

Abstract
To better define the biologic function of membrane-bound CSF1 (mCSF1) in vivo, we have generated mCSF1 knockout (k/o) mice. Spinal bone density (BMD) was 15.9% higher in k/o mice compared to wild-type (wt) controls (P < 0.01) and total BMD was increased by 6.8% (P < 0.05). A higher mean femur BMD was also observed but did not reach statistical significance (6.9% P = NS). The osteoclastogenic potential of bone marrow isolated from mCSF1 k/o mice was reduced compared to wt marrow. There were no defects in osteoblast number or function suggesting that the basis for the high bone mass phenotype was reduced resorption. In addition to a skeletal phenotype, k/o mice had significantly elevated serum triglyceride levels (123 ± 7 vs. 88 ± 3.2 mg/dl; k/o vs. wt, P < 0.001), while serum cholesterol levels were similar (122 ± 6 vs. 116 ± 6 mg/dl; k/o vs. wt, P = NS). One month after surgery, 5-month-old k/o and wt female mice experienced the same degree of bone loss following ovariectomy (OVX). OVX induced a significant fourfold increase in the expression of the soluble CSF1 isoform (sCSF1) in the bones of wt mice while expression of mCSF1 was unchanged. These findings indicate that mCSF1 is essential for normal bone remodeling since, in its absence, BMD is increased. Membrane-bound CSF1 does not appear to be required for estrogen-deficiency bone loss while in contrast; our data suggest that sCSF1 could play a key role in this pathologic process. The reasons why mCSF1 k/o mice have hypertriglyceridemia are currently under study.
Keywords: CSF1 Isoform, Knock out, Estrogen deficiency, Osteoclasts, Hypertriglyceridemia
Introduction
Most secreted cytokines are synthesized as precursors that undergo intracellular post-translational processing to the mature, soluble form of the protein. These soluble molecules can act as autocrine, paracrine, and even endocrine signals, binding to cognate high-affinity receptors in target cells and initiating diverse intracellular events. As an exception to this general model, a few cytokines, as a consequence of alternative gene splicing, are synthesized as transmembrane proteins [1]. Available evidence suggests that soluble and membrane-bound isoforms of certain cytokines may have different biological activities. Thus, the soluble and membrane-bound isoforms of stem cell factor (SCF) show differences in intracellular signaling kinetics and in activation of effector cells [2–4].
CSF1, another hematopoietic growth factor with significant structural homology to SCF, also has both soluble (sCSF1) and membrane-bound (mCSF1) isoforms [1, 5, 6]. Like the SCF receptor, c-kit, the CSF1 receptor, c-fms is a receptor tyrosine kinase [1, 6]. Multiple human CSF1 mRNA species (4.0, 3.0, 2.3, 1.9 and 1.6 kb) are transcribed from the CSF1 gene [7–11] and molecular cloning of cDNAs derived from these transcripts has demonstrated that the size differences are due to alternative splicing in exon 6 and the alternative use of exons 9 and 10 [8–10]. Similar findings have been reported for the murine CSF1 gene [12, 13].
The different isoforms of CSF1 all bind to the same receptor but binding of the membrane form results in slower clearance compared to that of its soluble counterpart. TNF-alpha converting enzyme (TACE), which cleaves TNF, can also act on mCSF1 to produce the so called “shed” form of CSF1 [14]. Interestingly, TACE also cleaves the CSF1 receptor, c-fms, producing a soluble CSF1 receptor [15]. Matrix metalloproteinase-9 can also generate soluble c-fms but not the shed form of CSF1 [16].
Interleukin-34 (IL-34) was recently identified as a novel ligand for c-fms in addition to CSF1 [17]. IL-34 is a secreted dimeric protein with no homology to CSF1. Thus the CSF1/c-fms signaling family is more complex than previously thought with four ligands, mCSF1, sCSF1, shed CSF1, and IL-34 and possibly two receptors, transmembrane and soluble c-fms. It is not unreasonable to expect that interactions among these molecules mediate specific biological effects, and subserve different functions. Indeed recent findings have suggested that IL-34 and CSF1 bind to different domains of c-fms resulting in differing bioactivities and signal activation kinetics and signal strength [18]. With regard to the differing actions of mCSF1 and sCSF1, Friel et al. [19] reported that mCSF1 supported long-term proliferation of hematopoietic precursor cells as well as self-renewal of this population. In contrast, sCSF1 did not support a self-renewing population of progenitors but rather drove cells to differentiate into monocytes and macrophages. Douglass et al. [20] reported that mCSF1 promotes prolonged signal transduction in bone marrow-derived macrophages. Perhaps related to this latter observation, macrophages can kill mCSF1-positive tumor cells [21, 22] but not sCSF1 positive cells [23].
We have shown that human and mouse osteoblasts express mCSF1 in addition to sCSF1 and that PTH and TNF increase expression of mCSF1 mRNA and protein. We have also found that mCSF1 supports the formation of osteoclast-like cells in a dose-dependent manner in vitro and at physiologic concentrations acts synergistically with sCSF1 to induce osteoclast formation [24, 25]. Further, we have found that selective expression of mCSF1 alone in osteoblasts rescues the osteopetrotic phenotype of op/op mice [26]. Since osteoblast-derived sCSF1 does not induce osteoclast like cell (OCLs) formation in the absence of cell-cell contact, even if RANKL is added [27], it appears that mCSF1 may be required for normal osteoclastogenesis.
We recently reported that transgenic mice expressing human sCSF1, human mCSF1 or both (s/mCSF1) in osteoblasts are osteopenic compared to non-transgenic littermates. When analyzed by sex, sCSF1 and m/sCSF1 female animals but not mCSF1 female mice were found to have lower bone mass than their male littermates [28]. By breeding CSF1 isoform-selective transgenic mice to an op/op background, mice were generated in which a single CSF1 isoform was the only source of the cytokine (sCSF1op/op and mCSF1op/op). Interestingly, when compared to sham-OVX mice of the same genotype, OVX in sCSF1op/op mice led to a greater loss of spinal BMD (22.1%) than was seen in either mCSF1op/op mice (12.9%) or in wt animals (10.9%) [28]. These findings suggest that sCSF1 and mCSF1 serve non-redundant functions in bone and that sCSF1 may play a role in mediating estrogen-deficiency bone loss [28]. To begin to more rigorously define the role of the different CSF1 isoforms in bone, we engineered mice in which mCSF1 was selectively deleted.
Materials and methods
Generation and identification of mCSF1 k/o mice
The targeting construct for deletion of mCSF1 introduced a stop codon followed by a lox-p site into exon 6 at a position 5′ to the splice site for mCSF1. The stop codon was designed to include all three reading frames. A Neo cassette with flanking frt sites (lox-p-frt-Neo-frt) was introduced in intron 6 (Fig. 1a). The targeting vector was linearized by digestion with NotI and electroporated into the ES cell line W 9.5. Homologous recombinants were selected using G418 and identified by PCR (5′-primer: CGCGGTGGAGGCTATAACT; 3′-primer: GCTTGGCCACACTTAACACA). Positive clones underwent a second round of electroporation to introduce a Cre expression vector resulting in selective deletion of the splice acceptor site for the mCSF1 isoform as well as removal of the transmembrane domain. This generated a modified exon 6 that includes the splice acceptor site for sCSF1 followed by the proteolytic cleavage sites for sCSF1, followed by a stop codon. Thus, a mature functional sCSF1 is generated but no mCSF1. Screening after the second round of electroporation was accomplished by PCR (5′-primer: CCAAGCCTGATTGCAACTGCCTGT and 3′-primer TGACTAGCCTAGAACTCATCC). A positive ES clone from this second round of cloning was introduced into blastocysts. Chimeric mice with greater than 80% agouti coat were selected for out-breeding to C57BL/6 mice to yield founder animals. mCSF1 knock/out (k/o) mice were confirmed by PCR. The Yale Animal Care and Use Committee approved the use of animals in this study.
Fig. 1.

Strategy for generating conditional mCSF1 knock out mice. The targeting construct for deletion of mCSF1 introduces a stop codon followed by a lox-p site into exon 6 at a position 5′ to the splice acceptor site for mCSF1. The stop codon is designed to include all three reading frames. A Neo cassette with flanking frt sites (frt-Neo-frt-lox-p) is introduced into intron 6 (a). Excision of the Neo cassette with flp recombinase leads to a CSF1 allele that contains two lox-p sites, one in intron 6 and one, which is upstream of the splice acceptor site for mCSF1 (d). In animals bearing two of these floxed alleles (i.e. before cre-mediated recombination) both isoforms of CSF1 will be generated. Splicing to the mCSF1 acceptor site will yield a normal mCSF1 transcript (d). Splicing to the sCSF1 acceptor site will lead to generation of an mRNA that includes the sequences for the proteolyitc cleavage sites for sCSF1 followed by a stop codon. The protein product of this mRNA will yield normal mature sCSF1 since it contains the appropriate signals for proteolytic cleavage with the surrounding sequences (e). Recombination with cre leads to selective deletion of the splice acceptor site for the mCSF1 isoform as well as removal of the transmembrane domain (e). This leaves a modified exon 6 that includes the splice acceptor site for sCSF1 followed by the proteolytic sites for sCSF1, followed by a stop codon. Thus a mature functional sCSF-1 will be generated but no mCSF1 (e). b The wt allele. c The engineered allele before removal of the Neo cassette in vitro
Flow cytometry
Femurs and tibiae were isolated, the ends removed and the marrow cavity flushed with PBS buffer. The resultant suspension was centrifuged at 500×g for 5 min, the pelleted cells resuspended in erythorocyte lysis buffer and incubated at room temperature for 5 min. Cells were then counted using a hemocytometer. Cell survival rates ranged from 95 to 100% as determined by trypan blue exclusion. After counting, cells were centrifuged at 500×g for 5 min and resuspended in FACS staining buffer at a concentration of 10 × 106 cells/ml. All fluorescence-conjugated antibodies used in this study were purchased from eBioscience (San Diego, CA). After blocking with anti-Fc receptor (clone 2.4G2), F4/80-FITC (5 μg/ml) was added and the cells incubated for 30 min. After washing with staining buffer, cells were subjected to flow cytometry using a FACSCalibur (Becton Dickinson, NJ) and analyzed using Flowjo software (Tree Star Inc, San Carlos, CA). To determine mCSF1 expression in mononuclear bone marrow cells, the cells were re-suspended in PBS buffer containing 5% FBS and incubated with biotinylated mouse CSF1 antibody (BAF416, R&D Systems) for 45 min at 4°C. Cells were washed with PBS buffer containing 5% FBS, collected and stained with PE-conjugated avidin for 45 min at 4°C. Labeled cells were analyzed using a Coulter® Epics® Altra™ flow cytometer (Beckman Coulter, Fullerton, CA). The signal from 1 × 106 cells was measured. PE-conguated avidin without primary antibody was used as a control for background staining.
BMD measurements
In vivo BMD measurements were performed by dual-energy X-ray absorptiometry (DXA) using a PIXImus densitometer (Lunar Corporation, Madison, WI). Anesthetized mice (ketamine, 30 mg/kg body wt and xylazine, 3 mg/kg body wt given IP) were placed in the prone position and scans performed with a 1.270-mm-diameter collimator, 0.762-mm line spacing, 0.380-mm point resolution, and an acquisition time of 5 min. The spine window is a rectangle spanning a length of the spine from T1 to the beginning of the sacrum. The femur window encompasses the entire right femur of each mouse. The coefficient of variation for total body BMD is approximately 1.5%.
Bone histomorphometry
Histomorphometry was performed as previously reported [29–31]. At the time of sacrifice the tibiae were removed, stripped of soft tissue, and fixed in 70% ethanol. Tibiae were then dehydrated through graded ethanol, cleared in toluene, infiltrated with increasing concentrations of methylmethacrylate, and embedded in methylmethacrylate according to previously described methods [29, 30]. Analyses were performed on 5 μM thick sections stained with toluidine blue, pH 3.7 using a Nikon microscope interfaced with the Osteomeasure system software and hardware (Osteometrics, Atlanta, GA). Measurements were obtained in an area of cancellous bone that measured approximately 2.5 mm2, containing only secondary spongiosa, and located 0.5–2.5 mm distal to the epiphyseal growth cartilage. Longitudinal sections (5 μm thick) taken in the frontal plane through the cancellous bone of the proximal tibia were prepared with a Leica RM2165 microtome, mounted on chromalum coated glass slides, and stained with toluidine blue, pH 3.7. All indices were defined according to the American Society of Bone and Mineral Research histomorphometry nomenclature [32].
Co-culture of osteoblasts and osteoclast precursors
Osteoblasts were prepared by sequential collagenase-dispase digestion of neonatal calvariae isolated from k/o and wt mice as previously described [25, 33]. Cells were grown in alpha-MEM supplemented with 10% FBS, penicillin, streptomycin, L-glutamine and 20 mM HEPES (co-culture medium). Osteoblasts were allowed to attach overnight, followed by the addition of bone marrow cells (2 × 106 cells per well) prepared from either 6 to 8 week old k/o or wt mice. One milliliter of media, containing prostaglandin E2 (final concentration 10−6 M) and 1,25 (OH)2 D3 (final concentration 10−8 M), was added to each well and the media changed every other day. Cells were co-cultured for 7–9 days. The co-cultures were then fixed and stained for TRAP as previously described [30] and TRAP-positive multinucleated cells containing three or more nuclei counted as OCLs.
Determining the osteoclastogenic potential of bone marrow
To determine the effect of the genetic absence of mCSF1 on the marrow osteoclast progenitor pool, bone marrow was isolated from wt and k/o mice. Red blood cells were lysed on ice for 5 min. Mononuclear cells were centrifuged, washed with PBS, and seeded in phenol red-free alpha-MEM containing 10% FBS and CSF1 (final concentration, 100 ng/ml). Twenty-four hours later, non-adherent cells were harvested, layered onto an equivalent volume of Ficoll-Hypaque, and centrifuged at 707×g for 20 min. Cells at the interface were collected, washed in PBS twice, and plated at a final concentration of 1.3 × 106 cells/35-mm dish. Cells were cultured in phenol red-free alpha-MEM containing 10% FBS, 75 ng/ml CSF1, 75 ng/ml RANKL, 10 ng/ml TNF-α, and 50 μg/ml AICAR, which has been previously reported to induce osteoclastogenesis [34]. The medium was changed every 3 days. After 6 days in culture, cells were fixed and stained using a Sigma-Aldrich Acid Phosphatase kit.
Quantifying the number of nuclei per osteoclast
Freshly isolated osteoclasts from 1 day old wt and k/o mice were plated onto gridded coverslips for 1.5 h in alpha-MEM containing 10% FBS. The FBS concentration was then reduced to 2% for an additional 1.5 h. Coverslips were washed, and cells with three or more nuclei counted as osteoclasts. Eighteen k/o and eighteen wt osteoclasts were analyzed.
Ovariectomy
Ovariectomy (OVX) was accomplished through a paralumbar incision in anesthetized animals as previously reported [35]. The ovarian bursa opposite each ovarian hilum was incised, the hilum exposed and clamped and the ovary removed. Sham ovariectomy animals underwent anesthesia and the paralumbar incision without removal of the ovaries. Four weeks after ovariectomy, BMD was determined by DXA.
Quantifying transcript levels for sCSF1, mCSF1 and IL-34 in bone
Expression of mCSF1, sCSF1 and IL-34 transcripts in bone isolated from wt and k/o mice was determined by quantitative RT-PCR (qRT-PCR) using a DNA Engine Opticon 2 System from M.J. Research (Waltham, MA) and the Brilliant® QRT-PCR Master Mix Kit from Stratagene (Lo Jolla, CA). The thermal cycling conditions consisted of an initial 50°C for 30 min and a denaturation step at 95°C for 5 min, 40 cycles at 95°C for 30 s, 60°C for 1 min, and 72°C for 30 s. The primers were mCSF1: forward 5′-TTTGCTAAGTGCTCTAGCCGAG-3′, reverse, 5′-CCACTTCCACTTGTAGAACAGGAGGCC-3′; sCSF1: forward 5′-GCTTGAGGGCAAGAGAAGTACC-3′, reverse, 5′-ATCCTTTCTATACTGGCAG-3′; IL-34: forward 5′-CTTTGGGAAACGAGAATTTGGAGA-3′, reverse, 5′-GCAATCCTGTAGTTGATGGGGAAG-3′.
Results
Confirmation of isoform-selective deletion of mCSF1
To confirm that our engineering strategy resulted in isoform-selective deletion of mCSF1 in vivo, RNA was isolated from tibiae of 8-week-old wt and k/o mice. Isoform-specific qRT-PCR demonstrated the absence of mCSF1 transcripts in k/o mice with no significant difference in the level of expression of sCSF1 or IL-34 in k/o compared to wt mice. The expected amplicon sizes for IL-34, sCSF1 and mCSF1 are 141, 371 and 152 bp, respectively (Fig. 2).
Fig. 2.

Isoform specific RT-PCR of bone mRNA demonstrates the absence of mCSF1 transcripts in mCSF1 k/o mice. A 2% agarose gel was used for this analysis. MW markers are shown in the far right lane
To determine whether selective deletion of mCSF1 altered the expression of sCSF1 protein expression, neonatal calvariae osteoblasts were cultured from k/o mice or wt littermates. Conditioned media from these cells was analyzed by western blot to determine the level of sCSF1 expression. As an additional control, conditioned media from by MC3T3E1 cells were used in the same western analysis. As shown in Fig. 3, the amount of endogenous sCSF1 expressed by k/o cells was no different from that in wt cultures. In general, the primary cells secreted more sCSF1 than did the MC3T3E1 cell line. The slight difference in molecular weight probably reflects differences in post-translational glycosylation in primary cells vs. the cultured cell line. These data further establish that genetic deletion of mCSF1 in vivo did not alter expression of endogenous sCSF1.
Fig. 3.
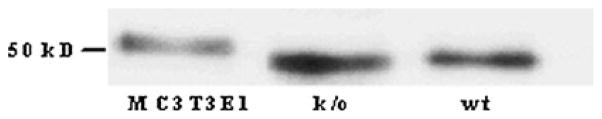
Western blot for sCSF1 using media conditioned by osteoblasts from either wt or k/o mice. Media conditioned for 24 h by MC3T3E1 cells (left lane), primary osteoblasts prepared from neonatal k/o mice (middle lane) or from wt littermates (right lane) was concentrated, desalted, and analyzed by Western blot using a polyclonal antibody to CSF1
To determine whether mCSF1 protein was functionally absent, we isolated macrophages from k/o mice and wt littermates and analyzed cell surface expression of mCSF1. Because mCSF1 is highly expressed by macrophages and these cells can be easily isolated as a homogenously pure population, they were chosen for analysis. In addition, these cells were freshly isolated from whole blood and therefore, did not need to be plated and trypsinized prior to analysis. Trypsinization could potentially lead to a loss of mCSF1 expression on the cell surface. As shown in Fig. 4, flow cytometric analysis of macrophages freshly isolated from wt littermates expressed mCSF1 as demonstrated by the histogram delimited by the black line. In contrast the k/o cells showed no staining above background.
Fig. 4.

Flow cytometric analysis demonstrating the absence of mCSF1 protein expression in macrophages isolated from the k/o mice. Macrophages prepared from k/o mice (tinted) or from wt littermates (black) were analyzed by flow cytometry using a polyclonal antibody to CSF1. There is no staining of the k/o cells
mCSF1 k/o animals have normal serum calcium values and normal circulating levels of blood cells
Mean serum calcium levels in wt and k/o animals were not significantly different (8.91 ± 0.1 vs. 9.0 ± 0.1 mg/dl; wt vs. k/o n = 7 per group, P = NS). Similarly, circulating levels of blood cells were similar in the wt and k/o mice. Mean values for hemoglobin were 15.0 versus 14.2 g/dl, for neutrophils: 1204 versus 629 mm3, for lymphocytes: 4587 versus 5949 mm3 and for monocytes: 58 versus 92 mm3; wt versus k/o respectively.
k/o animals have elevated serum triglycerides
Since mCSF1 is highly expressed on vascular endothelial cells, and macrophages play an important role in lipid metabolism, particularly in the vascular tree, we decided to determine whether the absence of mCSF1 would affect blood lipids, particularly since it has been previously reported that op/op mice (which lack both CSF1 isoforms) when bred to apoE deficient mice have an increase in serum cholesterol levels [36]. Interestingly, k/o animals had significantly elevated levels of serum triglycerides as compared to wt animals, (123 ± 7 vs. 88 ± 32 mg/dL, n = 10 vs. n = 8, k/o vs. wt; P = <0.01). In contrast, serum cholesterol levels were similar in the two groups (122 ± 6 vs. 116 ± 6 mg/dl, n = 10 vs. n = 8, k/o vs. wt; P = NS).
k/o mice have increased BMD
Twenty-two, 3-month old k/o and age- and sex-matched wt littermates were analyzed by DXA as previously described [37]. As shown in Fig. 5, k/o mice had a 15.9% greater mean spinal BMD than wt controls (P < 0.01). Total BMD was also significantly increased by 6.8% (P < 0.05). There was a similar trend in femoral BMD, which was 6.9% higher in the k/o animals, although this trend did not reach statistical significance. When this analysis was repeated in 2-month-old animals to determine if there was an age-dependent effect of deleting mCSF1, similar results were obtained. Spinal BMD was significantly increased by 8.9% (P < 0.01) and total BMD was increased by 7.8% (P < 0.01). As was the case with the 3-month old animals, femoral BMD was not significantly different in two groups of 2-month-old animals.
Fig. 5.

Femur spine and total body BMD determined by DXA (PIXImus) in 22 wt (12 male and 10 female) and 22 k/o (11 male and 11 female) mice. Mean values at the three sites were 734 ± 27 versus 784 ± 20; 552 ± 15 versus 641 ± 12 and 489 ± 12 versus 523 ± 6, g/cm2 × 103; wt versus ko for femur, spine and total body respectively; *P < 0.05, **P < 0.01 compared to wt. Results are expressed as M ± SEM
Bone histomorphometry
Since we had previously reported that mCSF1 could induce osteoclastogenesis in vitro, we anticipated that there would be a reduction in osteoclast number and/or activity in the k/o animal. Surprisingly, as shown in Table 1, there were no significant differences in osteoclast parameters in these animals although there was a trend towards lower numbers of osteoclasts in the k/o mice.
Table 1.
Bone histomorphometry in wild type and mCSF1 k/o mice
| N | Sex (M/F) | ObS/BS (%) | ObS/OS (%) | NOb/TAR (%) | NOb/Bpm (#) | OcS/BS (%) | NOc/TAR (#) | NOc/Bpm (#) | |
|---|---|---|---|---|---|---|---|---|---|
| k/o | 9 | 4/5 | 24.3 ± 2.4 | 66.4 ± 3.5 | 731.0 ± 35.4 | 43.0 ± 4.1 | 10.3 ± 1.2 | 75.3 ± 9.2 | 4.4 ± 0.6 |
| wt | 8 | 5/3 | 22.3 ± 3.0 | 63.0 ± 4.6 | 658.0 ± 74.3 | 41.7 ± 5.5 | 12.4 ± 1.5 | 85.9 ± 8.9 | 5.4 ± 0.8 |
Osteoblasts isolated from mCSF1 k/o mice fail to support normal osteoclastogenesis in vitro
In view of our earlier in vitro and in vivo work demonstrating a role for mCSF1 in osteoclastogenesis and the trend towards reduced numbers of osteoclasts in the k/o mice on histomorphometric analysis, we next investigated the ability of osteoblasts isolated from mCSF1 k/o mice to support osteoclastogenesis in vitro. Despite secreting normal amounts of sCSF1, as shown in Fig. 6, osteoblasts from k/o mice exhibited a reduced ability to support osteoclastogenesis. As shown in Fig. 6a, when k/o osteoblasts were co-cultured with wt bone marrow in the presence of 1,25(OH)2 D3 and prostaglandin PGE2, the number of osteoclast-like cells formed, as assessed by TRAcP, staining was significantly reduced when compared to co-cultures using wt osteoblasts and wt marrow. This is quantified in Fig. 6b, which shows that there was a significant reduction in the number of osteoclasts formed in co-cultures using osteoblasts from k/o mice and wt marrow. There was no significant difference in the number of osteoclasts formed when co-cultures were performed using wt osteoblasts and marrow from k/o mice as compared to the number of osteoclasts formed when wt marrow was cultured with wt osteoblasts (data not shown). These data clearly indicate that the defect resides in osteoblasts from the k/o mice.
Fig. 6.

a TRAP stain of co-cultures prepared using wt marrow and osteoblasts isolated from k/o (plates on the left) or wt (plates on the right) mice. b Numbers of TRAP-positive multinucleated OCLs formed in the co-cultures. The mean values were 95 ± 20 versus 32 ± 4 per well; wt vs. k/o, respectively. The results shown are the M ± SEM of three independent experiments with each determination performed in duplicate. **P < 0.01. c Numbers of TRAP-positive multinucleated OCLs generated for each genotype from osteoclast precursors cultured under osteoclastogenic conditions. d Mean values for the experiment shown panel C were 12.6 ± 0.8 versus 4.9 ± 0.4 cells per 130 mm2; n = 25 for both genotypes; ***P < 0.001. e Number of nuclei per cell; 8.3 ± 1.1 versus 5.8 ± 0.6, wt versus k/o; n = 18 for both genotypes; #P = 0.06
mCSF1 knock out mice have reduced marrow osteoclastogenic capacity
To further explore the mechanism underlying the impaired osteoclastogenesis in the k/o mice and to determine whether the genetic absence of mCSF1 in other cells impacts osteoclastogenesis in these animals, osteoclast precursors were isolated from wt and k/o animals and cultured in vitro in the absence of osteoblasts but in the presence of osteoclastogenic cytokines, as we have previously published [34]. As shown in Fig. 6c and d, not only were there fewer numbers of osteoclasts, but the number of large multinucleated cells generated was also significantly less when the k/o marrow was cultured. To determine the impact of adding AICAR to the cytokine mix used to drive osteoclast differentiation in these experiments, the studies were repeated it its presence and absence. The numbers of TRAP-positive multinucleated OCLs (per 130 mm2 area of culture dish) generated from wt and k/o osteoclast precursors cultured with AICAR, CSF1, RANKL, and TNF-α, were 12.6 ± 0.8 and 4.9 ± 0.4 cells respectively (P < 0.001). Without the addition of AICAR these numbers were 10.4 ± 0.6 vs. 3.5 ± 0.5 cells, respectively (P < 0.001). This suggests an intrinsic defect in the marrow of these animals. Whether this is a reflection of the long-standing absence of mCSF1 that affects osteoclast precursor pools or whether this is a cell autonomous effect is not yet clear. We have also quantified the number of nuclei in mature osteoclasts freshly isolated from k/o mice as compared to wt animals (Fig. 6e). Consistent with our in vitro data, there was a trend toward a fewer number of nuclei per cell in osteoclasts isolated from the k/o animals (P = 0.06).
Effect of OVX in k/o mice
To examine the impact of the genetic absence of mCSF1 on estrogen-deficiency bone loss, 19–22 week-old k/o and wt female mice underwent OVX or sham-OVX. The results from two independent experiments are summarized in Fig. 7 in which a total of 47 mice were studied. One month after surgery, femoral, spinal and total body BMD (determined by DXA) were reduced by 7.6, 12.6 and 5.0%, respectively in OVX-wt mice as compared to sham-OVX wt mice. OVX k/o mice showed very comparable 7.5, 12.3 and 4.4% reductions in femoral, spinal and total body BMD respectively compared to sham-OVX k/o mice. There were no significant differences in the extent of bone loss following OVX in k/o and wt mice.
Fig. 7.

Percent change in BMD in k/o and wt mice 1 month following OVX. BMD was reduced to similar extent in k/o and wt mice after OVX. The changes in femur, spine and total body BMD in wt mice compared so sham OVX wt mice were: −7.6, −12.6 and −5, respectively. Similar changes were observed in the OVX k/o mice compared to their sham OVX controls with declines of −7.5, −12.3 and −4.4% at the femur, spine and total body (P = NS for the difference by genotype at each site.)
OVX selectively up-regulates sCSF1 production in vivo
We have recently reported that estrogen-withdrawal selectively increases the expression of sCSF1 in primary murine osteoblasts isolated only from female neonatal pups while the level of mCSF1 expression is either unchanged or changed minimally [28]. We wondered if this might be the explanation for our finding in the OVX k/o animals and in particular whether estrogen withdrawal led to selective upregulation of sCSF1 in vivo. We therefore analyzed CSF-1 isoform expression after OVX in wt mice. Tibiae of OVX or sham-OVX 4 month-old wt mice were isolated 4 weeks after surgery, RNA isolated and analyzed by real-time PCR. Mean data from three separate experiments are shown in Fig. 8. There was significant and selective up-regulation of sCSF1 but not mCSF1 expression 4 weeks after estrogen withdrawal.
Fig. 8.

Effect of OVX on expression of the two CSF-1 isoforms in bone. Tibiae and femurs were isolated 1 month after surgery, the marrow was flushed, the bone snap frozen and pulverized while frozen, and RNA extracted. RNA was analyzed by isoform-specific real-time PCR. Mean fold induction in the sCSF1 isoform and the mCSF1 isoform were 4.0 ± 1.2 (*P < 0.05), and 1.4 ± 0.3 (P = NS)
Discussion
This is the first report of an isoform-selective deletion of any known cytokine with direct effects on skeletal metabolism. CSF1 has a critically important role in the skeleton but the role of the two CSF1 isoforms in bone remains poorly understood. Work from our lab has suggested different functions for these isoforms. In particular, we have reported that while targeted overexpression of mCSF1 in osteoblasts completely rescues the skeletal phenotype of the op/op mouse, the sCSF1 isoform does not [26]. We also reported that the mCSF1 transgenic mice, despite the normal skeletal phenotype, have a defect in reproductive efficiency [35]. In addition, we have reported that estrogen withdrawal selectively upregulates the sCSF1 isoform while not affecting the expression of mCSF1 in vitro [28]. In the aggregate, these data suggest but do not prove a non-redundant role for these two isoforms. Others have used a similar approach to ours in exploring the roles of the two CSF1 isoforms. Stanley and co-workers engineered op/op mice in which selective expression of mCSF1 or sCSF1 was controlled by a portion of the 5′ regulatory region of the CSF1 gene. In contrast to our findings, they reported that sCSF1 completely rescues the phenotype of the op/op mouse while mCSF1 does not [38, 39], although detailed histomorphometric analyses were not provided.
The most significant observations in the current study are that the selective deletion of mCSF1 does not affect hematopoiesis as judged by peripheral blood counts, but does significantly alter bone mass. Animals had higher total as well as spinal BMD. Total BMD is largely a cortical measurement, while spine BMD is primarily a trabecular determination, suggesting that there is a global effect on the skeleton in these animals. Although there was a trend towards higher femur BMD in both 2- and 3-month-old animals, this did not reach statistical significance. Our histomorphometric data do not provide significant insights into the mechanism by which this increase in bone mass occurs. However, the findings of our in vitro osteoclastogenesis assays support the conclusion that in the absence of mCSF1, there is a reduced efficiency of osteoclastogenesis. In particular, both the co-culture and osteoclast progenitor experiments demonstrated significantly less osteoclastogenic potential in the marrow of mCSF1 k/o mice compared to wt animals. It may be that in the resting state in vivo, this defect is less apparent, but under circumstances where osteoclastogenesis is augmented (such as in vitro conditions where osteoclastogenesis is driven by exogenous stimulators like 1,25(OH)2vitamin D and PGE2), this defect is more evident. It would be of interest to determine whether in vivo stimulation of osteoclastogenesis with hormones such as PTH or RANK ligand would uncover defects in osteoclastogenesis in these animals.
The finding of hypertriglyceridemia in the k/o mice is surprising. It is known that mCSF1 is highly expressed in vascular endothelial cells. This suggests that one of the important mechanisms by which macrophages home to the vascular tree may be through expression of mCSF1 in these cells. Whether the genetic absence of mCSF1 leads to altered lipid metabolism in the vascular tree and/or affects lipid metabolism in the liver is not clear from our data, but it is of interest that these animals had selective elevations in triglycerides with normal serum cholesterol measurements. This clearly warrants further study and may suggest mCSF1 as a target for treatment of atherosclerosis.
Previous studies have highlighted an important role for CSF1 in mediating estrogen-deficiency bone loss. Cenci et al. [40] reported that a neutralizing antibody to CSF1 markedly attenuated ovariectomy-induced bone loss in mice. This same group of investigators reported that estrogen withdrawal resulted in increased expression of sCSF1 in marrow stromal cells [40]. In contrast, Lea et al. [41, 42] have reported that the mCSF1 was selectively upregulated in the marrow of rats following ovariectomy. We have recently reported that estrogen withdrawal selectively upregulates sCSF1 in cultured primary mouse calvarial cells as well as in a murine osteoblast cell line [28].
The availability of the k/o mice has provided us with an opportunity to directly examine the contribution of this isoform to estrogen-deficiency bone loss. The data summarized in Fig. 7 clearly indicate that the genetic absence of mCSF1 does not attenuate estrogen-deficiency bone loss. When we examined the expression of sCSF1 in the bones of wt mice, we found that there was upregulation of sCSF1 following OVX. We have also recently reported that targeted transgenic over-expression of sCSF1 in op/op mice leads to exaggerated rates of bone loss following ovariectomy [28]. In the aggregate our findings and those of the Pacifici group cited above, support the hypothesis that sCSF1 is principally involved in estrogen-deficiency bone loss. The absolute importance of sCSF1 to estrogen-deficiency bone loss vis á vis other pro-resorptive cytokines, remains uncertain and will be best addressed by selective isoform deletion of sCSF1 in vivo.
In summary, we have found that selective deletion of mCSF1 in vivo leads to increased bone mass and reduced osteoclastogenic potential of marrow precursors. A surprising and as yet not well-understood finding is hypertriglyceridemia in these animals. The fact that our animals evidenced a skeletal phenotype despite normal levels of sCSF1 expression indicates that there are functions for mCSF1 that cannot be fully compensated for by the soluble isoform. It was recently reported that IL-34 is an endogenous ligand for c-fms [17]. Since levels of IL-34 are presumably unaffected in our k/o mice, given the unchanged transcript levels, one can conclude that IL-34 also cannot compensate for the genetic absence of mCSF1. These data together with our prior reports on differences in op/op mice engineered to selectively overexpress sCSF1 and mCSF1 in osteoblasts, demonstrate that there are non-redundant functions for these two isoforms in vivo.
Determining if there are differences in the cell signaling cascades entrained by mCSF1 and sCSF1 is an area that warrants further investigation and may help to explain the in vivo differences we have observed.
Acknowledgments
This work was supported by RO1 DE012459 (to KLI) as well as by the resources of the Yale Core Center for Musculoskeletal Disorders, a NIAMS-funded P30 Core Center award (P30AR46032).
Contributor Information
Gang-Qing Yao, Email: gang-qing.yao@yale.edu, Section of Comparative Medicine, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520-8016, USA.
Jian-Jun Wu, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT 06520, USA.
Nancy Troiano, Department of Orthopaedics, Yale University School of Medicine, New Haven, CT 06520, USA.
Mei-Ling Zhu, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT 06520, USA.
Xiao-Yan Xiao, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT 06520, USA.
Karl Insogna, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT 06520, USA.
References
- 1.Massague J, Pandiella A. Membrane-anchored growth factors. Annu Rev Biochem. 1993;62:515–541. doi: 10.1146/annurev.bi.62.070193.002503. [DOI] [PubMed] [Google Scholar]
- 2.Gommerman JL, Sittaro D, Klebasz NZ, Williams DA, Berger SA. Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood. 2000;96:3734–3742. [PubMed] [Google Scholar]
- 3.Caruana G, Ashman LK, Fujita J, Gonda TJ. Responses of the murine myeloid cell line FDC-P1 to soluble and membrane-bound forms of steel factor (SLF) Exp Hematol. 1993;21:761–768. [PubMed] [Google Scholar]
- 4.Friel J, Heberlein C, Itoh K, Ostertag W. Role of the stem cell factor (SCF) receptor and the alternative forms of its ligand (SCF) in the induction of long-term growth by stroma cells. Leukemia. 1997;11(Suppl 3):493–495. [PubMed] [Google Scholar]
- 5.Bazan JF. Genetic and structural homology of stem cell factor and macrophage colony-stimulating factor. Cell. 1991;65:9–10. doi: 10.1016/0092-8674(91)90401-j. [DOI] [PubMed] [Google Scholar]
- 6.Miyazawa K, Williams DA, Gotoh A, Nishimaki J, Broxmeyer HE, Toyama K. Membrane-bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c-kit gene-encoded protein than its soluble form. Blood. 1995;85:641–649. [PubMed] [Google Scholar]
- 7.Cerretti DP, Wignall J, Anderson D, Tushinski RJ, Gallis BM, Stya M, Gillis S, Urdal DL, Cosman D. Human macrophage-colony stimulating factor: alternative RNA and protein processing from a single gene. Mol Immunol. 1988;25:761–770. doi: 10.1016/0161-5890(88)90112-5. [DOI] [PubMed] [Google Scholar]
- 8.Kawasaki ES, Ladner MB, Wang AM, Van Arsdell J, Warren MK, Coyne MY, Schweickart VL, Lee MT, Wilson KJ, Boosman A, et al. Molecular cloning of a complementary DNA encoding human macrophage- specific colony-stimulating factor (CSF-1) Science. 1985;230:291–296. doi: 10.1126/science.2996129. [DOI] [PubMed] [Google Scholar]
- 9.Ladner MB, Martin GA, Noble JA, Nikoloff DM, Tal R, Kawasaki ES, White TJ. Human CSF-1: gene structure and alternative splicing of mRNA precursors. Embo J. 1987;6:2693–2698. doi: 10.1002/j.1460-2075.1987.tb02561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong GG, Temple PA, Leary AC, Witek-Giannotti JS, Yang YC, Ciarletta AB, Chung M, Murtha P, Kriz R, Kaufman RJ, et al. Human CSF-1: molecular cloning and expression of 4-kb cDNA encoding the human urinary protein. Science. 1987;235:1504–1508. doi: 10.1126/science.3493529. [DOI] [PubMed] [Google Scholar]
- 11.Pampfer S, Tabibzadeh S, Chuan FC, Pollard JW. Expression of colony-stimulating factor-1 (CSF-1) messenger RNA in human endometrial glands during the menstrual cycle: molecular cloning of a novel transcript that predicts a cell surface form of CSF-1. Mol Endocrinol. 1991;5:1931–1938. doi: 10.1210/mend-5-12-1931. [DOI] [PubMed] [Google Scholar]
- 12.Ladner MB, Martin GA, Noble JA, Wittman VP, Warren MK, McGrogan M, Stanley ER. cDNA cloning and expression of murine macrophage colony-stimulating factor from L929 cells. Proc Natl Acad Sci USA. 1988;85:6706–6710. doi: 10.1073/pnas.85.18.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawasaki ES, Ladner MB. Molecular biology of macrophage colony-stimulating factor. Immunol Ser. 1990;49:155–176. [PubMed] [Google Scholar]
- 14.Horiuchi K, Miyamoto T, Takaishi H, Hakozaki A, Kosaki N, Miyauchi Y, Furukawa M, Takito J, Kaneko H, Matsuzaki K, Morioka H, Blobel CP, Toyama Y. Cell surface colony-stimulating factor 1 can be cleaved by TNF-alpha converting enzyme or endocytosed in a clathrin-dependent manner. J Immunol. 2007;179:6715–6724. doi: 10.4049/jimmunol.179.10.6715. [DOI] [PubMed] [Google Scholar]
- 15.Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol. 2001;166:1583–1589. doi: 10.4049/jimmunol.166.3.1583. [DOI] [PubMed] [Google Scholar]
- 16.Rao Q, Zheng GG, Li G, Lin YM, Wu KF. Membrane-bound macrophage colony-stimulating factor mediated auto-juxtacrine downregulates matrix metalloproteinase-9 release on J6-1 leukemic cell. Exp Biol Med (Maywood, NJ) 2004;229:946–953. doi: 10.1177/153537020422900912. [DOI] [PubMed] [Google Scholar]
- 17.Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D, Hollenbaugh D, Linnemann T, Qin M, Wong J, Chu K, Doberstein SK, Williams LT. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 18.Chihara T, Suzu S, Hassan R, Chutiwitoonchai N, Hiyoshi M, Motoyoshi K, Kimura F, Okada S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010;17:1917–1927. doi: 10.1038/cdd.2010.60. [DOI] [PubMed] [Google Scholar]
- 19.Friel J, Heberlein C, Geldmacher M, Ostertag W. Diverse isoforms of colony-stimulating factor-1 have different effects on the development of stroma-dependent hematopoietic cells. J Cell Physiol. 2005;204:247–259. doi: 10.1002/jcp.20291. [DOI] [PubMed] [Google Scholar]
- 20.Douglass TG, Driggers L, Zhang JG, Hoa N, Delgado C, Williams CC, Dan Q, Sanchez R, Jeffes EW, Wepsic HT, Myers MP, Koths K, Jadus MR. Macrophage colony stimulating factor: not just for macrophages anymore! A gateway into complex biologies. Int Immunopharmacol. 2008;8:1354–1376. doi: 10.1016/j.intimp.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 21.Dan Q, Sanchez R, Delgado C, Wepsic HT, Morgan K, Chen Y, Jeffes EW, Lowell CA, Morgan TR, Jadus MR. Non-immunogenic murine hepatocellular carcinoma Hepa1-6 cells expressing the membrane form of macrophage colony stimulating factor are rejected in vivo and lead to CD8? T-cell immunity against the parental tumor. Mol Ther. 2001;4:427–437. doi: 10.1006/mthe.2001.0477. [DOI] [PubMed] [Google Scholar]
- 22.Jadus MR, Irwin MC, Irwin MR, Horansky RD, Sekhon S, Pepper KA, Kohn DB, Wepsic HT. Macrophages can recognize and kill tumor cells bearing the membrane isoform of macrophage colony-stimulating factor. Blood. 1996;87:5232–5241. [PubMed] [Google Scholar]
- 23.Graf MR, Jadus MR, Hiserodt JC, Wepsic HT, Granger GA. Development of systemic immunity to glioblastoma multiforme using tumor cells genetically engineered to express the membrane-associated isoform of macrophage colony-stimulating factor. J Immunol. 1999;163:5544–5551. [PubMed] [Google Scholar]
- 24.Yao GQ, Sun BH, Insogna KL, Weir EC. Nuclear factor-kappaB p50 is required for tumor necrosis factor-alpha-induced colony-stimulating factor-1 gene expression in osteoblasts. Endocrinology. 2000;141:2914–2922. doi: 10.1210/endo.141.8.7592. [DOI] [PubMed] [Google Scholar]
- 25.Yao GQ, Sun B, Hammond EE, Spencer EN, Horowitz MC, Insogna KL, Weir EC. The cell-surface form of colony-stimulating factor-1 is regulated by osteotropic agents and supports formation of multinucleated osteoclast- like cells. J Biol Chem. 1998;273:4119–4128. doi: 10.1074/jbc.273.7.4119. [DOI] [PubMed] [Google Scholar]
- 26.Yao GQ, Wu JJ, Sun BH, Troiano N, Mitnick MA, Insogna K. The cell surface form of colony-stimulating factor-1 is biologically active in bone in vivo. Endocrinology. 2003;144:3677–3682. doi: 10.1210/en.2002-221071. [DOI] [PubMed] [Google Scholar]
- 27.Itoh K, Udagawa N, Matsuzaki K, Takami M, Amano H, Shinki T, Ueno Y, Takahashi N, Suda T. Importance of membrane- or matrix-associated forms of M-CSF and RANKL/ODF in osteoclastogenesis supported by SaOS-4/3 cells expressing recombinant PTH/PTHrP receptors. J Bone Miner Res. 2000;15:1766–1775. doi: 10.1359/jbmr.2000.15.9.1766. [DOI] [PubMed] [Google Scholar]
- 28.Yao GQ, Wu JJ, Ovadia S, Troiano N, Sun BH, Insogna K. Targeted overexpression of the two colony-stimulating factor-1 isoforms in osteoblasts differentially affects bone loss in ovariectomized mice. Am J Physiol. 2009;296:E714–E720. doi: 10.1152/ajpendo.90631.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Insogna KL, Stewart AF, Vignery AM, Weir EC, Namnum PA, Baron RE, Kirkwood JM, Deftos LM, Broadus AE. Biochemical and histomorphometric characterization of a rat model for humoral hypercalcemia of malignancy. Endocrinology. 1984;114:888–896. doi: 10.1210/endo-114-3-888. [DOI] [PubMed] [Google Scholar]
- 30.Baron R, Vignery A, Neff L, Silvergate A, Santa Maria A. Bone histomorphometry. In: Recker R, editor. Techniques and interpretation. CRC Press; Boca Raton: 1983. pp. 31–32. [Google Scholar]
- 31.Knopp E, Troiano N, Bouxsein M, Sun BH, Lostritto K, Gundberg C, Dziura J, Insogna K. The effect of aging on the skeletal response to intermittent treatment with parathyroid hormone. Endocrinology. 2005;146:1983–1990. doi: 10.1210/en.2004-0770. [DOI] [PubMed] [Google Scholar]
- 32.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 33.Insogna KL, Sahni M, Grey AB, Tanaka S, Horne WC, Neff L, Mitnick M, Levy JB, Baron R. Colony-stimulating factor-1 induces cytoskeletal reorganization and c- src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest. 1997;100:2476–2485. doi: 10.1172/JCI119790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itokowa T, Zhu ML, Troiano N, Bian J, Kawano T, Insogna K. Osteoclasts lacking Rac2 have defective chemotaxis and resorptive activity. Calcif Tissue Int. 2010;88:75–86. doi: 10.1007/s00223-010-9435-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovadia S, Insogna K, Yao GQ. The cell-surface isoform of colony stimulating factor 1 (CSF1) restores but does not completely normalize fecundity in CSF1-deficient mice. Biol Reprod. 2006;74:331–336. doi: 10.1095/biolreprod.105.045047. [DOI] [PubMed] [Google Scholar]
- 36.Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci USA. 1995;92:8264–8268. doi: 10.1073/pnas.92.18.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao GQ, Wu JJ, Troiano N, Insogna K. Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab. 29:141–148. doi: 10.1007/s00774-010-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryan GR, Dai XM, Dominguez MG, Tong W, Chuan F, Chisholm O, Russell RG, Pollard JW, Stanley ER. Rescue of the colony-stimulating factor 1 (CSF-1)-nullizygous mouse (Csf1(op)/Csf1(op)) phenotype with a CSF-1 transgene and identification of sites of local CSF-1 synthesis. Blood. 2001;98:74–84. doi: 10.1182/blood.v98.1.74. [DOI] [PubMed] [Google Scholar]
- 39.Dai XM, Zong XH, Sylvestre V, Stanley ER. Incomplete restoration of colony-stimulating factor 1 (CSF-1) function in CSF-1-deficient Csf1op/Csf1op mice by transgenic expression of cell surface CSF-1. Blood. 2004;103:1114–1123. doi: 10.1182/blood-2003-08-2739. [DOI] [PubMed] [Google Scholar]
- 40.Cenci S, Weitzmann MN, Gentile MA, Aisa MC, Pacifici R. M-CSF neutralization and egr-1 deficiency prevent ovariectomy-induced bone loss. J Clin Invest. 2000;105:1279–1287. doi: 10.1172/JCI8672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lea CK, Sarma U, Flanagan AM. Macrophage colony stimulating-factor transcripts are differentially regulated in rat bone-marrow by gender hormones. Endocrinology. 1999;140:273–279. doi: 10.1210/endo.140.1.6451. [DOI] [PubMed] [Google Scholar]
- 42.Sarma U, Edwards M, Motoyoshi K, Flanagan AM. Inhibition of bone resorption by 17beta-estradiol in human bone marrow cultures. J Cell Physiol. 1998;175:99–108. doi: 10.1002/(SICI)1097-4652(199804)175:1<99::AID-JCP11>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]