

Significance
Phosphodiesterase 4 (PDE4) is an essential contributor to intracellular signaling and an important drug target. We have used protein engineering, biochemistry, and X-ray crystallography to elucidate how its conserved N-terminal regions regulate its activity. Our results show that a helical segment in the regulatory domain of one subunit crosses over to occlude the catalytic site of the other subunit of the homodimer. The structure suggests a strategy for the design of inhibitors that target specific splice variants of this enzyme. By mapping onto our structure all the mutations in PDE4D that underlie the rare human genetic disorder of acrodysostosis, we present a rationale for why they could lead to a dysregulation of PDE4D activity.
Keywords: PDE4, cAMP hydrolysis, acrodysostosis, UCR1/2, phosphodiesterase regulation
Abstract
Phosphodiesterase 4 (PDE4) is an essential contributor to intracellular signaling and an important drug target. The four members of this enzyme family (PDE4A to -D) are functional dimers in which each subunit contains two upstream conserved regions (UCR), UCR1 and -2, which precede the C-terminal catalytic domain. Alternative promoters, transcriptional start sites, and mRNA splicing lead to the existence of over 25 variants of PDE4, broadly classified as long, short, and supershort forms. We report the X-ray crystal structure of long form PDE4B containing UCR1, UCR2, and the catalytic domain, crystallized as a dimer in which a disulfide bond cross-links cysteines engineered into UCR2 and the catalytic domain. Biochemical and mass spectrometric analyses showed that the UCR2-catalytic domain interaction occurs in trans, and established that this interaction regulates the catalytic activity of PDE4. By elucidating the key structural determinants of dimerization, we show that only long forms of PDE4 can be regulated by this mechanism. The results also provide a structural basis for the long-standing observation of high- and low-affinity binding sites for the prototypic inhibitor rolipram.
The diverse, pivotal roles of cAMP and cGMP (1) in mammalian intracellular signaling include acting as “second messengers” of signals delivered through G protein-coupled receptors (2), regulating the opening and closing of ligand-gated ion-channels (3), activating guanine-nucleotide exchange factors (4), and activating kinase signaling cascades (5). Responsibility for degrading these 3′,5′-cyclic nucleotides, and thereby limiting the duration and amplitude of the signals that they convey, falls to enzymes of the intracellular phosphodiesterase family (PDEs). The significance of these enzymes in cellular control makes them important targets of medicinal discovery, and maximizing our understanding of their respective structures and activities is an urgent priority (6).
Mammalian PDEs are encoded by 21 genes and divided into 11 families (PDE1 to -11) defined by their amino acid sequences, with supporting distinctions based on pharmacologic and kinetic properties. Among the most intriguing of these families is PDE4, the four members (PDE4A to -D) of which have high selectivity for cAMP over cGMP as substrate and are characteristically sensitive to inhibition by rolipram. PDE4 inhibition has been pursued as a therapeutic target for over 30 y, since the first discovery of the potential efficacy of rolipram in treating clinical depression (7). Two different PDE4 inhibitors, roflumilast (Daxas) and apremilast (Otezla), are Food and Drug Administration-approved therapies for exacerbations of chronic obstructive pulmonary disease and psoriatic arthritis, respectively, and several others are in clinical trials for a variety of indications.
Because of alternative promoters/start sites and variable mRNA splicing, transcription from the four PDE4 genes results in the expression of more than 25 different isoforms of PDE4. Each isoform has a unique N-terminal region that determines its specific subcellular localization by mediating interactions with scaffolding proteins. The isoforms are further classified into long, short, and supershort forms based on the presence or absence of two upstream conserved regions (UCRs, known as UCR1 and UCR2). All isoforms share a highly conserved catalytic domain, so that a relatively invariant phosphodiesterase catalytic function is associated with variable ancillary features that confer specific biological potential on each isoform. These modulations may be delivered through interactions with other proteins, but also through mechanisms internal to the PDE4 isoform itself.
Long splice variants contain both UCR1 and UCR2, short variants lack UCR1, and the supershort forms of PDE4 additionally lack part of UCR2 (Fig. 1A). The extent to which UCRs are present determines critical functional differences between the isoforms. Phosphorylation by protein kinase A (PKA) at a conserved site on UCR1 activates all long PDE4 isoforms (8, 9). Mutation and deletion studies have shown that long forms of PDE4 are dimeric, with key dimerization interactions mediated by UCR1 and UCR2 (10, 11), and that the C-terminal half of UCR2 could play a negative regulatory role (12–16). Access to details of these different mechanisms of control would advance our capacity to design medicines that act selectively among the PDE4 isoforms, and should also cast light on their specific intracellular functions.
Fig. 1.

Domain organization of long, short, and supershort PDE4 splice variants. (A) UCR1 and -2 (red and green, respectively), and the catalytic domain (teal) are illustrated as rectangles. The variable N-terminal region is represented as a thick gray line. Internal loop regions (LR1 and LR2) and the isoform specific C-terminal region are shown as a black line. Long-form PDE4s contain a PKA phosphorylation site (S133 in PDE4B1) located in UCR1. PDE4B, -C, and -D also contain an ERK phosphorylation site (S659 in PDE4B). (B) The PDE4Bcryst construct contains PDE4B1 residues 122–736. The first and last residue of the modeled peptides, and the positions of the engineered cysteine mutations (C267 and C610), are indicated. The regions of PDE4Bcryst that are not included in the crystallographic model are shown as dashed lines.
Crystal structures are available for the catalytic domains of all of the isoforms of PDE4 (17), but they provide no insight into the structural basis for dimerization or the regulation of catalytic activity. The only structural information on a UCR comes from the crystal structures of truncated constructs of PDE4B and -4D in which the last 32-residue section of UCR2 is attached to the catalytic domain by an engineered linker (18). In these structures, the C-terminal part of UCR2 adopts an α-helical conformation that interacts with the catalytic domain at the entrance to the catalytic site, further supporting a regulatory function of UCR2. The linker is not visible in the electron density maps, and it cannot be determined whether the two interacting domains are from the same polypeptide (cis interaction) or from different polypeptides (trans interaction). This is clearly a fundamental question in PDE4 enzymology, bearing on the internal governance of the enzyme’s activity as modulated by internal cooperativity, interactions with other proteins, and other potential allosteric effectors.
In this study, an approach that combines protein engineering with biochemical, enzymological and crystallographic analyses is used to show that the UCR2 of one subunit of dimeric PDE4B1 crosses over to regulate the catalytic activity of the other subunit in a trans interaction. The results include a new structure of a large fragment of PDE4B1 (residues 122–736) (Fig. 1B) that contains both UCR1 and UCR2, providing improved understanding of how the full-length PDE4 dimer is organized and how its regulatory and catalytic domains interact. This first crystal structure of a long-form PDE4 also shows that dimerization is mediated by interactions between the UCR1 and UCR2 and is essential for regulation of enzymatic activity. These interactions are unique to long splice variants of PDE4, and inaccessible to short or supershort forms. We note also that these structural results explain the dysregulation of PDE4D activity created by point mutations associated with the rare genetic disorder of acrodysostosis (19, 20).
Results
Generation of an Interdomain Disulfide Cross-Linked PDE4 Dimer.
The crystal structures of certain truncated PDE4B and PDE4D constructs show that the helical C-terminal region of UCR2 has an intimate interaction with the active site located in the catalytic domain of each enzyme (18). These results account for important features of PDE4 inhibition, but leave unclear whether the active site receives contact from UCR2 in a cis or trans manner and the extent to which the interactions occur in solution in full-length enzyme in the absence of a small-molecule inhibitor.
We addressed these questions by expressing in insect cells a nearly full-length PDE4B1 in which UCR2 could be locked into position adjacent to the active site by formation of a disulfide bond. Noting that the γ-oxygens of Ser267 of UCR2 and Ser610 of the catalytic domain are only 4.4 Å apart in the crystal structure (PDB ID code 3G45), we mutated both residues to cysteine, hypothesizing that this should result in spontaneous disulfide bond formation if these domains have the same interaction in solution as in the crystal. The two Ser-to-Cys mutations were introduced into a truncated PDE4B1 construct containing residues 122–736, with the seven native cysteines of that sequence simultaneously being mutated to alanines to minimize the potential for complexity. This construct, which begins at the N terminus of UCR1, was selected from among those tested as the longest one that could be expressed and purified with minimal degradation. Longer constructs that included the variable N-terminal region suffered extensive N-terminal proteolytic degradation when overexpressed in insect cells. As our eventual goal was to obtain a crystal structure, we also introduced Ser-to-Ala mutations at the known PKA and ERK phosphorylation sites (Ser133, Ser554, Ser559, Ser561), to avoid having to deal with mixtures of phosphorylated and unphosphorylated protein during crystallization.
Because the two newly introduced cysteines became disulfide-linked once the protein was removed from the reducing cellular environment (see below), this strategically designed protein offered both biochemical and (when crystallized) direct structural routes to resolving the nature of the normally noncovalent interdomain interaction.
Characterization of Engineered PDE4 Construct.
This engineered construct of PDE4, designated PDE4Bcryst (Fig. 1B), was stable, dimeric, and resistant to aggregation. On gel filtration, both the reduced and cross-linked forms of PDE4Bcryst had elution profiles that were essentially superimposable on the profile of unmutated PDE4B1 (122–736), further indicating that the engineered enzyme was correctly folded (Fig. 2A). When treated with reducing agent, it had specific activity for cAMP hydrolysis comparable to that of wild-type PDE4B1 (Fig. 2B).
Fig. 2.
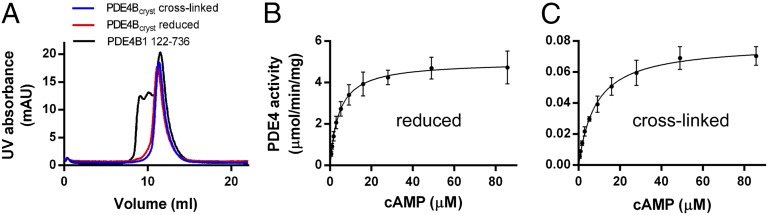
PDE4Bcryst is dimeric, free of aggregates and fully active when reduced. (A) Superimposed UV absorption elution profiles of cross-linked and reduced PDE4Bcryst and unmutated PDE4B1 (122–736). The major peak elutes at a position corresponding to a PDE4 dimer. Michaelis–Menten kinetics plots for PDE4Bcryst (B) reduced (specific activity 5.0 ± 0.2 µmol⋅min−1·mg−1, Km 4.3 ± 0.5 µM) and (C) directly after purification, without addition of reducing agent (specific activity 0.078 ± 0.002 µmol⋅min−1·mg−1, Km 8.4 ± 0.8 µM). Data shown are mean ± SEM from three independent experiments.
UCR2 of One Subunit Interacts with the Catalytic Domain of the Other Subunit in Long-Form PDE4.
In SDS/PAGE run under nonreducing conditions, PDE4Bcryst migrated as a 148-kDa dimeric species, but addition of DTT to the sample buffer caused the protein to migrate at its monomeric mass of 74 kDa (Fig. 3). This finding was consistent with one or two interchain disulfide bonds in the dimer. LC-MS peptide mapping of PDE4Bcryst digested at pH 2 with pepsin and analyzed before and after neutralization and reduction with DTT demonstrated the presence of the intended Cys267–Cys610 cross-link, and no evidence of any other disulfide bond or free cysteine could be found (Figs. S1–S3). The flexible specificity of pepsin caused a variety of cross-linked species to be detected, all containing the 267–610 disulfide (Table S1). Short (PDE4B2) and supershort (PDE4B5) isoforms containing the same engineered cysteines showed an insignificant amount of cross-linked protein as determined by SDS/PAGE (Fig. 3).
Fig. 3.

UCR2–catalytic domain interaction is in trans in dimeric, long-form PDE4B. (A) Stained SDS/PAGE gel and (B) Western blot with an anti-PDE4B antibody showing that PDE4Bcryst runs as a dimer (148 kDa) under nonreducing conditions and as a monomer when DTT is added to the loading buffer. PDE4B2 (S95C/S438C) and PDE4B5 (S34C/S377C) migrate mostly as monomers (59 kDa and 54 kDa, respectively) either with or without the addition of DTT, indicating limited interchain disulfide cross-linking in these proteins.
UCR2 Regulates Catalytic Activity in Long-Form PDE4.
In the absence of reducing treatment, PDE4Bcryst had a specific activity for cAMP hydrolysis of 0.078 ± 0.002 μmol⋅min−1·mg−1 (Fig. 2B). Treatment with reducing agent increased this to 5.0 ± 0.2 μmol⋅min−1·mg−1, a value in the range reported for recombinant wild-type full-length PDE4 isoforms (21).
Crystal Structure of a Long-Form PDE4.
X-ray diffraction data collected from a single crystal of PDE4Bcryst were processed to 2.6 Å resolution. The crystal belongs to space group P3221 with one dimer in the asymmetric unit. The structure was solved by molecular replacement, using the structure of truncated PDE4B (PDB ID code 3G45) as a search model. Because of the lack of electron density, the following segments of the polypeptide are not part of the final refined model: N-terminal portion of UCR1 (122–165), LR1 (188–210), LR2 (283–327), and the C-terminal segment (658–736). Secondary structure prediction algorithms predict all these regions to be largely unstructured, leading us to believe that they are highly flexible and likely to be disordered in any crystal structure. The final model was refined to good crystallographic statistics and stereochemistry (Table 1).
Table 1.
Data collection and refinement statistics
| Data collection and refinement | PDE4Bcryst | PDE4Bcryst–rolipram |
| Data collection | ||
| Space group | P3221 | P3221 |
| Cell dimensions | ||
| a, b, c (Å) | 137.83, 137.83, 141.74 | 137.40, 137.40, 142.98 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 91.30–2.58 (2.59–2.58)* | 142.98–3.22(3.23–3.22) |
| Rmerge | 0.033 (0.417) | 0.178(0.476) |
| I/σI | 18.5 (2.1) | 12.0(2.5) |
| Completeness (%) | 98.6 (56.2) | 91.8(73.0) |
| Redundancy | 4.8 (2.0) | 6.6(2.8) |
| Refinement | ||
| Resolution (Å) | 91.30–2.58 (2.65–2.58) | 119.00–3.22(3.36–3.22) |
| No. reflections | 48601 (3027) | 23620(2420) |
| Rwork/Rfree | 0.185/0.217 | 0.197/0.231 |
| No. atoms | ||
| Protein | 6773 | 6669 |
| Ligand/ion | 9 | 47 |
| Water | 342 | |
| B-factors | ||
| Protein | 53.85 | 72.51 |
| Ligand/ion | 51.54 | 57.92 |
| Water | 49.71 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (°) | 1.05 | 1.11 |
Values in parentheses are for highest-resolution shell.
The disulfide bond between the two cysteine mutations (S267C and S610C) is clearly visible in the electron density map (Fig. 4). The PDE4B dimer is mushroom-shaped, with the dimerization domains forming the stalk of the mushroom and the two catalytic domains forming the head. The catalytic sites are on opposite faces of the assembly, related by a twofold axis of symmetry that runs approximately down the long axis of the stalk. The dimerization domain (Fig. 5) consists of residues from the C terminus of UCR1 (169–187) and the N terminus of UCR2 (211–236), which form an antiparallel helix pair connected via salt bridges between Arg177–Glu234 and Arg180–Asp227. Biochemical pull-down studies and yeast two-hybrid analyses previously implicated three out of these four residues in interdomain contacts in PDE4D3 (Arg98, Arg101, and Asp149) (22). Helix pairs from each subunit associate in a tight four-helix bundle, with a core of conserved hydrophobic residues from UCR1 (Phe169, Leu173, Leu176, Val179, and Phe183) and UCR2 (Leu226, Leu230, and Leu233). With a buried surface area of ∼1,300 Å2, this is the largest intersubunit interface in the dimer, and explains why both UCRs are necessary for dimerization. Remarkably, with only secondary structure predictions as a guide, Richter and Conti showed (10) that mutating the corresponding hydrophobic residues in recombinant PDE4D3 to alanines completely disrupts dimer formation, whereas mutation of the polar residues described above as forming intrasubunit salt bridges does not disrupt dimerization. This finding gives confidence in the correctness of the dimer structure presented here. Except for Phe183, which is a valine in PDE4D, the polar and hydrophobic residues mentioned above are conserved in all PDE4 family members (Fig. S4), suggesting a common dimeric core structure for the long forms of PDE4A to -D.
Fig. 4.

Cartoon representation of X-ray crystallographic model of PDE4Bcryst. Two orthogonal views of the PDE4Bcryst dimer are shown. One subunit is colored as per the color scheme in Fig. 1 with unmodeled segments indicated by dashed lines, and the other subunit is colored gray with a transparent van der Waals surface. The engineered disulfide cross-link is shown (Inset), with a portion of the (2Fo-Fc) electron density map contoured at 1.0σ level around the disulfide bond.
Fig. 5.

UCR1 and 2 both participate in the dimer interface. Two orthogonal views of the dimerization domain, with the conserved hydrophobic residues that stabilize the four-helix bundle shown in ball-and-stick representation, and the conserved polar residues that make intrasubunit salt bridges in stick representation.
The second and third helices of UCR2 (residues 239–264 and 266–279) form a helix-turn-helix motif that acts as the autoinhibitory domain. This domain forms an extensive interface with the catalytic domain, with the third helix directly occluding the active site. This is the second largest intersubunit interface in the crystal structure, with a buried surface area of ∼950 Å2. On the basis of the biochemical evidence presented earlier, we can confidently assert that this interaction is between UCR2 of one subunit and the catalytic domain of the other, even though the linker connecting the two domains is not visible in the electron density map. The 42 amino acid linker, if modeled as an unstructured, extended polypeptide, is long enough to connect UCR2 to the catalytic domain (Fig. 4).
The dimer interface between the two catalytic domains is the smallest intersubunit interface, with a buried surface area of ∼700 Å2, and is predominantly stabilized by electrostatic interactions between Asp471 and Arg507 of the opposing domains. The same interface is present between two crystal symmetry-related molecules in the truncated PDE4B structure reported by Burgin et al. (18), and in crystal structures of the isolated catalytic domains of PDE4B and PDE4D (23, 24).
As noted earlier, regions of the polypeptide predicted to be unstructured were not visible in the electron density map. This observation, coupled with the fact that the crystals took weeks to appear in the crystallization drops, led us to investigate if the protein underwent unplanned in situ proteolytic cleavage during crystallization. SDS/PAGE of dissolved crystals confirmed this suspicion (Fig. S5). Following this notion, a batch of PDE4Bcryst free of protease contamination was subjected to limited proteolysis with a panel of proteases, in an attempt to reproduce these crystals. Treatment with pronase yielded a protein sample that did crystallize in the same conditions as the structure described above, with identical space group and unit cell dimensions. The nonproteolyzed protein failed to crystallize, suggesting that removal of these flexible termini and loop regions is necessary for crystallization.
Structure of Bound Rolipram Identifies the Basis of the High-Affinity Rolipram Binding Site.
PDE4 literature has multiple references to high-affinity (1–50 nM) and low-affinity (500–1,000 nM) rolipram binding sites (25, 26). More recently, it has generally been accepted that these represent two different conformational states of the enzyme (27). We determined the structure of a PDE4B–rolipram complex, obtained by transferring crystals of the cross-linked dimer into a solution containing rolipram. Initially, on the assumption that the disulfide-linked regulatory helix may block access of rolipram to the active site, reducing agent was included in the rolipram solution. This was unnecessary because the structures from crystals soaked in rolipram solution with or without reducing agent were identical, with clear electron density in both structures for the inhibitor bound at the active site (Fig. S6). The binding mode of rolipram in the catalytic pocket is very similar to that seen in previous structures of rolipram bound to the isolated catalytic domain (24), except that it sits slightly deeper in the pocket and has a small rotation of the cyclopentyl group (Fig. 6). However, in the structure presented here, rolipram makes two significant additional interactions with the regulatory helix. The hydrophobic cyclopentyl group is now completely buried in a hydrophobic pocket, whereas it was partially exposed to solvent in the catalytic domain structure. Tyr274 from the regulatory helix forms one wall of this pocket, and the tyrosine hydroxyl also makes a hydrogen bond with the amide carbonyl oxygen on rolipram. These additional interactions, which contribute to high-affinity rolipram binding, are only possible in dimeric PDE4 where the autoinhibitory domain of one subunit can interact with the catalytic domain of the other subunit.
Fig. 6.

High- and low-affinity binding modes of rolipram. The structures of rolipram (purple sticks) bound to (A) PDE4Bcryst and to (B) PDE4B catalytic domain (PDB ID code 1XMY) were superimposed by minimizing the rmsd of the Cα atoms of the catalytic domains, and are shown here in the same orientation in two separate figures for clarity. Hydrogen bonding interactions are shown as dashed lines, with distances labeled in angstroms. Both structures show a hydrogen-bond with the conserved glutamine at the base of the substrate-binding pocket. In the high-affinity binding mode (A), Tyr274 from the regulatory helix forms one wall of the hydrophobic pocket occupied by the cyclopentyl group, and its hydroxyl oxygen forms a hydrogen bond with the amide carbonyl oxygen in rolipram. Both these interactions are absent in the low-affinity binding mode shown in B.
An isothermal titration calorimetry (ITC) binding-affinity measurement of (R,S) rolipram for reduced PDE4Bcryst could be fit to a single binding site model with a Kd of 55 nM (Fig. 7A). Rolipram binds with comparable stoichiometry (0.9) and slightly weaker Kd (101 nM) to the cross-linked form of PDE4Bcryst (Table S2). The similarity in favorable enthalpic and unfavorable entropic terms observed for both forms is consistent with the above observation that the crystal structures with rolipram obtained in the presence or absence of reducing agent are superimposable.
Fig. 7.

PDE4Bcryst rolipram binding and inhibition. (A) ITC data for reduced PDE4Bcryst titration with rolipram (Kd = 55 nM). (B) Rolipram IC50 titration for reduced PDE4Bcryst (Hill slope = 1.13 ± 0.12, Ki = 15.98 ± 1.68 nM). Note that both experiments were carried out using a racemic mixture of (R,S) rolipram. Data are mean ± SEM from three independent experiments.
Rolipram displays complex inhibition kinetics toward PDE4 (28). The rolipram dose–response curve generated with PDE4Bcryst reduced immediately before the experiment has a Hill coefficient of ∼1 and a Ki of 16 nM (Fig. 7B). PDE4B1 (residues 122–736) without the engineered disulfide and native long form PDE4 proteins have a propensity to form higher-order oligomers that migrate in the void volume (>600 kDa) in size-exclusion chromatography (SEC) (Fig. 2A). Aggregated long-form PDE4 has the same or higher specific activity compared with the purified dimer peak from SEC. In addition, the SEC void volume fraction binds rolipram with low affinity. This finding, along with PDE4Bcryst being resistant to aggregation, suggests that the nonspecific aggregation of long-form PDE4 can be traced to the regulatory domain. The historical observation that both high-affinity and low-affinity rolipram binding sites are present in long-form PDE4 samples can be explained by dimeric PDE4 being contaminated with higher-order oligomers, which invariably would lead to shallow binding affinity curves. Another possibility is that native long-form PDE4 is continuously shifting between multiple conformational states, leading to the observation of complex binding kinetics, whereas this engineered construct with the disulfide bridge captures only one of the states.
Mapping PDE4D Gene Mutations Found in Acrodysostosis Patients Provides a Structural Basis for Gain-of-Function.
Sixteen different point mutations in the PDE4D gene have been identified as responsible for the rare de novo genetic disorder of acrodysostosis, which is characterized by nasal hypoplasia, peripheral dysostosis, variable short stature, and intellectual impairment (19, 29–31). It is possible to map all but one of these mutation sites onto the PDE4Bcryst crystal structure (Fig. 8). The mutations (eight in UCR1, four in UCR2, and four in the catalytic domain) are broadly distributed over the PDE4D coding region, but mapping them onto the structure makes it apparent that they are all localized either at the interface between the autoinhibitory and catalytic domains, or at the “hinge” region connecting the dimerization domain to the autoinhibitory and catalytic domains. The only site that cannot be mapped on the structure corresponds to the PKA activation site (Ser190 in PDE4D, Ser133 in PDE4Bcryst). Based on the structural localization of the documented mutations, we speculate that they all affect the regulation of PDE4 activity by PKA, perhaps by making PDE4 insensitive to PKA regulation.
Fig. 8.

Acrodysostosis mutations map to the interface between regulatory and catalytic domains. Mutations in PDE4D that result in the rare genetic disorder of acrodysostosis are mapped to the equivalent (identical) residues in PDE4Bcryst. Apart from one mutation at the PKA phosphorylation site, all 12 other known mutation sites (orange) are either at the interface between the autoinhibitory domain and the catalytic domain or at the hinge region between the dimerization domain and the catalytic domain. For clarity, the sites have been numbered sequentially from N to C terminus in the figure, and listed explicitly in Table S3. The active site is visible just below the regulatory helix, and the Zn2+ and Mg2+ ions at the site are labeled.
Discussion
cAMP and cGMP are formed enzymatically from the precursor molecules ATP and GTP, each of which typically exists in eukaryotic cells at concentrations of several hundred micromolar or more (32). That ubiquitous, abundant precursors provide the feedstock for cell signaling systems of almost infinite subtlety represents a masterpiece of evolutionary economy. The capacity to use almost the minimum number of messenger molecules to control a huge number of systems relies on profoundly specific mechanisms for producing, detecting, and terminating the requisite signals only in the locations where they are required. This multidimensional array of messaging elements necessarily has very many components, among which a very significant group are the enzymes comprising the PDE superfamily.
The demonstrated possibilities of drug discovery against PDEs (33, 34) have lent significance to studies of their individual features and functions. PDE4 is a particularly apposite example, because further major progress has appeared for some time to be just slightly out of reach. Despite the advance delivered by Burgin et al. (18), who followed the pathfinding contributions of other groups (10, 15, 16, 22) in showing that a helical segment of UCR2 can be positioned to cover the active site, the apparent flexibility and internal mobility of long-form PDE4 enzymes has previously foiled efforts to place this localized interaction in the context of a full molecular structure. In particular, it has remained unknown whether the UCR2 segment contacting the active site originates in a cis or trans manner from the identical polypeptides comprising the homodimeric enzyme.
The present work used protein engineering to introduce a covalent bond that reduced drastically the number of conformational states available to a long-form PDE4 molecule. With all seven naturally occurring cysteine residues mutated to serine, the molecule was newly equipped with two nonnative cysteines that, if they became disulfide-linked, would lock the UCR2 C-terminal helical element into precisely the location atop the catalytic site that it occupies in the structures of Burgin et al. (18). Biochemical evidence alone gave strong indications that the designed disulfide had formed, with gel electrophoresis performed without and with reduction indicating that the helix-to-active site contact occurs in trans. Peptide mapping analysis fully supported the homogeneity and full formation of the cross-link.
The ultimate benefit of introducing this artificial constraint in PDE4 was to permit crystallization and structural analysis, to our knowledge for the first time, of a long-form PDE4 dimer. Because the crystallized protein contained an engineered element that constrained it, we first considered closely the question of how well the common features of the long form and earlier less-complete structures overlapped. The rmsd between the new structure and a representative structure from Burgin et al. (18) (PDB ID code 3G45) was 0.93 Å when calculated using all 370 Cα atoms common to the two structures. Most of the small difference between the two structures is localized to the second helix of UCR2 (Fig. S7). The catalytic domains by themselves superimpose even better, with an rmsd of 0.68 Å (335 Cα atoms). The small shift in the position of the UCR2 helix could partly be a consequence of the restraint introduced by the engineered disulfide, and partly because of the fact that the present structure contains all three helices of UCR2 and a helix of UCR1. Overall, this outcome supports the conclusion that the engineered structure corresponds very closely to one important conformational state available to the native form of the enzyme in the reducing intracellular environment. The extent to which this conformation is populated will be a major factor governing PDE4 activity, and will respond to a range of influences that includes phosphorylation.
Our data support the conclusion that the UCR2-catalytic domain interaction regulates PDE4 activity by limiting the access of substrate to the active site, or through altered product release, or both. Km for the substrate cAMP and Kd for the inhibitor rolipram are relatively unchanged (4 μM vs. 8 μM, cAMP; 55 nM vs. 101 nM, rolipram) between the reduced and cross-linked forms of the enzyme, whereas the catalytic rates differ by 60-fold. The observation that rolipram can be soaked into crystals of cross-linked enzyme implies that access to the site is not completely blocked even when UCR2 is covalently attached to the catalytic domain. It seems likely that native long-form PDE4 exists in an equilibrium of “open” and “closed” states, and that the cross-linked form of the enzyme seen in the crystal structure represents the closed state.
The activity of all long PDE4 isoforms is increased by two- to sixfold upon PKA-catalyzed phosphorylation of a single serine residue (Ser133 in PDE4B1) found at the N-terminal region of UCR1 within the PKA consensus Arg-Arg-Glu-Ser-Phe (8, 9). Because this region is not visible in the structure described herein, the exact interactions of the phosphorylated serine residue that result in activation of the enzyme cannot be visualized. It is, however, possible to model the N-terminal portion of UCR1 to be in the vicinity of the autoregulatory domain, perhaps interacting with one of several conserved arginine and lysine residues on that face of the second of the three helices formed by UCR2. We hypothesize that such an interaction would shift the equilibrium more toward the open state, resulting in activation of PDE4.
The PDE4Bcryst crystal structure also supports the conclusion from earlier mutation and truncation experiments (10, 11) that both UCR1 and UCR2 are required for dimerization. This result, along with the fact that no interdomain cross-linking is seen for the short (PDE4B2 S95C, S438C) and supershort (PDE4B5 S34C, S377C) splice variants, suggests that the dimerization domain is required for the regulatory and catalytic domain interaction to occur. Because the autoinhibitory interaction is in trans, and therefore only possible in dimeric PDE4, the implication is that only the activity of long isoforms is regulated in this manner. However, there are at least two other possible mechanisms of regulation that apply to short and supershort as well as long isoforms. First, in some crystal structures of the catalytic domain of PDE4B and PDE4D, a short helical segment of the C-terminal region (not visible in the PDE4Bcryst crystal structure), has been seen to interact with the catalytic site in a manner similar to the interaction of the regulatory helix seen in this structure [PDB ID codes 1F0J (35), 1XM6 (24), 3HMV (36), and 4MYQ (37)]. The short linker between the C terminus of the catalytic domain and the C-terminal helix requires that this interaction has to be within one subunit, and does not require dimerization. It has been proposed that this could be an alternative regulatory mechanism in monomeric PDE4 (18, 38). A second possibility, for which there is no direct evidence so far, is that interactions with specific binding partner proteins may facilitate dimerization and trans inhibition by UCR2 in short and supershort isoforms.
Well-defined electron density at the C terminus of the catalytic domain in this structure ends at Pro657, which is at the beginning of a consensus site for ERK phosphorylation (Pro-Xaa-Ser-Pro) that is common to PDE4B, -C, and -D. Phosphorylation at this site in long forms of PDE4 leads to inhibition when the N-terminal serine (PKA site) is not phosphorylated (39, 40). In this structure, as in the earlier structure by Burgin et al. (18), it is possible to model electrostatic interactions between Ser659 and a conserved arginine in UCR2 and a lysine in the catalytic domain, which would hold the autoregulatory domain in a closed conformation over the active site in a similar way that the disulfide cross-link does in PDE4Bcryst.
The observation that several PDEs (1, 2, 4–6, 10, 11) have tandem N-terminal regulatory domains has led to a proposal that their regulatory mechanisms share some similarities (41). The only other full-length PDE structure known, PDE2 (42), has features in common with the present structure. Both structures are dimeric, with dimerization mediated by the N-terminal domains, and are autoinhibited by steric obstruction of the catalytic site. Activation in both structures occurs as a result of conformational changes that are driven by binding events in the regulatory domains.
The fact that the activity of different splice variants of PDE4 may be regulated differently opens up the possibility of designing small molecule inhibitors specifically tailored to target a specific splice variant. For example, inhibitors of PDE4 that derive a high fraction of their binding energy from interactions with UCR2 would preferentially inhibit long splice variants of PDE4. The targeted activity of such inhibitors could allow them to have a better safety profile than the current generation of PDE4 inhibitors.
Materials and Methods
Construct Design, Protein Expression, and Purification.
A BAP-tag–tobacco etch virus protease cleavage site–FLAG-tag–thrombin protease cleavage site–sequence followed by insect cell codon optimized cDNA encoding residues 122–736 of human PDE4B1 (UniProt entry Q07343) was, along with biotin ligase (BirA), cloned into a pFastBac Dual vector. All native cysteines in addition to Ser133, Ser554, Ser559 and Ser661 were point-mutated to alanines, and Ser267 and Ser610 were point mutated to cysteines. The protein encoded by this construct is referred to as PDE4Bcryst. A recombinant baculovirus stock was generated in Spodoptera frugiperda Sf9 cells following the Bac-to-Bac Baculovirus Expression System protocol (Life Technologies). Protein expression was performed using S. frugiperda Sf21 cells harvested 72 h postinfection. The harvested cells were resuspended in 20 mM Tris, 300 mM NaCl, 5% (vol/vol) glycerol, 1 mM MgCl2, pH 8.0 (Buffer 1), with EDTA-free protease inhibitor tablets, and lysed with a homogenizer. After centrifugation, the lysate was batch affinity-purified with anti-FLAG M2 resin. Specifically bound proteins were eluted with 0.1 mg/mL FLAG peptide. The eluate was dephosphorylated with λ protein phosphatase, twofold diluted with 20 mM Tris, 5% (vol/vol) glycerol, 1 mM MgCl2, pH 8.0 (Buffer 2), and subjected to Q ion-exchange chromatography. Elution was performed in Buffer 2 with a 0.2–1 M NaCl gradient. The eluate was concentrated and purified on a size exclusion column equilibrated with 10 mM Tris, 100 mM NaCl, 5% (vol/vol) glycerol, 1 mM MgCl2, pH 7.5 (Buffer 3).
Insect cell codon-optimized cDNA of human PDE4B2 (UniProt ID Q07343-2) with an N-terminal hexahistidine-tag and a C-terminal FLAG-tag, and human PDE4B5 (UniProt ID Q07343-4) followed by a hexahistidine-tag were cloned into pFastBac Dual vectors. The serines corresponding to Ser267 and Ser610 in PDE4B1 (PDE4B2, Ser95 and Ser438; PDE4B5 Ser34 and Ser377) were point-mutated to cysteines. The lysis and anti-FLAG affinity purification procedures for PDE4B2 S95C, S438C were identical to those for PDE4Bcryst. The anti-FLAG resin eluate was dephosphorylated with λ-protein phosphatase, concentrated, and then purified on a size-exclusion column equilibrated with Buffer 3. For PDE4B5 S34C, S377C the lysis buffer contained 20 mM imidazole in addition to the Buffer 1 components listed above. Following lysate centrifugation, the protein was batch affinity-purified with Ni-NTA resin. Elution was done in Buffer 1 with a 20 to 250-mM imidazole gradient. Fractions that were judged by SDS/PAGE to contain PDE4B5 were pooled and, like PDE4Bcryst, subjected to dephosphorylation, ion exchange, and SEC.
Peptide Mapping by Liquid Chromatography-Mass Spectrometry.
A 20-μL sample of 5 μM PDE4Bcryst (0.7 μg/μL) was adjusted to pH 2 with 1 N HCl, treated with 0.3 μg of pepsin and then incubated at 37 °C overnight, after which it was diluted twofold with water. To reduce disulfide bonds in a portion of the digest, 8 μL of diluted peptic digest was treated with 2 μL of 1 M sodium bicarbonate containing 0.02 M DTT and the mixture was incubated at 37 °C. The original diluted digest and the DTT-treated fraction (5 μL of each) were then analyzed separately by LC-MS performed using a nanoACQUITY UPLC system (Waters) and a LTQ Orbitrap XL mass spectrometer (Thermo Scientific). HPLC was conducted at a flow rate of 300 nL/min using a gradient method with the following solvents: A, 0.1% formic acid; B, 0.1% formic acid in 80% acetonitrile, 10% trifluoroethanol. Sample injections were delivered to a (0.18 mm × 20 mm) ACQUITY UPLC PST C18 nanoACQUITY trap column, where the analytical column was a (0.075 mm × 100 mm) ACQUITY UPLC PST C18 nanoACQUITY column. Full MS scans were conducted in the Orbitrap (15,000 resolution) and MS/MS scans were conducted at ion trap resolution. Sequence coverage (Fig. S2) was surveyed using database searches run with Mascot (43) (Matrix Science) against an in-house database that included PDE4Bcryst. Search parameters included a 5-ppm mass window for peptide masses and a 0.6-Da window for fragment masses.
SDS/PAGE and Western Blot Analysis.
The purified proteins were mixed with LDS loading buffer with and without DTT. For stained gel, QuickBlue Stain (Boston Biologicals) was used. For Western blotting, a gel was transferred to a PVDF membrane and immunoblotted with primary anti-PDE4B antibody (HPA003005, Sigma-Aldrich), and secondary horseradish peroxidase-conjugated anti-rabbit IgG antibody (NA934V, GE Healthcare Bio-Sciences). Labeled bands were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Phosphodiesterase Activity, Rolipram Dose–Response, and ITC.
The cAMP hydrolysis activity assay to evaluate PDE activity was performed according to the manufacturer’s instructions with the PDE Light Assay Kit (Lonza). The luminescence was detected with an Infinite M1000 PRO microplate reader (Tecan) at room temperature. Substrate kinetics were run at 100 pM PDE4Bcryst and kinetic parameters were determined by nonlinear regression to the Michaelis–Menten equation: v = Vmax × [S]/(Km + [S]), where v represents the initial rate of cAMP hydrolysis at any given substrate concentration ([S]), Vmax is the maximal rate and Km is the Michaelis–Menten constant representing the substrate concentration that produces one-half Vmax. Dose–response titrations were run at 3.3 pM PDE4Bcryst and 8 µM cAMP, with varying (R,S)-rolipram concentrations using rolipram synthesized in-house. The rolipram IC50 was obtained by fitting the dose–response curve to a sigmoidal IC50 equation: (% inhibition) = bottom + (top – bottom)/[1 + ([rolipram]/IC50)^(–slope)], where bottom is the % inhibition value at the bottom plateau and top is the value at the top plateau. The slope term denotes the Hill coefficient and IC50 is the inhibitor concentration ([rolipram]) that results in 50% inhibition. Both analyses were done using GraphPad Prism 6 (GraphPad Software). Isothermal titration experiments were performed using a high precision AutoITC200 titration calorimeter (MicroCal). The PDE4Bcryst solution (8.0 μM) in the titration cell at 25 °C was titrated with 80 µM rolipram dissolved in the same buffer as PDE4Bcryst. For reduction, 5–10 mM TCEP was used. The affinity of rolipram for PDE4Bcryst is reported as the dissociation constant Kd (Kd = 1/Ka). The free energy of binding (ΔG) and entropy (ΔS) were obtained using the following relationships: ΔG = −RT lnKa = ΔH-TΔS, where R is the gas constant and T is the absolute temperature in kelvins.
Crystallization, Crystal Handling, and X-Ray Diffraction Data Collection.
A single crystal, grown by hanging-drop vapor diffusion in a droplet of equal volumes protein (10 mg/mL PDE4Bcryst) and precipitant solution (21% PEG3350, 0.2 M potassium iodide, 0.1 M Hepes pH 7.5), was cryo protected in precipitant solution containing 25% glycerol, mounted in a cryo loop, and flash-frozen in liquid nitrogen. For the PDE4Bcryst–rolipram complex structure, the precipitant solution was 20% (wt/vol) PEG3350 and 0.2 M potassium iodide. A single crystal was soaked in precipitant solution containing 1 mM (R,S)-rolipram, 2 mM DTT, and 20% glycerol for 15 min before being mounted and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at 100 K with radiation of wavelength 1.0 Å at the Advanced Photon Source at Argonne National Laboratory, beamline 17ID. The diffraction data were processed with autoPROC (44). Further data manipulations were carried out using the CCP4 program suite (45). The data statistics are summarized in Table 1.
Structure Solution, Model Building, and Refinement.
The initial phases for the PDE4Bcryst apo structure were generated by molecular replacement with the program PHASER (46) using the truncated PDE4B structure (PDB ID code 3G45) as a search model. Model building was conducted in COOT (47) and structure refinement was done with autoBUSTER (48). The refined model has 97.1% in the preferred and 2.9% in the allowed regions in the Ramachandran plot. For the PDE4Bcryst–rolipram complex structure, phases were generated by Fourier synthesis using the PDE4Bcryst apo structure as a model. The rolipram complex structure has 95.9% in the preferred, 3.1% in the allowed regions and 0.98% outliers in the Ramachandran plot. The refinement statistics are summarized in Table 1.
Coordinates and structure factors have been deposited in the Protein Data Bank, with accession codes 4WZI for the apo structure and 4X0F for the rolipram complex.
Protein Crystal Content Analysis.
PDE4Bcryst crystals were dissolved in LDS sample buffer and subjected to SDS/PAGE. The bands from a gel stained with InstantBlue (Expedeon) were excised for digestion and proteomic analysis by mass spectrometry. For N-terminal sequence analysis performed at Tufts University Core Facility, a gel was blotted to PVDF and Coomassie blue-stained bands were excised.
Pronase Proteolysis and Crystallization of PDE4Bcryst.
The PDE4Bcryst construct with an additional K147A mutation was used in the limited proteolysis experiments. We introduced this mutation after we observed cleavage C-terminal to K147 when thrombin was used to cleave off the affinity tag. PDE4Bcryst (K147A) was purified the same way as described before, except that the affinity purification tag was cleaved off with thrombin before gel filtration. Purified protein was incubated with 2 ng/µL pronase at 4 °C for 3 d, and then set up in crystallization drops. Crystals grew in exactly the same conditions as described before, with the same cell dimensions and space group.
Supplementary Material
Acknowledgments
We thank Xiayang Qiu for his sponsorship of this project and valuable comments on the manuscript, Kim Fennell for assistance with molecular biology, the staff of Tufts University Core Facility for N-terminal sequence analysis, and Caroline Silve (Université Paris-Sud) for valuable discussions on acrodysostosis mutations. Use of the Industrial Macromolecular Crystallography Association Collaborative Access Team beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4WZI and 4X0F).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1419906112/-/DCSupplemental.
References
- 1.Beavo JA, Brunton LL. Cyclic nucleotide research—Still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3(9):710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 2.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36(1):461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 3.Broillet M-C, Firestein S. Cyclic nucleotide-gated channels. Molecular mechanisms of activation. Ann N Y Acad Sci. 1999;868(1):730–740. doi: 10.1111/j.1749-6632.1999.tb11352.x. [DOI] [PubMed] [Google Scholar]
- 4.de Rooij J, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 5.Pidoux G, Taskén K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol. 2010;44(5):271–284. doi: 10.1677/JME-10-0010. [DOI] [PubMed] [Google Scholar]
- 6.Francis SH, Conti M, Houslay MD. Phosphodiesterases as Drug Targets. Springer; Berlin: 2011. [Google Scholar]
- 7.Zeller E, Stief HJ, Pflug B, Sastre-y-Hernández M. Results of a phase II study of the antidepressant effect of rolipram. Pharmacopsychiatry. 1984;17(6):188–190. doi: 10.1055/s-2007-1017435. [DOI] [PubMed] [Google Scholar]
- 8.Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem. 1996;271(28):16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 9.MacKenzie SJ, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1) Br J Pharmacol. 2002;136(3):421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter W, Conti M. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs) J Biol Chem. 2002;277(43):40212–40221. doi: 10.1074/jbc.M203585200. [DOI] [PubMed] [Google Scholar]
- 11.Xie M, et al. The upstream conserved regions (UCRs) mediate homo- and hetero-oligomerization of type 4 cyclic nucleotide phosphodiesterases (PDE4s) Biochem J. 2014;459(3):539–550. doi: 10.1042/BJ20131681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin SL, Swinnen JV, Conti M. Characterization of the structure of a low Km, rolipram-sensitive cAMP phosphodiesterase. Mapping of the catalytic domain. J Biol Chem. 1992;267(26):18929–18939. [PubMed] [Google Scholar]
- 13.Jacobitz S, McLaughlin MM, Livi GP, Burman M, Torphy TJ. Mapping the functional domains of human recombinant phosphodiesterase 4A: Structural requirements for catalytic activity and rolipram binding. Mol Pharmacol. 1996;50(4):891–899. [PubMed] [Google Scholar]
- 14.Kovala T, Sanwal BD, Ball EH. Recombinant expression of a type IV, cAMP-specific phosphodiesterase: characterization and structure-function studies of deletion mutants. Biochemistry. 1997;36(10):2968–2976. doi: 10.1021/bi9613483. [DOI] [PubMed] [Google Scholar]
- 15.Saldou N, et al. Comparison of recombinant human PDE4 isoforms: Interaction with substrate and inhibitors. Cell Signal. 1998;10(6):427–440. doi: 10.1016/s0898-6568(97)00169-1. [DOI] [PubMed] [Google Scholar]
- 16.Lim J, Pahlke G, Conti M. Activation of the cAMP-specific phosphodiesterase PDE4D3 by phosphorylation. Identification and function of an inhibitory domain. J Biol Chem. 1999;274(28):19677–19685. doi: 10.1074/jbc.274.28.19677. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, et al. Structures of the four subfamilies of phosphodiesterase-4 provide insight into the selectivity of their inhibitors. Biochem J. 2007;408(2):193–201. doi: 10.1042/BJ20070970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgin AB, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28(1):63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- 19.Lindstrand A, et al. Different mutations in PDE4D associated with developmental disorders with mirror phenotypes. J Med Genet. 2014;51(1):45–54. doi: 10.1136/jmedgenet-2013-101937. [DOI] [PubMed] [Google Scholar]
- 20.Lynch DC, et al. FORGE Canada Consortium Identification of novel mutations confirms PDE4D as a major gene causing acrodysostosis. Hum Mutat. 2013;34(1):97–102. doi: 10.1002/humu.22222. [DOI] [PubMed] [Google Scholar]
- 21.Salanova M, Jin SC, Conti M. Heterologous expression and purification of recombinant rolipram-sensitive cyclic AMP-specific phosphodiesterases. Methods. 1998;14(1):55–64. doi: 10.1006/meth.1997.0565. [DOI] [PubMed] [Google Scholar]
- 22.Beard MB, et al. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J Biol Chem. 2000;275(14):10349–10358. doi: 10.1074/jbc.275.14.10349. [DOI] [PubMed] [Google Scholar]
- 23.Lee ME, Markowitz J, Lee J-O, Lee H. Crystal structure of phosphodiesterase 4D and inhibitor complex. FEBS Lett. 2002;530(1-3):53–58. doi: 10.1016/s0014-5793(02)03396-3. [DOI] [PubMed] [Google Scholar]
- 24.Card GL, et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure. 2004;12(12):2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Huston E, et al. The human cyclic AMP-specific phosphodiesterase PDE-46 (HSPDE4A4B) expressed in transfected COS7 cells occurs as both particulate and cytosolic species that exhibit distinct kinetics of inhibition by the antidepressant rolipram. J Biol Chem. 1996;271(49):31334–31344. doi: 10.1074/jbc.271.49.31334. [DOI] [PubMed] [Google Scholar]
- 26.Bolger G, et al. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol Cell Biol. 1993;13(10):6558–6571. doi: 10.1128/mcb.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370(Pt 1):1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rocque WJ, et al. Human recombinant phosphodiesterase 4B2B binds (R)-rolipram at a single site with two affinities. Biochemistry. 1997;36(46):14250–14261. doi: 10.1021/bi971112e. [DOI] [PubMed] [Google Scholar]
- 29.Linglart A, et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. 2012;97(12):E2328–E2338. doi: 10.1210/jc.2012-2326. [DOI] [PubMed] [Google Scholar]
- 30.Michot C, et al. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012;90(4):740–745. doi: 10.1016/j.ajhg.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee H, et al. Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet. 2012;90(4):746–751. doi: 10.1016/j.ajhg.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140(1):1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 33.Houslay MD, Schafer P, Zhang KYJ. Keynote review: Phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10(22):1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 34.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: Targets for drug development. Nat Rev Drug Discov. 2006;5(8):660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 35.Xu RX, et al. Atomic structure of PDE4: Insights into phosphodiesterase mechanism and specificity. Science. 2000;288(5472):1822–1825. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]
- 36.Kranz M, et al. Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg Med Chem. 2009;17(14):5336–5341. doi: 10.1016/j.bmc.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 37.Fox D, 3rd, Burgin AB, Gurney ME. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell Signal. 2014;26(3):657–663. doi: 10.1016/j.cellsig.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houslay MD, Adams DR. Putting the lid on phosphodiesterase 4. Nat Biotechnol. 2010;28(1):38–40. doi: 10.1038/nbt0110-38. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18(4):893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baillie GS, MacKenzie SJ, McPhee I, Houslay MD. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br J Pharmacol. 2000;131(4):811–819. doi: 10.1038/sj.bjp.0703636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banjac A, Kurz U, Schultz JE. The regulatory tandem domains of human phosphodiesterases 1 and 4 regulate a cyanobacterial adenylyl cyclase. Cell Signal. 2012;24(8):1479–1484. doi: 10.1016/j.cellsig.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 42.Pandit J, Forman MD, Fennell KF, Dillman KS, Menniti FS. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc Natl Acad Sci USA. 2009;106(43):18225–18230. doi: 10.1073/pnas.0907635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 44.Vonrhein C, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey S. Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bricogne G, et al. BUSTER version 2.11.5. Global Phasing Ltd.; Cambridge, UK: 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.