-6 |
| Nuclear division | 24 | 3.0 | 4.7e-6 |
| Cell cycle process | 43 | 2.1 | 7.0e-6 |
| Chromosome segregation | 13 | 4.4 | 3.1e-5 |
Those p-values passed Bonferroni-correction for testing of 826 distinct Gene-ontology biology processes.
Importantly, 80% of the miRNA-mRNA co-expression pairs in CHD cases were inversely correlated (Figure 2C), whereas only 30% of the miRNA-mRNA co-expression pairs in controls were inversely correlated (Figure 2D). The directional shift in miRNA-mRNA co-expression between CHD cases and controls was highly significant (p<1e-16 by proportions test26). This major shift toward negative correlation in CHD cases, coupled with our finding that CHD miRNA differential signatures were all upregulated in cases, suggested that the widespread upregulation of miRNAs in cases might either play an important role in the pathogenesis of CHD by inhibiting mRNA expression levels of genes protective of CHD, or function reactively to inhibit CHD-related mRNA expression. When we adjusted for whole blood cell counts, we observed even more striking differences in the correlated miRNA-mRNA pairs between CHD cases (85% inversely correlated) and controls (15% inversely correlated).
We further defined 1472 case-specific and 1325 control-specific miRNA-mRNA co-expression pairs, by requiring the miRNA-mRNA pairs to be co-expressed in one group at FDR<0.05 but not in the other, and to show significant miRNA-mRNA interaction with regard to CHD status at FDR<0.2 (Supplementary Table VI). Gene ontology enrichment analysis (Table 3) revealed that the case-specific co-expression set is enriched for genes involved in intracellular transport (BF p=2.6e-4) and gene silencing by RNA (BF p=0.01). In contrast, the control-specific co-expression set is enriched for genes involved in the M phase of the mitotic cell cycle (BF p=1.6e-3) and nuclear division (BF p=3.9e-3). We hypothesize that these case-specific and control-specific co-expression pairs reflect the different miRNA-mRNA regulatory patterns observed in pathological (i.e., CHD) and physiological (i.e., control) conditions, and therefore, might be relevant to the pathogenesis of CHD.
Validation of the differential miRNA-mRNA co-expression patterns between CHD cases and controls
In order to validate the observation of a higher proportion of inversely correlated miRNA-mRNA pairs in cases vs. controls, we utilized two approaches: 1) a re-sampling strategy within the case-control study, and 2) replication in separate CHD cases and controls in the FHS (n=63 cases and 1000 controls) that did not overlap with the original case-control sets.
Validation through resampling
We randomly sampled 80% of CHD cases and controls to calculate miRNA-mRNA co-expression and repeated this process 100 times. For each resampling set, we identified significant miRNA-mRNA co-expression pairs at FDR<0.05 (see miRNA and mRNA correlation analysis), and compared the average proportion of inversely correlated miRNA-mRNA pairs separately in CHD cases and controls. The average proportion of inversely correlated miRNA-mRNA pairs was 0.76 in CHD cases and 0.25 in controls (p=2.2e-16 by Student’s T-test; Figure 3A). Therefore, the observed trend was robust in subpopulations of the case-control study and was not driven by spurious subsets of individuals.
Figure 3. Comparison of the proportion of inversely correlated miRNA-mRNA co-expression pairs between CHD cases and controls.
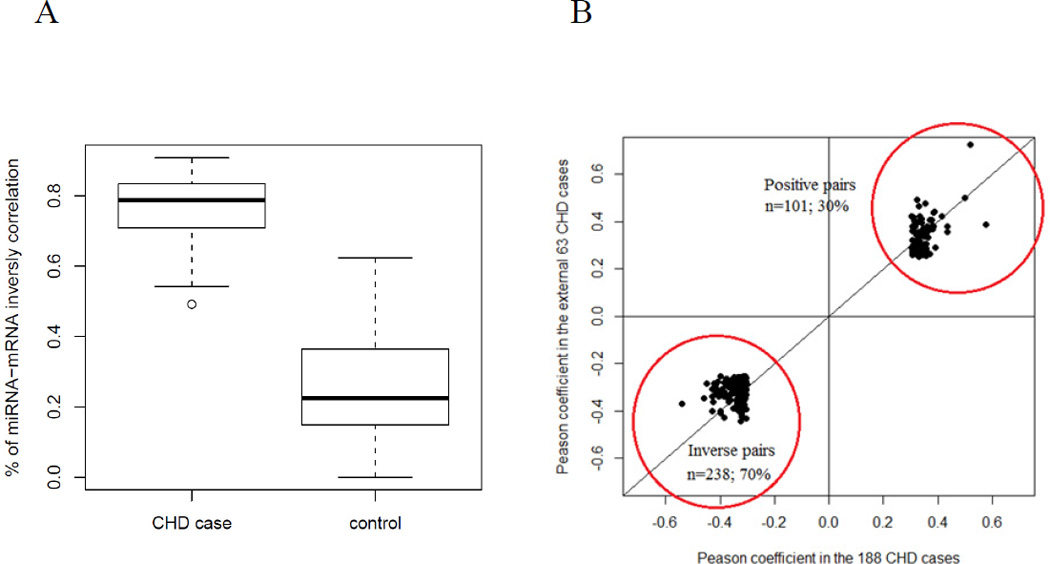
A) A re-sampling replication strategy confirmed the predominance of inversely correlated miRNA-mRNA pairs is in CHD cases, with the average proportion of negatively correlated pairs being 0.76 in CHD cases and 0.25 in controls (p=2.2e-16 by T-test). B) In an independent validation set of 63 CHD cases, 101 (30%) positively and 238 (70%) negatively correlated miRNA-mRNA pairs were observed at p<0.05.
Independent validation
We were unable to identify any external cohorts with miRNA and mRNA expression data for CHD cases and controls. However, through additional follow up, we identified 63 additional prevalent CHD cases and 1000 controls in the FHS. These individuals did not overlap with those included in our case-control study, and were used as independent replication samples. The clinical characteristics of these replication samples are provided in Supplementary Table VII.
In order to validate the observed higher proportion of positively correlated miRNA-mRNA pairs in controls (vs. cases), we selected 1000 individuals free of CHD who did not share familial relatedness. At FDR cutoffs of 1e-3 (4701 pairs) and 1e-4 (1487 pairs), with the number of co-expression pairs at a scale comparable to those in the case-control study, the proportions of positively correlated miRNA-mRNA pairs were 66% and 73%, respectively. We also found that the Pearson correlation coefficients of the miRNA-mRNA pairs among the 186 controls from our case-control study were highly consistent with those in the 1000 replication controls at p<1e-16 by linear regression testing. These results support the reproducibility of the observed trend of positive correlation of miRNA-mRNA co-expression pairs among controls.
Due to the small number of CHD cases (n=63) in the replication set, statistical power was low to detect significant miRNA-mRNA correlations. In fact, only five significant miRNA-mRNA co-expression pairs in the additional CHD cases were identified at FDR<0.05, precluding a direct comparison of significant miRNA-mRNA co-expression in this replication set with that from the 186 CHD cases in the case-control study using the same statistical cutoff. As an alternative strategy, we examined whether the directionality (positive or negative) of the miRNA-mRNA co-expression pairs identified in the original 186 CHD cases at FDR<0.05 was consistent in the 63 replication CHD cases. The Pearson coefficients of the miRNA-mRNA pairs between the 186 CHD cases and the 63 replication CHD cases were found to be highly consistent (p<1e-16 by linear regression testing). Among the pairs that passed p<0.05 and showed consistency in directionality in the replication CHD set, the proportion of inversely correlated mRNA-miRNA pairs was 70% (101 positively and 238 inversely correlated pairs; Figure 3B). These results support the original miRNA-mRNA co-expression pattern observed in the CHD cases.
Target relationships between miRNAs and mRNAs within the co-expression pairs
We overlapped the miRNA-mRNA co-expression pairs identified in our study with the miRNA-mRNA target pairs curated from six miRNA databases27–32 and found that 10% of the co-expression pairs overlapped with the predicted target pairs. We further investigated whether cis-eSNPs of the mRNAs in the co-expression pairs disrupt miRNA target sites. Of the 57,847 cis-mRNA-eSNPs, 34,195 could be mapped to mRNAs within the 9267 miRNAs-mRNAs co-expression pairs. For each eSNP, we extracted a genomic sequence of 13 bases surrounding the SNP. Then, we used TargetScan33, 34 to scan each sequence for potential miRNA target sites of the corresponding co-expressed miRNA. We identified 134 miRNA target site eSNPs for 172 predicted miRNA-mRNA target associations that also were co-expression pairs (Supplementary Table VIII). Among the cis-mRNA-eSNPs located in miRNA target sites, 11 were mapped to miR-423-5p, miR-425-3p, miR-502-3p and miR-505-5p, which appeared among the top CHD miRNA signatures (Table 1). These results support the regulator-target relationships for a subset of miRNA-mRNA co-expression pairs. However, co-expression between miRNA and mRNA does not have to be a regulator-target relationship in nature, but may simply reflect co-regulation of functionally related miRNAs and mRNAs by internal or external factors. Indeed, the gene ontology enrichment analysis (Table 3) revealed that the case- and control-specific miRNA-mRNA co-expressed pairs are enriched for genes involved in specific biological functions and pathways.
Testing whether the CHD-associated miRNA signatures and miRNA-mRNA co-expression pairs are potentially causal for CHD
The above analyses identified differentially expressed miRNA signatures of CHD, and CHD case-specific or control-specific miRNA-mRNA co-expression pairs. We used SNP set enrichment analysis 35 to test if any of these three gene sets are putatively causal for CHD by showing enrichment with low p value CHD SNPs from the Coronary ARteryDIsease Genome wide Replication and Meta-analysis Consortium (CARDIoGRAM) GWAS1. If the representative miR-eSNPs from a gene set showed CHD genetic risk enrichment, we termed the gene set “putatively causal via miRNAs”, and if the representative mRNA eSNPs of a gene set showed CHD genetic signal enrichment, we termed the gene set “putatively causal via mRNAs”.
When only the 123 cis-miR-eSNPs identified in the current case-control study were used in the analysis, there were no mRNA or miRNA sets found to be “putatively causal via miRNAs” (data not shown), which could result from a lack of statistical power when the number of eSNPs is small. To address this possibility, we repeated this analysis by utilizing another miR-eSNP resource consisting of approximately 6000 cis-miR-eSNPs that we identified from whole blood derived miRNA from 5329 FHS participants (details in the Methods). All the cis-miR-eSNPs in the case-control pairs were also identified as cis-miR-eSNPs in the larger sample of 5329 participants, supporting the reliability of the cis-miR-eSNPs. When the ~6000 cis-miR-eSNPs were used, the representative cis-miR-eSNPs from case-specific miRNA-mRNA co-expression pairs showed significant enrichment for CHD GWAS SNPs using both the KS test (p=0.03) and Fisher’s exact test (p=3.3e-6) (Table 4), supporting the hypothesis that CHD-related genetic variations may first perturb the expression levels of miRNAs in the case-specific miRNA-mRNA co-expression pairs, which in turn is associated with CHD. When examining individual miRNAs within the case-specific co-expression pairs and their cis-miR-eSNPs in relation to CHD, we found 172 cis-miR-eSNPs at p<0.05 in the CARDIoGRAM CHD GWAS1 (Supplementary Table IX).
Table 4.
SNP Set Enrichment Test to Infer Causal Relationship between miRNA Sets and CHD
| CARDIoGRAM GWAS miR-eSNP |
CARDIoGRAM GWAS mRNA eSNP |
|||
|---|---|---|---|---|
| miRNA / mRNA sets | KS* p Value | Fisher† p Value | KS* p Value | Fisher† p Value |
| CHD miRNA signatures | 0.98 | 1 | - | - |
| case-specific miRNA-mRNA co-expression pairs | 0.03 | 3.3e-6 | 0.48 | 0.17 |
| control-specific miRNA-mRNA co-expression pairs | 0.15 | 3.5e-3 | 2.1e-23 | 7.3e-59 |
KS indicated Kolmogorov-Smirnov test.
Fisher indicated Fisher’s exact test.
In contrast, the control-specific miRNA-mRNA co-expression pairs were found to be “putatively causal via mRNAs” (p=2.1e-23 by KS test and p=7.2e-59 by Fisher’s exact test) (Table 4). Among genes in the control-specific miRNA-mRNA co-expression pairs, 1129 eSNPs for 130 mRNAs were associated with CHD at p<0.05 in CARDIoGRAM 1 (194 eSNPs of 11 genes were associated with CHD at p<1e-3, Supplementary Table X). These results suggest that the differential miRNA-mRNA co-expression patterns between CHD cases and controls might be driven by eSNPs that disturb either miRNA or mRNA expression.
Association of miRNAs with a previously identified gene co-expression module tested causal for CHD
In our previous study, we identified a CHD causal mRNA co-expression module using systems network approaches7. The genes in the CHD causal module showed a co-expression pattern in controls, but the co-expression was disrupted in CHD cases. This CHD mRNA module was enriched for genes related to B-cell activation and differentiation processes, and also tested putatively causal for lipid regulation and CHD. In addition, we identified 63 key drivers that appeared to regulate the module genes.
In order to test whether any miRNAs are related to the CHD mRNA module, we retrieved the first principle component (PC) of this module, and correlated the PC with every miRNA expression trait. Three miRNAs (miR-1275, miR-365a-3p and miR-150-5p) showed significant correlation with the module PC at FDR<0.2 (Table 5), suggesting that these three miRNAs may participate in B-cell activation and differentiation processes along with the module genes. Eight genes within the module and their key drivers were predicted mRNA targets of these three miRNAs31, among which five genes (ARHGAP24, BTLA, DENND5B, MOBKL2B, and POU2AF1) are potential miR-150-5p targets31 and three genes (PAX5, POU2AF1, and BTLA) are related to B-cell developmental regulation 36, 37.
Table 5.
miRNAs Correlated with CHD mRNA Causal Module and Their Predicted Target Genes
| miRNA | PC* correlation p-value |
Predicted Target Gene† |
Gene Description |
|---|---|---|---|
| miR-1275 | 8.2e-6 | KLHL14‡ | kelch-like 14 |
| PAX5 | paired box 5 | ||
| miR-365a-3p | 1.3e-3 | ARHGAP24 | Rho GTPase activating protein 24 |
| FAM3C‡ | family with sequence similarity 3, member C | ||
| miR-150-5p | 1.6e-3 | ARHGAP24 | Rho GTPase activating protein 24 |
| BTLA‡ | B and T lymphocyte associated | ||
| DENND5B | DENN/MADD domain containing 5B | ||
| MOBKL2B‡ | MOB1, Mps One Binder kinase activator-like 2B | ||
| POU2AF1‡ | POU class 2 associating factor 1 |
In addition, nine differentially expressed miRNA signatures of CHD (e.g. miR-1275, miR-589-3p, and miR-324-5p) were predicted to target 17 mRNAs within the CHD causal module or KDs (Supplementary Table XI). The role of miR-1275 in the context of CHD is particularly interesting, because this miRNA was strongly correlated with the CHD mRNA causal module (p=8.2e-6) and also showed significant differential expression in relation to CHD status (p=0.009).
Discussion
Although the importance of miRNAs in cardiovascular development and disease is becoming increasingly recognized, the relations of miRNAs to CHD have been analyzed only in a few studies 12–15, 22. In this study, genome-wide genotyping and miRNA and mRNA expression profiling analysis were performed on 186 CHD case-control pairs. Using systems biology approaches, we explored the genetic architecture of miRNA regulation and investigated differential miRNA expression, and differential miRNA-mRNA correlation between cases and controls, seeking to delineate gene regulatory networks capturing complex DNA-miRNA-mRNA relationships that may contribute to CHD. To our knowledge, our study is the first comprehensive interrogation of CHD to integrate multidimensional molecular data including genetic variation, mRNA, and miRNA in a large, community-based cohort. Our study yielded important evidence supporting genetic control of miRNAs as well as an important role of miRNAs in CHD development.
In order to dissect the genetic elements and mechanisms governing miRNA expression, we conducted expression quantitative trait locus (eQTL) analysis to identify miR-eSNPs. In contrast to more extensive investigations of mRNA eSNPs in multiple tissues such as blood16, 38, adipose16 and liver17, there are relatively few studies of miR-eSNPs and most involve small sample sizes19, 20, 39. For example, one study using umbilical cord blood from 180 newborns identified only 12 cis-miR-eSNPs at FDR<0.520. In another investigation, no cis-miR-eSNPs were identified in 176 lymphoblastoid cell lines from CEU and YRI cohorts19. Our study represents the largest sample size (N=361) studied to date and we identified 123 cis-miR-eSNPs for 13 miRNAs at FDR<0.05. Two of the cis-miR-eSNPs (for miR-100 and miR-125) that we detected overlap with those identified by Borel et al.20, reflecting independent external replication. All 123 cis-miR-eSNPs were replicated in a larger study of >5000 individuals from the FHS. Our results provide additional evidence that miRNA expression is partly under genetic control.
Our study also provides multiple lines of evidence in support of a critical role of miRNAs in the pathogenesis of CHD. First, our study identified 15 differentially expressed miRNAs in whole blood, all of which were upregulated in CHD cases. In a previous study, Bidzhekov et al. identified 23 differentially expressed miRNAs in atherosclerotic plaques compared to healthy arteries, twenty of which displayed upregulation patterns in atherosclerotic plaques22. Therefore, the observed miRNA upregulation patterns in CHD were consistent between our study and that of Bidzhekov, suggesting that miRNAs may play an important role in CHD. A similar miRNA upregulation pattern has also been observed in heart failure (HF) studies. In an miRNA microarray analysis by Thum et al., 67 miRNAs were upregulated >1.5-fold in failing left ventricles versus normal hearts, whereas 43 miRNAs were downregulated >1.5-fold12. In two additional studies of HF using plasma samples, Tijsen et al. reported that 14 of the 16 differentially expressed miRNAs were upregulated in HF patients, and seven of the 14 upregulated miRNAs were validated in an independent HF case-control study 13. In a study by Goren et al., more down-regulated miRNAs (n=13) than upregulated miRNAs (n=10) were reported40. Nevertheless, all 4 replicated miRNA (miRNA-423-5p, miRNA-320a, miRNA-22, and miRNA-92b) were upregulated in the HF group. These comparisons highlight a general trend of increased miRNA expression in CHD cases that we observed in our study and that is largely consistent with previous studies.
Second, we detected striking differences in the significant miRNA-mRNA associations (at FDR<0.05) between CHD cases and controls, with 80% of miRNA-mRNA pairs being inversely correlated in CHD cases, compared with 30% of miRNA-mRNA pairs being inversely correlated in controls. These patterns were replicated in independent sets of CHD cases and controls. Recently, Thum et al.12 used bioinformatics methods to predict the binding sites of upregulated miRNAs in heart failure cases. They found that downregulated mRNAs had more binding sites for the upregulated miRNAs in heart failure cases12. Their results suggest that upregulated miRNAs might suppress mRNA expression in heart failure. Consistent with this hypothesis, our results support the notion that suppression of mRNAs by miRNAs may also be an important feature in CHD, given the upregulation of miRNAs and the inverse correlation of miRNA-mRNA pairs in CHD cases but not in controls in our study. We hypothesize that striking differences in miRNA-mRNA co-expression patterns between cases and controls represent a useful “transcriptomic biomarker” of CHD.
Third, by integrating the miR-eSNPs and mRNA eSNPs identified from our study as well SNPs as from the CARDIoGRAM CHD GWAS1 into our analysis, we show that disruptions of miRNA-mRNA co-expression are likely causal for CHD. In this study, we used SNP set enrichment analysis to examine whether a particular miRNA-mRNA co-expression set is significantly enriched for eSNPs that demonstrate low p value association with CHD in the CARDIoGRAM GWAS1. Based on the central dogma, genetics is upstream of gene/miRNA expression and CHD, but unlikely to be altered by CHD. Finding significant enrichment of CHD genetic signals in a given miRNA-mRNA co-expression set, therefore, supports a putative causal role of the miRNA-mRNA pairs. The genetic enrichment signals seen for the case-specific and control-specific co-expression pairs provide evidence that they are putatively causal for CHD, whereas the miRNA signature may not play a causal role in CHD and are likely to be reactive to CHD. The control-specific miRNA-mRNA co-expression pairs (enriched for cell cycle processes) tested “putatively causal” for CHD when mRNA eSNPs but not miR-eSNPs were used. Cell cycle pathways have also been recently implicated to be causally associated with coronary artery disease in a separate study41. Our observation suggests that tight miRNA-mRNA co-expression patterns that are necessary for normal cell cycle processes may be disrupted in CHD cases. It is possible that this disruption of cell cycle regulation is causal for CHD and that the alteration of miRNA-mRNA co-expression is led by eSNPs regulating mRNA expression rather than eSNPs controlling miRNA expression. On the other hand, the case-specific miRNA-mRNA co-expression pairs were enriched for genes involved in “gene silencing by RNA” and showed evidence of “putative causality” via miRNAs. These results support the possibility of a large shift toward negative miRNA-mRNA correlation in CHD cases due to RNA-based regulatory control.
Fourth, we linked miRNAs to a previously identified gene co-expression module that is enriched for genes involved in B cell functions and enriched for genetic risk polymorphisms associated with CHD7. Although macrophages and T cells, but not B cells, are prominent in atherosclerotic lesions in the artery wall42,43–46, several studies reported that deficiency of B cells either increases44, 45 or decreases46 atherosclerosis. A recent study by Hilgendorf et al. found that a subset of B cells, termed innate response activator (IRA) B cells, accumulate in the spleen, bone marrow and lymph nodes in Apoe−/− mice that also display severe inflammation and atherosclerosis, although they did not detect IRA B cells in the aorta[7]. These results suggest that IRA B cells do not affect lesions locally but promote atherogenesis in the artery wall through systemic activation of innate immune response 47, 48. Our B cell related findings come from whole blood, and also support a systemic role of B cells in atherosclerosis. In the current study, we correlated miRNA expression with the first principle component of the CHD gene co-expression module and identified 3 miRNAs correlated with this module. Among these 3 miRNAs, miR-150 has been previously reported to be dysregulated in the setting of myocardial infarction 15 and to be involved in B cell development49, 50. Zhou et al. reported that miR-150 participates in early B cell development by blocking the transition from the pro-B to the pre-B stage50. Xiao et al. reported that miR-150 controls B-cell differentiation by targeting c-Myb49. In our analysis, five genes within the CHD causal module were predicted targets of miR-150, among which two genes, BTLA and POU2AF1, are involved in B cell development. These lines of evidence support the hypothesis that a subset of miRNAs mediate gene regulation in the previously identified CHD causal module and the notion that B cell centered adaptive immune function plays an important role in CHD. Specifically, perturbations of particular miRNAs such as miR-150 may lead to dysregulation of B cell functions and in turn promote atherogenesis and CHD. Future experimental testing of this proposed mechanism is warranted.
Although our study is the largest to examine the interrelations among genetic variation and miRNA and mRNA expression, we acknowledge several limitations. First, our investigation focused on whole blood derived RNA and further studies in individual cell types and other CHD relevant tissues are necessary to determine the tissue- or cell type-specific genetic control of miRNAs and the precise roles of such genetic control in the gene regulatory networks pertaining to CHD. Nevertheless, as multiple blood constituents such as oxidized lipids, immune cells, and platelets play important roles in plaque accumulation, rupture, and thrombus formation in the vessel wall51, blood has strong pathological relevance to CHD. Indeed, previous whole blood profiling studies of CHD3–6 have revealed important biological insights. In addition, for epidemiological studies, accessing internal tissues and cell types is challenging and peripheral whole blood is the most readily available tissue. In our study, the miRNA and mRNA profiling data generated from whole blood mainly reflect the immune and inflammatory processes captured by the various types of leukocytes. We speculate that the large-scale gene regulatory network remodeling we have observed in whole blood from CHD cases represents systemic perturbations in the immune system in both the circulation and the CHD-related tissues. Second, additional molecular interactions, e.g. miRNA-miRNA52, could be incorporated by utilizing more complicated network analysis models. Last, although our systems biology analysis revealed scores of novel findings and we provided strong evidence from the literature and from validation analyses in independent CHD cases and controls, downstream experimental validation of the potential mechanisms is necessary, but is out of the scope of our current investigation.
In summary, we systematically analyzed the multidimensional relations between genetic variation, miRNAs, and mRNAs. We further analyzed miRNA expression profiling and miRNA-mRNA interactomic changes associated with CHD. CHD GWAS1 results were integrated into our systems analyses to identify the directionality of effects and to infer causality. We observed significant upregulation of 15 miRNAs in CHD cases and a genome-wide miRNA-mRNA correlation shift from positive in controls to negative in CHD cases. We provide evidence supporting the hypothesis that disruption of miRNA-mRNA co-expression by eSNPs that regulate the expression levels of mRNAs or miRNAs may contribute to the development of CHD. We also identified several miRNAs (e.g. miR-150) that potentially regulate an mRNA module involved in B cell-centered adaptive immune function and that tested causal for CHD. Overall, our findings support an important role of miRNAs in CHD pathogenesis.
Supplementary Material
Significance.
This study systematically characterized the complex genomic architecture of CHD by integrating whole blood miRNA and mRNA expression with genetic variation in 186 CHD cases and 186 controls. We identified a large number of correlated miRNA-mRNA pairs and genetic loci that appear to regulate miRNA levels. We also detected a large difference in miRNA-mRNA associations between CHD cases and controls, as demonstrated by a significantly higher proportion of inversely correlated miRNA-mRNA pairs in cases vs. controls (80% vs. 30%; p<1e-16), suggesting a genome-wide shift in the regulatory structure of the transcriptome in CHD. Further, we found that the differentially co-expressed miRNA-mRNA pairs showed enrichment for CHD risk genetic variants affecting both miRNA and mRNA expression levels, implicating a putatively causal role in CHD. These findings provide novel evidence that miRNAs are important regulators of biological processes involved in CHD via genetic control and via their tight co-expression with mRNAs.
Acknowledgements
D. L. and X. Y. designed, directed, and supervised the project. D. L. M. L, and J. F. were responsible for funding of the project. J. F. and K. T. directed and supervised the miRNA experiment. T. H., X. Y. and D. L. drafted the manuscript. P. C. organized the experiment material and data exchange. All authors participated in revising and editing the manuscripts. All authors have read and approved the final version of the manuscript. We thank Xiaoyan Yin, Chunyu Liu and Xiaoling Zhang for advice and discussion.
This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD. (http://biowulf.nih.gov).
Source of Funding
The FHS is funded by National Institutes of Health contract N01-HC-25195. The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health and NIH contract N01-HC-25195. The analytical component of this project was funded by the Division of Intramural Research and KL2RR031981 (David D. McManus), National Heart, Lung, and Blood Institute, and the Center for Information Technology, National Institutes of Health, Bethesda, MD. X. Y. is funded by the American Heart Association Scientist Development Grant and the Leducq Foundation.
Nonstandard Abbreviations and Acronym
- BF
Bonferroni correction
- CARDIoGRAM
Coronary ARteryDIsease Genome wide Replication and Meta-analysis Consortium
- CHD
Coronary heart disease
- eSNP
Expression-associated single-nucleotide polymorphism
- eQTL
Expression quantitative trait locus
- FDR
False discovery rate
- FHS
Framingham Heart Study
- GWAS
Genome-wide association studies
- KS
Kolmogorov-Smirnov test
Footnotes
Disclosures
None.
References
- 1.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Donnell CJ, Nabel EG. Genomics of cardiovascular disease. N Engl J Med. 2011;365:2098–2109. doi: 10.1056/NEJMra1105239. [DOI] [PubMed] [Google Scholar]
- 3.Joehanes R, Ying S, Huan T, et al. Gene expression signatures of coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:1418–1426. doi: 10.1161/ATVBAHA.112.301169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinnaeve PR, Donahue MP, Grass P, Seo D, Vonderscher J, Chibout SD, Kraus WE, Sketch M, Jr, Nelson C, Ginsburg GS, Goldschmidt-Clermont PJ, Granger CB. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One. 2009;4:e7037. doi: 10.1371/journal.pone.0007037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wingrove JA, Daniels SE, Sehnert AJ, Tingley W, Elashoff MR, Rosenberg S, Buellesfeld L, Grube E, Newby LK, Ginsburg GS, Kraus WE. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet. 2008;1:31–38. doi: 10.1161/CIRCGENETICS.108.782730. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg S, Elashoff MR, Beineke P, et al. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Ann Intern Med. 2010;153:425–434. doi: 10.7326/0003-4819-153-7-201010050-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huan T, Zhang B, Wang Z, et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:1427–1434. doi: 10.1161/ATVBAHA.112.300112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee R, Feinbaum R, Ambros V. A short history of a short rna. Cell. 2004;116:S89–S92. doi: 10.1016/s0092-8674(04)00035-2. 81 p following S96. [DOI] [PubMed] [Google Scholar]
- 9.Lee RC, Ambros V. An extensive class of small rnas in caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 10.Cordes KR, Srivastava D. Microrna regulation of cardiovascular development. Circ Res. 2009;104:724–732. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small EM, Olson EN. Pervasive roles of micrornas in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. Micrornas in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 13.Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE, Pinto YM. Mir423-5p as a circulating biomarker for heart failure. Circ Res. 2010;106:1035–1039. doi: 10.1161/CIRCRESAHA.110.218297. [DOI] [PubMed] [Google Scholar]
- 14.Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW., 2nd Reciprocal regulation of myocardial micrornas and messenger rna in human cardiomyopathy and reversal of the microrna signature by biomechanical support. Circulation. 2009;119:1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bostjancic E, Zidar N, Glavac D. Microrna microarray expression profiling in human myocardial infarction. Dis Markers. 2009;27:255–268. doi: 10.3233/DMA-2009-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 17.Schadt EE, Molony C, Chudin E, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Zhang B, Molony C, et al. Systematic genetic and genomic analysis of cytochrome p450 enzyme activities in human liver. Genome research. 2010;20:1020–1036. doi: 10.1101/gr.103341.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gamazon ER, Ziliak D, Im HK, LaCroix B, Park DS, Cox NJ, Huang RS. Genetic architecture of microrna expression: Implications for the transcriptome and complex traits. Am J Hum Genet. 2012;90:1046–1063. doi: 10.1016/j.ajhg.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borel C, Deutsch S, Letourneau A, Migliavacca E, Montgomery SB, Dimas AS, Vejnar CE, Attar H, Gagnebin M, Gehrig C, Falconnet E, Dupre Y, Dermitzakis ET, Antonarakis SE. Identification of cis- and trans-regulatory variation modulating microrna expression levels in human fibroblasts. Genome research. 2011;21:68–73. doi: 10.1101/gr.109371.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan BM, Robles AI, Harris CC. Genetic variation in microrna networks: The implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bidzhekov K, Gan L, Denecke B, Rostalsky A, Hristov M, Koeppel TA, Zernecke A, Weber C. Microrna expression signatures and parallels between monocyte subsets and atherosclerotic plaque in humans. Thromb Haemost. 2012;107:619–625. doi: 10.1160/TH11-09-0607. [DOI] [PubMed] [Google Scholar]
- 23.Ren J, Zhang J, Xu N, Han G, Geng Q, Song J, Li S, Zhao J, Chen H. Signature of circulating micrornas as potential biomarkers in vulnerable coronary artery disease. PLoS One. 2013;8:e80738. doi: 10.1371/journal.pone.0080738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sondermeijer BM, Bakker A, Halliani A, et al. Platelets in patients with premature coronary artery disease exhibit upregulation of mirna340* and mirna624*. PLoS One. 2011;6:e25946. doi: 10.1371/journal.pone.0025946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gauvreau K. Hypothesis testing - proportions. Circulation. 2006;114:1545–1548. doi: 10.1161/CIRCULATIONAHA.105.586487. [DOI] [PubMed] [Google Scholar]
- 27.Hsu SD, Lin FM, Wu WY, et al. Mirtarbase: A database curates experimentally validated microrna-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N, Dalamagas T, Hatzigeorgiou AG. Tarbase 6.0: Capturing the exponential growth of mirna targets with experimental support. Nucleic Acids Res. 2012;40:D222–D229. doi: 10.1093/nar/gkr1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. Mirecords: An integrated resource for microrna-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. Mir2disease: A manually curated database for microrna deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microrna.Org resource: Targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microrna targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 34.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. American journal of human genetics. 2010;86:581–591. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 37.Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. David bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, Taylor J, Burnett E, Gut I, Farrall M, Lathrop GM, Abecasis GR, Cookson WO. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 39.Civelek M, Hagopian R, Pan C, Che N, Yang WP, Kayne PS, Saleem NK, Cederberg H, Kuusisto J, Gargalovic PS, Kirchgessner TG, Laakso M, Lusis AJ. Genetic regulation of human adipose microrna expression and its consequences for metabolic traits. Hum Mol Genet. 2013;22:3023–3037. doi: 10.1093/hmg/ddt159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goren Y, Kushnir M, Zafrir B, Tabak S, Lewis BS, Amir O. Serum levels of micrornas in patients with heart failure. Eur J Heart Fail. 2012;14:147–154. doi: 10.1093/eurjhf/hfr155. [DOI] [PubMed] [Google Scholar]
- 41.Mäkinen V-P, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segrè AV, Ghosh S, Vivar J. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS genetics. 2014;10:e1004502. doi: 10.1371/journal.pgen.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 43.Perry HM, McNamara CA. Refining the role of b cells in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1548–1549. doi: 10.1161/ATVBAHA.112.249235. [DOI] [PubMed] [Google Scholar]
- 44.Sage AP, Mallat Z. Multiple potential roles for b cells in atherosclerosis. Ann Med. 2014 doi: 10.3109/07853890.2014.900272. [DOI] [PubMed] [Google Scholar]
- 45.Perry HM, Oldham SN, Fahl SP, Que X, Gonen A, Harmon DB, Tsimikas S, Witztum JL, Bender TP, McNamara CA. Helix-loop-helix factor inhibitor of differentiation 3 regulates interleukin-5 expression and b-1a b cell proliferation. Arterioscler Thromb Vasc Biol. 2013;33:2771–2779. doi: 10.1161/ATVBAHA.113.302571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ait-Oufella H, Herbin O, Bouaziz JD, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207:1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilgendorf I, Theurl I, Gerhardt LM, et al. Innate response activator b cells aggravate atherosclerosis by stimulating t helper-1 adaptive immunity. Circulation. 2014;129:1677–1687. doi: 10.1161/CIRCULATIONAHA.113.006381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. 2014;114:1743–1756. doi: 10.1161/CIRCRESAHA.113.301145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. Mir-150 controls b cell differentiation by targeting the transcription factor c-myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. Mir-150, a microrna expressed in mature b and t cells, blocks early b cell development when expressed prematurely. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 52.Matkovich SJ, Hu Y, Dorn GW., 2nd Regulation of cardiac micrornas by cardiac micrornas. Circ Res. 2013;113:62–71. doi: 10.1161/CIRCRESAHA.113.300975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.