

Abstract
The direct functionalization of unactivated sp3 C–H bonds is still one of the most challenging problems facing synthetic organic chemists. The appeal of such transformations derives from their capacity to facilitate the construction of complex organic molecules via the coupling of simple and otherwise inert building blocks, without introducing extraneous functional groups. Despite notable recent efforts,1 the establishment of general and mild strategies for the engagement of sp3 C–H bonds in carbon–carbon bond forming reactions has proven difficult. Within this context, the discovery of chemical transformations that are able to directly functionalize allylic methyl, methylene, and methine carbons in a catalytic manner is a priority. While protocols for direct allylic C–H oxidation and amination have become widely established,2,3 the engagement of allylic substrates in carbon–carbon bond-forming reactions has thus far required the use of pre-functionalized coupling partners.4 In particular, the direct arylation of non-functionalized allylic systems would enable chemists to rapidly access a series of known pharmacophores, though a general solution to this longstanding challenge remains elusive. We describe herein the use of both photoredox and organic catalysis to accomplish the first mild, broadly effective direct allylic C–H arylation. This new C–C bond-forming reaction readily accommodates a broad range of alkene and electron-deficient arene reactants and has been used in the direct arylation of benzylic C–H bonds.
While the well-known Heck reaction can be employed to generate allyl-substituted aromatics with high levels of efficiency via π-addition, olefin transposition sequences, there are few known examples of direct allylic arylation via the functionalization of C–H bonds.5,6 Indeed, a literature survey reveals an isolated report on the Fe-catalysed coupling of Grignard reagents with simple olefins as the only transition metal-mediated allylic arylation reported to date.7 Given the state-of-the-art of allylic C–H arylation (or for allylic C–C bond formation in general), we presumed that a mild and widely applicable solution to this high-profile challenge would be well received by practitioners of chemical synthesis.
The rapidly growing field of visible light-mediated photoredox catalysis offers a valuable platform for the design or discovery of new synthetic transformations.8–10 The ability of photoredox catalysts to act as both strong oxidants and reductants upon irradiation with low-energy visible light has enabled the invention of a series of useful bond constructions, previously thought to be unattainable via conventional pathways. In our laboratory, the synergistic merger of visible light photoredox catalysis with organocatlysis has been instrumental in the development of a number of methods for the direct functionalization of unactivated sp3 C–H bonds.11–13 Using this powerful dual-catalysis paradigm, we recently disclosed the direct arylation of benzylic ethers via a C–H abstraction mechanism that proceeds through the heterocoupling of two catalytically generated radical species.14 With this mechanistic blueprint in hand, we recently considered whether the combination of photoredox catalysis and organocatalysis could provide a solution to the longstanding and more significant challenge of direct allylic arylation. Given that the allylic C–H bonds of simple alkenes are relatively weak (cyclohexene allylic C–H bond dissociation energy (BDE) = 83.2 kcal mol−1),15 we hypothesized that olefinic substrates would undergo hydrogen atom abstraction using our photoredox conditions to generate transient allylic radicals, which thereafter would participate in a hetero radical–radical coupling pathway with in situ-generated arene radical anions. A detailed mechanism for the envisioned fragment coupling is depicted in Fig. 2. Upon irradiation with low-energy visible light (e.g. 26 W compact fluorescent lamp (CFL)), the iridium complex Ir(ppy)3 (1) (ppy = 2-phenylpyridine) is known to undergo a metal-to-ligand charge-transfer (MLCT) and intersystem crossing (ISC) to generate the long-lived excited state IrIII species 2 (τ = 1900 ns),16 which is a strong reductant (E1/2IV/*III = −1.73 V vs. the saturated calomel electrode (SCE) in MeCN).16 It has long been established that the photoexcited state of this complex will readily undergo single-electron transfer (SET) with electron-deficient arenes such as 4-cyanopyridines and 1,4-dicyanobenzene (e.g. E1/2red = −1.61 V vs. SCE for 1,4-dicyanobenzene in MeCN)17 to generate a persistent arene radical anion along with the oxidized photocatalyst 3. Though the IrIV species 3 (E1/2IV/III = +0.77 V vs. SCE in MeCN)16 is not likely to be sufficiently oxidizing to directly oxidize a typical thiol (E1/2red = +1.12 V vs. SCE for butanethiol in MeCN),18 this event should be facilitated by the presence of a suitable base. The weakly acidic thiol (pKa 7.91 for methyl 2-mercaptoacetate in H2O)19 is deprotonated to yield the thiolate anion (E1/2red = −0.85 V vs. SCE for butanethiolate in MeCN),18 which is readily oxidized by the photocatalyst. Based on the reported BDEs for typical thiols and allylic C–H bonds, we reasoned that the electrophilic thiyl radical 5 would readily abstract an allylic hydrogen atom from the alkene substrate (cyclohexene allylic C–H BDE = 83.2 kcal mol−1 vs S–H BDE = 87.0 kcal mol−1)15,20 to provide allylic radical 9 along with regenerated organocatalyst 4. At this time, an intermolecular radical–radical coupling would serve to forge the new carbon–carbon bond, with the resulting pyridienyl or cyclohexadienyl anion undergoing rapid rearomatization via elimination of cyanide.
Figure 2. Proposed mechanism for the direct arylation of allylic C–H bonds via photoredox and organic catalysis.
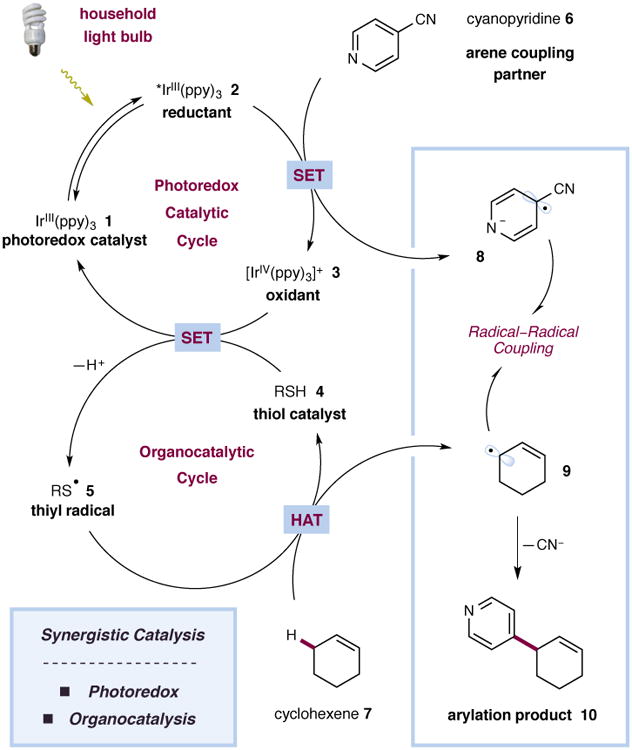
The catalytic cycle is initiated via excitation of photocatalyst 1 to give the excited state 2. Single-electron reduction of 4-cyanopyridine (6) generates the radical anion 8 along with oxidant 3. In the presence of a base, oxidant 3 is capable of oxidising the thiol catalyst 4 to give the thiyl radical 5 along with the regenerated photocatalyst 1. The thiyl radical 5 abstracts an allylic hydrogen atom from cyclohexene (7) to generate allylic radical 9. A radical–radical coupling and subsequent elimination of cyanide serve to construct the new C–C bond and form the arylation product 10.
We began our direct allylic C–H arylation studies with an evaluation of a range of photocatalysts, thiols, solvents, and bases in the presence of cyclohexene and 1,4-dicyanobenzene as representative coupling partners. To our delight, we found that the use of fluorescent light (2 × 26 W) in the presence of Ir(ppy)3, triisopropylsilanethiol, and K2CO3 enabled the desired aryl–allyl fragment coupling in excellent yield (87%) to furnish the desired arylcyclohexene. The necessity of each of the key reaction components – photocatalyst, thiol, and light – was demonstrated through a series of control experiments (see Supplementary Information). While trace amounts of product were formed in the absence of thiol and photocatalyst,5 no reaction was observed upon exclusion of light. It should be noted that these optimal conditions employ a standard household 26 W fluorescent light source and proceed readily at room temperature with low loadings of both the photocatalyst (1 mol%) and organocatalyst (5 mol%).
With these optimized conditions in hand, we next examined the scope of olefins that can be employed in this direct allylic arylation reaction. As shown in Fig. 3a, this simple arylation protocol permits the direct coupling of electron-deficient arenes with a wide range of unfunctionalized alkenes. Importantly, both cyclic and acyclic olefins are readily accommodated in this transformation. For example, a series of simple cyclic alkenes coupled with 1,4-dicyanobenzene in excellent yields (12–16, 71–87% yield). Notably, cyclic substrates bearing alkyl substituents also provide high levels of coupling efficiency, although the production of minor quantities of regioisomeric adducts can typically be detected due to the presence of secondary sites of hydrogen atom abstraction. Not surprisingly, installation of a tert-butyl group on the cyclohexene ring afforded only one major product, with only trace quantities of regioisomers being observed (17, 92% yield). Similarly useful levels of regioselectivity were obtained when α-pinene was employed as the olefinic substrate (18, 93% yield). The observed selectivity is attributed to the expectation that the electrophilic thiyl radical abstracts the most hydridic hydrogen atom and subsequent coupling of the radical proceeds through the least hindered secondary radical position.
Figure 3. Substrate scope for the direct allylic arylation reaction.

A range of alkenes are efficiently arylated under the standard reaction conditions (top, generalized reaction). The substrate scope includes both cyclic and acyclic alkenes (a). A range of arenes bearing electron withdrawing substituents can be employed as coupling partners under the standard conditions (b). Isolated yields are indicated below each entry. * Isomers observed; In all cases the major isomer is depicted. Yields refer to the combined yield of all isomers. Ratios of isomers where applicable: (±)-16 (2.2:1.0 E:Z), (±)-17 (>20:1), 18 (>10:1), (±)-19 (1.4:1.0), (±)-20 (4.9:1.0), (±)-21 (1.4:1.0), (±)-23 (1.1:1.0), (±)-24 (2.1:1.0), (±)-25 (>19:1), (±)-26 (∼1:1), (±)-28 (1.2:1.0), (±)-29 (1.1:1.0). ‡ Yield from silyl enol ether (83%), yield from 2-cyclohexen-1-ol (62%). † Additional thiol or base required; See Supplementary Information for experimental details.
Importantly, acyclic alkenes also serve as effective allyl coupling partners in this protocol while generally exhibiting high levels of regiocontrol with respect to the carbon–carbon bond forming step (19–23, 63–90% yield). For example, implementation of 2-pentene leads predominantly to the formation of the branched arylation product with excellent efficiency (20, 85% yield), with only trace quantities of linear arylation adducts arising from hydrogen atom abstraction of the terminal methylene group. Exposure of 2-methyl-2-pentene to these photoredox conditions afforded two arylation isomers in a 1.4:1 ratio: the major isomer arises from coupling of the allylic radical through the more nucleophilic tertiary substituted carbon, to generate a quaternary center with useful efficiency (21, 90% combined yield for both isomers). In contrast, the reaction with 2,2′-dimethyl-3-hexene yielded only a single product, resulting from coupling at the least hindered terminus of the allylic radical (22, 81% yield). With respect to medicinal chemistry applications it is important to note that heteroatoms can be incorporated into the alkene substrates with no deleterious effect on reaction efficiency (c.f. 19 and 23). Thus, coupling of 4-penten-1-ol afforded the desired arylation product in useful yield (23, 66% yield). Inclusion of a phenyl substituent on the alkene moiety resulted in a lower isolated yield, presumably owing to decreased hydridicity of the allylic C–H bonds (24, 76% yield); however, an analogous substrate bearing a 4-MeO substituent on the phenyl ring underwent efficient, regioselective coupling with 1,4-dicyanobenzene (25, 95% yield). Intriguingly, a naturally occurring heteroatom-substituted terpene could be employed directly to generate an arylated derivative in only one step and in good yield (26, 84% yield).
This new photoredox-organocatalytic methodology can also be applied to a one-step synthesis of β-aryl ketones. For example, when the TMS-silyl enol ether of cyclohexanone was exposed to these new arylation conditions, the resultant β-aryl ketone was isolated in high yield (27, 83% yield). Furthermore, direct reaction of the corresponding allylic alcohol (2-cyclohexen-1-ol) also furnished ketone 27 via regioselective hydrogen atom abstraction adjacent to the alcohol and subsequent coupling at the allylic β-enol position (see Supplementary Information). Finally, β,γ-unsaturated esters are viable substrates, affording a mixture of β,γ - and β,δ-unsaturated products (28, 81% yield).
We next sought to investigate the scope of the aromatic coupling partner in the arylation protocol. As shown in Fig. 3b, a range of electron-deficient arenes are well tolerated in this new photoredox protocol. Derivatives of dicyanobenzene, including those bearing ortho-substituents, readily coupled with the cyclohexenyl allylic radical to afford the corresponding arylation adducts in good to excellent yield (29–31, 50–83% yield). Moreover, extended aromatic systems, such as a biphenyl derivative, were also found to participate efficiently (32, 66% yield). Although the presence of one cyano-substituent was a requirement for the viability of the aryl coupling component, additional electron-withdrawing substituents, such as sulfones, may be readily incorporated without issue (33, 69% yield). Notably, a range of cyanopyridine derivatives underwent coupling with cyclohexene to afford the corresponding heteroarylation adducts with useful levels of efficiency (34–37, 70–84% yield). Additionally, halide substituents at the 2- and 3-positions of pyridine were also tolerated. In these cases it is important to note that the thiol catalyst did not participate in a non-productive pyridine addition SNAr pathway (an initial concern that did not prove to be valid for the 2-chloropyridine system). Finally, it was shown that a 7-azaindole derivative functions as a viable coupling partner (38, 70% yield), a notable finding with respect to potential applications of this technology in medicinal chemistry. Beyond regiocontrol, perhaps a more remarkable selectivity phenomenon that we have observed in these photoredox coupling studies is that mono-arylation adducts are observed exclusively, despite the fact that the allyl substituted aryl product contains a C–H bond that is far weaker than the allylic position of the starting material olefin. At the present time, we can envision two mechanistic scenarios that would account for this unique (and valuable) form of chemoselectivity. First, the doubly activated (benzylic and allylic) C–H bond of the product is significantly less hydridic than the starting material allylic C–H, and is thereby sufficiently less susceptible to hydrogen atom abstraction by the electrophilic thiyl radical of the organocatalyst (thereby governed by the inherent exchange rate constant). Alternatively, it is possible that hydrogen abstraction does indeed occur from the mono-arylation adduct; however, the resultant stabilized radical is insufficiently reactive to participate in a second radical–radical coupling event (a persistent radical effect), and hydrogen atom re-abstraction from solvent or thiol would reconstitute the initial arylation product. Given that we do not observe the formation of any olefin-transposed styrenyl side products, which would arise from the hydrogen atom quenching of a persistent benzylic–allylic radical species, we presume that the former, exchange constant-gated mechanism is operative.
We next sought to demonstrate the utility of this methodology for the preparation of high-value and versatile building blocks, and at the same time demonstrate the mild nature of the conditions employed (Fig. 4a). More specifically, we have found that boronic ester-substituted cyclohexenes are stable to these photoredox conditions and, more importantly, undergo regioselective arylation at the carbon center adjacent to the C–B bond (39, 73% yield). Moreover, incorporation of a boronic ester substituent on the arene coupling component can also be tolerated in this transformation (40, 60% yield).
Figure 4. Expanding the scope of the direct C–H arylation protocol.

Substrates bearing boronic esters substituents are tolerated, providing a means to rapidly access functionalized building blocks (a). The mild conditions allow for late-stage functionalization of advanced synthetic intermediates and bioactive natural products (b). Silyl ketene acetals are compatible with the reaction conditions, yielding β-aryl lactones (c). Arylation is not limited to allylic C–H bonds; benzylic C–H bonds can also be arylated (d). The reactivity is governed by bond strengths, with the weaker allylic bond undergoing exclusive functionalization in a direct competition experiment (e). * Isomers observed; In all cases the major isomer is depicted. Yields refer to the combined yield of all isomers. Ratios of isomers where applicable: (±)-39 (6:1), (±)-41 (>10:1). See Supplementary Information for experimental details.
Finally, a central advantage of this mild, visible light-mediated arylation protocol is the potential for the late-stage diversification of advanced, highly functionalized synthetic intermediates. As an illustration of this strategy, we subjected 5-pregnen-3β-ol-20-one, a complex biologically active molecule, to our standard reaction conditions. In the event, this compound underwent fragment coupling with 1,4-dicyanobenzene in a highly regioselective and diastereoselective fashion to deliver the aryl functionalized steroid framework 41 in good yield (Fig. 4b).
An overarching goal of this research program is to define a widely applicable mode of catalytic activation based on well-established physical properties (i.e. BDEs, hydrogen atom transfer (HAT) exchange constants, oxidation potentials), which permits activation of any given substrate in a predictable manner. Although the primary focus of this work has been the direct arylation of allylic C–H bonds, we reasoned that this generic activation mode could be extended to encompass a diverse menu of substrates. In theory, any substrate possessing hydridic C–H bonds of an appropriate strength (∼80–90 kcal mol−1) should have the potential to serve as a viable coupling partner in this arylation manifold. Along these lines, we found that silyl ketene acetals are also readily arylated to produce the corresponding β-aryl lactones (Fig. 4c). It is important to consider that lactones represent a high-value synthon class that are inaccessible through contemporary enamine mediated β-arylation technologies.13 Moreover, this fragment coupling protocol can be extended to benzylic substrates such as ethylbenzene to generate the corresponding benzhydryl systems in excellent yields (Fig. 4d).
The predictable and highly useful nature of this hydrogen atom activation mode is exemplified in a direct competition experiment conducted with cyclohexene and ethylbenzene. As shown in Fig. 4e, when both olefinic and benzylic substrates were combined in the same vessel, only the product of allylic arylation was observed (78% yield) with no competitive formation of the benzylic arylation product to any degree. This is a striking result given that ethylbenzene is in fact a suitable substrate for this arylation protocol (see Fig. 4d). The exclusive formation of the allylic arylation product can be readily rationalised by consideration of the BDEs for the two substrates (cyclohexene allylic C–H BDE = 83.2 kcal mol−1 vs ethylbenzene benzylic C–H BDE = 85.4 kcal mol−1),15,21 along with an appreciation of the hydridic nature of the respective C–H bonds involved. On this basis, it can be readily anticipated that the thiyl radical will preferentially abstract a hydrogen atom from the allylic substrate, which possesses the weaker and more hydridic C–H bond than the benzylic position. It cannot be ruled out that some degree of benzylic radical formation may occur; however, a rapid hydrogen atom exchange with the cyclohexene substrate may predominate over the rate of productive radical–radical coupling.
In summary, we have developed a reaction manifold that permits the direct functionalization and arylation of allylic sp3 C–H bonds under mild and operationally simple conditions. This new C–C bond-forming process, which relies on the mechanistic merger of photoredox and thiol-based organic catalysis, readily accommodates a diverse range of alkene and electron-deficient arene coupling partners. These studies have also established the versatility of this activation mode for the direct arylation of both complex and sensitive olefin containing molecules.
Supplementary Material
Figure 1. The direct arylation of allylic C–H bonds via the synergistic merger of photoredox and organic catalysis.

Arylation of allylic bonds is generally accomplished via transition metal-catalyzed couplings with pre-functionalized substrates (a). Installation of heteroatoms via direct allylic C–H functionalization is widely precedented (b). Direct C–H arylation is proposed via the synergistic merger of photoredox and organic catalysis (c).
Acknowledgments
Financial support was provided by NIHGMS (R01 GM103558-03) and kind gifts from Merck and Amgen. J.D.C. thanks the Marie-Curie Actions for an International Outgoing Fellowship.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions J.D.C. performed and analyzed experiments. J.D.C. and D.W.C.M. designed experiments to develop this reaction and probe its utility, and also prepared this manuscript.
Author Information. The authors declare no competing financial interests.
References
- 1.He J, et al. Ligand-controlled C(sp3)–H arylation and olefination in synthesis of unnatural chiral α-amino acids. Science. 2014;343:1216–1220. doi: 10.1126/science.1249198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eames J, Watkinson M. Catalytic allylic oxidation of alkenes using an asymmetric Kharasch-Sosnovsky reaction. Angew Chem Int Ed. 2001;40:3567–3571. doi: 10.1002/1521-3773(20011001)40:19<3567::AID-ANIE3567>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 3.Johannsen M, Jørgensen KA. Allylic Amination. Chem Rev. 1998;98:1689–1708. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]
- 4.Trost BM, Van Vranken DL. Asymmetric transition metal-catalyzed allylic alkylations. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 5.Borg RM, Arnold DR, Cameron TS. Radical ions in photochemistry. 15. The photosubstitution reaction between dicyanobenzenes and alkyl olefins. Can J Chem. 1984;62:1785–1802. [Google Scholar]
- 6.Inoue M, Hoshikawa T. Photoinduced direct 4-pyridination of C(sp3)–H bonds. Chem Sci. 2013;4:3118–3123. [Google Scholar]
- 7.Sekine M, Ilies L, Nakamura E. Iron-catalyzed allylic arylation of olefins via C(sp3)–H activation under mild conditions. Org Lett. 2013;15:714–717. doi: 10.1021/ol400056z. [DOI] [PubMed] [Google Scholar]
- 8.Narayanam JMR, Stephenson CRJ. Visible light photoredox catalysis: applications in organic synthesis. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 9.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz DM, Yoon TP. Solar synthesis: Prospects in visible light photocatalysis. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicewicz DA, MacMillan DWC. Merging photoredox catalysis with organocatalysis: The direct asymmetric alkylation of aldehydes. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagib DA, Scott ME, MacMillan DWC. Enantioselective α-trifluoromethylation of aldehydes via photoredox organocatalysis. J Am Chem Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Photoredox activation for the direct β-arylation of ketones and aldehydes. Science. 2013;339:1593–1596. doi: 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qvortrup K, Rankic DA, MacMillan DWC. A general strategy for organocatalytic activation of C–H bonds via photoredox catalysis: Direct arylation of benzylic ethers. J Am Chem Soc. 2014;136:626–629. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khursan SL, Mikhailov DA, Yanborisov VM, Borisov DI. AM1 Calculations of bond dissociation energies. Allylic and benzylic C–H bonds. React Kinet Catal Lett. 1997;61:91–95. [Google Scholar]
- 16.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Photochemistry and photophysics of coordination compounds: Iridium. Top Curr Chem. 2007;281:143–203. [Google Scholar]
- 17.Mori Y, Sakaguchi Y, Hayashi H. Magnetic field effects on chemical reactions of biradical ion pairs in homogeneous fluid solvents. J Phys Chem A. 2000;104:4896–4905. [Google Scholar]
- 18.Ogawa KA, Boydston AJ. Organocatalyzed anodic oxidation of aldehydes to thioesters. Org Lett. 2014;16:1928–1931. doi: 10.1021/ol500459x. [DOI] [PubMed] [Google Scholar]
- 19.Jencks WP, Salvesen K. Equilibrium deuterium isotope effects on the ionization of thiol acids. J Am Chem Soc. 1971;93:4433–4436. [Google Scholar]
- 20.Denisov E, Chatgilialoglu C, Shestakov A, Denisova T. Rate constants and transition-state geometry of reactions of alkyl, alkoxyl and peroxyl radicals with thiols. Int J Chem Kinet. 2009;41:284–293. [Google Scholar]
- 21.McMillen DF, Golden DM. Hydrocarbon bond dissociation energies. Ann Rev Phys Chem. 1982;33:493–532. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.