

Abstract
The primary hyperoxalurias (PH), types 1–3, are disorders of glyoxylate metabolism that result in increased oxalate production and calcium oxalate stone formation. The breakdown of trans-4-hydroxy-L-proline (Hyp) from endogenous and dietary sources of collagen makes a significant contribution to the cellular glyoxylate pool. Proline dehydrogenase 2 (PRODH2), historically known as hydroxyproline oxidase, is the first step in the hydroxyproline catabolic pathway and represents a drug target to reduce the glyoxylate and oxalate burden of PH patients. This study is the first report of the expression, purification, and biochemical characterization of human PRODH2. Evaluation of a panel of N-terminal and C-terminal truncation variants indicated that residues 157–515 contain the catalytic core with one FAD molecule. The 12-fold higher kcat/Km value of 0.93 M−1·s−1 for Hyp over Pro demonstrates the preference for Hyp as substrate. Moreover, an anaerobic titration determined a Kd value of 125 μM for Hyp, a value ~1600-fold lower than the Km value. A survey of ubiquinone analogues revealed that menadione, duroquinone, and CoQ1 reacted more efficiently than oxygen as the terminal electron acceptor during catalysis. Taken together, these data and the slow reactivity with sodium sulfite support that PRODH2 functions as a dehydrogenase and most likely utilizes CoQ10 as the terminal electron acceptor in vivo. Thus, we propose that the name of PRODH2 be changed to hydroxyproline dehydrogenase (HYPDH). Three Hyp analogues were also identified to inhibit the activity of HYPDH, representing the first steps toward the development of a novel approach to treat all forms of PH.
Keywords: flavin, glyoxylate, hydroxyproline metabolism, hydroxyproline oxidase, oxalate
INTRODUCTION
The primary hyperoxalurias (PH) are rare, monogenic disorders of glyoxylate metabolism that result in an increase in endogenous oxalate synthesis and the formation of calcium-oxalate kidney stones [1,2]. Over time, progressive renal inflammation and interstitial fibrosis from progressive nephrocalcinosis, recurrent urolithiasis, and secondary complications, such as urinary tract infections, can cause decreased renal function, systemic oxalate deposition, and end-stage renal failure [3–5].
There are three types of PH. PH1 results from mutations in peroxisomal alanine:glyoxylate aminotransferase (AGT; the AGXT gene product) (Figure 1) [6]. PH2 results from mutations in glyoxylate reductase (GR; the GRHPR gene product) [7,8]. PH3 results from inactivating mutations in 4-hydroxy-2-oxoglutarate aldolase (HOGA; the HOGA1 gene product) [9–12]. Current treatments for PH include increased fluid intake and treatment with alkali citrate to increase the solubility of calcium oxalate in the urine [13,14]. Double liver/kidney transplants have been shown to help PH1 patients [13,15]. For some PH1 patients, the administration of pyridoxine hydrochloride, a vitamin B6 precursor for pyridoxal-5′-phosphate, restores AGT function by correctly targeting the protein to the peroxisome [16,17]. Transplants and gene therapy are not an appropriate treatment for PH2 and PH3 patients given the ubiquitous expression of GR and the decreased severity of the disease, respectively [14,18,19]. Because of such a limited scope of treatments for PH1, PH2 and PH3, new therapies that reduce the glyoxylate and oxalate burden in PH patients need to be developed.
Figure 1. Hydroxyproline and glyoxylate metabolism.
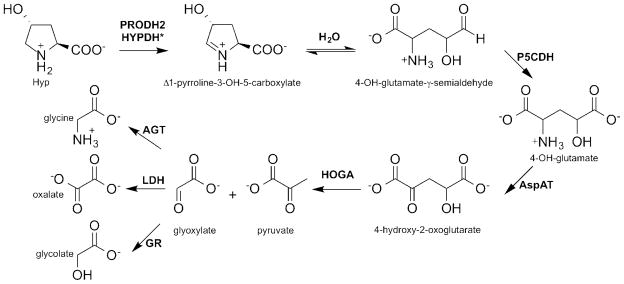
The breakdown of Hyp to glyoxylate requires the action of four enzymes: proline dehydrogenase 2 (PRODH2), Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH), aspartate aminotransferase (AspAT), and 4-hydroxy-2-oxoglutarate aldolase (HOGA). Glyoxylate can then be converted into either glycine by AGT or glycolate by GR. In individuals with PH, the pool of glyoxylate is large enough to allow LDH to convert it into oxalate. *Based upon the results of this study, we propose that the naming of the PRODH2 protein be changed to hydroxyproline dehydrogenase (HYPDH).
The metabolism of trans-4-hydroxy-L-proline (Hyp) makes a significant contribution to the pool of glyoxylate within the body [20]. Hyp is obtained from the diet, through the consumption of collagen, and endogenous collagen turnover, which is estimated to be 2–3 g daily resulting in 300–450 mg of Hyp [21]. Free Hyp cannot be reincorporated into peptides, and the majority of Hyp is metabolized by the liver and kidneys [22].
The Hyp catabolic pathway was originally characterized using bovine and mouse liver and kidney extracts, and the pathway was confirmed by our own group in HepG2 cells, using a metabolic tracer and mass spectrometry analysis [19,23]. The pathway involves four enzymatic steps within the mitochondria of the liver and kidneys (Figure 1). The first step involves the FAD-dependent oxidation of Hyp to Δ1-pyrroline-3-OH-5-carboxylate (3-OH-P5C) by an enzyme historically called hydroxyproline oxidase, also known as proline dehydrogenase 2 (PRODH2, accession number NM_021232, E.C. 1.5.5.2) [23]. This enzyme was localized to the inner mitochondrial membrane through the proteomic analysis of mouse liver cells [24]. By analogy to the Pro degradation pathway, the 3-OH-P5C intermediate undergoes a non-enzymatic hydrolysis to 4-hydroxy-glutamate-γ -semialdehyde [25]. The NAD+ -dependent enzyme Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH) converts the latter product into 4-hydroxy-glutamate (4-OH-Glu) [26]. Aspartate aminotransferase (AspAT) converts 4-OH-Glu into 4-hydroxy-2-oxoglutarate (HOG) using oxaloacetate [23]. HOGA cleaves HOG into glyoxylate and pyruvate [11,27]. Typically, glyoxylate is reduced to glycolate by GR while still in the mitochondria or once it enters the cytoplasm. Glycolate may then enter the peroxisome to be converted back into glyoxylate by glycolate oxidase (GO). In the peroxisome, glyoxylate is converted into glycine by AGT. In individuals with PH, the pool of glyoxylate is large enough to allow lactate dehydrogenase (LDH) to convert glyoxylate into oxalate, leading to the phenotype of elevated oxalate.
PRODH2 is a logical therapeutic target for the treatment of PH, for the following reasons. As the initial step in the Hyp metabolism pathway, PRODH2 inhibition would prevent the synthesis of glyoxylate and all preceding pathway intermediates. PRODH2 is expressed at the highest levels in the organs that are most affected by PH, the liver and kidneys [19]. PRODH2 and HOGA are unique to the pathway, while P5CDH and AspAT are ubiquitously expressed and involved in the proline catabolism pathway [19,26,28]. PH3 patient mutations in HOGA inactivate the enzyme and lead to a build-up of HOG in the blood and urine, which can inhibit GR and potentially contribute to a PH2-like phenotype [10,29]. Therefore, HOGA is also an inappropriate target. Further support for PRODH2 as a target is the phenotype of individuals with hydroxyprolinaemia, who lack PRODH2 activity. These otherwise healthy individuals are unable to degrade Hyp and excrete their excessive Hyp in urine [30–32]. Thus, the inhibition of PRODH2 has the potential to benefit all three types of PH patients by limiting the production of glyoxylate. However, there are no reports on the biochemical properties of human PRODH2.
In this study, we report the expression, purification, and biochemical characterization of a panel of human PRODH2 variants used to identify the catalytic core of the enzyme. The enzyme binds FAD as its cofactor, and kinetic analyses support that the enzyme strongly prefers Hyp over Pro as its substrate. The catalytic core has weak reactivity with sodium sulfite and oxygen and is able to utilize a variety of quinone analogues, which supports that ubiquinone, Coenzyme Q10, is used as the terminal electron acceptor in vivo. Altogether, these data support changing the name of the protein from PRODH2 to hydroxyproline dehydrogenase (HYPDH). Three inhibitors of the enzyme have also been identified, laying the foundation for developing novel therapeutics to block the hydroxyproline degradation pathway and reducing the glyoxylate burden in PH patients.
EXPERIMENTAL
Cloning
The full-length PRODH2 gene was obtained from OriGene and subcloned into the pET19b vector (Novagen) between the NdeI and BamHI restriction sites. This engineered construct also contained an intervening rhinovirus 3C protease cleavage site after the N-terminal His-tag and resulted in the full-length protein being generated, residues 1–536. A series of N-terminal truncation variants was generated in the same manner using the appropriate PCR primers, with the exception of the 147–515 and 165–515 variants. The 147–515 variant was manufactured to be codon optimized and subcloned into a pET15b vector with an engineered thrombin cleavage site between the His-tag and the gene by Genscript. The 165–515 variant was subcloned into the pET151 dTOPO with an engineered rhinovirus 3C protease cleavage site after the TEV protease site already contained within the vector. All C-terminal truncation variants were made by introducing a premature stop codon via site-directed mutagenesis; the DNA sequences of all expression clones were verified.
Expression and protein purification
Each expression construct was transformed into C41(DE3) Escherichia coli cells and grown in two 11 litre fermenters at 37°C. When the OD600 reached 0.8–1.0, expression was induced at 16°C, with the addition of 0.1 mM IPTG overnight. The cells were harvested by centrifugation and stored at −80°C. The cells were resuspended in 150 ml buffer A (20 mM HEPES, pH 7.9, 500 mM KCl, 5 mM imidazole 0.1%Triton X-100, 10% glycerol) containing 0.1 mM of the protease inhibitors PMSF and benzamidine, 1 mM MgCl2, 40 μg/ml Dnase I, 5 mM 2-mercaptoethanol, and 0.2 mM FAD. The cells were lysed using an Avestin EmulsiFlex-C3 cell homogenizer, and the supernatant loaded on to a 10 ml HisPur Cobalt Resin (Thermo Scientific) column. The column was washed with several column volumes of buffer B (20 mM HEPES, pH 7.9, 500 mM KCl, 5 mM imidazole) prior to a linear 5–500 mM imidazole gradient in buffer B. SDS/PAGE analysis was used, after this and all subsequent columns, to determine which samples to pool for the next column or storage. As described below, the percentages of flavin loading and enzymatic activity were assessed at this point to determine which constructs warranted further purification. The following detailed description is provided for those constructs that had significant flavin loading and activity.
For PRODH2 147–515, the post affinity column pool was dialysed overnight into 25 mM HEPES pH 8.0, 10% glycerol with the addition of 15 mM DTT and 0.5 mM EDTA. The following day, the solution was loaded on to a Q-Sepharose FF column (GE Healthcare) and eluted with a linear gradient to 1 M NaCl. To ensure complete loading of FAD and the removal of the His-tag, a 10-fold excess of FAD and biotinylated thrombin (Novagen) at 0.2 units·mg−1 of protein was added directly to the PRODH2 fractions and incubated overnight at 16°C. Removal of the His-tag was confirmed by SDS/PAGE analysis. Biotinylated thrombin was removed using streptavidin agarose beads, and the solution was concentrated using a Vivaspin 10000 MWCO concentrator, filtered and loaded on to a Superdex 200 column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM DTT, and 10%glycerol.
For PRODH2 157–515 and 165–515, the post affinity column pools were dialysed overnight into 25 mM HEPES pH 7.5, 10% glycerol with 15 mM DTT, and 0.5 mM EDTA at 4°C. Rhinovirus 3C protease was added directly to the pooled fractions at a ratio of 1 mg of protease to 15 mg of PRODH2. After confirmation of His-tag removal, the solution was loaded on to a Q-Sepharose FF column (GE Healthcare) and eluted with a linear gradient to 1 M NaCl. To ensure full loading of FAD and the removal of rhinovirus 3C protease, a 10-fold excess of FAD and 1 ml of buffer-washed Glutathione Sepharose beads (GE Healthcare) were added to the pooled fractions and incubated overnight at 4°C. The beads were removed using centrifugation, and the solution was concentrated, filtered and loaded on to a Superdex 200 column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM DTT.
After the Superdex 200 run for each construct, the fractions containing PRODH2 were pooled, concentrated, flash-frozen with liquid nitrogen, and stored at − 80°C until use. Protein concentration was determined using a Coomassie (Bradford) protein assay kit (Thermo Scientific). The flavin concentration was determined by measuring the absorbance at 450 nm (ε = 11300 M−1·cm−1) after denaturing the protein with 4 M guanidine hydrochloride.
Steady-state kinetic analyses
An assay utilizing o-aminobenzaldehyde (o-AB) was used to assess the activity for each PRODH2 variant. The product of the reaction, 3-OH-P5C, hydrolyses to 4-hydroxy-glutamate-γ -semialdehyde, which readily forms a covalent complex with o-AB. Adduct formation was monitored as an increase in absorbance at 443 nm (ε = 2590 M−1·cm−1) [33]. The 0.25 ml reaction mixtures contained 200 nM PRODH2 variant, 50 mM HEPES pH 7.5, 4 mM o-AB, and 200 μM Coenzyme Q1 (CoQ1) as the terminal electron acceptor. The steady-state kinetic parameters at 25°C were obtained for PRODH2 157–515 using Hyp (0–700 mM) and proline (0–750 mM) as substrates (Figure 2). The other stereoisomers of hydroxyproline (trans-4-OH-D-proline, cis-4-OH-L-proline, cis-4-OH-D-proline) were tested at a concentration of 500 mM. The activity of PRODH2 was also tested using air saturated buffers (~260 μM O2 at 25 °C) and Hyp (0–450 mM), without the presence of any other electron acceptor.
Figure 2.
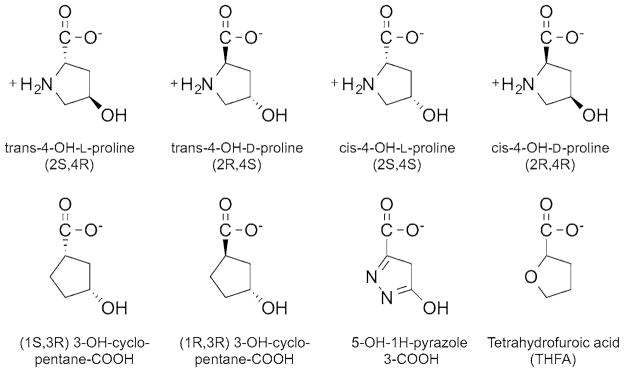
Congeners of trans-4-hydroxy-L-proline tested as substrates and inhibitors
The steady-state kinetic parameters were also determined for PRODH2 157–515 with different ubiquinone analogues (0–400 μM) using 600 mM Hyp. Duroquinone, menadione, CoQ1, CoQ2, and CoQ4 stock solutions were made using ethanol. Triton X-100 (468 μM) was included in reactions containing CoQ4. The final ethanol concentration of the reaction was less than 2%.
The reactions were repeated on several different days in triplicate, using fresh aliquots of each reagent. The kcat, Km, and kcat/Km values were determined for Hyp and the ubiquinone analogues using nonlinear regression in the enzyme kinetics module of SigmaPlot 12.0 (Systat Software). Since the rate of the reaction did not saturate, when using Pro as the substrate, the kcat/Km value for proline was determined using a linear regression fit.
Sulfite reactivity and anaerobic titration with Hyp
Sodium sulfite (0–600 μM) was added aerobically to 50 μM PRODH2 157–515 in 50 mM potassium phosphate buffer pH 7.6 at 25°C. The reaction with sodium sulfite was very slow; equilibration and stabilization of the absorbance spectrum required 6 h. The change in the absorbance at 455 nm was plotted and fit to a hyperbolic equation in SigmaPlot to determine the Kd value. To test the reversibility of the sulfite adduct, tetrahydrofuroic acid (THFA) was added at a final concentration of 1–5 mM and allowed to incubate for 30 min prior to re-acquiring the spectrum.
Hyp (0–800 μM) was anaerobically titrated into 32 μM PRODH2 157–515 in 50 mM potassium phosphate buffer pH 7.6 at 25°C, using an anaerobic gas train outfitted with 2 Oxiclear disposable gas purifier cartridges (Labclear), in order to remove any residual oxygen from the argon supply. After the addition of Hyp, the mixture was allowed to equilibrate for 10 min before the spectrum was recorded. The Kd value was determined in a similar manner to the sodium sulfite procedure, using the change in absorbance at 455 nm.
Inhibition studies
THFA (Sigma–Aldrich), 5-OH-1H-pyrazole-3-COOH (JR Medi-Chem), (1R, 3R) 3-OH-cyclopentane-COOH (RIH Chiragenics), and (1S, 3R) 3-OH-cyclopentane-COOH (RIH Chiragenics) (Figure 2) were tested as inhibitors using PRODH2 157–515 and the standard o-AB assay. The compounds were preincubated with the enzyme for 5 min prior to the addition of 600 mM Hyp. The concentration of each compound was varied up to 5 mM and plotted against the percentage of activity to determine the IC50 value, assuming single-site binding (SigmaPlot 12.0). Stocks of the compounds were made in water, with the exception of (1R, 3R) 3-OH-cyclopentane-COOH. This compound was diluted in DMSO; the final concentration of DMSO in the reaction was 3%. The 4-Hyp diastereomers, trans-4-OH-D-proline, cis-4-OH-L-proline, cis-4-OH-D-proline, were also tested for their ability to inhibit at a concentration of 5 mM.
RESULTS
Determination of the PRODH2 catalytic core
Initially, the purification of the full-length protein of PRODH2 was attempted, as the protein does not contain a canonical mitochondrial targeting sequence. The expression of full-length PRODH2 produced a low yield of aggregated protein with no activity or bound cofactor. Sequence alignments with bacterial PutA (proline utilization A) PRODHs and human PRODH1 were used to identify potential alternative translation start sites for the generation of a panel of PRODH2 variants truncated at the N-terminus (Table 1). It was found that variants with the larger truncations of the N-terminus had increased soluble expression and amount of bound FAD cofactor. However, these truncations still lacked enzymatic activity.
Table 1.
Constructs tested for PRODH2
| Construct | Solubility (%) | FAD loading (%) | kcat (s−1) |
|---|---|---|---|
| 1–536 | <1 | <1 | – |
| 77–536 | <1 | <1 | – |
| 94–536 | <1 | <1 | – |
| 128–536 | <10 | <1 | – |
| 147–536 | <50 | <1 | – |
| 157–536 | <50 | <1 | – |
| 165–536 | <50 | <1 | – |
| 173–536 | <50 | <1 | – |
| 1–515 | <1 | <1 | – |
| 147–515 | >90 | >96 | 0.22 ± 0.02 |
| 157–515 | >90 | >100 | 0.19 ± 0.01 |
| 165–515 | >90 | ~80 | – |
While the analysis of the N-terminal truncation variants was in progress, a report by Tallarita et al. identified that a C-terminal truncation was essential to generate the active form of human PRODH1 (called proline oxidase in this particular study), the enzyme specific for the first step of Pro degradation [34]. In order to determine the likely boundary between the catalytic core of PRODH2 and a possible C-terminal extension, a three-dimensional homology model of full-length PRODH2 was created using the I-TASSER server, using the structure of the PRODH domain of E. coli PutA (18.8% sequence identity, PDB 1TIW) as the reference model (Figure 3) [35–37]. In this model, the N-terminus (highlighted in dark blue, residues 1–156) wraps around the catalytic core an (αβ)8 barrel (cyan, residues 157–515). Like the PRODH domain, a peripheral C-terminal helix (red, residues 516–536) was identified. Using this information, C-terminal truncation variants lacking the last 21 residues were created in combination with an intact N-terminus and several N-terminal truncations (Table 1). Three of these constructs, 147–515, 157–515, and 165–515, were found to be expressed in a soluble form to high levels and to contain the FAD cofactor. However, only the PRODH2 variants 147–515 and 157–515 were found to be active. PRODH2 157–515 was the focus of further analysis, due to its high level of expression and its stability in the absence of glycerol. PRODH2 157–515 eluted from a calibrated Superdex 200 size-exclusion column at a volume consistent with a dimer in solution (data not shown).
Figure 3. Homology model of PRODH2 produced by I-TASSER.

The catalytic core is highlighted in light blue. The section of the N-terminus removed in the PRODH2 157–515 construct is coloured dark blue. The C-terminal truncation is coloured red. FAD was placed into the active site based upon the structure of the PRODH domain of E. coli PutA (PDB 3E2Q).
Kinetic analysis of substrates and ubiquinone analogues
The activity of PRODH2 157–515 was analysed with Hyp and Pro as substrates, using the standard o-AB assay that monitors the formation of the product at 443 nm. CoQ1, a common CoQ10 mimic, was held constant and in excess at 200 μM. PRODH2 157–515 displayed saturable, Michaelis–Menten kinetics with Hyp (Figure 4A). The kcat and Km values for Hyp were found to be 0.19 ± 0.01 s−1 and 200 ± 10 mM respectively, with kcat/Km being 0.93 ± 0.05 M−1·s−1. In contrast, Pro was unable to saturate PRODH2, despite going up to 750 mM Pro. Therefore, only the kcat/Km value of 0.075 ± 0.001 M−1·s−1 could be determined. This ~12-fold difference in the specificity constant supports that the preferred substrate for PRODH2 is Hyp. It was also found that PRODH2 does not react with the other diastereomers of Hyp (Figure 2), trans-4-OH-D-proline, cis-4-OH-L-proline, cis-4-OH-D-proline. In an effort to probe the ability of PRODH2 to use O2 as an electron acceptor, the kinetic parameters for Hyp turnover were also determined using air saturated buffer. The kcat, Km, and kcat/Km values were found to be 0.002 ± 0.001 s−1, 28 ± 6 mM, and 0.07 ± 0.02 M−1·s−1, respectively. Importantly, this kcat value is ~100-fold lower than when CoQ1 is used as the electron acceptor. Thus, PRODH2 is a poor oxidase.
Figure 4. Steady-state kinetic analysis of PRODH2.

(A) Rate versus substrate concentration plot for Hyp and L-proline. Reaction conditions: 50 mM HEPES pH 7.5, 200 nM PRODH2; 4 mM o-AB; 200 μM CoQ1, 25 °C. (B) Rate versus substrate concentration plot for the ubiquinone analogues menadione (▼), duroquinone (○), and CoQ1 (●). The same assay conditions were used holding the Hyp concentration constant at 600 mM.
The ability of other ubiquinone analogues to facilitate the reaction further supports that PRODH2 functions as a dehydrogenase. Of the ubiquinone analogues tested at a saturating concentration of Hyp (600 mM), only menadione, duroquinone, and CoQ1 (Figure 4B and Table 2) were found to support Hyp turnover. The Km values for the analogues were in the 25–143 μM range, and the kcat values were ~0.2–0.3 s−1, the latter being consistent with the values determined by varying the Hyp concentration (Figure 4A). The greatest catalytic efficiency was seen with menadione, but substrate inhibition was observed with a calculated Ki value of 306 ± 34 μM.
Table 2.
Kinetic parameters for ubiquinone analogues*
| Analogue | Km(μM) | kcat(s−1) | kcat/Km(M−1·s−1) | Ki(μM) |
|---|---|---|---|---|
| Duroquinone | 143 ± 18 | 0.27 ± 0.01 | 1900 ± 140 | |
| Menadione | 25 ± 3 | 0.28 ± 0.01 | 11000 ± 770 | 306 ± 34 |
| CoQ1 | 124 ± 19 | 0.19 ± 0.01 | 1500 ± 130 |
Reaction conditions: 50 mM HEPES pH 7.5, 4 mM o-AB, 200 nM PRODH2 157–515, 0–400 μM ubiquinone analogue, 600 mM Hyp.
Sulfite reactivity and anaerobic titration with Hyp
Typically, flavoproteins that are oxidases readily react with sulfite to form an N(5)-adduct with FAD, as indicated by a rapid change and equilibration of the flavin spectrum [38]. The spectrum of PRODH2 with the addition of sulfite also resembles the spectrum of reduced flavin (Figure 5A). However, adduct formation was slow, and the spectrum did not reach equilibrium for ~6 h (data not shown). The Kd for sulfite binding was determined to be 64.0 ± 7.7 μM (Figure 5A inset). The addition of 1–5 mM THFA, a proline analogue and known inhibitor of the bacterial PRODH enzymes, returned the flavin spectrum to the oxidized state (data not shown) [35]. Taken together with the kinetic studies with ubiquinone analogues, these data support that PRODH2 is a dehydrogenase. As such, we propose that the name of the protein be changed from PRODH2 to HYPDH.
Figure 5. Spectral properties of PRODH2 157–515 upon the addition of sodium sulfite and Hyp.

(A) Sodium sulfite binding 6 h after the aerobic addition of 0, 50, 100, 200, and 400 μM of sodium sulfite (labelled 1–5, respectively) to 50 μM PRODH2. Inset: change in absorbance at 455 nm plotted as a function of sulfite concentration. (B) Anaerobic titration 10 min after the addition of 0, 38, 75, 200, and 600 μM Hyp (labelled 1–5, respectively) to 32 μM PRODH2. Inset: change in absorbance at 455 nm plotted as a function of Hyp concentration.
Given the Km value of 200 mM for Hyp, the ability of the catalytic core of PRODH2 to bind Hyp was further assessed using an anaerobic titration of the flavin spectrum. The addition of Hyp (25–800 μM) readily reduced the FAD cofactor (Figure 5B). A Kd value of 125 ± 22 μM was determined by plotting the changes in absorbance at 455 nm (Figure 5B inset). Thus, the Kd value for Hyp is ~1600-fold lower than the Km value, lending additional confidence to the integrity of the recombinant enzyme and its ability to bind and convert Hyp into 3-OH-P5C.
Inhibition studies
Inhibitors of PRODH2/HYPDH activity are of interest, since they represent potential lead compounds to block glyoxylate formation in PH1, PH2, and PH3 patients. Several commercially available Pro and Hyp analogues (Figure 2) were tested. Of the compounds tested, (1S, 3R) 3-OH-cyclopentane carboxylic acid and the diastereomers of Hyp, trans-4-OH-D-proline, cis-4-OH-L-proline, and cis-4-OH-D-proline did not cause inhibition even up to 5 mM (data not shown). THFA, (1R, 3R) 3-OH-cyclopentane-COOH and 5-OH-1H-pyrazole-3-COOH were each found to be inhibitors of HYPDH activity (Figure 6). The inhibition curve for THFA showed a sharp transition, and the IC50 value was determined to be 1.5 ± 0.1 mM. The inhibition curves for (1R, 3R) 3-OH-cyclopentane carboxylic acid and 5-OH-1H-pyrazole-3-COOH were broader, resulting in IC50 values of 2.1 ± 0.1 and 2.9 ± 0.1 mM, respectively.
Figure 6. Inhibition curves for PRODH2 157–515.

THFA (●), (1R, 3R) 3-OH-cyclopentane-COOH (○), and 5-OH-1H-pyrazole-3-COOH (▲).
DISCUSSION
The premise of this study was to express, purify, and biochemically characterize human PRODH2, as there are no previous reports on the human protein. It is hoped that the data provided by this characterization will aid in the design of pharmaceutical compounds that will inhibit PRODH2 and its HYPDH activity. Inhibiting this novel drug target has the potential to ameliorate the high levels of glyoxylate and oxalate in all three forms of PH in patients.
Identification of catalytic core and assessment of substrate specificity
An evaluation of a panel of truncated PRODH2 variants (Table 1) for solubility, FAD loading efficiency, and enzymatic activity identified that the catalytic core of the enzyme contains residues 157–515. The further truncation of the protein to residue 165 resulted in a protein loaded with FAD but lacking in activity, suggesting that residues 157–164 influence substrate binding and catalysis. The role of the remaining residues at the N-terminus is not known, but these residues in the PutA protein from E. coli function as a DNA binding domain that serves as its own repressor [35,39]. The C-terminal portion of PutA encompasses a P5CDH domain, which catalyses the second step of Pro degradation. Interestingly, the crystal structure and biochemical analyses of the PutA enzyme from Bradyrhozobium japonicum and Geobacter sulfurreducens supports that product–substrate channelling occurs between the PRODH and P5CDH active sites [40,41]. In contrast, the C-terminus of PRODH2 does not contain a P5CDH domain, but the removal of the last 21 residues was necessary to generate functional protein, similar to the truncation required to obtain active PRODH1 [34]. At this time it is unclear what the role of the N- and C-terminal regions of PRODH2 are, if any. Interestingly, human PRODH1 also does not contain a C-terminal P5CDH domain. This observation suggests that both PRODH1 and PRODH2 might interact via a similar, intermolecular mechanism with P5CDH.
Human PRODH2 prefers Hyp over Pro as substrate, with a ~ 12-fold increase in catalytic efficiency, kcat/Km (Figure 4A). A comparison of the h(human)PRODH2 model to the crystal structure of the ECPutA (E. coli PutA) variant Y540S in complex with Hyp (Figure 7A), wild-type (WT) PutA in complex with Pro, and the associated kinetic data partially rationalizes the substrate preference of PRODH2 [42]. In the WT PutA-proline complex, Tyr540 projects toward the FAD moiety and would sterically clash with the 4-hydroxy group of Hyp. Mutation of Tyr540 to Ser, the residue present at position 485 of PRODH2, appears to enable Hyp to bind (Figure 7A). The majority of the other residues that encapsulate the Hyp molecule are conserved between the proteins: K329/K236 (EcPutA numbering first; hPRODH2 second), D370/D314, Y437/Y383, L513/L464, Y552/Y497, R555/R500 and R556/R501. The only other differences in this region, other than Y540/S485, are D285/A186 and S327/Q234. While the D285/A186 substitution in PRODH2 removes a charge and opens up the cavity, the S327/Q234 substitution introduces a larger side chain in the vicinity of the 4-hydroxy group of Hyp. Thus, it is clear that the active site of PRODH2 most likely contains structural changes that enable subtly different hydrophobic and hydrogen bonding interactions to preferentially select, bind and catalyse the oxidation of Hyp. Support for this notion comes from two observations. First, there was only a small increase in the kcat/Km value (3–11 M−1·s− 1) for Hyp for the Y540S mutant of PutA, while maintaining a 7-fold greater kcat/Km value for Pro [42]. Secondly, human PRODH2 is highly specific for Hyp and unable to interact with the other stereoisomers. Further structural and biochemical studies will be needed to test these hypotheses.
Figure 7. Comparisons of the human PRODH2 model active site to different ligand complexes of enzymes with PRODH domains.

(A) Hyp complex of the Y540S variant of E. coli PutA. The first residue number is that of E. coli PutA, while the second is for hPRODH2. The carbons atoms of E. coli PutA and hPRODH2 are coloured light blue and cyan, respectively. The * indicates the location of the Y540S mutation in Ec PutA. Portions of the FAD molecule have been omitted for clarity. PDB 3E2Q. (B) Menadione complex of GsPutA. Treatment of GsPutA with N-propargylglycine resulted in an imine linkage between the N5 atom of the reduced flavin (FAD*) and the Lys203 (not shown for clarity). The carbon atoms of GsPutA and hPRODH2 are coloured green and cyan, respectively. The first residue number indicated is that of GsPutA; for those cases where the positions of the side chains diverge, the residue label colour corresponds to the model in which they are contained. PDB 4NMF.
It is interesting to note that the Km value of 200 mM for Hyp with PRODH2 is slightly higher than that of the related PRODH enzymes of bacteria and Saccharomyces, which range from 100 to 150 mM [43–45]. One particular problem in this area is that the Km values appear to be highly dependent upon the reaction conditions, electron acceptor and spectral reporter used. Because of the differences in Km values and the necessity to truncate PRODH2 in order to obtain functional enzyme, we determined the ability of Hyp to bind to PRODH2 using an anaerobic titration (Figure 5B). The 1600-fold lower Kd value for Hyp supports that residues 157–515 contain the catalytic core of the enzyme.
Oxidase versus dehydrogenase
Upon the discovery of rat and bovine PRODH2 in the 1960s, the enzyme was labelled as hydroxyproline oxidase, without additional investigation into its oxygen reactivity. The ability and speed (s to min) of a flavoprotein to bind sulfite is indicative of oxygen being used as an electron acceptor [38]. PRODH2 binds sulfite much more tightly (Kd = 64.0 μM; Figure 5A) than human PRODH1 (10.4 mM) and the related Helicobacter hepaticus PutA (400 μM) [34, 45]. E. coli PutA and the PRODH homologue of Saccharomyces cerevisiae do not bind sulfite [43, 45]. The 6 h equilibration period for PRODH2 to form the sulfite adduct, however, suggests that the enzyme is not truly an oxidase. Further support for PRODH2 being a dehydrogenase comes from the ~100-fold lower kcat value when using buffers saturated with oxygen. Moreover, the ability of short chain ubiquinone analogues (Figure 4B, Table 2) to support PRODH2 catalysis suggests that the enzyme relies upon ubiquinone (CoQ10) in vivo as the electron acceptor. The inability to use the longer chain analogues, CoQ2 and CoQ4, is similar to the recombinant PRODH domain of E. coli PutA and suggests that interaction with the CoQ10 is dependent on interactions with the membrane [25].
A comparison of the binding of menadione bisulfite (MB) within the G. sulfurreducens PutA (GsPutA) to the hPRODH2 model (Figure 7B) provides additional support for PRODH2 functioning as a dehydrogenase in vivo [41]. The hydrophobic residues in GsPutA that surround MB are Tyr309, Leu385, Pro408, and Tyr418. These residues correspond to Tyr383, Leu464, Pro487, and Tyr497 in PRODH2, respectively. The sulfite moiety of MB interacts with Arg421 and Arg422 of GsPutA, but the corresponding residues, Arg500 and Arg521, of hPRODH2 do not. However, this is not surprising as MB was not present as part of the model generation process in I-TASSER (Figure 3). Therefore, the conservation of all residues surrounding the 1,4-naphthoquinone moiety supports that PRODH2 utilizes ubiquinone as an electron acceptor in vivo. Because of these observations, coupled with the slow binding of PRODH2 to sodium sulfite (Figure 4A), its poor ability to utilize oxygen relative to a variety of ubiquinone analogues, and the preference for Hyp as substrate, we propose that PRODH2 be renamed HYPDH.
Inhibitor studies
Inhibitors of PRODH2/HYPDH activity are of interest because blocking the activity of HYPDH is a potential avenue for the treatment of PH. It was found that the diastereomers of Hyp and (1S, 3R) 3-OH-cyclopentane-COOH did not inhibit HYPDH, while THFA, (1R, 3R) 3-OH-cyclopentane-COOH and 5-OH-1H-pyrazole-3-COOH were able to inhibit HYPDH activity (Figures 2 and 6) with the IC50 value of each compound being between 1.5 and 3.0 mM.
Based on these observations, hypotheses can be drawn on what features of each compound can be exploited to design the ideal inhibitor. Since HYPDH does not react with and is not inhibited by the diastereomers of Hyp and (1S, 3R) 3-OH-cyclopentane-COOH, the stereochemistry of the hydroxy group and its orientation related to the carboxylic group appear to be crucial for productive binding. The most effective of the three inhibitors identified was THFA. THFA is a proline analogue that is a known inhibitor of the PRODH enzymes [35]. The oxygen atom on the ring of THFA replaces the nitrogen atom in proline, and thus prevents the ring from opening during catalysis. Moreover, an electron rich atom at this position is important since (1R, 3R) 3-OH-cyclopentane-COOH, which lacks an electron rich atom in this position, has decreased potency, despite possessing the ideal stereochemistry for the hydroxy and carboxy groups. The least effective inhibitor, 5-OH-1H-pyrazole-3-COOH, appears to be a weaker inhibitor because of the presence of an extra nitrogen atom and the resulting planar nature of the ring system. The elucidation of the crystal structure of PRODH2 in the absence and presence of ligand will shed light on the validity of these hypotheses.
In summary, the catalytic core of human PRODH2 contains residues 157–515 and preferentially catalyses the oxidization of Hyp over proline. While PRODH2 can bind sodium sulfite slowly, its poor reactivity with oxygen and the ability to utilize a variety of ubiquinone analogues supports that the enzyme functions as a dehydrogenase in vivo. Further support for this notion comes from the conservation of residues that line the putative quinone binding site. Thus, the name of the PRODH2 protein should logically be changed to HYPDH. This study also identified three inhibitors of the enzyme. These data lay the foundation for the design of future inhibitors of HYPDH activity to treat PH patients.
Acknowledgments
We would like to thank Jill Clodfelter for her technical assistance and the ongoing support and encouragement from the Oxalosis and Hyperoxaluria Foundation.
FUNDING
This work was supported by the National Institutes of Health Grants [grant number DK083527 (to W.T.L./R.P.H.), grant numbers DK073732, DK054468 (to R.P.H.)]. Funding was also provided by the Oxalosis and Hyperoxaluaria Foundation (to W.T.L.) and the Comprehensive Cancer Center of Wake Forest University [grant number NCI CCSG (P30CA012197)].
Abbreviations
- AGT
alanine:glyoxylate aminotransferase
- AspAT
aspartate aminotransferase
- o-AB
o-aminobenzaldehyde
- CoQ1
Coenzyme Q1
- GR
glyoxylate reductase
- HOG
4-hydroxy-2-oxoglutarate
- HOGA
4-hydroxy-2-oxoglutarate aldolase
- HYPDH
hydroxyproline dehydrogenase
- Hyp
trans-4-hydroxy-L-proline
- LDH
lactate dehydrogenase
- MB
menadione bisulfite
- 4-OH-Glu
4-hydroxy-glutamate
- 3-OH-P5C
Δ1-pyrroline-3-OH-5-carboxylate
- P5CDH
Δ1-pyrroline-5-carboxylate dehydrogenase
- PH
primary hyperoxaluria
- PRODH
proline dehydrogenase
- PutA
proline utilization A
- THFA
tetrahydrofuroic acid
Footnotes
AUTHOR CONTRIBUTION
Candice Summitt, Lynnette Johnson, and Thomas Jönsson performed the research. Candice Summitt, Lynnette Johnson, Thomas Jönsson, Ross Holmes and W. Todd Lowther designed the research and analysed the data. Candice Summitt and W. Todd Lowther wrote the paper. Derek Parsonage, Ross Holmes, and W. Todd Lowther supervised the research. All authors approved the final version to be published.
References
- 1.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011 doi: 10.4061/2011/864580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol. 2006;21:1075–1081. doi: 10.1007/s00467-006-0124-4. [DOI] [PubMed] [Google Scholar]
- 4.Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18:986–991. doi: 10.1007/s00467-003-1234-x. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann G, Krieg T, Weber M, Sidhu H, Hoppe B. Unusual painful sclerotic plaques on the legs of a patient with late diagnosis of primary hyperoxaluria type I. Br J Dermatol. 2004;151:1104–1107. doi: 10.1111/j.1365-2133.2004.06247.x. [DOI] [PubMed] [Google Scholar]
- 6.Danpure C, Jennings P. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–34. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 7.Giafi C, Rumsby G. Kinetic analysis and tissue distribution of human D-glycerate dehydrogenase/glyoxylate reductase and its relevance to the diagnosis of primary hyperoxaluria type 2. Ann Clin Biochem. 1998;35:104–109. doi: 10.1177/000456329803500114. [DOI] [PubMed] [Google Scholar]
- 8.Webster KE, Ferree PM, Holmes RP, Cramer SD. Identification of missense, nonsense, and deletion mutations in the GRHPR gene in patients with primary hyperoxaluria type II (PH2) Hum Genet. 2000;107:176–185. doi: 10.1007/s004390000351. [DOI] [PubMed] [Google Scholar]
- 9.Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, Monico CG, Feinstein S, Ben-Shalom E, Magen D, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010;87:392–399. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riedel TJ, Knight J, Murray M, Milliner D, Holmes RP, Lowther WT. 4-Hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. BBA-Mol Basis Dis. 2012;1822:1544–1552. doi: 10.1016/j.bbadis.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riedel TJ, Johnson LC, Knight J, Hantgan RR, Holmes RP, Lowther WT. Structural and biochemical studies of human 4-hydroxy-2-oxoglutarate aldolase: implications for hydroxyproline metabolism in primary hyperoxaluria. PLoS One. 2011;6:e26021. doi: 10.1371/journal.pone.0026021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monico CG, Rossetti S, Belostotsky R, Cogal AG, Herges RM, Seide BM, Olson JB, Bergstrahl EJ, Williams HJ, Haley WE. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol. 2011;6:2289–2295. doi: 10.2215/CJN.02760311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck BB, Hoyer-Kuhn H, Gobel H, Habbig S, Hoppe B. Hyperoxaluria and systemic oxalosis: an update on current therapy and future directions. Expert Opin Inv Drug. 2013;22:117–129. doi: 10.1517/13543784.2013.741587. [DOI] [PubMed] [Google Scholar]
- 14.Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. BBA-Mol Basis Dis. 2012;1882:1453–1464. doi: 10.1016/j.bbadis.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Watts R, Danpure C, De Pauw L, Toussaint C. Combined liver–kidney and isolated liver transplantations for primary hyperoxaluria Type 1: the european experience. Nephrol Dial Transplant. 1991;6:502–511. doi: 10.1093/ndt/6.7.502. [DOI] [PubMed] [Google Scholar]
- 16.Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005;67:1704–1709. doi: 10.1111/j.1523-1755.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- 17.Fargue S, Rumsby G, Danpure CJ. Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta. 2013;1832:1776–1783. doi: 10.1016/j.bbadis.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Danpure CJ. Molecular etiology of primary hyperoxaluria type 1: new directions for treatment. Am J Nephrol. 2005;25:303–310. doi: 10.1159/000086362. [DOI] [PubMed] [Google Scholar]
- 19.Jiang J, Johnson LC, Knight J, Callahan MF, Riedel TJ, Holmes RP, Lowther WT. Metabolism of [13C5]hydroxyproline in vitro and in vivo: implications for primary hyperoxaluria. Am J Physiol Gastrointest Liver Physiol. 2012;302:G637–G643. doi: 10.1152/ajpgi.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney Int. 2006;70:1929–1934. doi: 10.1038/sj.ki.5001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kivirikko KI. Urinary excretion of hydroxyproline in health and disease. Int Rev Connect Tiss Res. 1970;5:93–163. doi: 10.1016/b978-0-12-363705-5.50008-7. [DOI] [PubMed] [Google Scholar]
- 22.Knight J, Easter LH, Neiberg R, Assimos DG, Holmes RP. Increased protein intake on controlled oxalate diets does not increase urinary oxalate excretion. Urol Res. 2009;37:63–68. doi: 10.1007/s00240-009-0170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 24.Da Cruz S, Xenarios I, Langridge J, Vilbois F, Parone PA, Martinou JC. Proteomic analysis of the mouse liver mitochondrial inner membrane. J Biol Chem. 2003;278:41566–41571. doi: 10.1074/jbc.M304940200. [DOI] [PubMed] [Google Scholar]
- 25.Moxley MA, Tanner JJ, Becker DF. Steady-state kinetic mechanism of the proline: ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch Biochem Biophys. 2011;516:113–120. doi: 10.1016/j.abb.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valle D, Goodman SI, Harris SC, Phang JM. Genetic evidence for a common enzyme catalyzing the second step in the degradation of proline and hydroxyproline. J Clin Invest. 1979;64:1365–1370. doi: 10.1172/JCI109593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldstone A, Adams E. Metabolism of γ-hydroxyglutamic acid. J Biol Chem. 1962;237:3476–3485. [PubMed] [Google Scholar]
- 28.Phang JM, Hu CA, Valle D. Disorders in proline and hydroxyproline metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. McGraw-Hill; New York: 2001. pp. 1821–1838. [Google Scholar]
- 29.Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III – a model for studying perturbations in glyoxylate metabolism. J Mol Med. 2012;90:1497–1504. doi: 10.1007/s00109-012-0930-z. [DOI] [PubMed] [Google Scholar]
- 30.Robinson M, Menzies I, Sloan I. Hydroxyprolinaemia with normal development. Arch Dis Child. 1980;55:484–486. doi: 10.1136/adc.55.6.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Efron ML, Bixby EM, Pryles CV. Hydroxyprolinemia. Ii A rare metabolic disease due to a deficiency of the enzyme “hydroxyproline oxidase”. N Engl J Med. 1965;272:1299–1309. doi: 10.1056/NEJM196506242722501. [DOI] [PubMed] [Google Scholar]
- 32.Pelkonen R, Kivirikko KI. Hydroxyprolinemia: an apparently harmless familial metabolic disorder. N Engl J Med. 1970;283:451–456. doi: 10.1056/NEJM197008272830903. [DOI] [PubMed] [Google Scholar]
- 33.Mezl VA, Knox WE. Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal Biochem. 1976;74:430–440. doi: 10.1016/0003-2697(76)90223-2. [DOI] [PubMed] [Google Scholar]
- 34.Tallarita E, Pollegioni L, Servi S, Molla G. Expression in Escherichia coli of the catalytic domain of human proline oxidase. Protein Expres Purif. 2012;82:345–351. doi: 10.1016/j.pep.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 35.Zhang M, White TA, Schuermann JP, Baban BA, Becker DF, Tanner JJ. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry. 2004;43:12539–12548. doi: 10.1021/bi048737e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massey V, Müller F, Feldberg R, Schuman M, Sullivan PA, Howell LG, Mayhew SG, Matthews RG, Foust GP. The reactivity of flavoproteins with sulfite. J Biol Chem. 1969;244:3999–4006. [PubMed] [Google Scholar]
- 39.Becker DF, Thomas EA. Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry. 2001;40:4714–4721. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- 40.Srivastava D, Schuermann JP, White TA, Krishnan N, Sanyal N, Hura GL, Tan A, Henzl MT, Becker DF, Tanner JJ. Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc Natl Acad Sci USA. 2010;107:2878–2883. doi: 10.1073/pnas.0906101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh H, Arentson BW, Becker DF, Tanner JJ. Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site. Proc Natl Acad Sci USA. 2014;111:3389–3394. doi: 10.1073/pnas.1321621111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostrander EL, Larson JD, Schuermann JP, Tanner JJ. A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry. 2009;48:951–959. doi: 10.1021/bi802094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wanduragala S, Sanyal N, Liang X, Becker DF. Purification and characterization of Put1p from Saccharomyces cerevisiae. Arch Biochem Biophys. 2010;498:136–142. doi: 10.1016/j.abb.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch Biochem Biophys. 2002;408:131–136. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]
- 45.Krishnan N, Becker DF. Oxygen reactivity of putA from helicobacter species and proline-linked oxidative stress. J Bacteriol. 2006;188:1227–1235. doi: 10.1128/JB.188.4.1227-1235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]