

Abstract
Excessive TGF-β signaling in epithelial cells, pericytes, or fibroblasts has been implicated in CKD. This list has recently been joined by endothelial cells (ECs) undergoing mesenchymal transition. Although several studies focused on the effects of ablating epithelial or fibroblast TGF-β signaling on development of fibrosis, there is a lack of information on ablating TGF-β signaling in the endothelium because this ablation causes embryonic lethality. We generated endothelium-specific heterozygous TGF-β receptor knockout (TβRIIendo+/−) mice to explore whether curtailed TGF-β signaling significantly modifies nephrosclerosis. These mice developed normally, but showed enhanced angiogenic potential compared with TβRIIendo+/+ mice under basal conditions. After induction of folic acid nephropathy or unilateral ureteral obstruction, TβRIIendo+/− mice exhibited less tubulointerstitial fibrosis, enhanced preservation of renal microvasculature, improvement in renal blood flow, and less tissue hypoxia than TβRIIendo+/+ counterparts. In addition, partial deletion of TβRII in the endothelium reduced endothelial-to-mesenchymal transition (EndoMT). TGF-β–induced canonical Smad2 signaling was reduced in TβRII+/− ECs; however, activin receptor-like kinase 1 (ALK1)–mediated Smad1/5 phosphorylation in TβRII+/− ECs remained unaffected. Furthermore, the S-endoglin/L-endoglin mRNA expression ratio was significantly lower in TβRII+/− ECs compared with TβRII+/+ ECs. These observations support the hypothesis that EndoMT contributes to renal fibrosis and curtailing endothelial TGF-β signals favors Smad1/5 proangiogenic programs and dictates increased angiogenic responses. Our data implicate endothelial TGF-β signaling and EndoMT in regulating angiogenic and fibrotic responses to injury.
Keywords: TGF-β receptor type II, microvasculature, Endoglin, ALK1, Smad1/5, renal fibrosis
CKD leading to end stage renal failure is associated with tissue scarring or fibrosis.1 For development of effective therapeutic strategies, it is essential to define the cellular and molecular mechanisms that modulate regeneration of kidney.2 There is strong evidence that TGF-β1 is a key mediator in the pathogenesis of renal fibrosis in both mouse models and human kidney diseases.1–3 Direct evidence for its role in renal fibrosis comes from studies demonstrating that mice overexpressing an active form of TGF-β1 in the liver develop both progressive liver and renal fibrosis.4 Although restoration of adequate microvascular supply is essential for renal regeneration after chronic injury, genetic studies have revealed the pivotal role of TGF-β signaling in angiogenesis, as well as vascular integrity.5 Incidentally, loss of peritubular capillaries, a hallmark of progressive renal disease in humans, plays a role in the etiology of interstitial fibrosis and tubular atrophy.6
TGF-β signal is initiated when the ligand binds to its cognate TGF-β type II receptor (TβRII) leading to phosphorylation of TβRI (also known as ALK5). The activated TβRI kinase phosphorylates canonical Smad2/3, which translocates to the nucleus and controls gene expression to inhibit endothelial proliferation, migration, and capillary tube formation.7 TGF-β can also bind to activin receptor-like kinase ALK1 exclusively in endothelial cells (ECs), which induces Smad1/5 to potentiate angiogenic programs. Phenotypic and molecular characterization of knockout mice for TGF-β signaling components have demonstrated their critical role in angiogenesis and cardiovascular system.8 Mice with ablation of endothelial TβRII or TβRI (ALK5) show vascular defects in the yolk sac and embryonic lethality as early as embryonic day 10.5. The embryos also have severe anemia and defective vasculogenesis,9,10 both signs of impaired differentiation of hemangioblast to hematopoietic stem cells and endothelial progenitor cells. Nguyen et al.11 recently showed that deletion of TβRII in ECs results in impaired blood vessel morphogenesis in the brain and intracerebral hemorrhage leading to embryonic lethality. The consequences of the partial ablation of TβRII in ECs remain unexplored. Collectively, these studies confirm that TGF-β signaling in ECs is essential for precise regulation of blood vessel assembly and consequently, normal embryonic development.
Although increasing evidence shows the role of TGF-β1 in vasculogenesis or renal inflammation and fibrosis, the role of EC TGF-β receptor signaling in the progression of kidney diseases remains unexplored. The link between endothelial dysfunction and progression of fibrosis awaits elucidation.
Several cell types have been proposed as initiators of tubulointerstitial fibrosis, including tubular epithelial cells, pericytes, fibroblasts, and more recently ECs, all of which can undergo mesenchymal transition to form myofibroblasts.12–16 Indeed, several studies focused on the ablation of epithelial TGF-β signaling or depletion of TβRII in kidney fibroblasts to alter the development of fibrosis.17 The same strategy has been reported for epithelial cells in the proximal tubule and collecting duct.18,19 However, there are no published data on its ablation in the endothelium partly because of the unavailability of endothelial TβRII knockout mice due to the embryonic lethality.9,10 To address the question of whether endothelial TGF-β signals contribute to renal fibrosis, we utilized mice with partial ablation of TβRII in this study (i.e., mice heterozygous for TβRII), and investigated the role of endothelial TGF-β signaling in renal fibrosis of the folic acid nephropathy (FAN) and unilateral ureteral obstruction (UUO) models. The data demonstrate that curtailing TGF-β signaling in vascular ECs is sufficient to reduce endothelial-to-mesenchymal transition (EndoMT), improve angiogenesis, and reduce fibrosis.
Results
Generation of Mice with Endothelial Deletion of TβRII
We intercrossed mice homozygous for the floxed TβRII allele with mice heterozygous for Cre-recombinase under the control of the Tie2-promoter to obtain Tie2-Cre;TβRIIflox/+, which were crossed with TβRIIflox/flox mice. Genotyped offspring from these intercrosses identified heterozygotes (Tie2-Cre;TβRIIflox/+) and wild-type pups (TβRIIflox/flox or TβRIIflox/+), whereas homozygote pups (Tie2-Cre; TβRIIflox/flox) were embryonic lethal, as previously reported.9–11 Percentages of these genotypes were found to be 32.3% each for TβRIIflox/flox and TβRIIflox/+ mice and 35.5% of Tie2-Cre;TβRIIflox/+ mice. Figure 1A shows results of genotyping PCR analysis of mice. Heterozygous mice (Tie2-Cre;TβRIIflox/+ hereafter referred to as TβRIIendo+/−) were viable, did not exhibit any phenotypic differences compared with control mice (TβRIIendo+/+), and showed no abnormalities in renal function at 6 weeks of age. We evaluated TβRII expression levels in kidney ECs isolated from TβRIIendo+/+ and TβRIIendo+/− mice by quantitative real-time PCR analysis. Figure 1B documents the expected significant reduction in mRNA expression of TβRII in isolated ECs from TβRIIendo+/− mice. When treated with TGF-β1 or TGF-β2 at a concentration of 5 ng/ml for 30 minutes, phosphorylation of Smad2 was reduced in ECs from TβRIIendo+/− mice (Figure 1C). To ensure endothelial origin of isolated ECs, we stained cells for the expression of endothelial markers and showed that >90% of cells expressed VE-cadherin (CD144) and platelet-EC adhesion molecules (CD31) (Supplemental Figure 1). These data provided essential information on mice with partial endothelial ablation of TβRII signaling, demonstrating their viability, as opposed to total ablation, and normal development under unstressed conditions.
Figure 1.

Characterization of endothelial TβRII heterozygote mice. (A) Genotyping of pups from TGF-βRIIFlox/Flox and Tie2-Cre: TGF-βRIIFlox/WT matings. Tail PCR genotyping analysis. The top panel shows detection of floxed and WT fragments. The bottom panel shows detection of Cre-transgene. (B) Real-time PCR analysis for TβRII mRNA expression in kidney ECs isolated from TβRIIendo+/+ and TβRIIendo+/− mice (n=4). *P<0.05, TβRII+/+ versus TβRII+/−. (C) Western blot for Smad2 signaling in kidney ECs. WT, wild type; Cont, control.
Partial Endothelial Ablation of TβRII Signaling Boosts Ex Vivo Angiogenic Potential and Favors L-Endoglin–Mediated Smad1/5 Signaling
We evaluated the angiogenic potential using an ex vivo aortic endothelial sprouting assay.20 Data demonstrated that thoracic aortic rings obtained from TβRIIendo+/− mice had accelerated sprouting in three-dimensional matrigel assays, which became clearly detectable by day 6 (Figure 2A). To prove that it was attributable to TGF-β signaling, we next examined the Smad1/5 profile in ECs isolated from kidneys of TβRIIendo+/+ and TβRIIendo+/− mice, because ALK1-induced Smad1/5 signaling is proangiogenic, as opposed to the Smad2/3 pathway, which inhibits proliferation and migration of ECs. Smad1/5 levels at baseline were slightly reduced in ECs from TβRIIendo+/− mice; nevertheless, after TGF-β1 or TGF-β2 treatment, phosphorylation of Smad1/5 was not impaired in ECs from TβRIIendo+/− mice (Figure 2B). The expression ratio of short/long (S/L) isoforms of the auxiliary TGF-β1 receptor endoglin was significantly lower in ECs from TβRIIendo+/− mice at baseline and after TGF-β1 treatment (Figure 2C). An increase in the S/L ratio of endoglin is known to be associated with senescence of ECs.21 Our observation of a reduced S/L ratio in ECs from TβRIIendo+/− mice suggests that L-endoglin–mediated Smad1/5 proangiogenic signaling is more pronounced and increases angiogenic response, as seen ex vivo in TβRIIendo+/− mice. This difference in angiogenic competence between TβRIIendo+/− and TβRIIendo+/+ mice could play a role in vivo after imposition of stress, as was examined in the next series of experiments.
Figure 2.

Functional characterization of TβRII heterozygote mice. (A) Images and quantification of ex vivo angiogenesis assays in three-dimensional matrigel using explant cultures of aortic ring segments at day 6 from TβRIIendo+/+ and TβRIIendo+/− mice (n=5). (B) Western blot for Smad1/5 signaling in kidney ECs isolated from TβRIIendo+/+ and TβRIIendo+/− mice. (C) Endoglin S/L mRNA expression ratios in kidney ECs. *P<0.05, TβRII+/+ versus TβRII+/− and TGF-β1 treated TβRII+/+ versus TβRII+/−. Cont, control.
Impaired Endothelial TβRII Signaling Improves Angiogenesis and Ameliorates Fibrotic Response in Chronic Kidney Injury
The FAN model was used to study the effects of acute and chronic injury in mice with partial deletion of TβRII in the endothelium. This model is advantageous for the study, because it is purely tubulopathic and does not affect ECs. Serum creatinine levels and urinary albumin/creatinine ratio were significantly increased in both TβRIIendo+/+ and TβRIIendo+/− mice 48 hours after folic acid (FA) injection (Supplemental Figure 2, B and C). Both groups of mice showed frequent attenuation and focal loss of brush borders, focal mild- to-moderate tubular dilation, simplification, and rare necrosis of tubular epithelium. Granular eosinophilic material was observed in tubular profiles suggesting necrotic debris and was more prominent in TβRIIendo+/− group (Supplemental Figure 2A).
In contrast with our observations in acute renal injury, during chronic phase of injury, interstitial fibrosis was significantly ameliorated in the TβRIIendo+/− +FA group compared with the TβRIIendo+/+ +FA group (Figure 3A). Consistent with these findings, expression of mRNA encoding collagen I and III was markedly increased in the TβRIIendo+/+ +FA group, whereas upregulation of these genes was ameliorated in the TβRIIendo+/− +FA group of mice (Figure 3C). In a UUO model, a similar observation of reduced fibrosis in TβRIIendo+/− mice was made (Figure 4B). Furthermore, the medullary region of UUO kidneys from TβRIIendo+/+ mice showed striking fibrosis, which was ameliorated in the TβRIIendo+/− group (Figure 4A). Consistent with this, α-smooth muscle actin (α-SMA) staining in the medulla was much more enhanced in UUO kidneys from TβRIIendo+/+ mice compared with TβRIIendo+/− mice (Figure 4C). Thoracic aortas were obtained from FAN mice to study their ex vivo angiogenic potential. Results showed that vessels obtained from mice heterozygous for endothelial TβRII have better angiogenic potential when challenged with FA compared with TβRIIendo+/+ mice, in which angiogenesis was found to be inhibited during FA-induced renal fibrosis (Figure 5).
Figure 3.
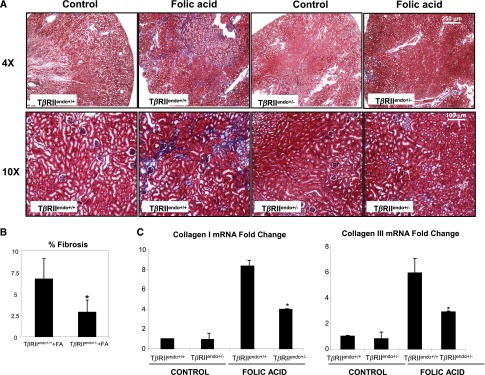
Fibrosis is reduced in TβRIIendo+/− mice with FA toxicity. (A) Representative images of Masson’s trichrome–stained kidney sections from 12-week-old TβRIIendo+/+ and TβRIIendo+/− mice treated with vehicle or FA 6 weeks after injection (n=5). (B) Quantification of fibrotic area (color quantification method). (C) Expression of collagen I and collagen III examined by quantitative real-time PCR. *P<0.05, treated TβRIIendo+/+ versus treated TβRIIendo+/−. Original magnification, ×4 and ×10.
Figure 4.

Fibrosis is reduced in TβRIIendo+/− UUO mice. (A) Representative images of Masson’s trichrome–stained UUO and contralateral kidney sections from TβRIIendo+/+ and TβRIIendo+/− mice (n=4). (B) Quantification of fibrotic area (color quantification method) *P<0.01, TβRIIendo+/+ UUO versus TβRIIendo+/− UUO. (C) Representative images for α-SMA staining in UUO and contralateral kidney sections from TβRIIendo+/+ and TβRIIendo+/− UUO mice. Original magnification, ×4 and ×10.
Figure 5.

Ex vivo angiogenesis assays in mice with FA nephrotoxicity. (A) Representative images of sprouting capillary cords in aortic explants at day 13. (B) Quantitative angiogenesis analysis in TβRIIendo+/+ or TβRIIendo+/− vehicle or FA-treated animals. *P<0.01 untreated versus treated TβRIIendo+/+ or untreated versus treated TβRIIendo+/−.
The impaired angiogenic response in TβRIIendo+/+ mice with FAN was associated with a significant loss of patent and functional capillaries in the kidney as judged by capillary density measurement using intravitally injected Lycopersicon esculentum (lectin) to label functional vessels, compared with CD31-positive total microvasculature in the kidneys (Figure 6A). Although CD31 staining was comparable in all groups of mice, lectin reporting only functional microvessels showed a dramatic decline in TβRIIendo+/+ mice with FAN, but TβRIIendo+/− mice with FAN had a much higher proportion of functional microvessels. The mismatch between patent and total vessels also suggests initiation of an active EndoMT program after renal injury and that curtailed TGF-β signaling in the endothelium may restrain EndoMT transition, as addressed in the next series of experiments.
Figure 6.

Microvascular density (CD31) and patency (lectin) in mice with FA nephrotoxicity. (A) Representative images for CD31 (green) and lectin (red) staining (n=5). (B) Average lengths (in micrometers) of CD31- or lectin-positive peritubular capillaries per image. (C) Ratios of average lengths of lectin- to CD31-positive capillaries (percentage) per image. *P<0.01, treated TβRIIendo+/+ versus TβRIIendo+/− lectin-positive capillary length; **P<0.001, control versus FA-treated TβRIIendo+/+ mice; #P<0.01, treated TβRIIendo+/+ versus TβRIIendo+/−; N.S., P=NS for control versus FA-treated TβRIIendo+/− mice. Original magnification, ×40. Per the journal style, P values were rounded to two decimal places.
In order to investigate the mechanisms underlying loss of patency of microvessels and to see whether this causes hypoperfusion and consequently tissue hypoxia, we performed laser-Doppler flowmetry of renal blood flow in the cortical and medullary regions of UUO kidneys. As shown in Figure 7, renal blood flow was impaired in the medulla of TβRIIendo+/+ mice, whereas flow in TβRIIendo+/− mice was significantly preserved. We also probed kidneys for formation of pimonidazole adducts to measure regions with <10 mmHg pO2 as represented in Figure 7C. A significant difference in staining was observed between both groups of mice with TβRIIendo+/+ mice displaying higher hypoxic regions compared with TβRIIendo+/− mice.
Figure 7.

Renal blood flow measurement and pimonidazole staining in kidneys of TβRIIendo+/+ and TβRIIendo+/− UUO mice. (A) Representative image scans from laser-Doppler flowmetry analysis of contralateral and UUO kidneys from TβRIIendo+/+ and TβRIIendo+/− mice (n=3). (B) Quantification of cortex and medullary renal blood flow in kidneys. *P<0.001, TβRIIendo+/+ and TβRIIendo+/− UUO mice. (C) Immunohistochemical staining for pimonidazole adducts in UUO kidneys of TβRIIendo+/+ and TβRIIendo+/− mice. Original magnification, ×10 and ×40.
Curtailed Endothelial TβRII Signaling Significantly Reduces EndoMT in FAN and UUO Injury
We investigated whether impaired TGF-β receptor signaling in the endothelium affects EndoMT processes during renal fibrosis. To identify myofibroblasts of endothelial origin, we performed double staining for α-SMA and CD31 in kidney sections of TβRIIendo+/+ and TβRIIendo+/− mice with and without FAN and in the UUO mice. Figure 8A shows representative images of α-SMA/CD31 double-positive cells in TβRIIendo+/+ mice treated with FA, which was significantly reduced in similarly treated TβRIIendo+/− mice. Figure 9A shows representative images of kidneys from UUO mice of both groups again attesting to the reduced numbers of α-SMA/CD31 double-positive cells in injured kidneys of TβRIIendo+/− mice. Percentages of α-SMA/CD31 double-positive cells among CD31-positive ECs were quantified to be 22.7% in FA-treated TβRIIendo+/+ mice and 10.1% in TβRIIendo+/− mice (n=5; P<0.01) (Figure 8B). These disparate numbers of EC conversion into myofibroblasts are consistent with the curtailed TGF-β signaling in TβRIIendo+/− mice and further support the notion that TGF-β is an ultimate trigger to induce EndoMT.22
Figure 8.

EndoMT in mice with FA nephrotoxicity. (A) Representative images for CD31 (green) and α-SMA (red) staining (n=5). Wild-type control kidneys shows costaining for CD31 and α-SMA only in vessels. These areas were excluded during quantitative analysis. (B) Quantitative analysis of CD31 plus α-SMA double-positive cells in TβRIIendo+/+ or TβRIIendo+/− vehicle- or FA-treated mice. *P<0.01; #P<0.01. Original magnification, ×60.
Figure 9.

EndoMT in UUO mice. (A) Representative images for CD31 (green) and α-SMA (red) staining (n=4). No primary antibody controls from CD31 staining and α-SMA staining are also shown. (B) Quantitative analysis of CD31 plus α-SMA double-positive cells in UUO kidneys of TβRIIendo+/+ and TβRIIendo+/− mice. *P<0.01. Original magnification, ×60.
To obtain more rigorous evidence that the above “snapshot” figures of endothelial-to-mesenchyme transiting cells faithfully reflect the proportion of myofibroblasts originating from ECs in TβRIIendo+/− mice, we costained kidney sections from mice with and without FA treatment for Cre-recombinase and α-SMA. Expression of Cre-transgene, as a stable marker during epithelial-to-mesenchymal transition in kidney fibrosis was recently demonstrated.23 This lineage tracing approach is valuable in our study because of its ability to positively identify ECs (because Cre-recombinase expression in TβRIIendo+/− mice is driven by the Tie2 promoter). In addition, it circumvents problems arising from the potential loss of endogenous endothelial markers, such as CD31, during EndoMT. Findings presented in Figure 10 demonstrate that the percentages of α-SMA/Cre double-positive cells among Cre-positive ECs were comparable with percentages of α-SMA/CD31 double-positive cells among CD31-positive ECs (Figure 8B). Moreover, in cultured renal ECs, staining for α-SMA–positive cells after TGF-β1 treatment was significantly reduced in TβRII+/− ECs (Figure 11C). These data demonstrate that EndoMT is suppressed approximately 2.0- to 2.5-fold in TβRIIendo+/− mice, a finding that may underlie the improved patency of renal microvasculature and reduced fibrotic response in the FAN model.
Figure 10.

EndoMT in TβRIIendo+/− mice with FA nephrotoxicity. (A) Representative images for Cre (red) and α-SMA (green) staining (n=5). Arrows indicate α-SMA–stained areas with and without Cre costaining. (B) Quantitative analysis of Cre plus α-SMA double-positive cells in TβRIIendo+/− vehicle or FA-treated mice. *P<0.01. Original magnification, ×40.
Figure 11.

Modulation of EndoMT in TβRII+/+ and TβRII+/− kidney ECs. (A) Immunofluorescence images for CD31 or α-SMA in cultured TβRII+/+ and TβRII+/− kidney ECs treated with TGF-β1 (5 ng/ml) for 6 days (n=5). The nuclei are stained with 4′,6-diamidino-2-phenylindole. (B) Quantitative analysis of α-SMA–positive cells per 100 cells. *P<0.002. Original magnification, ×10 for α-SMA; ×40 for CD31 and α-SMA.
Discussion
The data presented herein are based on a novel mouse model of curtailed TGF-β signaling in ECs, which is characterized by a normal phenotype under unstressed conditions, but exhibits a remarkable protection against fibrotic complications of FAN and UUO. This protection is related to the reduced level of endothelial transition toward the mesenchymal phenotype.
During progression of CKD, the most common finding in both animals and humans is upregulation of TGF-β1.2,3,24 Its signaling is usually profibrotic, but is not without exceptions. For instance, although conditional deletion of TβRII in proximal tubular epithelial cells inhibits tubulointerstitial fibrosis in kidneys with UUO,19 the opposite effect is observed when deletion targets epithelia of the collecting duct.18
ECs represent a unique target for TGF-β because they are the predominant cell type expressing an alternative receptor, ALK1.25 The downstream canonical ALK5 cascade of Smad 2/3 with its antiangiogenic program coexists with the ALK1-induced Smad 1/5 and its proangiogenic program of transcriptional activation.26–28 The main switch between these two pathways is provided by endoglin,7,29,30 which itself is thought to be under regulatory control by TGF-β.31,32 Under basal conditions, balance between both pathways exist through endoglin inhibition of Smad3 signaling and activation of Smad1.33 However, it is unknown whether prolonged and intense TGF-β signaling in ECs is associated with the predominant switch to ALK5 pathway. For this reason, attempts at manipulating endothelial TGF-β signaling under stress situations have ample rationale, as detailed below.
In chronic kidney injury, in contrast with acute renal injury, tubulointerstitial fibrosis is ameliorated when endothelial TGF-β signaling is curtailed. Distinct and opposing roles for TβRII in regulating renal fibrosis and inflammation were previously observed in mice with deletion of TβRII in proximal tubular epithelial cells,17 in accord with our observations in mice with endothelial TβRII impaired signaling. Of note, it is important to distinguish the effect of short- and long-term upregulation of TGF-β on cell biology, as exemplified by exhaustion of endoglin regulation of the ALK switch34 or by a switch from the L-endoglin isoform to the S-endoglin isoform, as shown in our study.
Recognition of the role played by EndoMT in kidney fibrosis has grown in recent years. Our model provides an important tool to test this emerging notion. Our comparative analysis of the total number of CD31-labeled microvessels with the actual number of patent perfused microvessels detected by an intravenously injected endothelial-specific lectin discloses a striking difference. It appears that in the kidney undergoing fibrosis, the total number of CD31-labeled vessels is an inaccurate indicator of the microvascular compromise. The decline in lectin-labeled microvessels much exceeds the marginal changes in CD31 labeling, indicating that the patency of circulatory beds is severely impaired with the lesser structural damage to the endothelial lining. These findings are supported by the fact that we observed changes in perfusion and hypoxia in injured kidneys (Figure 7). Moreover, as shown in Figures 8 and 9, many CD31-positive cells actually undergo EndoMT, thus masking the deteriorating microcirculation. On the basis of these considerations, we propose that (1) measuring microvascular patency is a better test to detect abnormalities in tissue perfusion than labeling CD31-positive cells, and (2) that the discrepancy between the total CD31-positive and lectin-positive microvessels is a direct consequence and measure of EndoMT.
The source of myofibroblasts in fibrotic kidneys has been a subject of intense interest. Studies have suggested that fibrocytes, local fibroblasts, epithelial cells, or vascular pericytes are a major source of myofibroblasts.12–16 Each of these sources has experimental limitations due to the lack of specific markers. In light of these issues, our observations that curtailing endothelial TGF-β signaling is sufficient to blunt progressive scarring of injured kidneys is supported by the fact that myofibroblasts can be derived from the capillary endothelium through EndoMT.16,35 Participation of EndoMT in renal fibrosis has been shown in three mouse models of CKD (including Alport’s syndrome, diabetic nephropathy, and chronic nitric oxide synthase inhibition16,35,36) and our findings add to this list that mice with curtailed endothelial TGF-β signaling have significantly blunted EndoMT during chronic renal injury. Data also imply that EndoMT contributes to renal fibrosis after prolonged, but not transient, injury.37 The better preservation of functional capillaries and improved angiogenic responses during kidney injury in our mouse model with curtailed endothelial TGF-β signaling represent additional evidence of subverted EndoMT and protection of these mice from developing fibrosis.
The data collectively document that partial ablation of TβRII in the endothelium reduces fibrosis during chronic kidney injury. Our results indicate the critical role of excessive endothelial TGF-β signaling in enhanced EndoMT, defective angiogenic response to injury, and development of fibrosis.
Concise Methods
Animals
Tie2-Cre mice were obtained from The Jackson Laboratory [B6.Cg-Tg(Tek-cre)1Ywa/J]. TβRIIflox/flox mice [strain name B6.129S6-Tgfbr2tm1Him], which have exon2 of TβRII flanked with lox-p sites as described by Chytil et al.,38 were obtained from the National Cancer Institute Mouse Repository in Maryland. All animal protocols were conducted in accordance with the National Institutes of Health (NIH) 1999 guidance and were approved by the Institutional Animal Care and Use Committee. Mice aged between 8 and 12 weeks were used in this study.
Genotyping
Genotyping was performed by tail DNA PCR analysis. Tail DNA was isolated using the REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich, St. Louis, MO). Primer sequences used for genotyping floxed TβRII allele were as follows: TβRII forward, 5′TAA ACA AGG TCC GGA GCC CA3′; and TβRII reverse, 5′ACT TCT GCA AGA GGT CCC CT3′. Tie2 transgene was detected using the following: forward, 5′GCG GTC TGG CAG TAA AAA CTA TC3′; and reverse, 5′ GTG AAA CAG CAT TGC TGT CAC TT3′. PCR products were analyzed with acrylamide gels.
Aortic Endothelial Sprouting Assay
Thoracic aortas were obtained from 12-week-old mice. Aortic rings sectioned with 1-mm intervals were embedded in three-dimensional matrigel in culture chambers. Newly formed capillary cords in explant cultures were imaged for 6–13 days. The number of capillary sprouts along the perimeter of each explant was counted by two independent observers blinded to the origin of explant cultures. Explants were imaged using an inverted fluorescence microscope (Nikon) equipped with a charge-coupled device camera (Hamamatsu Photonics). Quantitative angiogenesis assays were performed according to previously published protocols.39,40
Folate Model of Acute Renal Injury and Chronic Nephropathy
A single intraperitoneal injection of FA (250 μg/g body weight) was administered in 12-week-old mice, and kidneys and whole blood collected from FA-treated or vehicle-treated animals. Mice were injected with FA and euthanized at 48 hours (AKI phase) or at 6 weeks (chronic fibrotic phase). Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin. Paraffin sections (4-µm thick) were stained with hematoxylin and eosin and periodic acid–Schiff and were examined by a nephropathologist blinded to the origin of individual preparations. Paraffin sections were also stained with Masson’s trichrome and fibrosis was quantified using the NIH ImageJ program.
UUO
Mice were anesthetized with isoflurane and placed in the left lateral decubitus position. The left ureter was identified and ligated at the level of the lower pole of the kidney with 3-0 silk sutures through a left flank incision. Mice were euthanized after 2 weeks and kidneys were processed as described above. Contralateral kidneys were used as controls.
Microalbuminuria, Serum Creatinine, and BP Measurements
Urine albumin content was measured using mouse albumin-specific ELISA (Exocell Laboratories, PA) and creatinine was determined with a creatinine assay kit (Cayman Chemicals, Ann Arbor, MI). Proteinuria was expressed as the urinary albumin/creatinine ratio. Serum creatinine concentration was measured using a commercially available kit (Abcam, Inc., MA).
Isolation of Primary Renal ECs
ECs were isolated from kidneys dissected from 12-week-old TβRIIendo+/+ or TβRIIendo+/− mice, according to the previously published protocol.41 Perfused kidneys were diced and tissue fragments were then digested with Dispase II (0.8 U/ml; Roche, Mannheim) and Collagenase H (1 mg/ml; Roche) in DMEM/F12 medium containing 2% FBS and antibiotics at 37°C for 1 hour, after which the suspension was homogenized by trituration. The homogenate was filtered through 100-μm nylon mesh (Falcon; BD Biosciences, San Jose, CA) followed by 40-μm nylon mesh and pelleted by centrifugation (300g for 6 minutes). Cells were resuspended in 1% BSA in PBS and incubated with anti-rat Dynabeads (Invitrogen, Carlsbad, CA) coated with rat anti-mouse CD31 (BD Biosciences Pharmingen, San Diego, CA) at 4°C for 20 minutes, washed, magnetically separated and ECs were cultured on gelatin-coated plates in Iscove’s modified Dulbecco’s medium containing 15% FBS, 1% BSA, 0.01 mg/ml recombinant human insulin, 0.2% mg/ml human transferrin, 100 ng/ml each of recombinant mouse vascular endothelial growth factor, basic fibroblast growth factor, and epidermal growth factor (all from Life Technologies, Guilford, CT) at 37°C in a humidified 5% CO2 atmosphere. Cells were serum starved overnight and treated either with recombinant TGF-β1 or TGF-β2 (5 ng/ml) (R&D Systems, Minneapolis, MN) for 30 minutes for protein and 48 hours for RNA isolation.
Quantitative PCR Analyses
Total RNA was isolated using a Spinsmart RNA Mini purification kit from Denville Scientific Inc. (Metuchen, NJ). One microgram of RNA was reverse transcribed using the High-Capacity RNA-cDNA kit (Applied Biosystems, Foster City, CA). Real-time quantitative PCR was performed using Perfecta SYBR Green FastMix on a Stratagene MX3000P. The following primers were used: COL1 forward primer, 5′CTG CTG GCA AAG ATG GAG A3′; COL1 reverse primer, 5′ACC AGG AAG ACC CTG GAA TC3′; COL3 forward primer, 5′ CAA ATG GCA TCC CAG GAG3′; COL3 reverse primer, 5′CAT CTC GGC CAG GTT CTC3′; TβRII forward primer, 5′GTG AGA AGC CGC ATG AAG TC3′; TβRII reverse primer, 5′GGG ACT GCT GGT GGT GTA TT3′; S-ENDO forward primer, 5′-TGA GTA TCC CAA GCC TCC AGC CCA T-3′; S-ENDO reverse primer, 5′-CTG AGG GGC GTG GGT GAA GGT CAG; L-ENDO forward primer, 5′-GCA CTC TGG TAC ATC TAT TCT CAC ACA CGT GG-3′; and L-ENDO reverse primer, 5′-GGG CAC TAC GCC ATG CTG CTG GTG G-3′. Values were normalized for the abundance of the amplified 18s rRNA in each experiment. Fold change in gene expression was determined using the 2-ΔΔCT threshold cycle method as previously described.42
Detection of Patent Vessels
To visualize patent vasculature mice were injected with 100 μl of Texas Red-labeled L. esculentum lectin (1 mg/ml in 10 mm HEPES, 150 mm NaCl, pH 7.5; Sigma-Aldrich) via the tail vein 5 minutes before euthanasia.
Laser-Doppler Flowmetry of Renal Blood Flow
Renal blood flow was evaluated 14 days after UUO under xylazine/ketamine sedation. A midline incision was performed to access the kidneys. Cortical and medullary renal blood flow was measured using the laser-Doppler Perfusion Monitoring probes, image scanner, and accompanying software (PeriScan PIM II, PeriFlux system 5000, and LDPIwin version 2.6.1, respectively; PeriMed Instruments, PA). The correct positioning of laser-Doppler probes was verified after completion of each experiment.
Detection of Pimonidazole Adducts
To analyze renal oxygenation in UUO mice, mice received an intraperitoneal injection of 60 mg/kg pimonidazole HCL (Hypoxyprobe-1; Hypoxyprobe, Inc., Burlington, MA) 50 minutes before euthanasia. Pimonidazole immunostaining was performed using a Hypoxyprobe-1 Plus Kit (Hypoxyprobe, Inc.) on 4-μm thick paraffin sections.43
Immunofluorescence Staining
Kidneys were fixed in 4% paraformaldehyde overnight, transferred to PBS containing 30% sucrose overnight, embedded in O.C.T. (Tissue-Tek; Sakura Finetek, Inc., Torrance, CA), and cryosectioned (7-μm thick sections).44 Sections were blocked with PBS-BSA (1%) and stained with rat mAb to CD31 (PECAM-1; BD Biosciences Pharmingen) followed by AlexaFluor FITC-conjugated goat anti-rat (Invitrogen) secondary antibody, according to the manufacturer’s recommendation. Nuclei were stained with mounting fluid containing 4′,6-diamidino-2-phenylindole(Vector Laboratories, Burlingame, CA). Costaining of CD31 or Cre-recombinase and α-SMA on frozen sections was done by permeabilizing in 0.3% Triton×100 in PBS, blocking in PBS-BSA (1%) followed by staining with CD31 antibody or mouse mAb to Cre (Sigma-Aldrich) and rabbit polyclonal anti-α-SMA antibody (Abcam, Inc.) followed by AlexaFluor FITC–conjugated goat anti-rat or AlexaFluor 594–conjugated goat anti-mouse and AlexaFluor 594– or AlexaFluor 488–conjugated goat anti-rabbit (Invitrogen) secondary antibodies. Primary ECs were cultured on gelatin-coated eight-well culture slides (Lab-Tek Chamber Slides; Thermo Fisher Scientific, Vernon Hills, IL) and fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.3% Triton X-100 for 5 minutes, and blocked with PBS/1% BSA for 1 hour. Cells were incubated with primary anti-CD31, anti-CD144 (VE-Cadherin; BD Biosciences Pharmingen), or anti-α-SMA antibody, followed by incubation with secondary antibodies. Images were obtained using a compound Nikon microscope (TE-2000U) equipped with a Spot digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI). Quantitative analysis of lectin- and CD31-positive vessels was performed by measuring average lengths of capillaries using Metavue Software. Quantification of percentages of CD31-positive or Cre-positive and α-SMA–positive cells was performed using the grid method with images taken under ×40 or ×60 magnification. Results are presented as ratios of CD31 or Cre and α-SMA double-positive areas to CD31- or Cre-positive areas.
Western Blotting
Cells were washed with ice-cold PBS and lysed in radioimmunoprecipitation assay buffer. Lysates were clarified by centrifugation, and protein concentration was determined by the BCA protein assay (Thermo Fisher Scientific). Proteins were separated on 4%–20% SDS-PAGE gels (Bio-Rad) and transferred. Membranes were blocked and incubated with phospho Smad2, phospho Smad1/5, total Smad2, or total Smad1 antibodies (Cell Signaling Technologies, Danvers, MA) overnight at 4°C and secondary antibodies (GE Healthcare) for 1 hour at room temperature. The bands were visualized using a SuperSignal chemiluminescence kit (Thermo Fisher Scientific).
Statistical Analyses
All experiments were repeated at least three times. All values are expressed as the mean±SD. Data were analyzed using ANOVA with post hoc analysis for multiple group comparisons using Bonferroni method. P<0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
These studies were supported by grants from the NIH (DK54602, DK052783, and DK45462) and Westchester Artificial Kidney Foundation (to M.S.G.), as well as an intramural grant from New York Medical College (to S.X.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Microvascular Endothelial Cells Poised to Take Center Stage in Experimental Renal Fibrosis,” on pages 767–769.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013101137/-/DCSupplemental.
References
- 1.Harris RC, Neilson EG: Toward a unified theory of renal progression. Annu Rev Med 57: 365–380, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Wynn TA, Ramalingam TR: Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Böttinger EP, Bitzer M: TGF-beta signaling in renal disease. J Am Soc Nephrol 13: 2600–2610, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Böttinger EP, Klotman PE, Thorgeirsson SS: Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest 74: 991–1003, 1996 [PubMed] [Google Scholar]
- 5.Goumans MJ, Mummery C: Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44: 253–265, 2000 [PubMed] [Google Scholar]
- 6.Bohle A, Mackensen-Haen S, Wehrmann M: Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res 19: 191–195, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Lebrin F, Goumans MJ, Jonker L, Carvalho RLC, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P: Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J 23: 4018–4028, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ten Dijke P, Arthur HM: Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol 8: 857–869, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Carvalho RL, Itoh F, Goumans MJ, Lebrin F, Kato M, Takahashi S, Ema M, Itoh S, van Rooijen M, Bertolino P, Ten Dijke P, Mummery CL: Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice. J Cell Sci 120: 4269–4277, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS: Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development 133: 4585–4593, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Nguyen HL, Lee YJ, Shin J, Lee E, Park SO, McCarty JH, Oh SP: TGF-β signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest 91: 1554–1563, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Bertram JF: Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology (Carlton) 15: 507–512, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Strutz F, Zeisberg M: Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol 17: 2992–2998, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin SL, Kisseleva T, Brenner DA, Duffield JS: Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R: Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng XM, Huang XR, Xiao J, Chen HY, Zhong X, Chung ACK, Lan HY: Diverse roles of TGF-β receptor II in renal fibrosis and inflammation in vivo and in vitro. J Pathol 227: 175–188, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Gewin L, Bulus N, Mernaugh G, Moeckel G, Harris RC, Moses HL, Pozzi A, Zent R: TGF-beta receptor deletion in the renal collecting system exacerbates fibrosis. J Am Soc Nephrol 21: 1334–1343, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gewin L, Vadivelu S, Neelisetty S, Srichai MB, Paueksakon P, Pozzi A, Harris RC, Zent R: Deleting the TGF-β receptor attenuates acute proximal tubule injury. J Am Soc Nephrol 23: 2001–2011, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicosia RF, Zhu WH, Fogel E, Howson KM, Aplin AC: A new ex vivo model to study venous angiogenesis and arterio-venous anastomosis formation. J Vasc Res 42: 111–119, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Blanco FJ, Grande MT, Langa C, Oujo B, Velasco S, Rodriguez-Barbero A, Perez-Gomez E, Quintanilla M, López-Novoa JM, Bernabeu C: S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ Res 103: 1383–1392, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Arciniegas E, Sutton AB, Allen TD, Schor AM: Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J Cell Sci 103: 521–529, 1992 [DOI] [PubMed] [Google Scholar]
- 23.Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S, Baron R, Scadden D, Schipani E, Guarente L: SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Mol Med 5: 430–440, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roelen BAJ, van Rooijen MA, Mummery CL: Expression of ALK-1, a type 1 serine/threonine kinase receptor, coincides with sites of vasculogenesis and angiogenesis in early mouse development. Dev Dyn 209: 418–430, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P: Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell 12: 817–828, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P: Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21: 1743–1753, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray BN, Lee NY, How T, Blobe GC: ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 31: 435–441, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanco FJ, Bernabeu C: Alternative splicing in endothelial senescence: Role of the TGF-β co-receptor endoglin. In: Senescence, edited by Nagata T, Rijeka, Croatia, InTech, 2012, pp 499–518 [Google Scholar]
- 30.Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CPH, Bernabeu C: Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol 204: 574–584, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez-Peña A, Eleno N, Düwell A, Arévalo M, Pérez-Barriocanal F, Flores O, Docherty N, Bernabeu C, Letarte M, López-Novoa JM: Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension 40: 713–720, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Ota T, Fujii M, Sugizaki T, Ishii M, Miyazawa K, Aburatani H, Miyazono K: Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J Cell Physiol 193: 299–318, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R: Endoglin differentially modulates antagonistic transforming growth factor-beta1 and BMP-7 signaling. J Biol Chem 282: 13934–13943, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Gaengel K, Genové G, Armulik A, Betsholtz C: Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29: 630–638, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Li J, Qu X, Bertram JF: Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol 175: 1380–1388, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Riordan E, Mendelev N, Patschan S, Patschan D, Eskander J, Cohen-Gould L, Chander P, Goligorsky MS: Chronic NOS inhibition actuates endothelial-mesenchymal transformation. Am J Physiol Heart Circ Physiol 292: H285–H294, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Kanasaki K, Taduri G, Koya D: Diabetic nephropathy: The role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol (Lausanne) 4: 7, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chytil A, Magnuson MA, Wright CV, Moses HL: Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis 32: 73–75, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Brodsky S, Chen J, Lee A, Akassoglou K, Norman J, Goligorsky MS: Plasmin-dependent and -independent effects of plasminogen activators and inhibitor-1 on ex vivo angiogenesis. Am J Physiol Heart Circ Physiol 281: H1784–H1792, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Brodsky S, Li H, Hampel DJ, Miyata T, Weinstein T, Gafter U, Norman JT, Fine LG, Goligorsky MS: Delayed branching of endothelial capillary-like cords in glycated collagen I is mediated by early induction of PAI-1. Am J Physiol Renal Physiol 281: F71–F80, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Fang S, Wei J, Pentinmikko N, Leinonen H, Salven P: Generation of functional blood vessels from a single c-kit+ adult vascular endothelial stem cell. PLoS Biol 10: e1001407, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Stoessel A, Paliege A, Theilig F, Addabbo F, Ratliff B, Waschke J, Patschan D, Goligorsky MS, Bachmann S: Indolent course of tubulointerstitial disease in a mouse model of subpressor, low-dose nitric oxide synthase inhibition. Am J Physiol Renal Physiol 295: F717–F725, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Park HC, Addabbo F, Ni J, Pelger E, Li H, Plotkin M, Goligorsky MS: Kidney-derived mesenchymal stem cells contribute to vasculogenesis, angiogenesis and endothelial repair. Kidney Int 74: 879–889, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.