

Abstract
Phosphoinositides (PIs) are a group of key signaling and structural lipid molecules involved in a myriad of cellular processes. PI phosphatases, together with PI kinases, are responsible for the conversion of PIs between distinctive phosphorylation states. PI phosphatases are a large collection of enzymes that are evolved from at least two disparate ancestors. One group is distantly related to endonucleases, which applies divalent metal ions for phosphoryl transfer. The other group is related to protein tyrosine phosphatases, which contains a highly conserved active site motif Cys-X5-Arg (CX5R). In this review, we focus on structural insights to illustrate current understandings of the molecular mechanisms of each PI phosphatase family, with emphasis on their structural basis for substrate specificity determinants and catalytic mechanisms.
Keywords: PTEN, Sac1, myotubularin, OCRL, Lowe syndrome
1. Introduction
Phosphoinositides (PIs) are pivotal cellular regulators and play essential roles in a broad spectrum of cellular processes including defining intracellular organelle identity, cell signaling, proliferation, cytoskeleton organization, and membrane trafficking [1–4]. PIs are a collection of seven lipid derivatives that differ with regard to the presence or absence of phosphate groups at the 3′, 4′, and 5′ positions. The reversible phosphorylation of PIs is under tight spatial temporal control by a large number of PI kinases and phosphatases that are present throughout eukaryotic species.
In the past thirty years, significant advances have been made in the PI phosphatase field. With the completion of human genome and whole genome sequencing of many other species, genes that encode PI phosphatases have been well documented. Based on catalytic mechanisms, PI phosphatases can be categorized into two major classes.
One class is the inositol polyphosphate 5-phosphatases, which are Mg2+-dependent phosphatases comprising 10 mammalian [5] and 4 yeast members [6]. 5-phosphatases have a characteristic central catalytic domain that exhibits sequence homology to the apurinic/apyrimidic family of endonucleases [7]. Members of this class hydrolyze the D5 phosphate of the inositol ring of both soluble inositol polyphosphates and membrane-bound PIs [8].
The other class is characteristic of a highly conserved CX5R active site motif, of which the cysteine residue functions as the nucleophile while the arginine residue positions the scissile phosphate group. Based on their primary sequences, enzymes in this class can be further divided into four sub-families that include the Sac1 domain containing phosphatase, PTEN, myotubularin, and 4-phosphatase. In addition to the eukaryotic PI phosphatases, several bacterial pathogens also encode CX5R-based PI phosphatases. The SopB/SigD of Salmonella typhimurium [9] and its ortholog IpgD from Shigella flexneri [10] are PI phosphatases that play multiple roles in bacterial entry and/or intracellular survival [10, 11]. Recently, we identified two novel CX5R-based PI phosphatases, SidF and SidP from Legionella pneumophila that may play a role in the active controlling of the lipid composition of Legionella-containing vacuoles [12, 13]. Studies of these PI phosphatases are providing new insights into the mechanism of CX5R-dependent PI phosphatases.
Over the last decade, a wealth of structural information of PI phosphatases has accumulated. This review highlights structural insights into each family of PI phosphatases to illustrate the mechanism of substrate specificity, membrane targeting, and catalysis.
2. 5-phosphatases
2.1 The 5-phosphatase family
The inositol polyphosphate 5-phosphatases are a group of Mg2+-dependent phosphatases first characterized in studies on the metabolism of the inositol phosphate second messenger Ins(1,4,5)P3 [14, 15]. The first 5-phosphatase gene cloned in the early 90’s [16, 17] is named INPP5A or 5-phosphatase type I. It has a molecular weight of 43-kD and hydrolyses only the soluble inositol polyphosphates Ins(1,4,5)P3 and Ins(1,3,4,5)P4. Following these earlier studies, other 5-phosphatases were discovered. For example, the 75-kD INPP5B purified from human platelet was shown to have activity against PI(4,5)P2, and PI(3,4,5)P3 besides Ins(1,4,5)P3 and Ins(1,3,4,5)P4 [18, 19]. Subsequently, the human cDNA encoding INPP5B was cloned [20, 21]. At approximately the same time, the gene responsible for the human X-linked disease Oculocerebrorenal Syndrome of Lowe (OCRL) was mapped and shown to be highly homologous to INPP5B [22]. Like INPP5B, OCRL is a multidomain-containing phosphatase with a PH domain at the N-terminus [23] followed by a central 5-phosphatase catalytic domain, an ASH domain [24], and a C-terminal RhoGAP-like domain. Unlike INPP5B, which has a CAAX motif at its C-terminus [21], OCRL has two clathrin-binding motifs within its N-terminal PH domain and C-terminal RhoGAP-like domain, respectively (Fig. 1) [23, 25]. OCRL hydrolyzes PI(4,5)P2, PI(3,4,5)P3, and inositol polyphosphates Ins(1,4,5)P3 and Ins(1,3,4,5)P4, with a preference for PI(4,5)P2 [26, 27].
Fig. 1.

Schematic domain organization of 5-phosphatases. Abbreviations: 5-ptase, 5-phosphatase catalytic domain; PH, pleckstrin homology; ASH, ASPM-SPD2-Hydin; CB, clathrin binding; RBD, RNA binding domain; SH2, Src homology 2; SKICH, SKIP carboxyl homology.
The repertoire of 5-phosphatases continued to grow when synaptojanin 1 (SYNJ1) was discovered to be responsible for the hydrolysis of PI(4,5)P2 on synaptic vesicles at the nerve terminal [28]. SYNJ1 deletion mice develop neurological defects with accumulated clathrin-coated vesicles in nerve endings [29]. These findings provided strong evidence for the critical role of PIs in membrane trafficking in addition to their classical roles as second messengers in signal transduction [30]. Along this line, a ubiquitously expressed SYNJ1 homologue, SYNJ2 was identified [31]. Besides the 5-phosphatase domain, SYNJ1 and SYNJ2 share an N-terminal Sac1 domain, which was later found to be a member of a new family of PI phosphatases [32]. Human SYNJ1 and SYNJ2, but not yeast synaptojanin homologues, also contain an RNA binding domain (protein data bank ID: 2DNR; 1UFW) after their central 5-phosphatase domain (Fig. 1). The large C-terminal portion in both SYNJs is a divergent proline-rich region (PRD), which contains a string of peptide motifs that mediate bindings with a variety of endocytic proteins such as amphiphysin [28] and endophilin [33, 34]. Biochemical characterization showed that the N-terminal Sac1 domain of synaptojanins predominantly dephosphorylates PI(3)P and PI(4)P, whereas the 5-phosphatase domain dephosphorylates PI(4,5)P2 and PI(3,4,5)P3 at the 5′ position of the inositol ring [28, 29, 32, 35]. Abnormalities of these two phosphatases have been linked to several neuronal diseases. For example, overexpression of SYNJ1 resulting from trisomic duplication may contribute to brain dysfunction in mouse models of Down’s syndrome [36, 37] and bipolar disorder [38, 39]. In addition, a mutation in the Sac1 domain is linked with early-onset progressive Parkinsonism [40]. In a mouse mutant strain called Mozart, a mutation in SYNJ2 is implicated in progressive hearing loss [41].
The fact that synaptojanins possess both the Sac1 and the 5-phosphatase domains leads to a fascinating speculation that the two catalytic domains may function in a concerted way. In the concerted model, the 5-phosphatase domain converts PI(4,5)P2 to PI(4)P, which is efficiently diffused to the Sac1 domain for further dephosphorylation to phosphatidylinositol (PtdIns) without accumulation of the intermediate en route. This processive mode of hydrolysis may permit the rapid inactivation of PI(4,5)P2 following endocytosis, thereby avoiding mis-targeting of endocytic transport factors to intracellular membranes.
The other 5-phosphatase family member SHIP1 plays an important role in immune response. It was first identified as a 145-kD tyrosine phosphorylated protein from haematopoietic cells upon cytokine stimulation [42]. SHIP1 was then cloned and shown to hydrolyze PI(3,4,5)P3 and Ins(1,3,4,5)P4 in vitro [43–45]. The close homologue of SHIP1, SHIP2, plays a role in the negative regulation of insulin signaling [46] and serves as a potential drug target for obesity. Interestingly, SHIP2 hydrolyses Ins(1,3,4,5)P4, PI(3,5)P2, and PI(3,4,5)P3, but not PI(4,5)P2 [47, 48]. Both SHIP1 and SHIP2 have an SH2 domain at their N-terminus, a central 5-phosphatase catalytic domain, and a C-terminal PRD. SHIP2, but not SHIP1, also has a SAM (sterile alpha motif) domain at its C-terminus (Fig. 1).
Other 5-phosphatases, including PIPP (INPP5J), SKIP (INPP5K), and Pharbin (INPP5E), are less well studied. INPP5E has an N-terminal PRD, a central 5-phosphatase catalytic domain, and a C-terminal CAAX motif. INPP5E preferentially hydrolyses PI(3,4,5)P3 and PI(4,5)P2 [49, 50]and is linked to PI signaling in the primary cilium and mutations in INPP5E cause cilium signaling defects, which lead to a human genetic disease, called Joubert syndrome [51–53]. SKIP (INPP5K) is a skeletal muscle and kidney enriched 5-phosphatase, which contains an N-terminal 5-phosphatase domain and a unique SKICH domain at the C-terminus [54, 55]. SKIP decreases PI(3,4,5)P3 levels upon insulin stimulation and SKIP knockout mice show increased insulin sensitivity [56]. The last human 5-phosphatase is PIPP, which has two PRD domains at its N-and C-terminus and a central catalytic domain followed by a SKICH domain [57]. PIPP can hydrolyze Ins(1,4,5)P3, Ins(1,3,4,5)P4, PI(4,5)P2, and PI(3,4,5)P3 in vitro [57]. However, it is shown to be a novel negative regulator of PI 3-kinase pathway by hydrolyzing PI(3,4,5)P3 in vivo [58].
With the completion of genome projects of human and other organisms, ten 5-phosphatases have been identified in mammals and five in yeast [8]. In addition to their catalytic domain of approximately 300 amino acids, all 5-phosphatases contain a variety of domains or motifs that determine their cellular localization and/or regulate their activity (Fig. 1). Crystal structures of 5-phosphatase catalytic domains from several members have been determined [59–61]. These structures provide insights into the molecular basis for catalysis, substrate recognition, membrane interaction, as well as human genetic diseases.
2.2 Crystal structure of the PI-5-phosphatase catalytic domain
The first crystal structure of the 5-phosphatase domain was determined from Schizosaccharomyces pombe synaptojanin [59]. The structure reveals that the core of the 5-phosphatase domain consists of two layered β-sheets with one containing 5 β-strands and 6 for the other. These two sheets stack on each other in a parallel way forming a stable core by burying a large number of hydrophobic residues in between. The α-helices form two α-helical layers on each side of the central β-sheet core. The catalytic site created by loops and several α-helices lies at the top edge of the two layered β-sheets (Fig 2A). Although it has no detectable sequence homology, the 5-phosphatase shows an extensive structural similarity to endonucleases, such as the well-studied apurinic/apyrimidic (AP) endonuclease APE1 [62]. Structural comparison with APE1, together with recent crystal structures of 5-phosphatase/lipid complexes [60], has shed light on the molecular basis for the catalytic function of 5-phosphatases.
Fig. 2.

5-phosphatase structure and catalytic mechanism. (A) Ribbon diagram of the catalytic domain of INPP5B with bound diC8 PI(4)P (in sticks) and a Mg2+ ion (in green sphere) (PDB ID: 3MTC). (B) Zoom-in view of the membrane insertion motif (MIM) of INPP5B. MIM is colored in gold and enriched with hydrophobic and cationic residues. (C) Zoom-in view of the interactions between INPP5B with the head-group of PI(4)P. Residues Y502, K503, K516, and R518 form a cationic pocket P4 interacting motif (P4IM) for the recognition of the D4 phosphate group. The D1 phosphate group is bonded by N379 and K380. The Mg2+ ion is coordinated by N275 and E303. Residues directly involved in hydrolysis (D447, N449, D524, and H549) are labeled and shown in sticks. (D) Schematic model of 5-phosphatase catalytic mechanism. The reaction starts with the activation of a water molecule by the general base D447. The nucleophilic attack by this water molecule results in a pentavalent transition state of the scissile phosphate. The negative charge on the scissile phosphate is stabilized by H549 and the Mg2+ ion. Finally, the 5′ hydroxyl group of the lipid is protonated by a proton relayed from D447.
2.3 Membrane binding
Most of the 5-phosphatases prefer to act on membrane-embedded PI substrates. Therefore, hydrophobic membrane interacting motifs (MIM) on the catalytic domain are often required to mediate the interaction between the protein and membrane bilayer. The 5-phosphatase domain of INPP5B in complex with diC8 PI(4)P reveals exemplary structural features for membrane docking (Fig. 2B) [60]. In the complex structure, the aliphatic chains of diC8 PI(4)P pack against two α-helices enriched with hydrophobic residues (colored gold in Fig. 2B). These hydrophobic residues are likely to penetrate into the hydrophobic lipid bilayer to access the substrate. In addition to hydrophobic residues, several cationic residues, including His314, Lys308, Arg376, and Arg410, may contribute to the electrostatic interactions with negatively charged lipid head groups. Therefore, both hydrophobic and electrostatic interactions may facilitate the anchoring of the 5-phosphatase domain on membrane interface and may enhance the processivity of PI hydrolysis. It is interesting to note that although the primary sequence of these two α-helices is variable in other 5-phosphatases, the hydrophobic nature of these residues is conserved. The differences in the membrane binding region imply a differential preference for membrane with unique physical properties. This may explain the observed enzyme sensitivities to fatty acid composition and membrane curvature [26, 63].
2.4 Substrate specificity
Most of the 5-phosphatases selectively hydrolyze the D5 phosphate from either PI(4,5)P2 or PI(3,4,5)P3. This preference suggests that the D4 phosphate is the key group for enzyme recognition. The complex structure of the diC8 PI(4)P bound INPP5B catalytic domain reveals four highly conserved residues within a long loop, forming a so-called P4IM (P4-interacting-motif) to accommodate the D4 phosphate [60]. Within the P4IM, three cationic residues, K503, K516, and R518 interact with the D4 phosphate through intensive electrostatic and hydrogen bonding interactions. The hydroxyl group of Y502 also makes a hydrogen bond with the D4 phosphate (Fig. 2C). Thus, these four residues function as the key specificity determinants for D4-phosphate binding. As a deviation from this basic structural feature for substrate selectivity, the P4IM loop in SHIP2 and SHIP1 is seven residues longer than in INPP5B. Upon binding of a competitive inhibitor, the P4IM loop folds over the inhibitor and an arginine residue shifts to a position allowing direct binding with the D3 phosphate on the inositol ring [61]. This new structural feature in SHIP2 and SHIP1 may explain the preferred specificity of these two enzymes towards PI(3,4,5)P3 vs. PI(4,5)P2. The complex structure of INPP5B with diC8 PI(4)P also highlights the binding site for the D1 phosphate group, which is recognized by Asn379 and Lys380 through direct hydrogen bonding. The specific recognition by the P4IM and the D1 phosphate binding site aligns the substrate in a way that allows the D5 phosphate to be positioned in the catalytic site (Fig. 2C).
2.5 Catalytic mechanism
Structural similarity between 5-phosphatases and endonucleases and the similar chemical nature of the scissile bond suggest a conserved catalytic mechanism between these two families of enzymes [7, 59]. Recent comprehensive studies combining structural, biochemical, and computational analyses have led to a unified mechanism for AP nucleases [64]. A similar mechanism can be applied to PI-5-phosphatases. Serveral key residues, as well as a magnesium ion, required for phosphoryl cleavage have been identified based on the diC8 PI(4)P bound INPP5B catalytic domain structure [60] (Fig. 2D). Invariable residues N449 and D447 coordinate and activate a water molecule with D447 acting as a general base. The activated water molecule attacks the D5 phosphate to form a pentavalent phosphate intermediate. The highly negatively charged intermediate is stabilized by the Mg2+, which is coordinated by invariable residues E303 and N275, as well as several first hydration shell water molecules. The protonation of the departing PI product is not well understood, however, one possible mechanism is that the cleaved phosphate group may relay the proton from the protonated D524 (Fig. 2D).
2.6 Human disease mutations associated with the 5-phosphatase domain
Structural studies on these enzymes have shed light on many missense mutations in 5-phosphatases that are associated with several human hereditary diseases [59, 65]. Human genetic studies have documented a large number of missense mutations within the 5-phosphatase domain of OCRL. These are responsible for the Lowe syndrome, as well as the Dent 2 disease, which has a less severe symptom compared to the Lowe syndrome [66]. Most of the missense mutations found in Lowe syndrome patients are clustered either in the hydrophobic core of the catalytic domain or at the catalytic site. These mutations may cause the instability of the protein or directly impair the enzymatic activity. Mutations found in the Dent 2 patients tend to be on the surface around the catalytic site, which may have less impact on the enzymatic activity [65].
3. The Sac1 family of phosphatases
3.1 The Sac1 family
The founding member of this family Sac1 was originally identified in yeast by two independent genetic screens searching for modifiers of actin cytoskeleton defects and of trans-Golgi network exocytic failure caused by inactivation of Sec14p, respectively [67–69]. Subsequently, the Sac1 domain was found to share considerable homology with mammalian synaptojanin [28]. The Sac1 domain comprises approximately 500 amino acids with a highly conserved CX5R motif [70] and was demonstrated to possess a PI phosphatase activity [32]. With the completed genomes of several species, all genes that contain the Sac1 domain have been identified. There are five Sac1 domain-containing proteins in both human and yeast (Fig. 3).
Fig. 3.

Schematic domain organization of the phosphatases in the Sac1 family. Abbreviations: TM, transmembrane motif; hSac2, homology of Sac2.
Sac1 is a 67kD type II membrane protein that localizes to both ER and Golgi apparatus [71, 72]. In yeast, loss of function of Sac1 causes a broad range of cellular defects [6] such as disorganization of the actin cytoskeleton [67], inositol auxotrophy [71], cold sensitivity for growth [67], multiple drug sensitivities [73], vacuolar function [74, 75], cell wall maintenance [76], and ATP uptake into the ER [77]. Sac1 mutants in Drosophila die as embryos with defects in dorsal closure [78]. Mouse strains deficient in Sac1 are embryonic lethal [79, 80]. Knockdown of Sac1 expression in mammalian cells results in disorganization of Golgi membranes and mitotic spindles [79, 80]. These findings suggest an essential role of Sac1 in multicellular organisms. In vitro studies have demonstrated that Sac1 dephosphorylates a number of PIs, including PI(3)P, PI(4)P, and PI(3,5)P2 [32]. In vivo, genetic ablation of Sac1 activity in yeast results in a nearly 10-fold increase in the steady state levels of PI(4)P with little effect on PI(4,5)P2 [32, 72, 81], suggesting that yeast Sac1p is a major enzyme for PI(4)P degradation in vivo.
Sac2 is also named INPP5F. Besides its N-terminal Sac1 domain, Sac2 has a unique domain named hSac2 (homology of Sac2) with unknown function and a C-terminal proline-rich domain (Fig. 3). Initial cloning and characterization show that this enzyme exhibits 5-phosphatase activity specific for PI(4,5)P2 and PI(3,4,5)P3 [82]. Along this line, Sac2 is shown to hydrolyze PI(3,4,5)P3 and thereby inactivates Akt signaling, which eventually leads to the attenuation of heart hypotrophy [83]. Moreover, Sac2 deficient mice display abnormal fetal gene reactivation and increased susceptibility to stress induced heart hypertrophy. On the other hand, Sac2 transgenic mice prevent the animal from developing these symptoms [84].
Fig. 5.
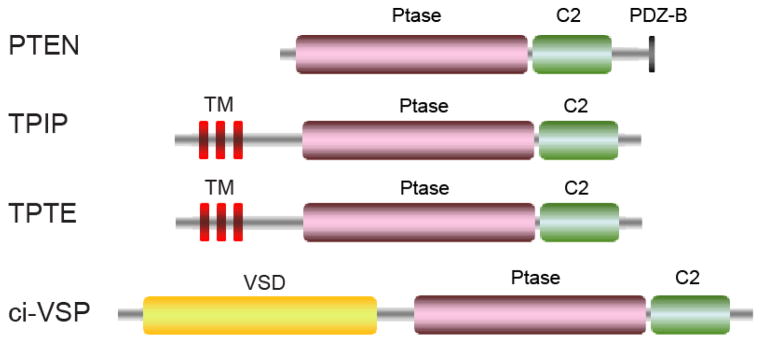
Schematic domain organization of the phosphatases in the PTEN family. Abbreviations: PDZ-B, PDZ domain binding sequence; VSD, Voltage sensing domain.
Sac3, also known as Fig4p, was originally identified in a screen for genes induced by mating pheromone in Saccharomyces. cerevisiae [85]. Sac3 is a PI-5-phosphatase that specifically hydrolyzes PI(3,5)P2 to generate PI(3)P both in vitro and in vivo [86–88]. Interestingly, Sac3/Fig4p forms a complex with a PI(3)P-5-kinase Fab1p and a scaffold protein Vac14p. This complex is also conserved in mammals and is responsible for the acute regulation of subcellular levels of PI(3,5)P2 [89–92]. Genetic mutations affecting the function of this complex lead to a number of diseases, including Charcot-Marie-Tooth (CMT) type 4J, a subset of amyotrophic lateral sclerosis (ALS) in human, and neurodegeneration in the pale tremor mouse [93–95].
Fig. 4.

Representative structures of CX5R motif-containing PI phosphatases. (A) Ribbon diagram of the crystal structure of Sac1 (PDB ID: 3LWT). Sac1 has two tightly packed domains, the SacN (cyan) and the catalytic domain (labeled as Ptase in grey). The conserved structural core is colored in pink and the catalytic P-loop in red. (B) Zoom-in view of the structural core (in pink), which is conserved in all CX5R-based PI phosphatases. (C) Molecular surface of the catalytic site of Sac1. The surface is colored based on electrostatic potential with positively charged region in blue (+4 kcal/electron) and negatively charged surface in red (−4 kcal/electron). Sac1 is in the same orientation in (A) and (C). (D)–(F) Crystal structure of PTEN (PDB ID: 1D5R). The C2 domain is shown in yellow. (G)–(I) Crystal structure of MTMR2 (PDB ID: 1ZVR). The PH-GRAM domain is shown in green. (J)–(L) Crystal structure of SidF (PDB ID: 4FYG).
3.2 Crystal structure of Sac1
The Sac1 domain of yeast Sac1 is the only crystal structure solved in this family to this day [96]. The structure reveals that the Sac1 domain is composed of two closely packed subdomains: a novel N-terminal sub-domain and the catalytic sub-domain. The N-domain has a unique fold comprised of three layers of β-sheets, and one long and three short α-helices. The function of the N-domain is still unclear, but it may mediate protein-protein interactions that may direct Sac1-mediated dephosphorylation of PI(4)P on a specific membrane compartment [97]. Indeed, a recent report showed that the N-domain of Sac1 interacts with Vps74 for the localized phosphatase activity within the Golgi apparatus [98].
The catalytic domain consists of a nine-stranded β-sheet flanked by five α-helices with two and three on each side of the β-sheet (Fig. 4A). Conserved among all CX5R motif-based phosphatases, Sac1 has a central structural core formed by four parallel β-strands and one α-helix. The loop (P-loop) connecting this α-helix and one of the β-strands harbors the catalytic CX5R motif (Fig. 4B). The other conserved structural feature among all CX5R motif-based phosphatases is the overall surface charge distribution. The surface hosting the catalytic CX5R motif is largely positively charged (Fig. 4C). On this surface, several flexible loops enriched with hydrophobic residues surround the catalytic site and likely facilitate the association of the enzyme with the anionic membrane bilayer.
Compared with the structure of PTEN [99] or myotubularins [100, 101], one difference is evident. The catalytic P-loop of Sac1 assumes a unique conformation in that the catalytic cysteine (C392) is oriented away from the conserved R398 in the CX5R motif. This difference suggests that a conformational change of the catalytic P-loop may be required to achieve full enzymatic activity (Figs. 4B and E). Sac1 is found to be an allosteric enzyme and its activity can be stimulated by anionic phospholipids [102]. However, comprehensive understanding of the catalytic mechanism requires co-crystal structures of Sac1 with bound substrate or activator.
Sac1 is localized on the endoplasmic reticulum (ER) and Golgi apparatus via its two C-terminal transmembrane motifs. An intriguing question remains in the field as to whether Sac1 hydrolyzes PI(4)P on the same membrane that it anchors (cis model) or on a different membrane compartment (trans model). Genetic data have shown that inactivation of Sac1p leads to a specific increase in the cellular level of PI(4)P and the bulk of accumulated PI(4)P is generated by the phosphoinositol-4 kinase Stt4 at the plasma membrane [74, 75]. Sac1 has been proposed to control the PI(4)P levels at the plasma membrane (PM) at the ER-PM contact sites in a trans mode [103]. The presence of a long linker between the catalytic domain and the first transmembrane motif allows the enzyme to overcome the gap between ER and PM at the contact sites and hydrolyze substrates in trans. Recent studies also suggest that Sac1 can hydrolyze PI(4)P in cis on the ER membrane to facilitate the sterol/PI(4)P exchange between ER and Golgi apparatus [104]. The exact mode of how Sac1 functions is still under debate. More delicate experiments are needed to dissect the mechanism. However, it is possible that Sac1 can hydrolyze its substrates both in cis and in trans depending on the presence of its associated regulatory proteins.
4. PTEN
4.1 The PTEN phosphatase family
PTEN (phosphatase and tensin homolog located on chromosome TEN) was first identified as a tumor suppressor gene located on chromosome 10q23 [105, 106]. PTEN is one of the most frequently mutated genes found in diverse human cancers [105–109] and in hereditary cancer predisposition syndromes, such as Cowden [110, 111]. PTEN contains the CX5R phosphatase catalytic signature motif and is initially reported to function as a protein tyrosine phosphatase [107]. It was later demonstrated that the principal catalytic function of PTEN is to dephosphorylate the D3 phosphate of PI(3,4,5)P3 [112], thereby antagonizing the PI 3-kinase signaling pathway [113]. Following its initial identification and characterization, extensive research on its physiological roles has been done in various cell cultures and animal models [114]. Results from these studies have implicated PTEN in a variety of cellular processes such as cell polarity, adhesion, migration, survival, metabolism, immune response, cell cycle progression, and DNA repair [115].
PTEN is composed of an N-terminal phosphatase domain, followed by a C2 domain, which contributes to membrane binding and phosphatase activation [116]. PTEN also has a C-terminal tail region that contains multiple phosphorylation sites and a PDZ domain-binding sequence (Fig. 5). Bioinformatic analysis revealed several distinct PTEN-related genes. The testis specific TPTE (transmembrane phosphatase with tensin homology) is localized to the PM but lacks PI phosphatase activity [117, 118]. TPIP (TPTE and PTEN homologous inositol lipid phosphatase), which comprises several splice isoforms and displays similar 3-phosphatase activity compared with PTEN [118]. Interestingly, another TPIP ortholog in the marine invertebrate, Ciona intestinales, is a voltage-sensitive phosphatase (VSP) [119]. VSP consists of a transmembrane voltage sensing domain (VSD) and a cytoplasmic PTEN-related phosphatase domain (Fig. 5), which couples the phosphatase action directly to membrane potential changes. However, unlike PTEN, VSP hydrolyzes the D5 phosphate from PI(4,5)P2 and PI(3,4,5)P3 [120, 121]. Structural studies of PTEN [99] and several VSPs [122, 123] have provided significant insights into their substrate specificity and mutations in the context of various human diseases.
4.2 Crystal structure of PTEN
The crystal structure of PTEN was solved with the phosphatase domain and the C2 domain in tandem [99]. The phosphatase assembles in an overall fold similar to protein tyrosine phosphatases with a central five-stranded β-sheet sandwiched by 6 α-helices (Fig. 4D). Compared with other proteins or PI phosphatases, the core of the phosphatase domain, which contains four parallel β-strands and one α-helix with the catalytic P-loop connecting one of the β-strands and the α-helix, is conserved (Fig. 4E). The catalytic P-loop containing the CX5R motif resides in a deep and wide pocket at the center of the phosphatase domain. This pocket is surrounded by several functional loops including a so-called “TI” loop (named after the conserved threonine and isoleucine residues) and a “WPD” loop, where the Asp residue functions as both a general acid and base during catalysis. The catalytic pocket is highly positively charged (Fig. 4F), which is a critical determinant for the binding of heavily negatively charged substrates. The structure also reveals the presence of a C2 domain. The C2 domain consists of a core β-sandwich made up of two antiparallel β-sheets, and two short α-helices near the interface. The phosphatase and the C2 domains are tightly packed against each other sharing an extensive interface enriched with hydrophobic and aromatic residues, and an interdomain hydrogen-bonding network. Mutations affecting this interface are frequently observed in cancer [124].
4.3 Substrate specificity and membrane binding
The structure of PTEN, as well as recently determined VSP structures, unraveled several structural determinants for the remarkable substrate specificity of PTEN family phosphatases. PTEN prefers to hydrolyze the D3 phosphate of PI(3,4,5)P3. To accommodate the bulky headgroup of PI(3,4,5)P3, PTEN has evolved to possess a wide and deep catalytic pocket. This enlarged pocket is created by a conserved 4-residue insertion within the “TI” loop. Moreover, the catalytic site of PTEN is more positively charged than that of Sac1 or MTM as demonstrated by the calculated electrostatic potentials (Fig. 4F), which is in agreement with its preference for PI(3,4,5)P3, the most negatively charged PIs species. Contrary to PTEN, the other family member, VSP prefers to hydrolyze the D5 phosphate on PI(4,5)P2 and PI(3,4,5)P3 [120, 121]. This discrepancy is at least partially explained by a residue substitution within the catalytic CX5R motif (a G365 in VSP, while A126 in PTEN) since the G365A mutation abolishes the activity of VSP against PI(4,5)P2 [120]. Structures of the PTEN family enzymes also revealed the role of the C2 domain in membrane association. The C2 domain of PTEN has two protruding solvent exposed loops clustered with 9 basic and two hydrophobic residues on the same face as and in close proximity to the active site. These two loops are likely inserted into the lipid bilayer, thereby contributing to the activation of the phosphatase domain [99].
In spite of the intense biochemical and structural studies of PTEN family phosphatases, the exact molecular mechanisms for substrate determination, particularly the distinct preference for the scissile phosphate group by PTEN and VSP, remain to be established. Future work with complex structures of enzymes with their cognate lipid substrates or products is necessary to answer these intriguing questions.
5. Myotubularins
5.1 The myotubularin family of phosphatases
The myotubularin family consists of 15 members that are named MTM1 and MTMRs 1–14. Myotubularins are only found in eukaryotes and are highly conserved from yeast to human. The first member of this family, MTM1, was identified as gene mutated in patients with a congenital muscle disorder called X-linked myotubular myopathy [125]. The protein sequence of MTM1 shares similarity with protein tyrosine phosphatases (PTPs), but exhibits poor activity towards phosphoprotein substrates [126]. The enzymatic activity of MTM1 was further characterized by two independent studies which demonstrated that MTM1 is a PI-3-phosphatase that hydrolyzes PI(3)P and PI(3,5)P2 [127, 128]. With complete genome sequencing, 14 myotubularin-related (MTMR) genes are identified to date. MTM1 and MTMRs constitute the largest family of CX5R dependent PI phosphatases.
Members of the myotubularin family share a common functional core, which encompasses the PI-binding PH-GRAM (Pleckstrin homology-glucosyltransferases, rab-like GTPase activators and myotubularin) domain [129–131] and the catalytic PTP (Protein tyrosine phosphatase) domain (Fig. 6). Most myotubularins contain a CC (coiled-coil) region, which is important for homodimerization and/or heterodimerization [132]. In addition, MTM1 and MTMR1/2 have a PDZ-binding domain at the C-terminal end to mediate protein-protein interactions. Other individual family members, MTMR3/4 have a C-terminal FYVE domain [133], while MTMR5/13 contain a DENN [134] and a PH domain at the N-and C-terminus, respectively (Fig. 6). Interestingly, the PTP phosphatase domains of some members are catalytically inactive. Hence, the MTM family of phosphatases can be divided into both catalytically active and inactive members. Similar to MTM1, the catalytically active members (MTMR2-4, MTMR7-8, and MTMR14) contain the intact canonic CX5R motif and preferentially hydrolyze PI(3)P and PI(3,5)P2. However, the rest of members in the family (MTMR5 and MTMR9-13) contain inherent missense substitutions at the conserved Cys and Arg residues within the CX5R motif and thus are catalytically inactive [135, 136]. Despite lacking enzymatic activity, these catalytically-dead phosphatases can heterodimerize with the active homologs thus may act in conjunction with active MTMRs to regulate, perhaps, the localization and activity of the active MTMR [132, 137, 138].
Fig. 6.

Schematic domain organization of the phosphatases in the myotubularin family. Abbreviations: PH-GRAM, Pleckstrin homology-glucosyltransferases, rab-like GTPase activators and myotubularin; CC, coiled-coil; FYVE: Fab1, YOTB, Vac1, and EEA1; Dead Ptase, catalytically-inactive phosphatase; DENN, Differentially expressed in neoplastic versus normal cells.
Crystal structures of human MTMR2 in its apo form and in complex with its substrates have been determined. The structural information has significantly advanced our understanding of the catalytic mechanism as well as the pathological mutations associated with human diseases for this family of phosphatases.
5.2 Crystal structure of myotubularin phosphatases
The crystal structure of human MTMR2 [100] reveals that the PH-GRAM domain adopts the PH domain architectural fold of 7 β-strands sealed on one side by a C-terminal α-helix. It is connected to the phosphatase domain via a 20-residue linker and is directly apposed to the phosphatase domain to form a compact globular structure (Fig. 4G). However, a recent structure of MTMR6 (PDB ID: 2YF0; Moche et al., unpublished) reveals that the PH-GRAM domain is completely displaced from the phosphatase domain. This difference suggests that at least in some myotubularins, the PH-GRAM domain has an independent role to the phosphatase domain. The function of the PH-GRAM is likely to facilitate the membrane association via binding with PIs by flexible solvent accessible loops (loops connecting β1/β2, β3/β4, and β6/β7). The phosphatase domain (~375 residues) is composed of a flat 7-stranded β-sheet surrounded by 13 α-helices. Similar to other CX5R motif-based phosphatases, the catalytic core consists of 4 parallel β-strands, one α-helix, and a loop containing the catalytic CX5R motif connecting between one of the β-strands and the α-helix (Fig. 4H). Conserved in all phosphatases acting at the membrane interface, the structure also reveals a highly positively charged surface encompassing the active site pocket (Fig. 4I).
5.3 Substrate specificity and catalytic mechanism
The complex structures of MTMR2 with its substrates PI(3)P and PI(3,5)P2 revealed the structural component for membrane interaction [101, 139] (Fig. 7A). The aliphatic lipid acyl chains of the substrate are packed against helix α6, which contains a number of surface exposed hydrophobic residues and two lysine residues (Fig. 7B). This α-helix is highly solvent exposed and projects above the substrate binding pocket. The structure and chemical nature of α6 may allow it to partially insert into the membrane bilayer while membrane-embedded substrates are being hydrolyzed.
Fig. 7.

Crystal structure of MTMR2, a representative member of the myotubularin family. (A) Ribbon diagram of the structure of MTMR2 with bound diC4 PI(3,5)P2 (in sticks) (PDB ID: 1ZVR). (B) Zoom-in view of the MTMR2 MIM, which is colored in gold and enriched with hydrophobic residues. (C) Zoom-in view of the interactions between MTMR2 with the head-group of PI(3,5)P2. R459 and R463 form the recognition site for the D5 phosphate. The bulky residues W421 and R463 form hydrogen bond with the D4 hydroxyl group and prohibit the binding of D4 hydroxyl phosphorylated substrates due to steric clash. Residues N330, N355, S418, and R423 interact with the D1 phosphate.
The complex structures also highlighted structural features for substrate selectivity [101, 139]. MTMR2 has a unique D5 phosphate recognition site, where R459 and R463 engage with the D5 phosphate of PI(3,5)P2 through extensive salt bridges as well as hydrogen bonding interactions (Fig. 7C). However, the D5 phosphate recognition site is not involved in the binding of PI(3)P. The recognition of PI(3)P is through two other key structural features. First, residues N330, N355, S418, and R423 form another site to accommodate the D1 phosphate of the substrate by making hydrogen bonds with the D1 phosphate. Second, the side chains of R463 and particularly the bulky W421 are close to and form hydrogen bonds with the 4′ hydroxyl group of the inositol ring. These two residues prohibit the binding of any PIs phosphorylated at the D4 position due to a potential steric clash with the D4 phosphate. Together, these structural features provide not only the selectivity but also the proper positioning of the D3 phosphate of the substrate. The D3 phosphate makes seven hydrogen bonds with the amides of the P-loop and the guanidinium side chain of R423 and is readily attacked by the catalytic cysteine. Due to the conservation of the catalytic motif, it is plausible to propose a similar catalytic mechanism for all CX5R-based phosphatases including myotubularins. The detailed mechanism is illustrated in Fig. 8D.
Fig. 8.

Crystal structure of the bacterial CX5R motif-based PI phosphatase SidF. (A) Ribbon diagram of the structure of SidF with bound diC4 PI(3,4)P2 (in sticks) (PDB ID: 4FYG). (B) Zoom-in view of the SidF MIM (in gold). (C) Zoom-in view of the interactions between SidF with the head-group of PI(3,4)P2. Residues H233, K740, K646, S647, and K717 form a strong cationic pocket for the specific docking to the D4 phosphate of the substrate. Residue N419 forms a hydrogen bond with D1 phosphate. Like other CX5R phosphatases, the scissile phosphate is positioned close to the catalytic cysteine by interactions with the P-loop main-chain amide groups and R651. (D) Schematic model of the general catalytic mechanism for all CX5R containing PI phosphatases, using SidF as an example. The reaction has two steps. In the first step, the catalytic cysteine attacks the scissile phosphate to break the bond between the phosphate and hydroxyl group of the lipid. D650 functions as the general acid to donate a proton to the leaving hydroxyl, resulting in a phospho-cysteine intermediate. In the second step, D650 now functions as the general base to activate a water molecule. The activated water molecule hydrolyzes the phospho-cysteine intermediate to restore the enzyme.
6. 4-phosphatases
The first member of this unique CX5R motif-based phosphatase family was demonstrated to hydrolyze specifically the D4 phosphate of PI(3,4)P2 with proteins purified from rat brain [140]. Subsequently, two mammalian genes encoding PI(3,4)P2 4-phosphatases, INPP4A and INPP4B were cloned [141, 142]. INPP4A and INPP4B share an overall 37% sequence identity and a similar domain structure of an N-terminal C2 domain, a large unknown central domain, and a C-terminal catalytic domain [143]. INPP4A is shown to be a negative regulator of Akt signaling [144]. Mutations in this gene in two different mouse models lead to neurodegeneration, suggesting a critical role of INPP4A in neuronal function [145, 146]. INPP4B is also shown to reduce Akt activation and accumulating evidence suggests INPP4B is a tumor suppressor in human cancers [143]. The 4-phosphatase catalytic domain is found to share sequence homology with two other enzymes, P-Rex1 and P-Rex2 [147]. Both P-Rex1 and P-Rex2 have a RacGEF activity mediated by their N-terminal DH-PH domain. However, neither of the 4-phosphatase homology domains exhibits 4-phosphatase activity [143, 147].
Unfortunately, crystal structures of any members in this family are still unavailable. It is intriguing to address how this group of enzymes achieve their unique substrate specificity by hydrolyzing the D4 phosphate only from PI(3,4)P2. Due to their essential role in mammals and links to human diseases, elucidation of the protein structure of this family is warranted.
7. Bacterial PI-phosphatases
7.1 PI-phosphatases encoded by bacterial pathogens
It has been increasingly recognized that modulation of host PI signaling and metabolism is critical for the pathogenicity of many human pathogens [148, 149]. One strategy to exploit host PI pathways by several bacterial pathogens is to inject PI metabolizing enzymes into the host. Many of these enzymes, particularly PI phosphatases have been documented. The enterobacteria Salmonella enterica, which causes food-borne gastroenteritis and typhoid fever in human, delivers a PI phosphatase SigD/SopB into the host and is essential for enteropathogenicity [9]. SopB is shown to have sequence homology with mammalian inositol polyphosphate 4-phosphatases and has a broad spectrum of substrate specificity [150, 151]. Shigella flexneri, the causative agent of human dysentery injects a SopB ortholog, IpgD into the host cells. IpgD prefers to hydrolyze PI(4,5)P2 to generate PI(5)P [10] leading to membrane blebbing and actin filament remodeling and promotes the entry of the bacterium. Mycobaterium tuberculosis, the causative agent for tuberculosis, secretes two PI phosphatases SapM and MptpB. SapM hydrolyzes PI(3)P and inhibits phagosome-late endosome fusion in vitro, and inhibits phagosomal maturation [152]. MptpB was first reported as a protein tyrosine phosphatase [153] and was later demonstrated to preferentially hydrolyze PI(3)P and PI(3,5)P2 [154]. The marine bacterium, Vibrio parahaemolyticus, which causes gastroenteritis in humans, encodes a PI-5-phosphatase-like effector VPA0450 [155]. VPA0450 hydrolyzes the D5 phosphate from the plasma membrane PI(4,5)P2 causing plasma membrane blebbing and cell lysis [155].
Two novel CX5R-based PI phosphatases, named SidF and SidP were recently reported in Legionella pneumophila, the causative agent of Legionnaires’ disease [12, 13]. Both SidF and SidP are PI-3-phosphatases with different substrate specificity. SidF specifically hydrolyzes the D3 phosphate of PI(3,4)P2 and PI(3,4,5)P3 [12], while SidP hydrolyzes PI(3)P and PI(3,5)P2 [13]. Through the concerted action of these two PI phosphatases, Legionella may be able to change the lipid identity of the Legionella-containing vacuoles (LCV), thus establishing an amenable niche for bacterial intracellular propagation. Structural studies of SidF and SidP provided a structural paradigm for bacterial CX5R-based PI phosphatases and shed light on the molecular mechanisms of this new family of phosphatases.
7.2 Crystal structures of SidF and SidP
SidF and SidP have unique primary sequences. Except for the sequence near the catalytic core, SidF and SidP lack detectable sequence homology with other known PI phosphatases. However, the 3D structures of the phosphatase catalytic domains of SidF and SidP are highly similar with each other. Both SidF and SidP consist of a central 10–11 pleated β-sheet sandwiched within 18–19 α-helices (Fig. 4J). The catalytic core of these two enzymes, which contains four parallel β strands and one α-helix with the catalytic P-loop connecting between one of β strands and the α-helix, are conserved among all CX5R motif-based phosphatases (Fig. 4K). In SidF, the catalytic site falls in a groove, which nearly bisects the catalytic domain. This grove is highly positively charged and may facilitate the loading of lipid substrate to the catalytic site (Fig. 4L). The determination of SidF with a lipid substrate molecule diC4 PI(3,4)P2 highlighted the structural basis for membrane interaction, substrate recognition, and catalytic mechanism [12] (Fig 8A).
7.3 Membrane binding
The complex structure of SidF with diC4 PI(3,4)P2 immediately reveals that the acyl chains of the substrate interface with two hydrophobic loops (colored in gold in Fig. 8B). These two hydrophobic loops are likely to penetrate into the hydrophobic lipid bilayer while the substrate is loaded to the catalytic site. In addition to hydrophobic residues, two cationic residues (R366, R422) may contribute to the electrostatic interactions with negatively charged lipid head groups. Thus, similar to 5-phosphatases, both hydrophobic and electrostatic interactions facilitate the stable association of the enzyme with membrane bilayers to enhance the processivity of catalysis.
7.4 Substrate specificity
The complex structure of SidF with diC4 PI(3,4)P2 delineates a cationic pocket mainly formed by residues H233, K646, S647, K717, and K740 for the recognition of the D4 phosphate of PI(3,4)P2 [12] (Fig. 8C). These residues distribute rather dispersedly in primary sequence, but cluster together to form a pocket critical for substrate selectivity. Similar to the P4IM described in 5-phosphatase, extensive hydrogen bonds and electrostatic interactions are involved to accommodate the D4 phosphate. On the other hand, the binding of D1 phosphate group is mediated by R651 through salt bridge and N419 through hydrogen bond interactions. Besides the two phosphate groups, a hydrogen bond between the 2′ hydroxyl group of the inositol ring with the side chain of D650 also plays a key role in orienting the substrate at the catalytic site. These specific interactions allow the positioning of the scissile D3 phosphate to be close to the catalytic residue C645.
7.5 Catalytic mechanism
The catalytic mechanism is conserved among all CX5R-based phosphatases, including the well-studied protein tyrosine phosphatases [156, 157]. In the case of SidF, the D3 scissile phosphate is held in a position close to the Sγ-atom of C645 for nucleophilic attack through intensive polar interactions with five main-chain amide groups of the catalytic P-loop and with the guanidinium group of the CX5R arginine. The reaction can be divided into two steps (Fig. 8D). The first step is the formation of a phospho-cysteine intermediate. The cleavage starts with a nucleophilic attack by the deprotonated Sγ-atom of C645. The breakage of the scissile P-O bond between the D3 phosphate and the inositol ring is likely facilitated by D650, which protonates the 3′ hydroxyl as a general acid. As a result, the transient intermediate, phospho-cysteine is formed. The second step is the hydrolysis of the cysteinyl-phosphate intermediate. The hydrolysis requires a water molecule, which is likely activated by the same D650. This aspartic acid residue acts as a general base to accept a proton from the attacking water molecule (Fig. 8D).
8. Conclusion remarks
Over the past years, our understanding of the structural basis for the function of each family of PI phosphatases has expanded considerably. Although the fundamental structural fold and the catalytic mechanism are apparently different between the two major classes of PI phosphatases, some structural features are common among them. First, PI phosphatases usually have a membrane interaction motif near the catalytic site, which not only plays a role in anchoring the enzyme to the membrane interface but also in discriminating specific membrane bilayers in terms of membrane curvature or lipid composition. Second, for phosphatases that hydrolyze PIs with di-or tri-phosphate groups, a cationic pocket is usually present adjacent to the catalytic site to provide substrate selectivity by the recognition with the non-scissile phosphate group on the inositol ring. Despite the substantial progress, paramount questions still remain unaddressed. The 4-phosphatase family still lacks a representative structure to explain their unique D4 phosphatase activity against PI(3,4)P2. How the substrate specificity is achieved by members in the Sac1 and PTEN families is still uncertain due to the lack of complex structures of the enzymes with their cognate substrates. Moreover, many PI phosphatases contain regulatory domains besides the catalytic domain. How these regulatory domains function in cellular targeting or direct control of the enzyme activity requires more investigation. Further structural work pursuing these questions will certainly enhance our knowledge about the molecular basis of PI phosphatases and provide a framework for the understanding of human genetic diseases and the development of potential therapeutics.
Highlights.
Phosphoinositide phosphatases can be divided into two classes.
The two classes of enzymes have distinct structural fold and catalytic mechanism.
PI phosphatases use a hydrophobic motif for membrane insertion.
A cationic pocket adjacent to the catalytic site determines substrate specificity.
Acknowledgments
This work was supported by the Harry Samuel Mann Award (F.H.) and National Institutes of Health (NIH) Grants R01-GM094347 (Y.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Odorizzi G, Babst M, Emr SD. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem Sci. 2000;25:229–235. doi: 10.1016/s0968-0004(00)01543-7. [DOI] [PubMed] [Google Scholar]
- 2.De Matteis MA, Godi A. PI-loting membrane traffic. Nat Cell Biol. 2004;6:487–492. doi: 10.1038/ncb0604-487. [DOI] [PubMed] [Google Scholar]
- 3.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 4.Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell CA, Gurung R, Kong AM, Dyson JM, Tan A, Ooms LM. Inositol polyphosphate 5-phosphatases: lipid phosphatases with flair. IUBMB Life. 2002;53:25–36. doi: 10.1080/15216540210815. [DOI] [PubMed] [Google Scholar]
- 6.Strahl T, Thorner J. Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1771:353–404. doi: 10.1016/j.bbalip.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whisstock JC, Romero S, Gurung R, Nandurkar H, Ooms LM, Bottomley SP, Mitchell CA. The inositol polyphosphate 5-phosphatases and the apurinic/apyrimidinic base excision repair endonucleases share a common mechanism for catalysis. J Biol Chem. 2000;275:37055–37061. doi: 10.1074/jbc.M006244200. [DOI] [PubMed] [Google Scholar]
- 8.Astle MV, Seaton G, Davies EM, Fedele CG, Rahman P, Arsala L, Mitchell CA. Regulation of phosphoinositide signaling by the inositol polyphosphate 5-phosphatases. IUBMB Life. 2006;58:451–456. doi: 10.1080/15216540600871159. [DOI] [PubMed] [Google Scholar]
- 9.Galyov EE, Wood MW, Rosqvist R, Mullan PB, Watson PR, Hedges S, Wallis TS. A secreted effector protein of Salmonella dublin is translocated into eukaryotic cells and mediates inflammation and fluid secretion in infected ileal mucosa. Mol Microbiol. 1997;25:903–912. doi: 10.1111/j.1365-2958.1997.mmi525.x. [DOI] [PubMed] [Google Scholar]
- 10.Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J. 2002;21:5069–5078. doi: 10.1093/emboj/cdf522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakowski MA, Braun V, Brumell JH. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic. 2008;9:2022–2031. doi: 10.1111/j.1600-0854.2008.00827.x. [DOI] [PubMed] [Google Scholar]
- 12.Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci U S A. 2012;109:13567–13572. doi: 10.1073/pnas.1207903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toulabi L, Wu X, Cheng Y, Mao Y. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem. 2013;288:24518–24527. doi: 10.1074/jbc.M113.474239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majerus PW, Connolly TM, Deckmyn H, Ross TS, Bross TE, Ishii H, Bansal VS, Wilson DB. The metabolism of phosphoinositide-derived messenger molecules. Science. 1986;234:1519–1526. doi: 10.1126/science.3024320. [DOI] [PubMed] [Google Scholar]
- 15.Connolly TM, Bross TE, Majerus PW. Isolation of a phosphomonoesterase from human platelets that specifically hydrolyzes the 5-phosphate of inositol 1,4,5-trisphosphate. J Biol Chem. 1985;260:7868–7874. [PubMed] [Google Scholar]
- 16.Laxminarayan KM, Chan BK, Tetaz T, Bird PI, Mitchell CA. Characterization of a cDNA encoding the 43-kDa membrane-associated inositol-polyphosphate 5-phosphatase. J Biol Chem. 1994;269:17305–17310. [PubMed] [Google Scholar]
- 17.De Smedt F, Verjans B, Mailleux P, Erneux C. Cloning and expression of human brain type I inositol 1,4,5-trisphosphate 5-phosphatase. High levels of mRNA in cerebellar Purkinje cells. FEBS Lett. 1994;347:69–72. doi: 10.1016/0014-5793(94)00509-5. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell CA, Connolly TM, Majerus PW. Identification and isolation of a 75-kDa inositol polyphosphate-5-phosphatase from human platelets. J Biol Chem. 1989;264:8873–8877. [PubMed] [Google Scholar]
- 19.Matzaris M, Jackson SP, Laxminarayan KM, Speed CJ, Mitchell CA. Identification and characterization of the phosphatidylinositol-(4, 5)-bisphosphate 5-phosphatase in human platelets. J Biol Chem. 1994;269:3397–3402. [PubMed] [Google Scholar]
- 20.Ross TS, Jefferson AB, Mitchell CA, Majerus PW. Cloning and expression of human 75-kDa inositol polyphosphate-5-phosphatase. J Biol Chem. 1991;266:20283–20289. [PubMed] [Google Scholar]
- 21.Jefferson AB, Majerus PW. Properties of type II inositol polyphosphate 5-phosphatase. J Biol Chem. 1995;270:9370–9377. doi: 10.1074/jbc.270.16.9370. [DOI] [PubMed] [Google Scholar]
- 22.Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, Mcinnes RR, Nussbaum RL. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358:239–242. doi: 10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- 23.Mao Y, Balkin DM, Zoncu R, Erdmann KS, Tomasini L, Hu F, Jin MM, Hodsdon ME, De Camilli P. A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. Embo J. 2009;28:1831–1842. doi: 10.1038/emboj.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdmann KS, Mao Y, Mccrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, De Camilli P. A role of the lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell. 2007;13:377–390. doi: 10.1016/j.devcel.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowe M. Structure and function of the Lowe syndrome protein OCRL1. Traffic. 2005;6:711–719. doi: 10.1111/j.1600-0854.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- 26.Schmid AC, Wise HM, Mitchell CA, Nussbaum R, Woscholski R. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004;576:9–13. doi: 10.1016/j.febslet.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Jefferson AB, Auethavekiat V, Majerus PW. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1995;92:4853–4856. doi: 10.1073/pnas.92.11.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mcpherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P. A presynaptic inositol-5-phosphatase. Nature. 1996;379:353–357. doi: 10.1038/379353a0. [DOI] [PubMed] [Google Scholar]
- 29.Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, Mccormick DA, De Camilli P. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99:179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- 30.De Camilli P, Emr SD, Mcpherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- 31.Nemoto Y, Arribas M, Haffner C, Decamilli P. Synaptojanin 2, a novel synaptojanin isoform with a distinct targeting domain and expression pattern. J Biol Chem. 1997;272:30817–30821. doi: 10.1074/jbc.272.49.30817. [DOI] [PubMed] [Google Scholar]
- 32.Guo S, Stolz LE, Lemrow SM, York JD. SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem. 1999;274:12990–12995. doi: 10.1074/jbc.274.19.12990. [DOI] [PubMed] [Google Scholar]
- 33.Ringstad N, Nemoto Y, De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci U S A. 1997;94:8569–8574. doi: 10.1073/pnas.94.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Heuvel E, Bell AW, Ramjaun AR, Wong K, Sossin WS, Mcpherson PS. Identification of the major synaptojanin-binding proteins in brain. J Biol Chem. 1997;272:8710–8716. doi: 10.1074/jbc.272.13.8710. [DOI] [PubMed] [Google Scholar]
- 35.Woscholski R, Finan PM, Radley E, Totty NF, Sterling AE, Hsuan JJ, Waterfield MD, Parker PJ. Synaptojanin is the major constitutively active phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase in rodent brain. J Biol Chem. 1997;272:9625–9628. doi: 10.1074/jbc.272.15.9625. [DOI] [PubMed] [Google Scholar]
- 36.Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci U S A. 2008;105:9415–9420. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S. Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 2002;24:67–72. doi: 10.1016/s0387-7604(01)00405-3. [DOI] [PubMed] [Google Scholar]
- 38.Saito T, Guan F, Papolos DF, Lau S, Klein M, Fann CS, Lachman HM. Mutation analysis of SYNJ1: a possible candidate gene for chromosome 21q22-linked bipolar disorder. Molecular psychiatry. 2001;6:387–395. doi: 10.1038/sj.mp.4000871. [DOI] [PubMed] [Google Scholar]
- 39.Stopkova P, Vevera J, Paclt I, Zukov I, Lachman HM. Analysis of SYNJ1, a candidate gene for 21q22 linked bipolar disorder: a replication study. Psychiatry Res. 2004;127:157–161. doi: 10.1016/j.psychres.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisan-Ruiz C. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manji SS, Williams LH, Miller KA, Ooms LM, Bahlo M, Mitchell CA, Dahl HH. A mutation in synaptojanin 2 causes progressive hearing loss in the ENU-mutagenised mouse strain Mozart. PLoS One. 2011;6:e17607. doi: 10.1371/journal.pone.0017607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu L, Damen JE, Cutler RL, Krystal G. Multiple cytokines stimulate the binding of a common 145-kilodalton protein to Shc at the Grb2 recognition site of Shc. Mol Cell Biol. 1994;14:6926–6935. doi: 10.1128/mcb.14.10.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ware MD, Rosten P, Damen JE, Liu L, Humphries RK, Krystal G. Cloning and characterization of human SHIP, the 145-kD inositol 5-phosphatase that associates with SHC after cytokine stimulation. Blood. 1996;88:2833–2840. [PubMed] [Google Scholar]
- 45.Rohrschneider LR, Fuller JF, Wolf I, Liu Y, Lucas DM. Structure, function, and biology of SHIP proteins. Genes Dev. 2000;14:505–520. [PubMed] [Google Scholar]
- 46.Wada T, Sasaoka T, Funaki M, Hori H, Murakami S, Ishiki M, Haruta T, Asano T, Ogawa W, Ishihara H, Kobayashi M. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol Cell Biol. 2001;21:1633–1646. doi: 10.1128/MCB.21.5.1633-1646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chi Y, Zhou B, Wang WQ, Chung SK, Kwon YU, Ahn YH, Chang YT, Tsujishita Y, Hurley JH, Zhang ZY. Comparative mechanistic and substrate specificity study of inositol polyphosphate 5-phosphatase Schizosaccharomyces pombe Synaptojanin and SHIP2. J Biol Chem. 2004;279:44987–44995. doi: 10.1074/jbc.M406416200. [DOI] [PubMed] [Google Scholar]
- 48.Pesesse X, Moreau C, Drayer AL, Woscholski R, Parker P, Erneux C. The SH2 domain containing inositol 5-phosphatase SHIP2 displays phosphatidylinositol 3,4,5-trisphosphate and inositol 1,3,4,5-tetrakisphosphate 5-phosphatase activity. FEBS Lett. 1998;437:301–303. doi: 10.1016/s0014-5793(98)01255-1. [DOI] [PubMed] [Google Scholar]
- 49.Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem. 2000;275:20110–20116. doi: 10.1074/jbc.M910119199. [DOI] [PubMed] [Google Scholar]
- 50.Kong AM, Speed CJ, O’malley CJ, Layton MJ, Meehan T, Loveland KL, Cheema S, Ooms LM, Mitchell CA. Cloning and characterization of a 72-kDa inositol-polyphosphate 5-phosphatase localized to the Golgi network. J Biol Chem. 2000;275:24052–24064. doi: 10.1074/jbc.M000874200. [DOI] [PubMed] [Google Scholar]
- 51.Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, Schurmans S, Dallapiccola B, Majerus PW, Valente EM, Gleeson JG. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compere P, Schiffmann SN, Gergely F, Riley JH, Perez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 53.Conduit SE, Dyson JM, Mitchell CA. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett. 2012;586:2846–2857. doi: 10.1016/j.febslet.2012.07.037. [DOI] [PubMed] [Google Scholar]
- 54.Ijuin T, Mochizuki Y, Fukami K, Funaki M, Asano T, Takenawa T. Identification and characterization of a novel inositol polyphosphate 5-phosphatase. J Biol Chem. 2000;275:10870–10875. doi: 10.1074/jbc.275.15.10870. [DOI] [PubMed] [Google Scholar]
- 55.Gurung R, Tan A, Ooms LM, Mcgrath MJ, Huysmans RD, Munday AD, Prescott M, Whisstock JC, Mitchell CA. Identification of a novel domain in two mammalian inositol-polyphosphate 5-phosphatases that mediates membrane ruffle localization. The inositol 5-phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J Biol Chem. 2003;278:11376–11385. doi: 10.1074/jbc.M209991200. [DOI] [PubMed] [Google Scholar]
- 56.Ijuin T, Yu YE, Mizutani K, Pao A, Tateya S, Tamori Y, Bradley A, Takenawa T. Increased insulin action in SKIP heterozygous knockout mice. Mol Cell Biol. 2008;28:5184–5195. doi: 10.1128/MCB.01990-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mochizuki Y, Takenawa T. Novel inositol polyphosphate 5-phosphatase localizes at membrane ruffles. J Biol Chem. 1999;274:36790–36795. doi: 10.1074/jbc.274.51.36790. [DOI] [PubMed] [Google Scholar]
- 58.Ooms LM, Fedele CG, Astle MV, Ivetac I, Cheung V, Pearson RB, Layton MJ, Forrai A, Nandurkar HH, Mitchell CA. The inositol polyphosphate 5-phosphatase, PIPP, Is a novel regulator of phosphoinositide 3-kinase-dependent neurite elongation. Mol Biol Cell. 2006;17:607–622. doi: 10.1091/mbc.E05-05-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsujishita Y, Guo S, Stolz LE, York JD, Hurley JH. Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell. 2001;105:379–389. doi: 10.1016/s0092-8674(01)00326-9. [DOI] [PubMed] [Google Scholar]
- 60.Tresaugues L, Silvander C, Flodin S, Welin M, Nyman T, Graslund S, Hammarstrom M, Berglund H, Nordlund P. Structural basis for phosphoinositide substrate recognition, catalysis, and membrane interactions in human inositol polyphosphate 5-phosphatases. Structure. 2014;22:744–755. doi: 10.1016/j.str.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 61.Mills SJ, Persson C, Cozier G, Thomas MP, Tresaugues L, Erneux C, Riley AM, Nordlund P, Potter BV. A synthetic polyphosphoinositide headgroup surrogate in complex with SHIP2 provides a rationale for drug discovery. ACS Chem Biol. 2012;7:822–828. doi: 10.1021/cb200494d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 63.Chang-Ileto B, Frere SG, Chan RB, Voronov SV, Roux A, Di Paolo G. Synaptojanin 1-mediated PI(4,5)P2 hydrolysis is modulated by membrane curvature and facilitates membrane fission. Dev Cell. 2011;20:206–218. doi: 10.1016/j.devcel.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsutakawa SE, Shin DS, Mol CD, Izumi T, Arvai AS, Mantha AK, Szczesny B, Ivanov IN, Hosfield DJ, Maiti B, Pique ME, Frankel KA, Hitomi K, Cunningham RP, Mitra S, Tainer JA. Conserved structural chemistry for incision activity in structurally non-homologous apurinic/apyrimidinic endonuclease APE1 and endonuclease IV DNA repair enzymes. J Biol Chem. 2013;288:8445–8455. doi: 10.1074/jbc.M112.422774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pirruccello M, De Camilli P. Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem Sci. 2012;37:134–143. doi: 10.1016/j.tibs.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoopes RR, Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–267. doi: 10.1086/427887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Novick P, Osmond BC, Botstein D. Suppressors of yeast actin mutations. Genetics. 1989;121:659–674. doi: 10.1093/genetics/121.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cleves AE, Novick PJ, Bankaitis VA. Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J Cell Biol. 1989;109:2939–2950. doi: 10.1083/jcb.109.6.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bankaitis VA, Aitken JR, Cleves AE, Dowhan W. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature. 1990;347:561–562. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- 70.Hughes WE, Cooke FT, Parker PJ. Sac phosphatase domain proteins. Biochem J. 2000;350(Pt 2):337–352. [PMC free article] [PubMed] [Google Scholar]
- 71.Whitters EA, Cleves AE, Mcgee TP, Skinner HB, Bankaitis VA. SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J Cell Biol. 1993;122:79–94. doi: 10.1083/jcb.122.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nemoto Y, Kearns BG, Wenk MR, Chen H, Mori K, Alb JG, Jr, De Camilli P, Bankaitis VA. Functional characterization of a mammalian Sac1 and mutants exhibiting substrate-specific defects in phosphoinositide phosphatase activity. J Biol Chem. 2000;275:34293–34305. doi: 10.1074/jbc.M003923200. [DOI] [PubMed] [Google Scholar]
- 73.Hughes WE, Pocklington MJ, Orr E, Paddon CJ. Mutations in the Saccharomyces cerevisiae gene SAC1 cause multiple drug sensitivity. Yeast. 1999;15:1111–1124. doi: 10.1002/(SICI)1097-0061(199908)15:11<1111::AID-YEA440>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 74.Tahirovic S, Schorr M, Mayinger P. Regulation of intracellular phosphatidylinositol-4-phosphate by the Sac1 lipid phosphatase. Traffic. 2005;6:116–130. doi: 10.1111/j.1600-0854.2004.00255.x. [DOI] [PubMed] [Google Scholar]
- 75.Foti M, Audhya A, Emr SD. Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell. 2001;12:2396–2411. doi: 10.1091/mbc.12.8.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schorr M, Then A, Tahirovic S, Hug N, Mayinger P. The phosphoinositide phosphatase Sac1p controls trafficking of the yeast Chs3p chitin synthase. Curr Biol. 2001;11:1421–1426. doi: 10.1016/s0960-9822(01)00449-3. [DOI] [PubMed] [Google Scholar]
- 77.Kochendorfer KU, Then AR, Kearns BG, Bankaitis VA, Mayinger P. Sac1p plays a crucial role in microsomal ATP transport, which is distinct from its function in Golgi phospholipid metabolism. Embo J. 1999;18:1506–1515. doi: 10.1093/emboj/18.6.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei HC, Sanny J, Shu H, Baillie DL, Brill JA, Price JV, Harden N. The Sac1 lipid phosphatase regulates cell shape change and the JNK cascade during dorsal closure in Drosophila. Curr Biol. 2003;13:1882–1887. doi: 10.1016/j.cub.2003.09.056. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y, Boukhelifa M, Tribble E, Morin-Kensicki E, Uetrecht A, Bear JE, Bankaitis VA. The Sac1 phosphoinositide phosphatase regulates Golgi membrane morphology and mitotic spindle organization in mammals. Mol Biol Cell. 2008;19:3080–3096. doi: 10.1091/mbc.E07-12-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, Boukhelifa M, Tribble E, Bankaitis VA. Functional studies of the mammalian Sac1 phosphoinositide phosphatase. Adv Enzyme Regul. 2009;49:75–86. doi: 10.1016/j.advenzreg.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rivas MP, Kearns BG, Xie Z, Guo S, Sekar MC, Hosaka K, Kagiwada S, York JD, Bankaitis VA. Pleiotropic alterations in lipid metabolism in yeast sac1 mutants: relationship to “bypass Sec14p” and inositol auxotrophy. Mol Biol Cell. 1999;10:2235–2250. doi: 10.1091/mbc.10.7.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minagawa T, Ijuin T, Mochizuki Y, Takenawa T. Identification and characterization of a sac domain-containing phosphoinositide 5-phosphatase. J Biol Chem. 2001;276:22011–22015. doi: 10.1074/jbc.M101579200. [DOI] [PubMed] [Google Scholar]
- 83.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 84.Zhu W, Trivedi CM, Zhou D, Yuan L, Lu MM, Epstein JA. Inpp5f is a polyphosphoinositide phosphatase that regulates cardiac hypertrophic responsiveness. Circ Res. 2009;105:1240–1247. doi: 10.1161/CIRCRESAHA.109.208785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Erdman S, Lin L, Malczynski M, Snyder M. Pheromone-regulated genes required for yeast mating differentiation. J Cell Biol. 1998;140:461–483. doi: 10.1083/jcb.140.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006;5:723–731. doi: 10.1128/EC.5.4.723-731.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rudge SA, Anderson DM, Emr SD. Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15:24–36. doi: 10.1091/mbc.E03-05-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Botelho RJ, Efe JA, Teis D, Emr SD. Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol Biol Cell. 2008;19:4273–4286. doi: 10.1091/mbc.E08-04-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sbrissa D, Ikonomov OC, Fenner H, Shisheva A. ArPIKfyve homomeric and heteromeric interactions scaffold PIKfyve and Sac3 in a complex to promote PIKfyve activity and functionality. J Mol Biol. 2008;384:766–779. doi: 10.1016/j.jmb.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sbrissa D, Ikonomov OC, Fu Z, Ijuin T, Gruenberg J, Takenawa T, Shisheva A. Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem. 2007;282:23878–23891. doi: 10.1074/jbc.M611678200. [DOI] [PubMed] [Google Scholar]
- 92.Jin N, Chow CY, Liu L, Zolov SN, Bronson R, Davisson M, Petersen JL, Zhang Y, Park S, Duex JE, Goldowitz D, Meisler MH, Weisman LS. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. Embo J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, Mckenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Zolov SN, Chow CY, Slutsky SG, Richardson SC, Piper RC, Yang B, Nau JJ, Westrick RJ, Morrison SJ, Meisler MH, Weisman LS. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A. 2007;104:17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manford A, Xia T, Saxena AK, Stefan C, Hu F, Emr SD, Mao Y. Crystal structure of the yeast Sac1: implications for its phosphoinositide phosphatase function. Embo J. 2010;29:1489–1498. doi: 10.1038/emboj.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wood CS, Hung CS, Huoh YS, Mousley CJ, Stefan CJ, Bankaitis V, Ferguson KM, Burd CG. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol Biol Cell. 2012;23:2527–2536. doi: 10.1091/mbc.E12-01-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cai Y, Deng Y, Horenkamp F, Reinisch KM, Burd CG. Sac1-Vps74 structure reveals a mechanism to terminate phosphoinositide signaling in the Golgi apparatus. J Cell Biol. 2014;206:485–491. doi: 10.1083/jcb.201404041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 100.Begley MJ, Taylor GS, Kim SA, Veine DM, Dixon JE, Stuckey JA. Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Mol Cell. 2003;12:1391–1402. doi: 10.1016/s1097-2765(03)00486-6. [DOI] [PubMed] [Google Scholar]
- 101.Begley MJ, Taylor GS, Brock MA, Ghosh P, Woods VL, Dixon JE. Molecular basis for substrate recognition by MTMR2, a myotubularin family phosphoinositide phosphatase. Proc Natl Acad Sci U S A. 2006;103:927–932. doi: 10.1073/pnas.0510006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhong S, Hsu F, Stefan CJ, Wu X, Patel A, Cosgrove MS, Mao Y. Allosteric activation of the phosphoinositide phosphatase Sac1 by anionic phospholipids. Biochemistry. 2012;51:3170–3177. doi: 10.1021/bi300086c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stefan CJ, Manford AG, Baird D, Yamada-Hanff J, Mao Y, Emr SD. Osh Proteins Regulate Phosphoinositide Metabolism at ER-Plasma Membrane Contact Sites. Cell. 2011;144:389–401. doi: 10.1016/j.cell.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 104.Mesmin B, Bigay J, Moser Von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 105.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, Mccombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 106.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 107.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res. 1997;57:4736–4738. [PubMed] [Google Scholar]
- 109.Cairns P, Evron E, Okami K, Halachmi N, Esteller M, Herman JG, Bose S, Wang SI, Parsons R, Sidransky D. Point mutation and homozygous deletion of PTEN/MMAC1 in primary bladder cancers. Oncogene. 1998;16:3215–3218. doi: 10.1038/sj.onc.1201855. [DOI] [PubMed] [Google Scholar]
- 110.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 111.Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–585. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- 112.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 113.Stambolic V, Suzuki A, De La Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 114.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 115.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 116.Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen H, Rossier C, Morris MA, Scott HS, Gos A, Bairoch A, Antonarakis SE. A testis-specific gene, TPTE, encodes a putative transmembrane tyrosine phosphatase and maps to the pericentromeric region of human chromosomes 21 and 13, and to chromosomes 15, 22, and Y. Hum Genet. 1999;105:399–409. doi: 10.1007/s004390051122. [DOI] [PubMed] [Google Scholar]
- 118.Walker SM, Downes CP, Leslie NR. TPIP: a novel phosphoinositide 3-phosphatase. Biochem J. 2001;360:277–283. doi: 10.1042/0264-6021:3600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 120.Iwasaki H, Murata Y, Kim Y, Hossain MI, Worby CA, Dixon JE, Mccormack T, Sasaki T, Okamura Y. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2008;105:7970–7975. doi: 10.1073/pnas.0803936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murata Y, Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol. 2007;583:875–889. doi: 10.1113/jphysiol.2007.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Matsuda M, Takeshita K, Kurokawa T, Sakata S, Suzuki M, Yamashita E, Okamura Y, Nakagawa A. Crystal structure of the cytoplasmic phosphatase and tensin homolog (PTEN)-like region of Ciona intestinalis voltage-sensing phosphatase provides insight into substrate specificity and redox regulation of the phosphoinositide phosphatase activity. J Biol Chem. 2011;286:23368–23377. doi: 10.1074/jbc.M110.214361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu L, Kohout SC, Xu Q, Muller S, Kimberlin CR, Isacoff EY, Minor DL., Jr A glutamate switch controls voltage-sensitive phosphatase function. Nat Struct Mol Biol. 2012;19:633–641. doi: 10.1038/nsmb.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 125.Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, Coy JF, Klauck SM, Poustka A, Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 126.Laporte J, Blondeau F, Buj-Bello A, Tentler D, Kretz C, Dahl N, Mandel JL. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet. 1998;7:1703–1712. doi: 10.1093/hmg/7.11.1703. [DOI] [PubMed] [Google Scholar]
- 127.Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, Mandel JL. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. 2000;9:2223–2229. doi: 10.1093/oxfordjournals.hmg.a018913. [DOI] [PubMed] [Google Scholar]
- 128.Taylor GS, Maehama T, Dixon JE. Inaugural article: myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2000;97:8910–8915. doi: 10.1073/pnas.160255697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choudhury P, Srivastava S, Li Z, Ko K, Albaqumi M, Narayan K, Coetzee WA, Lemmon MA, Skolnik EY. Specificity of the myotubularin family of phosphatidylinositol-3-phosphatase is determined by the PH/GRAM domain. J Biol Chem. 2006;281:31762–31769. doi: 10.1074/jbc.M606344200. [DOI] [PubMed] [Google Scholar]
- 130.Tsujita K, Itoh T, Ijuin T, Yamamoto A, Shisheva A, Laporte J, Takenawa T. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817–13824. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- 131.Doerks T, Strauss M, Brendel M, Bork P. GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biochem Sci. 2000;25:483–485. doi: 10.1016/s0968-0004(00)01664-9. [DOI] [PubMed] [Google Scholar]
- 132.Lorenzo O, Urbe S, Clague MJ. Systematic analysis of myotubularins: heteromeric interactions, subcellular localisation and endosome related functions. J Cell Sci. 2006;119:2953–2959. doi: 10.1242/jcs.03040. [DOI] [PubMed] [Google Scholar]
- 133.Lorenzo O, Urbe S, Clague MJ. Analysis of phosphoinositide binding domain properties within the myotubularin-related protein MTMR3. J Cell Sci. 2005;118:2005–2012. doi: 10.1242/jcs.02325. [DOI] [PubMed] [Google Scholar]
- 134.Marat AL, Dokainish H, Mcpherson PS. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 2011;286:13791–13800. doi: 10.1074/jbc.R110.217067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Laporte J, Bedez F, Bolino A, Mandel JL. Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum Mol Genet. 2003;12(Spec No 2):R285–292. doi: 10.1093/hmg/ddg273. [DOI] [PubMed] [Google Scholar]
- 136.Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med. 2012;18:317–327. doi: 10.1016/j.molmed.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 137.Kim SA, Vacratsis PO, Firestein R, Cleary ML, Dixon JE. Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A. 2003;100:4492–4497. doi: 10.1073/pnas.0431052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mochizuki Y, Majerus PW. Characterization of myotubularin-related protein 7 and its binding partner, myotubularin-related protein 9. Proc Natl Acad Sci U S A. 2003;100:9768–9773. doi: 10.1073/pnas.1333958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Begley MJ, Dixon JE. The structure and regulation of myotubularin phosphatases. Curr Opin Struct Biol. 2005;15:614–620. doi: 10.1016/j.sbi.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 140.Norris FA, Majerus PW. Hydrolysis of phosphatidylinositol 3,4-bisphosphate by inositol polyphosphate 4-phosphatase isolated by affinity elution chromatography. J Biol Chem. 1994;269:8716–8720. [PubMed] [Google Scholar]
- 141.Norris FA, Auethavekiat V, Majerus PW. The isolation and characterization of cDNA encoding human and rat brain inositol polyphosphate 4-phosphatase. J Biol Chem. 1995;270:16128–16133. doi: 10.1074/jbc.270.27.16128. [DOI] [PubMed] [Google Scholar]
- 142.Norris FA, Atkins RC, Majerus PW. The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J Biol Chem. 1997;272:23859–23864. doi: 10.1074/jbc.272.38.23859. [DOI] [PubMed] [Google Scholar]
- 143.Rynkiewicz NK, Liu HJ, Balamatsias D, Mitchell CA. INPP4A/INPP4B and P-Rex proteins: related but different? Adv Biol Regul. 2012;52:265–279. doi: 10.1016/j.advenzreg.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 144.Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, Whisstock JC, Rowe T, Majerus PW, Mitchell CA. The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol Biol Cell. 2005;16:2218–2233. doi: 10.1091/mbc.E04-09-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sasaki J, Kofuji S, Itoh R, Momiyama T, Takayama K, Murakami H, Chida S, Tsuya Y, Takasuga S, Eguchi S, Asanuma K, Horie Y, Miura K, Davies EM, Mitchell C, Yamazaki M, Hirai H, Takenawa T, Suzuki A, Sasaki T. The PtdIns(3,4)P(2) phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature. 2010;465:497–501. doi: 10.1038/nature09023. [DOI] [PubMed] [Google Scholar]
- 146.Nystuen A, Legare ME, Shultz LD, Frankel WN. A null mutation in inositol polyphosphate 4-phosphatase type I causes selective neuronal loss in weeble mutant mice. Neuron. 2001;32:203–212. doi: 10.1016/s0896-6273(01)00468-8. [DOI] [PubMed] [Google Scholar]
- 147.Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3-and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 148.Payrastre B, Gaits-Iacovoni F, Sansonetti P, Tronchere H. Phosphoinositides and cellular pathogens. Subcell Biochem. 2012;59:363–388. doi: 10.1007/978-94-007-3015-1_12. [DOI] [PubMed] [Google Scholar]
- 149.Pizarro-Cerda J, Cossart P. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat Cell Biol. 2004;6:1026–1033. doi: 10.1038/ncb1104-1026. [DOI] [PubMed] [Google Scholar]
- 150.Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci U S A. 1998;95:14057–14059. doi: 10.1073/pnas.95.24.14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marcus SL, Wenk MR, Steele-Mortimer O, Finlay BB. A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett. 2001;494:201–207. doi: 10.1016/s0014-5793(01)02356-0. [DOI] [PubMed] [Google Scholar]
- 152.Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102:4033–4038. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Koul A, Choidas A, Treder M, Tyagi AK, Drlica K, Singh Y, Ullrich A. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J Bacteriol. 2000;182:5425–5432. doi: 10.1128/jb.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beresford N, Patel S, Armstrong J, Szoor B, Fordham-Skelton AP, Tabernero L. MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem J. 2007;406:13–18. doi: 10.1042/BJ20070670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Broberg CA, Zhang L, Gonzalez H, Laskowski-Arce MA, Orth K. A Vibrio effector protein is an inositol phosphatase and disrupts host cell membrane integrity. Science. 2010;329:1660–1662. doi: 10.1126/science.1192850. [DOI] [PubMed] [Google Scholar]
- 156.Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 157.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]