

1. Introduction
1.1. Introduction to Mammalian DNA Base Modifications
Genetic information is encoded by the four bases adenine (A), guanine (G), cytosine (C), and thymine (T). Base pairing through hydrogen bonding between the cognate pairs A-T and C-G together within the base stack of the DNA double helix provides the molecular basis for the genetic code.1 It is evident that there are other molecular mechanisms for encoding function within DNA. The major groove and the minor groove each exhibit a hydrogen bonding pattern that enables the primary sequence of the DNA double helix to be read, without being unwound, which is important for sequence-dependent events such as the binding of transcription factors. Furthermore, there are enzyme-dependent chemical modifications to the canonical bases that have the potential to dynamically alter the structure, recognition and function of DNA. Examples of naturally occurring DNA base modifications are shown in Figure 1. There are organisms whose genomes exhibit a substantial level of chemically modified bases, for example in bacteriophages, all or a major proportion of one of the four bases are commonly replaced by a modified base.2
Figure 1.

Structures of modified bases in phage DNAs: (A) α-putrescinylthymine, (B) 5-dihydroxypentyluracil, (C) α-glutamylthymine, and (D) 2-aminoadenine.
The biosynthetic pathway to modified bases in genomes can occur at the level of modified mononucleotides subsequently incorporated via polymerase-mediated DNA synthesis or post-DNA synthesis from the canonical bases within DNA.3 Functions of modified bases in the DNA of phages include protection from host and phage nucleases, signaling for transcription and replication of the DNA, and facilitating the packaging of the DNA.4
Eukaryotes would appear to comprise a smaller repertoire of DNA base modifications. 5-Methylcytosine (5mC) is the best-studied DNA base modification. It contains a methyl group at the 5-position of the cytosine base, which protrudes into the major groove of the DNA presenting a potential recognition site (or obstacle) for protein binding without changing the Watson–Crick base pairing. This chemical derivative of C has functional consequences for the cell, most notably in the control of gene expression. The study of heritable changes in gene expression mediated by dynamic changes in 5mC, without changes in the primary DNA sequence, is a major aspect of the field of epigenetics. Given that epigenetic changes are of vital importance to developmental biology and numerous areas of human disease, that include cancer and metabolic diseases, it is of great importance to elucidate the underlying mechanisms that cause and stem from the chemical modification of DNA bases.
In mammals, 5mC was first discovered in the late 1940s and has been found to play essential roles in maintaining cellular function and genomic stability, including processes such as the inactivation of one of the two X chromosomes in female mammals; genomic imprinting such that genes are expressed in a manner dependent on the parent-of-origin; and the silencing of moveable genetic elements called transposons.5 A family of enzymes called the DNA methyltransferases (DNMTs) are known to be responsible for the generation and maintenance of 5mC in genomes.6 The standard mechanism of 5mC formation involves initial nucleophilic attack of a cysteine residue in DNMT at the C6 position and nucleophilic attack by C5 on the methyl donor from S-adenosyl methionine (SAM), followed by elimination to restore aromaticity in the base (Figure 2).
Figure 2.

Mechanism of cytosine methylation by the DNMT and the cofactor SAM.
The function of methylation in mammals depends on the context of the modification within the genome. There is a strong positive correlation between gene silencing and methylation of regions rich in C-G diads called CpG islands (CGIs) near transcription start sites (TSS) and also the first exon within long-term silenced genes.5a Within gene bodies, there is a positive correlation between active transcription and gene body methylation on active X chromosomes. Studies also suggest that DNA methylation in gene bodies could play a role in regulating alternative splicing.7
In the 1970s two papers suggested mammals contained very high levels of another cytosine modification, 5-hydroxymethylcytosine (5hmC); up to 25% of all C bases.8 However, others could not corroborate these results9 and 5hmC had been widely viewed as a potential DNA damage product.10 In 2009, two studies were published in Science demonstrating the presence of 5hmC, in mouse brain and embryonic stem (ES) cells.11 Furthermore, Tahiliani et al. showed that the ten-11 translocation 1 (TET1), a 2-oxoglutarate (2-OG) and Fe(II)-dependent dioxygenase, could catalyze the conversion of 5mC to 5hmC (Figure 3).11b
Figure 3.

Mechanism of 2-oxoglutarate (2-OG) and Fe(II)-catalyzed TET oxidation.
Genome-wide experiments have since mapped the location of 5hmC to promoter regions, transcription start sites, and gene bodies. In ES cells, 5hmC is also enriched at developmental genes that are poised for changes in transcriptional activity.12
In 2011, 5-formylcytosine (5fC) was detected in mouse ES cells and brain cortex and 5-carboxycytosine (5caC) in mouse ES cells by thin layer chromatography and tandem liquid chromatography–mass spectrometry.13 Quantification by mass spectrometry of DNA digested into nucleosides showed that the genomic DNA of ES cells contained 5fC at levels of around 0.2% relative to G and 5caC at 10-fold lower levels than 5fC.13 In mammalian brain tissues, levels of 5fC were found to be 2–3 and 5caC 3–4 orders of magnitude lower than 5hmC.14 Several studies have mapped the locations of 5fC and 5caC in the genomes of mouse ES cells.15 Furthermore, single base resolution 5fC sequencing methods have enabled single base resolution genomic maps of 5mC, 5hmC, and 5fC in embryonic stem cells.15c,16
The discovery of 5hmC, 5fC, and 5caC, in mammalian DNA has raised the need to elucidate their function. A popular hypothesis is that such oxidized cytosine modifications constitute part of the pathways that lead to active DNA demethylation.
There are several proposed pathways for demethylation; one mechanism suggests the iterative oxidation of 5mC by the TET family enzymes, followed by base excision repair or deformylation/decarboxylation. A potential mechanism for active demethylation is through the thymine DNA glycosylase (TDG) enzyme, which can excise both 5fC and 5caC from DNA but does not remove 5mC or 5hmC.13c,17 Following this base excision the abasic site would be repaired by the base excision repair (BER) pathway.18 It is also possible that a decarboxylase enzyme could directly remove the carboxylic acid group from 5caC (Figure 4).19
Figure 4.

Potential pathways for DNA demethylation.
It is clear from work during the past five years that the enzyme-mediated chemical modification of cytosine in DNA has emerged as an important area of scientific investigation. The focus of this review will be to discuss the chemical methodologies that have been created and explored to detect, measure, and elucidate cytosine derivatives in the genomic DNA from living systems.
2. Genome-Wide Profiling Methods
The decoding of DNA falls within the general scope of chemical structure elucidation and has been naturally enabled by the creation and application of chemical approaches. Decoding the sequence of the four canonical DNA bases was first made widely accessible in the late 1970s with two independent chemical approaches from Maxam and Gilbert20 and from Sanger.21 The Sanger sequencing approach was optimized, automated and employed to decode the 3 billion base human genome reference sequence via the Human Genome Project. The Solexa/Illumina sequencing approach originated from the Balasubramanian and Klenerman laboratories in the late 1990s22 and has been developed23 and optimized to a level where the past five years have progressively shown that very high capacity (human genome scale) sequencing experiments are routinely possible in relatively small laboratories. While the advent of widely accessible large scale sequencing has had an impact on genetics and genomics, these advances also hold the potential to decode and help elucidate noncanonical DNA bases in the genomes of organisms.
2.1. Restriction Endonucleases
2.1.1. Restriction Endonuclease Detection of 5mC
Restriction enzymes recognize short DNA sequences present in double stranded DNA and cleave the phosphodiester backbone of both strands by direct hydrolysis or through a covalent enzyme intermediate (Figure 5).24 This reaction forms two fragments of double stranded DNA. This DNA cleavage reaction can be blocked in the presence of modifications to the DNA bases in the recognition site.25 Absolute quantitation of the levels of modified bases at a specific restriction site can be obtained by using two restriction enzymes that cleave at the same site, but where only one can cleave in the presence of a specific DNA base modification.26 This difference occurs due to the capacity of enzymes to recognize the DNA sequences when a methyl group is present in the major groove. The modification can be quantified by measuring the difference in how many times a specific site has been cut with each restriction enzyme. This method is regularly used with quantitative polymerase chain reaction (qPCR) to measure specific restriction sites.
Figure 5.

Mechanism of DNA strand cleavage by restriction endonucleases. (A) Cleavage of the DNA into two strands is performed by hydrolysis of the phosphate backbone. (B) Cleavage occurs via an enzyme-DNA intermediate.
The two restriction enzymes regularly used to detect 5mC are HpaII and MspI, which both cut at the same DNA sequence; CCGG. This sequence is ideal as it contains a CpG dinucleotide, which is where the majority of 5mC resides in mammals.27HpaII is methylation-sensitive and will only cut a CCGG sequence that does not contain 5mC, whereas MspI is methylation-insensitive and will cut a CCGG sequence with or without 5mC (Figure 6).
Figure 6.

Detection of 5mC and 5hmC with restriction endonucleases. 5mC and 5hmC can be distinguished at an HpaII/MspI restriction digestion site by comparing digestion reactions. HpaII only digests DNA that does not contain 5mC or 5hmC. MspI digests DNA that contains 5mC and 5hmC. Glucosylation of 5hmC (5gC) inhibits digestion with MspI. 5hmC can be distinguished by comparing glucosylation/MspI digestion and MspI only digestion. 5mC can be distinguished by comparing glucosylatin/MspI digestion and HpaII digestion.
2.1.2. Genome Wide Restriction Endonuclease Detection of 5mC
The two most frequently used restriction enzyme based techniques that are used to detect 5mC, on a genome wide scale, are the HpaII tiny fragment enrichment by ligation-mediated PCR (HELP) assay28 and Methyl-Seq (29)
The HELP assay was developed in 2006 and Methyl-Seq in 2009 and both rely on a comparison of genomic DNA after HpaII and MspI digestion. In both methods, genomic DNA is separately digested with HpaII and MspI, and then adapters are ligated to the ends of the digested DNA fragments. The library of MspI digested fragments represents the total population of sites, as MspI digests both 5mC and C, whereas the HpaII library represents a subset of these sites as HpaII only digests C.
The HELP assay uses fluorescently labeled primers to amplify each adapted library using PCR. Different fluorophores are applied to the HpaII and MspI libraries. A DNA microarray is then used that contains the sequences of specific genomic regions of interest. The method was developed using a DNA microarray that detects 1339 sites in the mouse genome, which represents a total of 6.2 Mbp.28 The presence of a CCGG site results in a fluorescent signal being detected in the MspI library. When a site is fully methylated no fluorescent signal is detected in the HpaII library, however if there is partial or no methylation a fluorescent signal will be detected. A HpaII/MspI ratio is then calculated for each genomic region to give relative quantification at a large number of genomic loci simultaneously.
In Methyl-Seq, following ligation of adapters to the HpaII and MspI libraries, each is sequenced using next generation sequence technologies. This creates millions of short genomic reads all starting at CCGG sites. Sites that are only sequenced in the MspI library are fully methylated and are called as “methylated”. When sites are present in the HpaII library there is either partial or no methylation, and they are called as “unmethylated”. The initial publication demonstrated this method could be used to assay 90 000 regions in the human genome.29
Partially methylated regions are undetectable in Methyl-Seq, as they are labeled as “unmethylated”, whereas there is relative quantification from the HELP assay. However, new DNA arrays are needed for each new region of interest in the HELP assay, whereas in Methyl-Seq a much larger quantity of sites are analyzed without the need for DNA microarray development.
2.1.3. Restriction Endonuclease Detection of 5hmC
With the recent discovery of 5hmC in the mammalian genome,11 there has been growing interest in developing techniques to detect this base to enable the elucidation of its function. Restriction endonuclease methods developed to quantitatively detect 5hmC in the genome rely heavily on the βGT enzyme found in T4 bacteriophage.26a βGT adds a glucose moiety to the primary alcohol on the hydroxymethyl group of 5hmC while present in double stranded DNA.30 As the primary alcohol group of 5hmC is present in the major groove of the double stranded DNA, this enzymes functionalizes the DNA major groove with a glucose moiety that consequently alters the recognition potential at that site.
The most commonly used method involves designing primers for quantitative PCR analysis of a specific region of interest in the genome that contains a single restriction site for the enzyme used.26aMspI will cleave DNA with C, 5mC, or 5hmC in its restriction site, but when glucosylated 5hmC is present in the restriction site, MspI will no longer cut the DNA.26a The levels of 5hmC can be determined by performing quantitative PCR on undigested, digested, and glucosylated then digested DNA. Quantifying the difference between each digestion then gives the percentage of 5hmC at that restriction site. HpaII does not cleave 5mC or 5hmC DNA and can be used in parallel to the above method to determine the levels of both 5mC and 5hmC at the same site. Thus, when comparing this HpaII data with that obtained for 5hmC alone, levels of C, 5mC, and 5hmC can be obtained through the differences (Figure 6).
Along with the creation of methods to detect 5mC and 5hmC using HpaII and MspI and βGT, there has also been the development of novel families of enzymes, PvuRts1I31 and MspJI,32 that only digest 5hmC or glucosylated 5hmC, and not C or 5mC. These enzymes offer the ability to directly detect 5hmC modifications on a genome wide scale.33
2.1.4. Restriction Endonuclease Detection of 5fC and 5caC
Following the discovery of 5fC and 5caC little has been done to detect them using restriction endonucleases or find out how previous enzymes interact with them. Little research has been carried out to use restriction enzymes to map 5fC and 5caC in the genome. One study has indicated that MspI could not digest synthetic DNA that contained 5fC or 5caC;13a however, this has not been taken further to look at genome-wide levels. There is a need for robust data on the specificity/discrimination of these restriction enzymes on all cytosine modifications, before such methods can be widely used with confidence.
2.1.5. Advantages and Disadvantages of Restriction Endonucleases
Restriction endonucleases provide a simple and relatively low cost way of accurately quantifying modified bases at single restriction sites. These methods do not detect modified bases at single base resolution, as a modification present at any position at its cut site can block digestion. Using PCR to achieve absolute quantification at many genomic sites in parallel can become very time-consuming, as separate PCR reactions must be run for each site. However, restriction endonuclease techniques are now available to detect 5hmC at a genome wide scale, albeit with only relative quantification. It will be of great interest to combine the HELP assay and Methyl-Seq approaches to also detect 5hmC, using the βGT enzyme that inhibits MspI digestion of 5hmC.
2.2. Chemical Based Profiling
DNA immunoprecipitation sequencing (DIP-Seq) is a technique that uses a probe (protein or small molecule) that noncovalently or covalently recognizes a DNA feature of interest that can be isolated by affinity enrichment of fragmented genomic DNA and then characterized by high throughput DNA sequencing. For example, methylated DIP-seq (MeDIP-Seq) uses an antibody that binds and enriches for methylated DNA fragments from genomic DNA. The enriched fragments are decoded by sequencing and the sequences are then computationally aligned and “stacked” against the reference genome to provide a genome-wide profile of methylation sites. The resolution of this approach is a function of the fragment size of the prepared DNA library34 (Figure 7). A similar antibody-based approach has been developed for mapping 5hmC, called hMeDIP-Seq.35 Trypanosomes contain a protein, called JBP1, which binds to glucosylated 5-hydroxymethyluracil. It was shown that JBP1 also binds glucosylated 5hmC and then used as an antibody to map 5hmC.36
Figure 7.
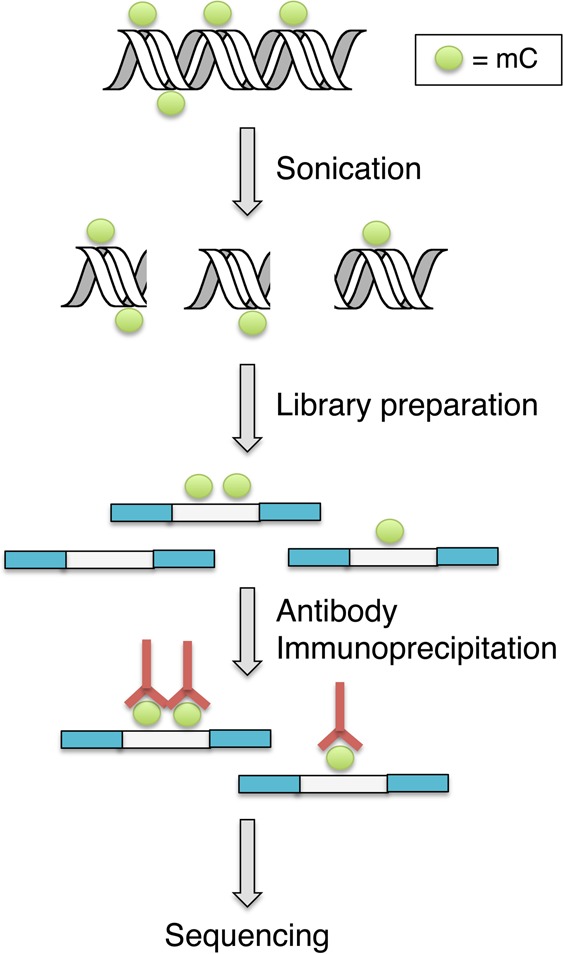
Schematic overview of the MeDIP sequencing procedure.
Although protein-based enrichment methods are widely used to map DNA modifications, this technique highly depends on the quality of the antibody used. Low specificity of the antibody for targeted modifications or cross-reactivity with off-target sites results in high background noise. In order to overcome these issues alternative chemical profiling methods were developed (Figure 8). The first chemical profiling method reported for 5hmC, hmC-seal, was developed by Song et al.37 The method exploits the use of a β-glucosyltransferase (βGT) that can transfer a 6-N3-glucose onto the hydroxyl moiety of 5hmC. Subsequent copper-free click chemistry attaches dibenzocyclooctyne-modified biotin to the base. The biotin–streptavidin interaction can either be used to quantify 5hmC with avidin-horseradish peroxidase (HRP) or efficiently enrich for 5hmC containing fragments with streptavidin-coated beads.
Figure 8.

Summary of methods that exploit the chemical functionality of modified bases for genome-wide profiling.
Another chemical enrichment method for 5hmC termed GLIB (glucosylation, periodate oxidation, and biotinylation) used glucosylated 5hmC that was subsequently treated with sodium periodate, which oxidatively cleaved the vicinal diols in glucose to yield a dialdehyde.12a The aldehydes were then reacted with a hydroxylamine-biotin probe. In the case of 5fC, the chemical reactivity of the aldehyde moiety on the modified bases itself was exploited by chemoselective reaction with a hydroxylamine-biotin probe to perform the first genome wide mapping of this modification.15a Fragmented genomic DNA containing 5fC from mouse ES cells was reacted to the probe and pulled-down with streptavidin-coated beads to enrich for 5fC-containing DNA fragments that are subsequently decoded by sequencing. Song et al. extended their hmC-seal method in order to enrich for 5fC containing DNA (fC-seal method).15c Therefore, they first blocked 5hmC with unmodified UDP-Glc using βGT. Subsequently they reduced 5fC to 5hmC using sodium borohydride and then glucosylated the newly generated 5hmC with an azide-modified glucose. The azide was clicked to a biotin containing probe using copper-free click chemistry, which in turn allowed the pulldown of 5fC containing fragments. A joint chemical-antibody approach has also been deployed to detect 5hmC in vivo. Bisulfite treatment of DNA was used to convert all the 5hmC to a stable cytosine-5-methylsulfonate adduct (CMS), then an antibody was used to detect these chemically modified 5hmC bases.12a Furthermore, it has been demonstrated that DNMTs can be used to tag small molecules onto 5hmC,38 and this reaction could be used to map 5hmC.
With regards to 5caC, it can also be captured by using 1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide hydrochloride (EDC)-catalyzed amide bond formation between the carboxyl group of 5caC and a biotin modified amine.39 It remains to be seen if the labeling sensitivity and selectivity of this method is sufficient to apply it to genomic DNA, given the very low abundance of 5caC in genomic DNA.
2.3. Chemical Single Base Sequencing Methods
2.3.1. Maxam and Gilbert
The Maxam and Gilbert sequencing method uses 5′-radiolabeled DNA that undergoes four different chemical treatments generating base-selective strand breakages.20 The Gs are methylated by dimethylsulfate, the purines (A and G) are depurinated using formic acid, and the pyrimidines (C and T) are hydrolyzed using hydrazine. The addition of high salt and hydrazine hydrolyses C only. The DNA backbone is subsequently cleaved at the sites where the bases have been reacted, using hot piperidine. Electrophoresis of these fragments generates a sequencing ladder corresponding to reading 5′ to 3′ on the DNA (Figure 9).
Figure 9.
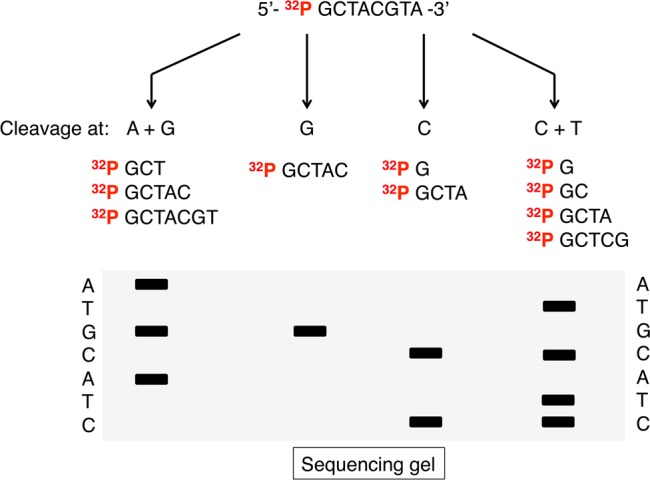
Principle of the Maxam–Gilbert DNA sequencing.
Aspects of this sequencing method allow it to also be used to sequence methylcytosine in DNA.40 The reaction of hydrazine (which leads to cleavage at C and T) with 5mC is inefficient and therefore does not introduce a strand cleavage. This results in a gap in the sequencing pattern. This gap together with the identification of G on the complementary strand determines the location of 5mC in the DNA sequence.40
A modified Maxam and Gilbert sequencing method uses different chemicals for the selective detection of 5-methylcytosine in DNA sequences.41N-Sodio-N-chloro-p-nitrobenzenesulfonamide and N-sodio-N-bromo-m-nitrobenzenesulfonamide display differential reactivity toward C and 5mC in that only N-sodio-N-chloro-p-nitrobenzenesulfonamide showed high selectivity toward C producing a cleavage at C sites upon hot piperidine treatment. Treatment with N-sodio-N-bromo-m-nitrobenzenesulfonamide generated two products with cleavage at the C and 5mC sites (Figure 10).
Figure 10.

Treatment of cytosine and 5mC with N-sodio-N-chloro-p-nitrobenzenesulfonamide and N-sodio-N-bromo-m-nitrobenzenesulfonamide result in different reaction products.
By combining the results obtained using both of these compounds, the authors claim the accurate identification of 5mC residues in DNA sequences. When this method was combined with the use of β-glucosyltransferase, the introduction of a glucose moiety to the hydroxyl group of 5hmC could be used to distinguish 5mC from 5hmC and C.
Two more methods are available that can supplement the Maxam and Gilbert sequencing method for the interrogation of cytosine modification in DNA. The first one exploits the selective detection of 5mC by using uracil DNA glycosylase (UDG).42 Bisulfite treated DNA is subsequently treated with UDG to initiate uracil elimination followed by DNA cleavage in alkaline conditions. As 5mC is resistant to conversion to U by bisulfite, cleavage can only be observed at C sites. The second method uses hot alkali treatment, thereby selectively cleaving the DNA at sites of 5fC and 5caC.43 While these sequencing methods work well on short synthetic DNA strands and could potentially be applied for the development of probes to study genomic samples, Maxam and Gilbert type sequencing is rather time-consuming and cumbersome compared to modern sequencing approaches and so this approach may not be suitable for the routine genome-wide study of epigenetic modifications.
Münzel et al. described a chemical method to discriminate between C and 5mC.44 The chemical reagent O-allylhydroxylamine, in contrast to bisulfite, does not exploit reactivity differences but gives different reaction products with cytosine and 5mC (Figure 11).
Figure 11.
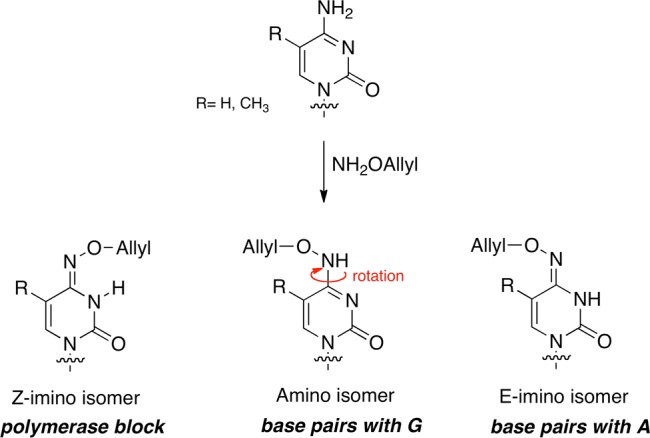
Reaction of cytosine and 5mC with O-allylhydroxylamine results in the formation of the E- or Z-isomer that are in equilibrium via the amino isomer.
The reagent forms a stable mutagenic adduct with cytosine, which can exist in two oxime-type configurations, E or Z, which in turn are in equilibrium via the amino isomeric form. The amino tautomer effectively base pairs as C, whereas the E-imino isomer will pair as T and the Z-imino isomer interferes with the base pairing causing a polymerase stalling. Which of the isomer is formed depends on the steric hindrance between the O-allyl chain and the functional group on 5-position of the cytosine. In case of C the allylhydroxylamine adduct switches into the E-isomeric form, which generates C to T transition mutations that can easily be detected by sequencing. In contrast, the 5mC-adduct adopts exclusively the Z-isomeric form, which causes the polymerase to stop. A limitation of this method is that it does not distinguish distinguish between 5mC and 5hmC. The detection principle is based on sterics and 5hmC imposes an even larger steric strain and therefore it is not possible to distinguish 5mC and 5hmC after incubation with O–allylhydroxylamine.
2.3.2. Bisulfite Sequencing of 5mC
Bisulfite sequencing (BS-Seq) has been regarded as the gold standard for 5mC detection and therefore widely used to detect 5mC at single base resolution in a large variety of cell types and disease models.45 In BS-Seq the bisulfite mediates and overall hydrolytic deamination of C to U, but does not alter 5mC.46 Following DNA sequencing, all of the Cs in the DNA that were deaminated will read as Ts, so any remaining Cs are assumed to have come from 5mC, which does not deaminate during the bisulfite treatment.
The deamination reaction of C to U with bisulfite was first observed in 1970.47 The bisulfite anion adds across the C5–6 double bond of C at acidic pH, to generate an adduct, which has lost aromaticity of the base and undergoes hydroysis with loss of ammonia. The resulting uracil bisulfite adduct rearomatises to form uracil upon an increase in pH (Figure 12). This reaction requires the single stranded form of DNA (ssDNA), owing to the inaccessibility of the C5–6 double bond to the bisulfite anion in the double helix.48
Figure 12.

Mechanism of sodium bisulfite deamination of cytosine to uracil through the addition of sodium bisulfite across the C5–6 double bond of C, followed by deamination of the cytosine bisulfite adduct. The uracil bisulfite adduct can then be worked up using alkaline conditions.
This bisulfite deamination of C to U is highly pH sensitive; the optimal pH for the overall reaction, adduct formation and deamination is between 5.0 and 5.3.49 When increasing the pH above 5.3 there is a sharp decrease in cytosine bisulfite adduct formation due to the dissociation of the bisulfite anion to the sulfite conjugate base.49 The deamination rate also decreases above pH 5.3 as the N3 unprotonated bisulfite adduct deaminates at 1% of the rate of the N3 protonated adduct. However, deamination of the bisulfite adduct is base-catalyzed, so the rate also decreases at pH values below 5.0 due to the protonation of the most effective catalytic species, sulphite.49 There is a positive linear relationship between concentrations of bisulfite and reaction rate.49
Further studies in 1980 demonstrated that bisulfite reacted slower with 5mC than with C, due to inhibition of the adduct formation from the electronic effect of the methyl group.50 This difference in reactivity between C and 5mC is the basis of BS-Seq (46)
Bisulfite treatment of DNA causes a degree of DNA degradation, and for a long time the mechanism was thought to be through depurination of A and G due to protonation at low pH.51 However, it was later discovered that the true degradation mechanism involves depyrimidination of the bisulfite adduct with C, while no degradation was observed with A, G, or T as no bisulfite adduct forms.51 Once depyrimidination has occurred to form an abasic site, DNA strand scission (degradation) will occur in basic conditions,20 such as those in the bisulfite work up (Figure 13). Hydroquinone has been used as an additive52 to protect DNA from degradation and commercial BS-Seq products contain “DNA protect buffers”; however, there has been no definitive examination of their effectiveness.
Figure 13.

Proposed mechanism of bisulfite DNA strand cleavage from depyrimination of the uracil bisulfite adduct following deamination from the cytosine bisulfite adduct. Figure adapted from ref (51).
When analyzing genomic DNA samples it is usually the case that there are multiple copies of the same genetic sequence due to the extraction of DNA from more than one cell, unless working on the single cell level. Each copy of the same genetic sequence can contain different cytosine modifications, as epigenetic states are dynamic. This means that when analyzing a population of DNA samples, what can be quantified is the percentage of sites that contain each modification at the same genomic location, at a given time point (e.g., if 50% of the sequencing reads show a C at a given site in BS-Seq, this would suggest 50% of the cell population exhibits 5mC at that site). BS-Seq has been used to gain this quantitative map of 5mC across whole genomes of many plants and mammals, at single base resolution.45 An adaptation to this method has been developed, reduced representative bisulfite sequencing (RRBS-Seq), that can fraction the genome into only biologically relevant CGIs, genomic regions that contain a high percentage of CpG dinucleotides.7e RRBS-Seq works by enzymatically digesting the genome with MspI followed by removal of the undigested DNA resulting in enrichment of CpG sites. Due to this enrichment, RRBS-Seq allows the same depth of coverage (number of times each site is sequenced) of whole genomic sequencing but with less sequencing, as regions that have a low percentage of CpG sites will not be sequenced.
BS-Seq has also been used to jointly map 5mC with histone modifications, ChIP-bisulfite-sequencing (ChIP-BS-Seq)53 and bisulfite-treated chromatin immunoprecipitated DNA (BisChiP-Seq).54 Both of these methods involve initially enriching the genome for DNA located around histones of interest by chromatin immunoprecipitation of genomic DNA with antibodies targeted at histone modifications of interest. BS-Seq is then carried out on this enriched DNA to obtain a map of 5mC in these regions. This allows the direct analysis of methylation status at regions of the genome that coincide with specific histone modifications. The drawback of these techniques is that by enriching the DNA using antibodies there is no longer absolute quantification of the DNA methylation status. Furthermore, they are both reliant on the availability and specificity of histone modification antibodies.
A further adaptation of BS-Seq has been made to simultaneously map methylation status and nucleosome positions by a method termed nucleosome occupancy and methylome sequencing (NOMe-Seq).55 This method uses a GpC methyltransferase (M.CviPI) that will methylate all of the cytosines present in GpC context outside nucleosomes. BS-Seq of this GpC methylated DNA can then be used to detect regions of unmethylated cytosines in GpC context, as they will convert to Us, and relate to the position of nucleosomes. All of the cytosines in GpC context outside of nucleosomes are methylated and will still read as a C. Furthermore, it is possible to detect the natural cytosine CpG methylation status of DNA around each nucleosome. It would be of great interest to combine ChIP-BS-Seq/BisChiP-Seq with NOMe-Seq to generate a joint map of 5mC with specific histone modifications along with the exact position of each nucleosome.
2.3.3. Detection of 5hmC with Bisulfite
The realization that 5hmC exists in mammalian DNA has revealed an important shortcoming of BS-Seq treatment of 5hmC with bisulfite results in a stable cytosine-5-methylsulfonate adduct (CMS) that, like 5mC, does not undergo deamination and is therefore read as C during sequencing data.5c,56 Thus, 5mC and 5hmC are indistinguishable by sequencing that follows bisulfite conversion, and therefore all reported examples of BS-Seq methylation analysis have actually been measuring the contributions from the sum of 5mC plus 5hmC, rather than the true 5mC level, which may confound the interpretation of the data in some cases. Another potential issue is that the CMS adduct, when present at high density can stall common DNA polymerases.56
Resolving 5mC and 5hmC in sequencing data is important given that each modification may have a distinct role in biology.57 Two distinct methods, oxidative bisulfite sequencing (oxBS-Seq)16a and TET-assisted bisulfite sequencing (TAB-Seq),16b have been invented to quantitatively sequence 5hmC at single base resolution in genomic DNA. Both methods unequivocally resolve 5mC from 5hmC during bisulfite treatment.
The oxBS-Seq approach exploits the observation that reaction of bisulfite with 5fC leads to deformylation and deamination. Thus, oxBS-Seq comprises a selective and quantitative chemical oxidation of 5hmC to 5fC in genomic DNA using potassium perruthenate.16a,58 The resulting 5fC is subsequently, efficiently transformed to U with bisulfite treatment. In oxBS-Seq, only 5mC will read as a C, giving a direct read out for the level and position of 5mC in a DNA sequence. 5hmC can be identified as the difference between oxBS-Seq and BS-Seq, where 5mC and 5hmC read as a C (Figure 14). OxBS-Seq has been used, in combination with targeting (RRBS-Seq), to generate a single base resolution map of 5mC and 5hmC status of CpG islands in mouse embryonic stem cells (mESCs).16a
Figure 14.
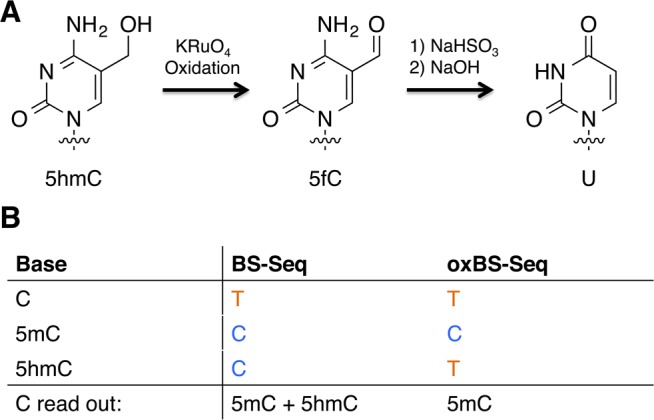
Reaction scheme and sequencing output for oxBS-Seq (A) 5hmC is oxidized to 5fC by potassium perruthenate, which is then deaminated by sodium bisulfite. (B) 5mC is the only base to read as a C in oxBS-Seq 5hmC can be distinguished as the difference between the read out of C bases from BS-Seq and oxBS-Seq.
TAB-Seq uses the β-glucosyltransferase enzyme to modify 5hmC present in the genome, and a recombinant mouse TET1 enzyme to oxidize the 5mC to 5caC.16b,59 Prior glucosylation of the 5hmC protects it from oxidation by the TET1 enzyme. Reaction of DNA with bisulfite cause decarboxylation and deamination of 5caC to form uracil, leaving the glucosylated 5hmC unconverted. TAB-Seq therefore gives a direct read out of 5hmC. 5mC can be identified as the difference between TAB-Seq and BS-Seq (Figure 15).
Figure 15.

Reaction scheme and sequencing output for TAB-Seq (A) 5hmC is blocked from further reaction by glucosylation by βGT. 5mC is then oxidized to 5caC with TET1 oxidase, which is then deaminated by sodium bisulfite. (B) 5hmC is the only base to read as a C in TAB-Seq 5mC can be distinguished as the difference between the read out of C bases from BS-Seq and TAB-Seq.
TAB-Seq has been used to generate a high-resolution map of 5hmC across the whole genome of mESCs.16b The researchers detected sites that contained high levels of 5hmC throughout the entire genome. β-Glucosyltransferase was shown to exhibit inefficiencies when glucosylating 5hmCpGs when another 5hmC is within 4 bp,16b which may pose difficulties for sites with multiple 5hmCs in close proximity.
The deformylation of 5fC by reaction with bisulfite had not previously been described, prior to oxBS-Seq, however the decarboxylation of 5-carboxyuridine (analogous to 5caC) was observed previously in 1969.60 The mechanism of decarboxylation of 5caC is thought to go through a single addition of bisulfite to the C5–6 double bond, which breaks the aromaticity of base, and then decarboxylative elimination leading to the desulfonation (Figure 16A). The mechanism of deformylation of 5fC has been proposed to go through a double addition of bisulfite to 5fC, across the C5–6 double bond and the aldehyde, which are well documented in the literature.61 This bis-adduct could then deformylate and desulfonate to cytosine (Figure 16B).
Figure 16.

Potential mechanisms of decarboxylation of 5caC (A) and deformylation of 5fC (B) by sodium bisulfite.
2.3.4. Detection of 5fC with Bisulfite
As discussed, bisulfite causes 5fC to deformylate and deaminates to form U, thus naturally occurring 5fC is indistinguishable from C in BS-sequencing and does not interfere with the detection of 5mC, unlike 5hmC. However, 5fC cannot be directly identified using BS-Seq.
Two chemical methods, 5fC-assisted bisulfite sequencing (fCAB-Seq)15c and reduced bisulfite sequencing (redBS-Seq),16c have been invented to quantitatively sequence 5fC at single base resolution in genomic DNA. Both methods function by exploiting chemistry to block the conversion of 5fC to U during bisulfite treatment.
In fCAB-Seq a substituted hydroxylamine is reacted with the formyl group of 5fC to form an oxime.15c This oxime is not susceptible to hydrolytic deamination to U during bisulfite treatment. Therefore, by subtracting the data obtained from BS-Seq, where C and 5fC read as a U, from data obtained by fCAB-Seq where only C reads as a U and 5fC reads as a C, 5fC can be identified as the difference (Figure 17).
Figure 17.
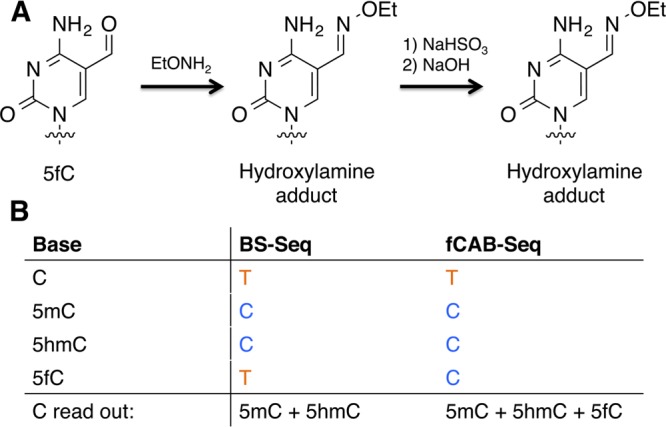
Reaction scheme and sequencing output for fCAB-Seq (A) 5fC is coupled to a hydroxylamine, which is then resistant to deamination by sodium bisulfite. (B) 5fC can be distinguished as the difference between the read out of C bases from BS-Seq and fCAB-Seq.
fCAB-Seq has been used to detect 5fC status at single base resolution of several targeted regions in mouse genomic DNA. Sequencing was carried out on wild type mESC genomic DNA, along with mESC DNA from cells where the TDG enzyme, thought to be responsible for the removal of 5fC, has been knocked down.
In redBS-Seq sodium borohydride is used to reduce 5fC to 5hmC in genomic DNA.16c Given 5hmC is read as a C during sequencing that following bisulfite reaction the reduced 5fC will no longer deaminate to U during bisulfite treatment. By subtracting the data obtained from BS-Seq, where C and 5fC are read as a U, from data obtained by redBS-Seq where only C reads as a U and 5fC reads as a C, 5fC can be identified as the difference (Figure 18).
Figure 18.
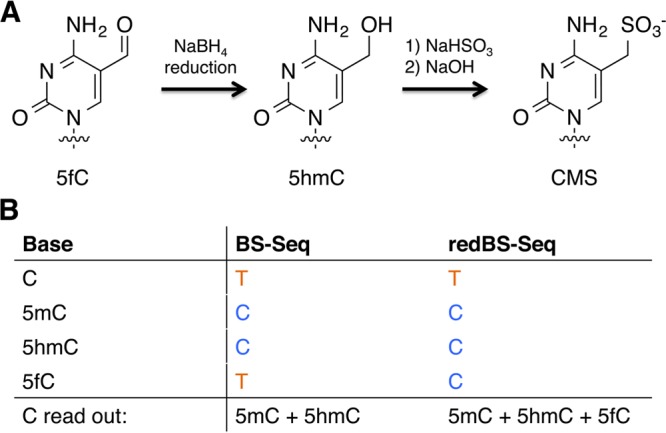
Reaction scheme and sequencing output for redBS-Seq (A) 5fC is reduced to 5hmC with sodium borohydride, which is then resistant to deamination by sodium bisulfite. (B) 5fC can be distinguished as the difference between the read out of C bases from BS-Seq and redBS-Seq.
By combining RRBS-Seq with redBS-Seq, a single base resolution map of 5fC at CpG sites across the mESC genome was generated. Furthermore, this method was employed in parallel with oxBS-Seq to generate a high resolution map of 5mC, 5hmC and 5fCs.
2.3.5. Detection of 5caC with Bisulfite
Along with the discovery of 5fC was the discovery of 5caC at levels ten times lower than 5fC in genomic DNA.13a 5caC deaminates during bisulfite treatment to form U, like 5fC (Figure 16), so naturally occurring 5caC is indistinguishable from C and 5fC.
One method has been published to detect 5caC in DNA, termed chemical modification-assisted bisulfite sequencing (CAB-Seq).39 In CAB-Seq, 5caC is converted to an amide by reaction with EDC and a primary amine. It was demonstrated that amide-derivatives of 5caC inhibit the conversion to U during bisulfite treatment, and are therefore read as a C. This method could potentially be used to detect 5caC by subtracting the BS-Seq data, where C, 5fC and 5caC read as a U, from this CAB-Seq method, where 5caC reads as a C (Figure 19).
Figure 19.
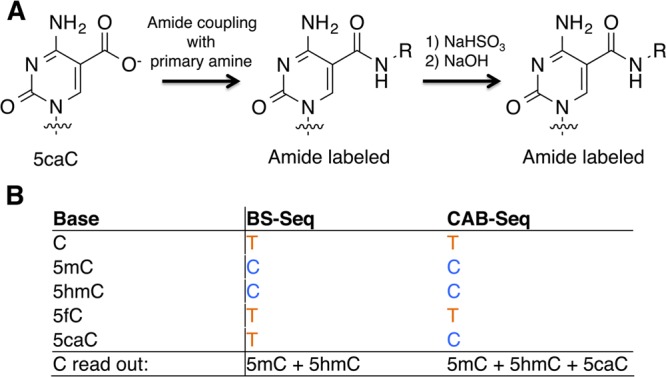
Reaction scheme and sequencing output for CAB-Seq. (A) 5caC is coupled to a primary amine that is then resistant to deamination by sodium bisulfite. (B) 5caC can be distinguished as the difference between the read out of C bases from BS-Seq and CAB-Seq.
The CAB-Seq method has been demonstrated on synthetic DNA with qualitative sequencing technologies. It would be of great interest to explore how quantitatively CAB-seq can measure the level of 5caC in DNA and at specific locations in the genome. While global genomic levels of 5caC are extremely low, it will be important to address if there are sites where 5caC is abundant as is the case with 5fC.15c,16c
3. Single Molecule Sequencing
Sequence analysis of modified cytosines by bisulfite-based methods has been hugely enabled by the advent of low cost, high-throughput (sometimes called “Next Generation”) sequencing on platforms such as the Solexa/Illumina system.22,23 Generally, such approaches have been applied on genomic DNA derived from populations of cells, thereby providing an average representation from the cell population. Single cell analysis via bisulfite sequencing can be achieved by careful and efficient manipulation of the genomic DNA isolated from a single cell.62 There are also single molecule sequencing approaches at various stages of development, that have the potential to directly detect modifications to DNA bases and decode genomes from single cells without prior amplification steps.
3.1. SMRT Sequencing
One approach for the single molecule sequencing of modified bases is to exploit the pausing of a polymerase due to the presence of chemical tags; this has been demonstrated for the detection of 5hmC in single-molecule real-time sequencing (SMRT).63 SMRT DNA sequencing is a single molecule sequencing technology, whereby the continuous incorporation of phospholinked nucleotides by a DNA polymerase is detected as fluorescent pulses. The kinetics of nucleotide incorporation is dependent on the nature of the bases and typically the polymerase incorporation rate at the modified base position is slower. 5mC and 5hmC have a similar low kinetic signature, which makes it is difficult to distinguish between them and nonmodified C.63b However, 5fC and 5caC have a greater signal than 5mC and 5hmC and, through oxidiation of 5mC with the TET enzymes, have been used to detect 5mC.64 In order to sequence 5hmC in a genomic DNA sample with high confidence, Song et al. combined the selective chemical labeling of 5hmC and SMRT sequencing technology.65 Therefore, 5hmC was glucosylated using β-glucosyltransferase. Then a cleavable biotin-containing disulfide linker was clicked onto the azide group (Figure 20).
Figure 20.

Principle of 5hmC SMRT sequencing.
After enrichment of 5hmC containing DNA strands, the fragments were released from the streptavidin beads by DTT treatment and tested for kinetic signatures during SMRT sequencing. This method represents the first example of a single molecule sequencing method being employed to detect 5hmC at single base resolution. In principle, this approach could enable sequencing of modified bases in long reads (>10 kbp).
3.2. Nanopore Sequencing
Protein or solid state nanopores, which contain pores that allow single stranded DNA to pass through, have the potential to sequence DNA.66 The nanopore sequencing concept involves the measurement of the current passing through a pore as DNA translocates the pore. Each different base gives a distinct current signature when moving through a nanopore, which provides the basis for decoding the base sequence.66b Early attempts suggest it might be feasible to use such nanopores to discriminate 5mC and 5hmC (in addition to G, C, T, and A) in DNA in the future.67 By chemically altering the primary alcohol of 5hmC it is possible to create more distinct current signature to sequence 5hmC in synthetic DNA.68
4. Elucidating DNA Modifications in the Future
There have been considerable advances in the creation of chemical and enzymatic methods that enable the detection of modifications of cytosine bases in genomic DNA. It is now possible to decode 5mC, 5hmC, 5fC, and 5caC in addition to G, C, A, and T in DNA at single base resolution. When coupled with the recent (and ongoing) transformations in DNA sequencing technologies, it is practical to carry out such analysis on whole human (and other species’) genomes. Collectively these methods will pave the way to understand the role of modified cytosines in nature and ultimately the exploitation of this knowledge in medicine, agriculture, and biotechnology.
Acknowledgments
We thank the Biotechnology and Biological Sciences Research Council for a studentship to M.J.B. E.A.R is a Herchel Smith Fellow. S.B. is a Senior Investigator of The Wellcome Trust and the Balasubramanian group is core-funded by Cancer Research U.K.
Glossary
Abbreviations
- 2-OG
2-oxyglutarate
- 5caC
5-carboxycytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- A
adenine
- BisChiP-Seq
bisulfite-treated chromatin immunoprecipitated DNA
- BS-Seq
bisulfite sequencing
- C
cytosine
- CAB-Seq
chemical modification-assisted bisulfite sequencing
- CGIs
CpG islands
- ChIP-BS-Seq
ChIP-bisulfite-sequencing
- CMS
cytosine-5-methylsulfonate
- DIP-Seq
DNA immunoprecipitation sequencing
- DNA
deoxyribonucleic acid
- DNMTs
DNA methyltransferases
- EDC
1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide hydrochloride
- ES
embryonic stem
- fCAB-Seq
5fC-assisted bisulfite sequencing
- G
guanine
- GLIB
glucosylation, periodate oxidation and biotinylation
- HELP
HpaII tiny fragment enrichment by ligation-mediated PCR
- hMeDIP-Seq
hydroxymethylated DIP-Seq
- HRP
horseradish peroxidase
- MeDIP-Seq
methylated DIP-Seq
- mESC
mouse ES cells
- NOMe-Seq
nucleosome occupancy and methylome sequencing
- oxBS-Seq
oxidative bisulfite sequencing
- qPCR
quantitative polymerase chain reaction
- redBS-Seq
reduced bisulfite sequencing
- RRBS-Seq
reduced representative bisulfite sequencing
- SAM
S-adenosyl methionine
- SMRT
single-molecule real-time
- ssDNA
single stranded DNA
- T
thymine
- TAB-Seq
TET-assisted bisulfite sequencing
- TET
ten-11 translocation
- TSS
transcription start site
- UDG
uracil DNA glycosylase
- UDP
uridine diphosphate
- βGT
β-glucosyltransferase
Biographies

Michael J. Booth obtained his M.Chem. degree at the University of Southampton. He received his Ph.D. in Chemistry from the University of Cambridge under the supervision of Professor Shankar Balasubramanian, where he developed sequencing techniques for modified cytosine bases. He is currently based at the University of Oxford in the laboratory of Professor Hagan Bayley, where he is a Junior Research Fellow at Merton College.

Eun-Ang Raiber obtained her Ph.D. in organic chemistry from the University of Salford and Paterson Institute for Cancer Research, in Manchester, U.K., and has worked as a postdoctoral researcher at Harvard University and University College London. Dr. Raiber is currently a senior postdoctoral research associate in Balasubramanian Laboratory at the University of Cambridge.

Shankar Balasubramanian is the Herchel Smith Professor of Medicinal Chemistry at the University of Cambridge. He directs research laboratories in the Department of Chemistry and also the Cancer Research U.K. Cambridge Institute. He received his Ph.D. from the University of Cambridge in 1991, then spent two years as a postdoctoral fellow at The Pennsylvania State University. He has been a member of the faculty in Cambridge since 1994. Balasubramamian’s research is on the chemical biology of nucleic acids and the genome and has included an approach for genome sequencing, the study of G-quadruplex nucleic acids, and modified bases in DNA.
Author Present Address
⊥ Chemistry Research Laboratory, University of Oxford, Mansfield Road, OX1 3TA United Kingdom.
Author Contributions
∥ Contributed equally. The manuscript was written by M.J.B., E.A.R., and S.B.
The authors declare the following competing financial interest(s): M.J.B. and S.B. are co-inventors on a published U.S. patent for redBS-Seq and oxBS-Seq (publication number WO/2013/017853). M.J.B. and S.B. are shareholders in and M.J.B. a consultant for Cambridge Epigenetix, Ltd. S.B. is an advisor to and shareholder of Illumina, Inc.
References
- Watson J. D.; Crick F. H. Nature 1953, 171, 737. [DOI] [PubMed] [Google Scholar]
- Warren R. A. Annu. Rev. Microbiol. 1980, 34, 137. [DOI] [PubMed] [Google Scholar]
- Walker M. S.; Mandel M. J. Virol. 1978, 25, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fukasawa T. J. Mol. Biol. 1964, 9, 525. [DOI] [PubMed] [Google Scholar]; b Goldblum A.; Perahia D.; Pullman A. FEBS Lett. 1978, 91, 213. [DOI] [PubMed] [Google Scholar]; c Mathews C. K.Reproduction of Large Virulent Bacteriophages; Springer: New York, 1977. [Google Scholar]
- a Bird A. Genes Dev. 2002, 16, 6. [DOI] [PubMed] [Google Scholar]; b Deaton A. M.; Bird A. Genes Dev. 2011, 25, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wyatt G. R. Nature 1950, 166, 237. [DOI] [PubMed] [Google Scholar]; d Jones P. A. Nat. Rev. Genet. 2012, 13, 484. [DOI] [PubMed] [Google Scholar]; e Meilinger D.; Fellinger K.; Bultmann S.; Rothbauer U.; Bonapace I. M.; Klinkert W. E.; Spada F.; Leonhardt H. EMBO Rep. 2009, 10, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Okano M.; Bell D. W.; Haber D. A.; Li E. Cell 1999, 99, 247. [DOI] [PubMed] [Google Scholar]; b Suetake I.; Shinozaki F.; Miyagawa J.; Takeshima H.; Tajima S. J. Biol. Chem. 2004, 279, 27816. [DOI] [PubMed] [Google Scholar]; c Bostick M.; Kim J. K.; Esteve P. O.; Clark A.; Pradhan S.; Jacobsen S. E. Science 2007, 317, 1760. [DOI] [PubMed] [Google Scholar]; d Sharif J.; Muto M.; Takebayashi S.; Suetake I.; Iwamatsu A.; Endo T. A.; Shinga J.; Mizutani-Koseki Y.; Toyoda T.; Okamura K.; Tajima S.; Mitsuya K.; Okano M.; Koseki H. Nature 2007, 450, 908. [DOI] [PubMed] [Google Scholar]
- a Chodavarapu R. K.; Feng S.; Bernatavichute Y. V.; Chen P. Y.; Stroud H.; Yu Y.; Hetzel J. A.; Kuo F.; Kim J.; Cokus S. J.; Casero D.; Bernal M.; Huijser P.; Clark A. T.; Kramer U.; Merchant S. S.; Zhang X.; Jacobsen S. E.; Pellegrini M. Nature 2010, 466, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Laurent L.; Wong E.; Li G.; Huynh T.; Tsirigos A.; Ong C. T.; Low H. M.; Kin Sung K. W.; Rigoutsos I.; Loring J.; Wei C. L. Genome Res. 2010, 20, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Maunakea A. K.; Nagarajan R. P.; Bilenky M.; Ballinger T. J.; D’Souza C.; Fouse S. D.; Johnson B. E.; Hong C.; Nielsen C.; Zhao Y.; Turecki G.; Delaney A.; Varhol R.; Thiessen N.; Shchors K.; Heine V. M.; Rowitch D. H.; Xing X.; Fiore C.; Schillebeeckx M.; Jones S. J.; Haussler D.; Marra M. A.; Hirst M.; Wang T.; Costello J. F. Nature 2010, 466, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hellman A.; Chess A. Science 2007, 315, 1141. [DOI] [PubMed] [Google Scholar]; e Meissner A.; Mikkelsen T. S.; Gu H.; Wernig M.; Hanna J.; Sivachenko A.; Zhang X.; Bernstein B. E.; Nusbaum C.; Jaffe D. B.; Gnirke A.; Jaenisch R.; Lander E. S. Nature 2008, 454, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Sharp A. J.; Stathaki E.; Migliavacca E.; Brahmachary M.; Montgomery S. B.; Dupre Y.; Antonarakis S. E. Genome Res. 2011, 21, 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Penn N. W.; Suwalski R.; O’Riley C.; Bojanowski K.; Yura R. Biochem. J. 1972, 126, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Penn N. W. Biochem. J. 1976, 155, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothari R. M.; Shankar V. J. Mol. Evol. 1976, 7, 325. [DOI] [PubMed] [Google Scholar]
- Privat E.; Sowers L. C. Chem. Res. Toxicol. 1996, 9, 745. [DOI] [PubMed] [Google Scholar]
- a Kriaucionis S.; Heintz N. Science 2009, 324, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. Science 2009, 324, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pastor W. A.; Pape U. J.; Huang Y.; Henderson H. R.; Lister R.; Ko M.; McLoughlin E. M.; Brudno Y.; Mahapatra S.; Kapranov P.; Tahiliani M.; Daley G. Q.; Liu X. S.; Ecker J. R.; Milos P. M.; Agarwal S.; Rao A. Nature 2011, 473, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Williams K.; Christensen J.; Pedersen M. T.; Johansen J. V.; Cloos P. A.; Rappsilber J.; Helin K. Nature 2011, 473, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wu H.; D’Alessio A. C.; Ito S.; Wang Z.; Cui K.; Zhao K.; Sun Y. E.; Zhang Y. Genes Dev. 2011, 25, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Xu Y.; Wu F.; Tan L.; Kong L.; Xiong L.; Deng J.; Barbera A. J.; Zheng L.; Zhang H.; Huang S.; Min J.; Nicholson T.; Chen T.; Xu G.; Shi Y.; Zhang K.; Shi Y. G. Mol. Cell 2011, 42, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ito S.; Shen L.; Dai Q.; Wu S. C.; Collins L. B.; Swenberg J. A.; He C.; Zhang Y. Science 2011, 333, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pfaffeneder T.; Hackner B.; Truss M.; Munzel M.; Muller M.; Deiml C. A.; Hagemeier C.; Carell T. Angew. Chem., Int. Ed. Engl. 2011, 50, 7008. [DOI] [PubMed] [Google Scholar]; c He Y. F.; Li B. Z.; Li Z.; Liu P.; Wang Y.; Tang Q.; Ding J.; Jia Y.; Chen Z.; Li L.; Sun Y.; Li X.; Dai Q.; Song C. X.; Zhang K.; He C.; Xu G. L. Science 2011, 333, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Wang J.; Su Y.; Guerrero C.; Zeng Y.; Mitra D.; Brooks P. J.; Fisher D. E.; Song H.; Wang Y. Nucleic Acids Res. 2013, 41, 6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Raiber E. A.; Beraldi D.; Ficz G.; Burgess H. E.; Branco M. R.; Murat P.; Oxley D.; Booth M. J.; Reik W.; Balasubramanian S. Genome Biol. 2012, 13, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shen L.; Wu H.; Diep D.; Yamaguchi S.; D’Alessio A. C.; Fung H. L.; Zhang K.; Zhang Y. Cell 2013, 153, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Song C. X.; Szulwach K. E.; Dai Q.; Fu Y.; Mao S. Q.; Lin L.; Street C.; Li Y.; Poidevin M.; Wu H.; Gao J.; Liu P.; Li L.; Xu G. L.; Jin P.; He C. Cell 2013, 153, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Booth M. J.; Branco M. R.; Ficz G.; Oxley D.; Krueger F.; Reik W.; Balasubramanian S. Science 2012, 336, 934. [DOI] [PubMed] [Google Scholar]; b Yu M.; Hon G. C.; Szulwach K. E.; Song C. X.; Zhang L.; Kim A.; Li X.; Dai Q.; Shen Y.; Park B.; Min J. H.; Jin P.; Ren B.; He C. Cell 2012, 149, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Booth M. J.; Marsico G.; Bachman M.; Beraldi D.; Balasubramanian S. Nat. Chem. 2014, 6, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Maiti A.; Drohat A. C. J. Biol. Chem. 2011, 286, 35334. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hashimoto H.; Hong S.; Bhagwat A. S.; Zhang X.; Cheng X. Nucleic Acids Res. 2012, 40, 10203. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang L.; Lu X.; Lu J.; Liang H.; Dai Q.; Xu G. L.; Luo C.; Jiang H.; He C. Nat. Chem. Biol. 2012, 8, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M.; Reik W.; Henikoff S. Trends Genet. 2009, 25, 82. [DOI] [PubMed] [Google Scholar]
- Schiesser S.; Hackner B.; Pfaffeneder T.; Muller M.; Hagemeier C.; Truss M.; Carell T. Angew. Chem., Int. Ed. Engl. 2012, 51, 6516. [DOI] [PubMed] [Google Scholar]
- Maxam A. M.; Gilbert W. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F.; Nicklen S.; Coulson A. R. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Balasubramanian S. Angew. Chem., Int. Ed. Engl. 2011, 50, 12406. [DOI] [PubMed] [Google Scholar]; b Balasubramanian S. Chem. Commun. 2011, 47, 7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley D. R.; Balasubramanian S.; Swerdlow H. P.; Smith G. P.; Milton J.; Brown C. G.; Hall K. P.; Evers D. J.; Barnes C. L.; Bignell H. R.; Boutell J. M.; Bryant J.; Carter R. J.; Keira Cheetham R.; Cox A. J.; Ellis D. J.; Flatbush M. R.; Gormley N. A.; Humphray S. J.; Irving L. J.; Karbelashvili M. S.; Kirk S. M.; Li H.; Liu X.; Maisinger K. S.; Murray L. J.; Obradovic B.; Ost T.; Parkinson M. L.; Pratt M. R.; Rasolonjatovo I. M.; Reed M. T.; Rigatti R.; Rodighiero C.; Ross M. T.; Sabot A.; Sankar S. V.; Scally A.; Schroth G. P.; Smith M. E.; Smith V. P.; Spiridou A.; Torrance P. E.; Tzonev S. S.; Vermaas E. H.; Walter K.; Wu X.; Zhang L.; Alam M. D.; Anastasi C.; Aniebo I. C.; Bailey D. M.; Bancarz I. R.; Banerjee S.; Barbour S. G.; Baybayan P. A.; Benoit V. A.; Benson K. F.; Bevis C.; Black P. J.; Boodhun A.; Brennan J. S.; Bridgham J. A.; Brown R. C.; Brown A. A.; Buermann D. H.; Bundu A. A.; Burrows J. C.; Carter N. P.; Castillo N.; Chiara E. C. M.; Chang S.; Neil Cooley R.; Crake N. R.; Dada O. O.; Diakoumakos K. D.; Dominguez-Fernandez B.; Earnshaw D. J.; Egbujor U. C.; Elmore D. W.; Etchin S. S.; Ewan M. R.; Fedurco M.; Fraser L. J.; Fuentes Fajardo K. V.; Scott Furey W.; George D.; Gietzen K. J.; Goddard C. P.; Golda G. S.; Granieri P. A.; Green D. E.; Gustafson D. L.; Hansen N. F.; Harnish K.; Haudenschild C. D.; Heyer N. I.; Hims M. M.; Ho J. T.; Horgan A. M. Nature 2008, 456, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A.; Jeltsch A. Nucleic Acids Res. 2001, 29, 3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waalwijk C.; Flavell R. A. Nucleic Acids Res. 1978, 5, 3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kinney S. M.; Chin H. G.; Vaisvila R.; Bitinaite J.; Zheng Y.; Esteve P. O.; Feng S.; Stroud H.; Jacobsen S. E.; Pradhan S. J. Biol. Chem. 2011, 286, 24685. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Szwagierczak A.; Brachmann A.; Schmidt C. S.; Bultmann S.; Leonhardt H.; Spada F. Nucleic Acids Res. 2011, 39, 5149. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Song C. X.; Yu M.; Dai Q.; He C. Bioorg. Med. Chem. Lett. 2011, 21, 5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth R. S.; Bird A. P. FEBS Lett. 2009, 583, 1713. [DOI] [PubMed] [Google Scholar]
- Khulan B.; Thompson R. F.; Ye K.; Fazzari M. J.; Suzuki M.; Stasiek E.; Figueroa M. E.; Glass J. L.; Chen Q.; Montagna C.; Hatchwell E.; Selzer R. R.; Richmond T. A.; Green R. D.; Melnick A.; Greally J. M. Genome Res. 2006, 16, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner A. L.; Johnson D. S.; Kim S. W.; Valouev A.; Reddy T. E.; Neff N. F.; Anton E.; Medina C.; Nguyen L.; Chiao E.; Oyolu C. B.; Schroth G. P.; Absher D. M.; Baker J. C.; Myers R. M. Genome Res. 2009, 19, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kornberg S. R.; Zimmerman S. B.; Kornberg A. J. Biol. Chem. 1961, 236, 1487. [PubMed] [Google Scholar]; b Terragni J.; Bitinaite J.; Zheng Y.; Pradhan S. Biochemistry 2012, 51, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Guan S.; Quimby A.; Cohen-Karni D.; Pradhan S.; Wilson G.; Roberts R. J.; Zhu Z.; Zheng Y. Nucleic Acids Res. 2011, 39, 9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Karni D.; Xu D.; Apone L.; Fomenkov A.; Sun Z.; Davis P. J.; Kinney S. R.; Yamada-Mabuchi M.; Xu S. Y.; Davis T.; Pradhan S.; Roberts R. J.; Zheng Y. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Khare T.; Pai S.; Koncevicius K.; Pal M.; Kriukiene E.; Liutkeviciute Z.; Irimia M.; Jia P.; Ptak C.; Xia M.; Tice R.; Tochigi M.; Morera S.; Nazarians A.; Belsham D.; Wong A. H.; Blencowe B. J.; Wang S. C.; Kapranov P.; Kustra R.; Labrie V.; Klimasauskas S.; Petronis A. Nat. Struct. Mol. Biol. 2012, 19, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gao F.; Xia Y.; Wang J.; Luo H.; Gao Z.; Han X.; Zhang J.; Huang X.; Yao Y.; Lu H.; Yi N.; Zhou B.; Lin Z.; Wen B.; Zhang X.; Yang H.; Wang J. Epigenetics 2013, 8, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sun Z.; Terragni J.; Borgaro J. G.; Liu Y.; Yu L.; Guan S.; Wang H.; Sun D.; Cheng X.; Zhu Z.; Pradhan S.; Zheng Y. Cell Rep. 2013, 3, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Weber M.; Davies J. J.; Wittig D.; Oakeley E. J.; Haase M.; Lam W. L.; Schubeler D. Nat. Genet. 2005, 37, 853. [DOI] [PubMed] [Google Scholar]; b Pomraning K. R.; Smith K. M.; Freitag M. Methods 2009, 47, 142. [DOI] [PubMed] [Google Scholar]
- Ficz G.; Branco M. R.; Seisenberger S.; Santos F.; Krueger F.; Hore T. A.; Marques C. J.; Andrews S.; Reik W. Nature 2011, 473, 398. [DOI] [PubMed] [Google Scholar]
- Robertson A. B.; Dahl J. A.; Vagbo C. B.; Tripathi P.; Krokan H. E.; Klungland A. Nucleic Acids Res. 2011, 39, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C. X.; Szulwach K. E.; Fu Y.; Dai Q.; Yi C.; Li X.; Li Y.; Chen C. H.; Zhang W.; Jian X.; Wang J.; Zhang L.; Looney T. J.; Zhang B.; Godley L. A.; Hicks L. M.; Lahn B. T.; Jin P.; He C. Nat. Biotechnol. 2011, 29, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liutkeviciute Z.; Kriukiene E.; Grigaityte I.; Masevicius V.; Klimasauskas S. Angew. Chem., Int. Ed. Engl. 2011, 50, 2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Song C. X.; Szulwach K.; Wang Z.; Weidenbacher P.; Jin P.; He C. J. Am. Chem. Soc. 2013, 135, 9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori H.; Tomizawa J. I.; Maxam A. M. Nucleic Acids Res. 1978, 5, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Hong T.; Tang T.; Zhai Q.; Xing X.; Mao W.; Zheng X.; Xu L.; Wu J.; Weng X.; Wang S.; Tian T.; Yuan B.; Huang B.; Zhuang L.; Zhou X. J. Am. Chem. Soc. 2013, 135, 1240. [DOI] [PubMed] [Google Scholar]
- Huang R.; Wang J.; Mao W.; Fu B.; Xing X.; Guo G.; Zhou X. Talanta 2013, 117, 445. [DOI] [PubMed] [Google Scholar]
- Tian T.; Zhang X.; Fu B.; Long Y.; Peng S.; Wang S.; Zhou X.; Zhou X. Chem. Commun. 2013, 49, 9968. [DOI] [PubMed] [Google Scholar]
- Munzel M.; Lercher L.; Muller M.; Carell T. Nucleic Acids Res. 2010, 38, e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lister R.; O’Malley R. C.; Tonti-Filippini J.; Gregory B. D.; Berry C. C.; Millar A. H.; Ecker J. R. Cell 2008, 133, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lister R.; Ecker J. R. Genome Res. 2009, 19, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M.; McDonald L. E.; Millar D. S.; Collis C. M.; Watt F.; Grigg G. W.; Molloy P. L.; Paul C. L. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hayatsu H.; Wataya Y.; Kai K.; Iida S. Biochemistry 1970, 9, 2858. [DOI] [PubMed] [Google Scholar]; b Shapiro R.; Servis R. E.; Welcher M. J. Am. Chem. Soc. 1970, 92, 422. [Google Scholar]
- Shapiro R.; Braverman B.; Louis J. B.; Servis R. E. J. Biol. Chem. 1973, 248, 4060. [PubMed] [Google Scholar]
- Shapiro R.; Difate V.; Welcher M. J. Am. Chem. Soc. 1974, 96, 906. [DOI] [PubMed] [Google Scholar]
- Wang R. Y.; Gehrke C. W.; Ehrlich M. Nucleic Acids Res. 1980, 8, 4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K.; Okamoto A. Bioorg. Med. Chem. Lett. 2007, 17, 1912. [DOI] [PubMed] [Google Scholar]
- Hayatsu H. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008, 84, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman A. B.; Gu H.; Bartels S. J.; Zhang Y.; Matarese F.; Simmer F.; Marks H.; Bock C.; Gnirke A.; Meissner A.; Stunnenberg H. G. Genome Res. 2012, 22, 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statham A. L.; Robinson M. D.; Song J. Z.; Coolen M. W.; Stirzaker C.; Clark S. J. Genome Res. 2012, 22, 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J. S.; Kelly T. K.; De Carvalho D. D.; Taberlay P. C.; Liang G.; Jones P. A. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Pastor W. A.; Shen Y.; Tahiliani M.; Liu D. R.; Rao A. PloS One 2010, 5, e8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. R.; Ficz G.; Reik W. Nat. Rev. Genet. 2012, 13, 7. [DOI] [PubMed] [Google Scholar]
- Booth M. J.; Ost T. W.; Beraldi D.; Bell N. M.; Branco M. R.; Reik W.; Balasubramanian S. Nat. Protoc. 2013, 8, 1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Hon G. C.; Szulwach K. E.; Song C. X.; Jin P.; Ren B.; He C. Nat. Protoc. 2012, 7, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono K.; Asahi K.; Suzuki S. J. Am. Chem. Soc. 1969, 91, 7490. [DOI] [PubMed] [Google Scholar]
- Johnson T. J.; Jones R. A. Tetrahedron 1978, 34, 547. [Google Scholar]
- Guo H.; Zhu P.; Wu X.; Li X.; Wen L.; Tang F. Genome Res. 2013, 23, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Eid J.; Fehr A.; Gray J.; Luong K.; Lyle J.; Otto G.; Peluso P.; Rank D.; Baybayan P.; Bettman B.; Bibillo A.; Bjornson K.; Chaudhuri B.; Christians F.; Cicero R.; Clark S.; Dalal R.; Dewinter A.; Dixon J.; Foquet M.; Gaertner A.; Hardenbol P.; Heiner C.; Hester K.; Holden D.; Kearns G.; Kong X.; Kuse R.; Lacroix Y.; Lin S.; Lundquist P.; Ma C.; Marks P.; Maxham M.; Murphy D.; Park I.; Pham T.; Phillips M.; Roy J.; Sebra R.; Shen G.; Sorenson J.; Tomaney A.; Travers K.; Trulson M.; Vieceli J.; Wegener J.; Wu D.; Yang A.; Zaccarin D.; Zhao P.; Zhong F.; Korlach J.; Turner S. Science 2009, 323, 133. [DOI] [PubMed] [Google Scholar]; b Flusberg B. A.; Webster D. R.; Lee J. H.; Travers K. J.; Olivares E. C.; Clark T. A.; Korlach J.; Turner S. W. Nat. Methods 2010, 7, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T. A.; Lu X.; Luong K.; Dai Q.; Boitano M.; Turner S. W.; He C.; Korlach J. BMC Biol. 2013, 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C. X.; Clark T. A.; Lu X. Y.; Kislyuk A.; Dai Q.; Turner S. W.; He C.; Korlach J. Nat. Methods 2012, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Venkatesan B. M.; Bashir R. Nat. Nanotechnol. 2011, 6, 615. [DOI] [PubMed] [Google Scholar]; b Manrao E. A.; Derrington I. M.; Laszlo A. H.; Langford K. W.; Hopper M. K.; Gillgren N.; Pavlenok M.; Niederweis M.; Gundlach J. H. Nat. Biotechnol. 2012, 30, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wallace E. V.; Stoddart D.; Heron A. J.; Mikhailova E.; Maglia G.; Donohoe T. J.; Bayley H. Chem. Commun. 2010, 46, 8195. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Laszlo A. H.; Derrington I. M.; Brinkerhoff H.; Langford K. W.; Nova I. C.; Samson J. M.; Bartlett J. J.; Pavlenok M.; Gundlach J. H. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18904. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schreiber J.; Wescoe Z. L.; Abu-Shumays R.; Vivian J. T.; Baatar B.; Karplus K.; Akeson M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. W.; Gong L.; Bayley H. Angew. Chem., Int. Ed. Engl. 2013, 52, 4350. [DOI] [PubMed] [Google Scholar]