

Abstract
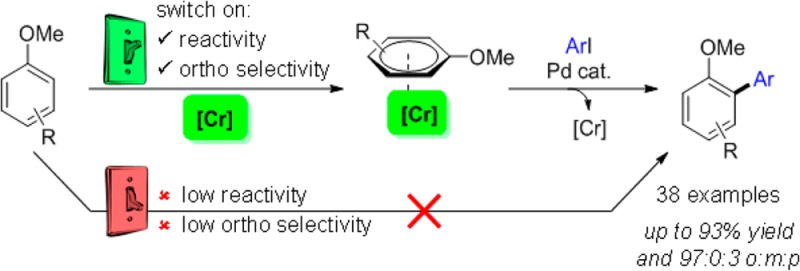
Current approaches to achieve site selectivity in the C–H activation of arenes involve the use of directing groups or highly electron-poor arenes. In contrast, simple arenes, such as anisole, are characterized by poor reactivity and selectivity. We report that π-complexation to a Cr(CO)3 unit enhances the reactivity of anisoles providing an unprecedented ortho-selective arylation. This mild methodology can be used for the late stage functionalization of bioactive compounds containing the anisole motif, allowing the construction of novel organic scaffolds with few synthetic steps.
Introduction
Over the past decade, C–H arylation has emerged as a powerful methodology for the synthesis of biaryls,1 important motifs in pharmaceuticals, agrochemicals, organic materials, and natural products.2 Arenes containing a wide variety directing groups can now be readily ortho-arylated.3 Some progress has also been made in recent years toward achieving selective meta- and para-arylation.4,5 However, C–H arylation of so-called simple arenes,6 that is arenes without a directing group, remains a challenge: (1) their low reactivity results in large amounts of the arene being generally required and (2) useful regioselectivity is rarely obtained, thus limiting these reactions to symmetrical substrates. The only exceptions are highly electron-deficient substrates, such as polyfluorobenzenes, where the more acidic C–H bonds can be readily activated.7 In this context, the efficient arylation of anisole, a ubiquitous motif in biologically active compounds (Scheme 1a),8 still represents a remarkable reactivity and selectivity challenge (Scheme 1b).9
Scheme 1. Regioselective Ortho-Arylation of Anisole via Arene–Metal π-Complexation.
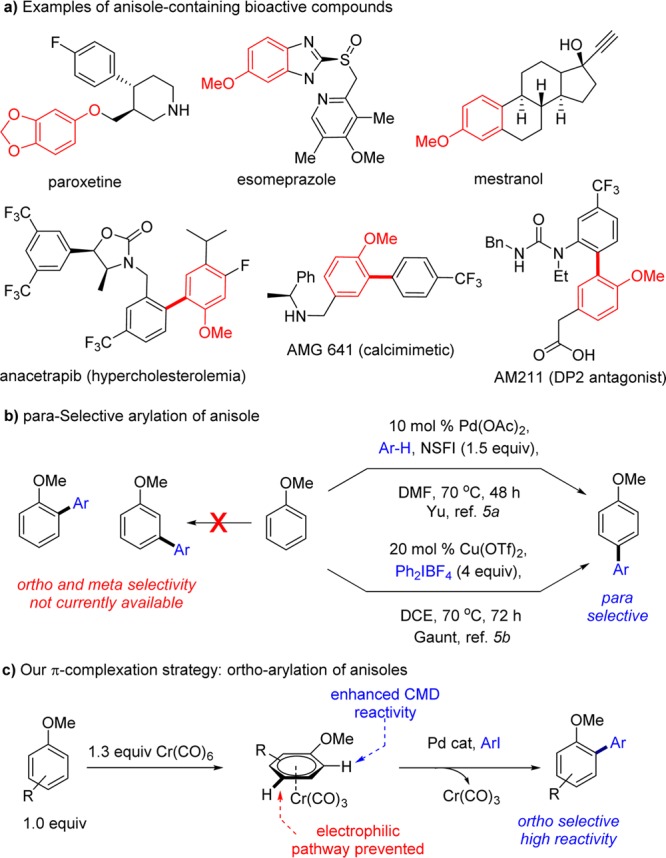
Fagnou’s initial report on the Pd-catalyzed direct arylation of anisole with para-bromotoluene6b highlighted its low reactivity and poor site selectivity (o:m:p 25:50:25). Subsequent reports on Pd-catalyzed oxidative couplings with anisole described the formation of mixtures of regioisomers with a preference for the para isomer.6d,9c,9f Conversely, Fe-,9i Rh-,9e and Ir-catalyzed9b,9g and metal-free couplings9h,9j show a slight preference for the ortho isomer (59:41 to 71:29 of o:m+p). Besides the overall lack of regioselectivity, all of these reported methodologies also require a large excess of anisole (10–100 equiv) and high reaction temperatures (100–180 °C). Groundbreaking developments were reported in 2011 by the groups of Yu5a and Gaunt,5b with two elegant examples of highly regioselective Pd- and Cu-catalyzed para-arylation reactions of anisoles (Scheme 1b). In both methods, the regioselectivity of arylation was associated with a sterics-biased electrophilic-type pathway. Here we report the first example of an ortho-selective direct arylation of anisoles (Scheme 1c),10 using a π-complexation strategy for the enhancement of reactivity and regioselectivity.
Recently, our group reported that π-complexation of a strongly electron-withdrawing Cr(CO)3 unit to fluorobenzene greatly enhances its reactivity toward C–H arylation via a proposed concerted metalation–deprotonation (CMD) pathway.11 We initially hypothesized that the increased reactivity was due to the formation of a highly electron-poor arene, resulting in weaker C–H bonds. However, computational studies indicated that a more facile out-of-plane bending of the C–H bond to adopt the CMD transition state geometry was the dominating factor. Thus, we reasoned that this effect may also operate on electron-rich arenes, allowing a previously inaccessible CMD pathway. Interestingly, computational studies by Fagnou and Gorelsky12 showed that, despite the general low reactivity of anisole, a CMD process would selectively proceed at the ortho position. Therefore, we hypothesized that π-complexation could have a triple effect on the reactivity of anisole: (1) enhance reactivity toward a CMD process, avoiding the need for using a large excess of the arene; (2) eliminate SEAr-type reactivity (and therefore para-reactivity), and (3) afford the CMD-preferred ortho-regioisomer.
Results and Discussion
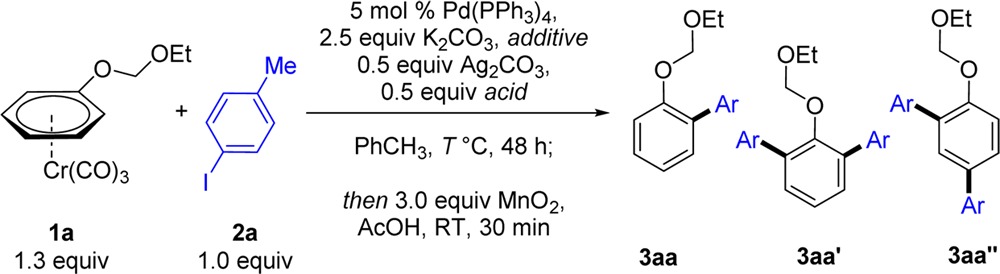
To test our hypothesis, we chose (ethoxymethoxy)benzene as a benchmark substrate (Table 1). Chromium complexes of anisoles are easily prepared, and 1a was obtained in high yield (81%) from reaction of 1.0 equiv of the arene with 1.3 equiv of Cr(CO)6. Initially, we tested the catalytic system previously developed in our group, followed by in situ demetalation (Table 1, entry 1). Gratifyingly, biaryl 3aa was obtained in good yield (58%) and excellent ortho-selectivity, confirming that π-complexation for enhancement of reactivity is not exclusive to electron-poor arenes. Screening of different carboxylic acid cocatalysts (Table 1, entries 1–4) showed that acids with low a pKa, such as p-NO2–C6H4–CO2H (pKa 3.4), provide no reactivity, while p-NMe2–C6H4–CO2H (pKa 4.9) can impart similar reactivity to that of 1-AdCO2H (pKa 5.0). We hypothesized that a more reactive catalytic system may be achieved by addition of an organic base soluble in toluene, which could assist in the deprotonation of 1-AdCO2H. Screening of basic additives (entries 5–7) showed that the highly hindered amine TMP (2,2,6,6-tetramethylpiperidine)13 provided 3aa in high yield and with excellent ortho-selectivity (o:m:p 95:0:5).14 The reaction proceeded in lower yields in the absence of K2CO3 or 1-AdCO2H, and not at all in the absence of Ag2CO3 (entries 9–11). Finally, a control experiment with 20 equiv of (ethoxymethoxy)benzene (entry 12) gave no yield of the C–H arylation product, highlighting the outstanding reactivity-enhancing effect imparted by Cr(CO)3.
Table 1. Optimization of the Direct Arylation of Complex 1a and 4-Iodotoluene (2a)a.
| entry | R–CO2H | additive (2 equiv) | T (°C) | 3aa:3aa′:3aa″ yield (%)b |
|---|---|---|---|---|
| 1 | 1-AdCO2H | – | 60 | 58:3:1 |
| 2 | PhCO2H | – | 60 | 34:1:1 |
| 3 | p-NO2–C6H4–CO2H | – | 60 | 0:0:0 |
| 4 | p-NMe2–C6H4–CO2H | – | 60 | 52:3:5 |
| 5 | 1-AdCO2H | piperidine | 60 | 0:0:0 |
| 6 | 1-AdCO2H | Et3N | 60 | 57:3:3 |
| 7 | 1-AdCO2H | TMP | 60 | 78:3:5 |
| 8 | 1-AdCO2H | TMP | 50 | 69:3:5 |
| 9c | 1-AdCO2H | TMP | 60 | 52:5:6 |
| 10 | – | TMP | 60 | 39:2:1 |
| 11d | 1-AdCO2H | TMP | 60 | 0:0:0 |
| 12e | 1-AdCO2H | TMP | 60 | 0:0:0 |
Reactions carried out on 0.1 mmol scale with respect to 2a.
Yield determined by 1H NMR of the crude using an internal standard.
No K2CO3 was added.
No Ag2CO3 was added.
20 equiv of (ethoxymethoxy)benzene were used instead of complex 1a.
With the optimal reaction conditions in hand, we set out to explore the generality of the method with respect to different substituted anisoles (Scheme 2).15 Varied alkyl substitution at the oxygen was possible, with the corresponding biaryls 3aa–3da obtained in high yields and with high ortho-selectivities, even when using a sterically hindered isopropyl substituent (3da). Para- and meta-substituted anisole complexes led to the corresponding biaryl products in excellent yields (3ea–3la) and complete regioselectivity. It is noteworthy that a strongly electron-withdrawing CO2Me para-substituent is compatible with the reaction, with the regioselectivity still being governed by the MeO group (3la). Furthermore, more electron-rich arenes, containing two MeO groups (3ma–3oa), were still highly reactive under the reaction conditions, strongly supporting our hypothesis that reactivity in this case does not correlate with electron density at the arene. Interestingly, ortho-substituted anisole, 1r, provided only a low yield of product 3ra. This may result from a C–OMe conformation with increased steric hindrance at the ortho-C–H bond. Indeed, while the MeO group in 1r would be projected toward the ortho-C–H bond, when this conformation is prevented via ring closure, as in dihydrobenzofuran (1s), high reactivity is restored. This effect was used to impart complete regioselectivity in the direct arylation of the unsymmetrical 1,4-hydroquinone derivative 1t.
Scheme 2. Scope of the Direct Arylation of Anisole–Cr(CO)3 Complexes 1a–t with 2a.
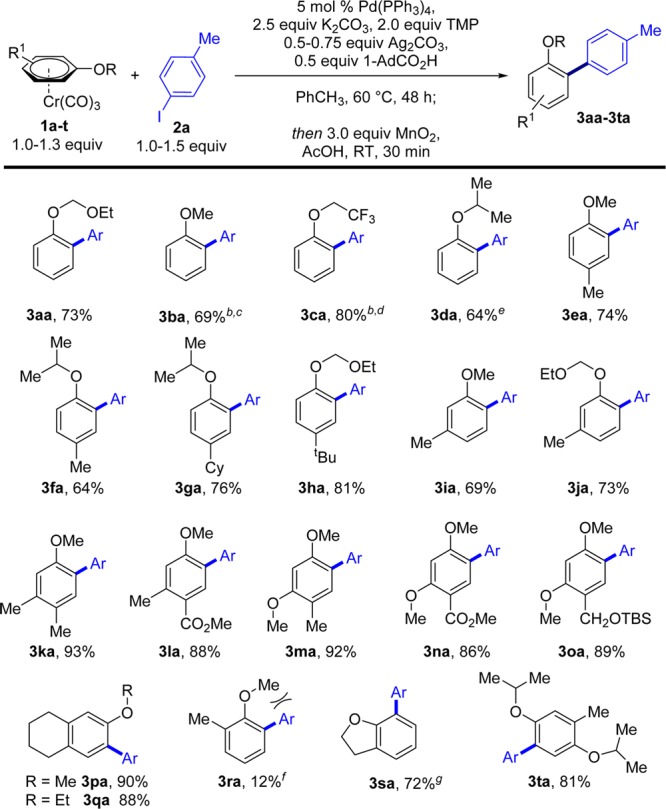
Reactions carried out on a 0.5 mmol scale with respect to the limiting reagent. Yields are of isolated product. b Performed with p-NMe2–C6H4–CO2H. c o,p-Bisarylated product 3ba′′ (3%) was also obtained. d o,p-Bisarylated product 3ca′′ (4%) was also obtained. e o,p-Bisarylated product 3da′′ (7%) was also obtained. f o,p-Bisarylated product 3ra′′ (3%) was also obtained. g Performed without TMP.
We then explored the compatibility of our reaction conditions with a variety of functional groups in the aryl iodide coupling partner. Our optimized conditions were applicable to a wide range of electron-donating and -withdrawing substituents in the ortho, meta, and para positions, affording the corresponding biaryl products 3ka–3ko in excellent yields (Scheme 3). In particular, the reaction is compatible with Cl and Br substituents (3ke, 3kf), which would allow for further Pd-catalyzed cross-couplings, as well as esters, ketones, and thioethers among other functionalities.
Scheme 3. Scope of the Direct Arylation of Complex 1k with Iodoarenes 2a–o.
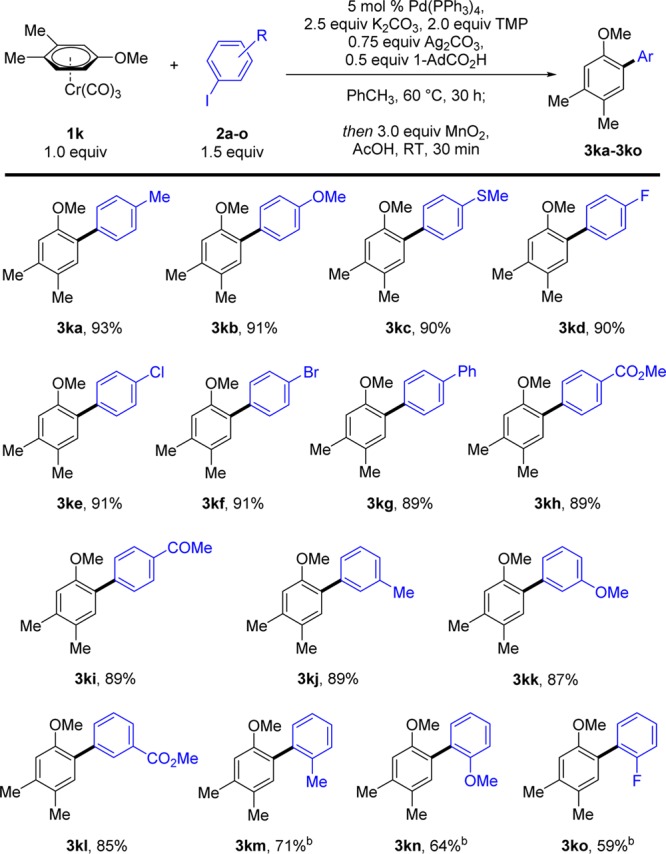
Reactions carried out on a 0.5 mmol scale with respect to the limiting reagent. Yields are of isolated product. b Performed with 2.0 equiv of 2 for 40 h.
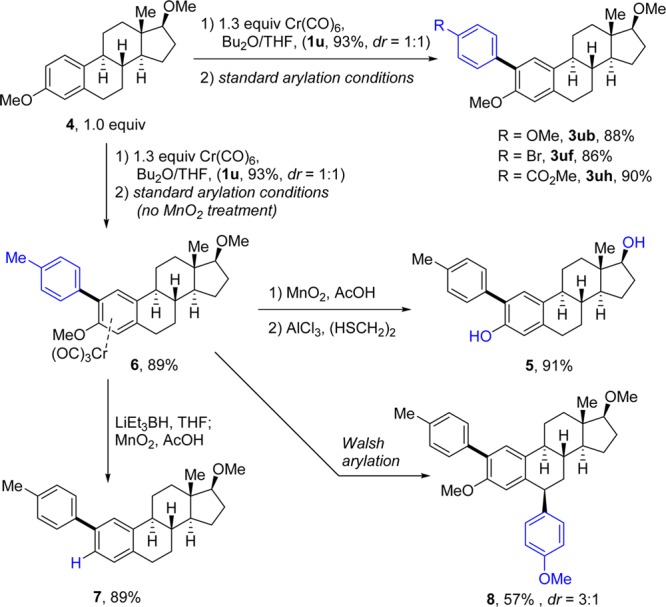
C–H functionalization is an attractive tool for late-stage functionalization of bioactive compounds.16 Steroidal derivatives possess broad spectrum utility in modern medicine and currently find wide application as adjuvants in cancer chemotherapy.17,18 We explored the applicability of our novel approach to the selective ortho-arylation of estradiol (Scheme 4). Cr-complexation of dimethylestrone (4) yielded complex 1u in 93% yield and a 1:1 diastereomeric mixture. Application of our methodology to this mixture with different iodoarenes provided the desired arylated adducts (3ub, 3uf, and 3uh) with complete regioselectivity and excellent yields. Treatment with AlCl3 allowed simultaneous demethylation of both hydroxyl groups, producing aryl-estradiol derivative 5 in excellent yield. Furthermore, the air-stable Cr-complexed biaryls could also be easily isolated in high yields, and the Cr(CO)3 unit was then utilized to control other functionalizations on the steroid core. Reaction of 6 with LiEt3BH allowed demethoxylation to steroid derivative 7.19 Application of Walsh’s benzylic arylation conditions to 6 allowed selective arylation at the less hindered benzylic position, forming 8 with good yield and diastereoselectivity.20
Scheme 4. Late-Stage Functionalization of Estradiol Derivatives via Metal–Arene π-Complexation.
Anisole is approximately 104 times less reactive than 1,3,5-trifluorobenzene under standard CMD-type reaction conditions.6b,7a Interestingly, competition experiments indicate that anisole complex 1b is 4.7 times more reactive than 1,3,5-trifluorobenzene, showcasing a 4 orders of magnitude enhancement of reactivity toward C–H arylation of anisole after complexation.21,22 A competition experiment between deuterated complex 1p-5,7-d2 and 1q highlighted the latter as 2.0 times more reactive, consistent with the KIE previously measured for complexed fluorobenzene,11 suggesting a similar reaction pathway is in operation.
Conclusions
In conclusion, we have demonstrated that π-complexation of a Cr(CO)3 unit to anisole-type arenes can “switch on” a highly ortho-selective Pd-catalyzed direct arylation process. Our method allows for the easy ortho-arylation of a range of (di)alkoxybenzenes. The high reactivity achieved with just 1 equiv of the arene under mild conditions (no strong acids/bases, 60 °C) makes this approach suitable for the late-stage functionalization of anisole-containing bioactive compounds. In addition, the Cr-unit can be used as a handle for further transformations on and around the arene, allowing for quick access to a variety of structures from a common precursor. This process is likely occurring via a concerted metalation–deprotonation type pathway, which is normally not accessible to electron-rich arenes.
Experimental Section
General Procedure A: Preparation of Arene Chromium Tricarbonyl Complexes (1)
A flame-dried round-bottom flask equipped with a magnetic stirrer and a reflux condenser was charged with Cr(CO)6 (6.50 mmol, 1.3 equiv), evacuated, and backfilled with Ar. The required O-substituted phenol (5.00 mmol, 1.0 equiv) was added to the flask, followed by the addition of anh. nBu2O and THF (9:1 v/v, 0.15 M). The resulting suspension was subjected to freeze–pump–thaw cycles (3 × 30 min) and then refluxed (external temperature 150 °C) for 48 h. The solution was then cooled down to room temperature and filtered through a short pad of silica. The silica pad was washed with Et2O (3 × 20 mL), and the organic layer was then concentrated in vacuo. Recrystallization from a cold mixture of hexane/Et2O 9:1 provided the arene tricarbonyl chromium complex 1.
(Ethoxymethoxy)benzene Tricarbonyl Chromium (1a, Table 1)
General procedure A was applied by using Cr(CO)6 (1.43 g, 6.5 mmol, 1.3 equiv) and (ethoxymethoxy)benzene (0.760 g, 5 mmol, 1.0 equiv). Recrystallization from cold hexane gave the title product 1a as a yellow solid in 81% yield (1.17 g, 4.06 mmol). 1H NMR (400 MHz, (CD3)2CO): δ (ppm) = 5.84 (app. t, J = 6.6 Hz, 2H), 5.54 (d, J = 6.4 Hz, 2H), 5.22–5.16 (m, 3H), 3.75 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, (CD3)2CO): δ (ppm) = 235.6, 143.2, 97.7, 95.4, 89.0, 83.2, 66.5, 16.0. IR: ν = 3096, 1949, 1843, 1531, 1226, 1082, 959 cm–1. Mp: 78–80 °C. HRMS EI+ m/z calcd C12H13CrO5: [M + H]+ 289.0163; found: [M + H]+ 289.0162.
General Procedure B: Direct Arylation of Arene Tricarbonyl Chromium Complexes 1 with Iodoarenes 2 (Excess of Chromium Tricarbonyl Complex)
To an oven-dried microwave 10 mL glass vial equipped with a round stirrer bar, the following reagents were added in this order: K2CO3 (172.5 mg, 1.25 mmol, 2.5 equiv), 1-AdCO2H (45.0 mg, 0.25 mmol, 0.5 equiv), Ag2CO3 (70 mg, 0.25 mmol, 0.5 equiv), Pd(PPh3)4 (5 mol %, 28.9 mg, 0.01 mmol), the required arene Cr(CO)3 complex 1 (0.65 mmol, 1.3 equiv), and iodoarene 2 (0.50 mmol, 1.0 equiv). PhCH3 (0.3 mL, 1.7 M) and 2,2,6,6-tetramethylpiperidine (170 μL, 1.00 mmol, 2.0 equiv) were added, and the glass vial was sealed with a disposable microwave cap. The resulting mixture was stirred for 48 h at 60 °C. The reaction was then cooled down, and AcOH (2 mL) was slowly added with moderate stirring. After 5 min, MnO2 (130 mg, 1.50 mmol, 3 equiv) was added in small portions and the black suspension was vigorously stirred for 30 min. The suspension was then loaded on a short silica plug (2 cm × 4 cm) and eluted with Et2O (30 mL) before concentrating in vacuo. Purification via flash chromatography column on silica gel provided the required biaryl product.
2-(Ethoxymethoxy)-4′-methyl-1,1′-biphenyl (3aa, Table 1)
General procedure B was applied with arene chromium complex 1a and 4-iodotoluene 2a. Flash chromatography (gradient 1–5% Et2O in hexane) that was performed prior to demetalation afforded the corresponding biaryl Cr(CO)3 complex. AcOH (2 mL) and MnO2 (130 mg, 1.5 mmol, 3 equiv) were then added, and the black suspension was vigorously stirred for 30 min. The suspension was then loaded on a short silica plug (2 cm × 4 cm) and eluted with Et2O (30 mL). Removal of solvent in vacuo afforded the title 3aa as a colorless oil in 73% yield (88.6 mg, 0.366 mmol). Crude 1H NMR of the reaction shows an isomer ratio o:o,o:o,p = 26:1:1.7 which corresponds to an o:m:p = 95:0:5 ratio. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.43 (d, J = 7.6 Hz, 2H), 7.35–7.20 (m, 5H), 7.07 (app t, J = 6.8 Hz, 1H), 5.16 (s, 2H), 3.66 (q, J = 7.2 Hz, 2H), 2.40 (s, 3H), 1.19 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ (ppm) = 154.6, 136.7, 135.9, 131.9, 131.0, 129.6, 128.9, 128.5, 122.2, 115.8, 93.9, 64.4, 21.3, 15.2. IR: ν = 2976, 1600, 1485, 1218, 1103, 993 cm–1. HRMS EI+ m/z calcd C16H21O2N: [M+NH4]+ 260.1645; found: [M+NH4]+ 260.1647.
General Procedure C: Direct Arylation of Arene Tricarbonyl Chromium Complexes 1 with Iodoarenes 2 (Excess of Iodoarene)
To an oven-dried microwave 10 mL glass vial equipped with a round stirrer bar, the following reagents were added in this order: K2CO3 (172.5 mg, 1.25 mmol, 2.5 equiv), 1-AdCO2H (45.0 mg, 0.250 mmol, 0.5 equiv), Ag2CO3 (105 mg, 0.375 mmol, 0.75 equiv), Pd(PPh3)4 (5 mol %, 28.9 mg, 0.010 mmol), the required arene Cr(CO)3 complex 1 (0.5 mmol, 1.0 equiv), and iodoarene 2 (0.75 mmol, 1.5 equiv). PhCH3 (0.3 mL, 1.7 M) and 2,2,6,6-tetramethylpiperidine (170 μL, 1 mmol, 2.0 equiv) were added, and the glass vial was sealed with a disposable microwave cap. The resulting mixture was stirred for 30 h at 60 °C. The reaction was then cooled down, and AcOH (2 mL) was slowly added with moderate stirring. After 5 min, MnO2 (130 mg, 1.5 mmol, 3 equiv) was added in small portions and the black suspension was vigorously stirred for 30 min. The suspension was then loaded on a short silica plug (2 cm × 4 cm) and eluted with Et2O (30 mL) before concentrating in vacuo. Purification via flash chromatography column on silica gel provided the required biaryl product.
2-Isopropoxy-4′,5-dimethyl-1,1′-biphenyl (3fa, Scheme 2)
General procedure C was applied with arene chromium complex 1f and 4-iodotoluene 2a. Flash chromatography (gradient 0.01–5% DCM in hexane) afforded the title product 3fa as colorless oil in 64% yield (77.1 mg, 0.321 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.49 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 7.6 Hz, 2H), 7.18 (d, J = 2.0 Hz, 1H), 7.09 (dd, J = 8.4, 2.0 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 4.35 (septet, J = 6.0 Hz, 1H), 2.42 (s, 3H), 2.36 (s, 3H), 1.25 (d, J = 6.0 Hz, 6H) . 13C NMR (101 MHz, CDCl3): δ (ppm) = 152.9, 136.3, 136.2, 132.2, 131.8, 130.6, 129.5, 128.9, 128.7, 116.2, 71.4, 22.2, 21.3, 20.7. IR: ν = 3021, 2975, 1492, 1230, 1110, 953 cm–1. HRMS EI+ m/z calcd C17H21O: [M + H]+ 241.1587; found: [M + H]+ 241.1587.
Acknowledgments
We are grateful to the European Research Council for a Starting Research Grant (I.L.), the Engineering and Physical Sciences Research Council (EPSRC), QMUL for a studentship (K.K.), and EPSRC National Mass Spectrometry Service (Swansea).
Supporting Information Available
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- For recent reviews on direct arylation reactions and Pd-catalyzed C–H activation methodologies, see:; a Alberico D.; Scott M. E.; Lautens M. Chem. Rev. 2007, 107, 174. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Vicente R.; Kapdi A. R. Angew. Chem., Int. Ed. 2009, 48, 9792. [DOI] [PubMed] [Google Scholar]; c Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Daugulis O.; Do H.-Q.; Shabashov D. Acc. Chem. Res. 2009, 42, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]; e McGlacken G. P.; Bateman L. M. Chem. Soc. Rev. 2009, 38, 2447. [DOI] [PubMed] [Google Scholar]; f Ashenhurst J. A. Chem. Soc. Rev. 2010, 39, 540. [DOI] [PubMed] [Google Scholar]; g Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Yu J.-Q., Shi Z., Eds. Topics in Current Chemistry: C–H Activation, 1st ed.; Springer: Berlin/Heidelberg, 2010. [Google Scholar]; i Wencel-Delord J.; Dröge T.; Liu F.; Glorius F. Chem. Soc. Rev. 2011, 40, 4740. [DOI] [PubMed] [Google Scholar]; j Hussain I.; Singh T. Adv. Synth. Catal. 2014, 356, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yamaguchi J.; Yamaguchi A. D.; Itami K. Angew. Chem., Int. Ed. 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]; b Chen D. Y.-K.; Youn S. W. Chem.—Eur. J. 2012, 18, 9452. [DOI] [PubMed] [Google Scholar]; c Facchetti A.; Vaccaro L.; Marrocchi A. Angew. Chem., Int. Ed. 2012, 51, 3520. [DOI] [PubMed] [Google Scholar]
- a Chiong H. A.; Pham Q.-N.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 9879. [DOI] [PubMed] [Google Scholar]; b Wang D.-H.; Mei T.-S.; Yu J.-Q. J. Am. Chem. Soc. 2008, 130, 17676. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nishikata T.; Abela A. R.; Huang S.; Lipshutz B. H. J. Am. Chem. Soc. 2010, 132, 4978. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cornella J.; Righi M.; Larrosa I. Angew. Chem., Int. Ed. 2011, 50, 9429. [DOI] [PubMed] [Google Scholar]; e Tredwell M. J.; Gulias M.; Bremeyer N. G.; Johansson C. C. C.; Collins B. S. L.; Gaunt M. J. Angew. Chem., Int. Ed. 2011, 50, 1076. [DOI] [PubMed] [Google Scholar]; f Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ackermann L.; Pospech J.; Potukuchi H. K. Org. Lett. 2012, 14, 2146. [DOI] [PubMed] [Google Scholar]; h Gulevich A. V.; Melkonyan F. S.; Sarkar D.; Gevorgyan V. J. Am. Chem. Soc. 2012, 134, 5528. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Pan F.; Lei Z.-Q.; Wang H.; Li H.; Sun J.; Shi Z.-J. Angew. Chem., Int. Ed. 2013, 52, 2063. [DOI] [PubMed] [Google Scholar]; j Aihara Y.; Chatani N. Chem. Sci. 2013, 4, 664. [Google Scholar]; k Arroniz C.; Ironmonger A.; Rassias G.; Larrosa I. Org. Lett. 2013, 15, 910. [DOI] [PubMed] [Google Scholar]; l Arroniz C.; Denis J. G.; Ironmonger A.; Rassias G.; Larrosa I. Chem. Sci. 2014, 5, 3509. [Google Scholar]
- For examples of meta-arylation:; a Phipps R. J.; Gaunt M. J. Science 2009, 323, 1593. [DOI] [PubMed] [Google Scholar]; b Duong H. A.; Gilligan R. E.; Cooke M. L.; Phipps R. J.; Gaunt M. J. Angew. Chem., Int. Ed. 2011, 50, 463. [DOI] [PubMed] [Google Scholar]; c Wan L.; Dastbaravardeh N.; Li G.; Yu J.-Q. J. Am. Chem. Soc. 2013, 135, 18056. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Luo J.; Preciado S.; Larrosa I. J. Am. Chem. Soc. 2014, 136, 4109. [DOI] [PubMed] [Google Scholar]
- For examples of para-arylation:; a Wang X.; Leow D.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 13864. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ciana C.-L.; Phipps R. J.; Brandt J. R.; Meyer F.-M.; Gaunt M. J. Angew. Chem., Int. Ed. 2011, 50, 458. [DOI] [PubMed] [Google Scholar]
- For a recent review on “undirected” C–H activation, see:; a Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Angew. Chem., Int. Ed. 2012, 51, 10236. [DOI] [PubMed] [Google Scholar]; For selected examples, see:; b Lafrance M.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 16496. [DOI] [PubMed] [Google Scholar]; c Stuart D. R.; Fagnou K. Science 2007, 316, 1172. [DOI] [PubMed] [Google Scholar]; d Yeung C. S.; Zhao X.; Borduas N.; Dong V. M. Chem. Sci. 2010, 1, 331. [Google Scholar]; e Liu W.; Cao H.; Lei A. Angew. Chem., Int. Ed. 2010, 49, 2004. [DOI] [PubMed] [Google Scholar]; f Wang X.; Leow D.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples, see:; a Lafrance M.; Rowley C. N.; Woo T. K.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 8754. [DOI] [PubMed] [Google Scholar]; b Wei Y.; Kan J.; Wang M.; Su N.; Hong M. Org. Lett. 2009, 11, 3346. [DOI] [PubMed] [Google Scholar]; c Sun Z.-M.; Zhang J.; Manan R. S.; Zhao P. J. Am. Chem. Soc. 2010, 132, 6935. [DOI] [PubMed] [Google Scholar]; d Rene O.; Fagnou K. Org. Lett. 2010, 12, 2116. [DOI] [PubMed] [Google Scholar]; e Chen F.; Min Q.-Q.; Zhang X. J. Org. Chem. 2012, 77, 2992. [DOI] [PubMed] [Google Scholar]; f Wang Y.-N.; Guo X.-Q.; Zhu X.-H.; Zhong R.; Cai L.-H.; Hou X.-F. Chem. Commun. 2012, 48, 10437. [DOI] [PubMed] [Google Scholar]
- a Bringmann G.; Gulder T.; Gulder T. A. M.; Breuning M. Chem. Rev. 2011, 111, 563. [DOI] [PubMed] [Google Scholar]; b Magano J.; Dunetz J. R. Chem. Rev. 2011, 111, 2177. [DOI] [PubMed] [Google Scholar]; For selected examples of ortho-arylanisoles with remarkable biological activity, see:; c Gutstein D. E.; Krishna R.; Johns D.; Surks H. K.; Dansky H. M.; Shah S.; Mitchel Y. B.; Arena J.; Wagner J. A. Clin. Pharmacol. Ther. 2012, 91, 109. [DOI] [PubMed] [Google Scholar]; d Lopez I.; Mendoza F. J.; Aguilera-Tejero E.; Perez J.; Guerrero F.; Martin D.; Rodriguez M. Kidney Int. 2008, 73, 300. [DOI] [PubMed] [Google Scholar]; e Bain G.; Lorrain D. S.; Stebbins K. J.; Broadhead A. R.; Santini A. M.; Prodanovich P.; Darlington J.; King C. D.; Lee C.; Baccei C.; Stearns B.; Troung Y.; Hutchinson J. H.; Prasit P.; Evans J. F. J. Pharmacol. Exp. Ther. 2011, 338, 290. [DOI] [PubMed] [Google Scholar]
- a Quesnelle C. A.; Familoni O. B.; Snieckus V. Synlett 1994, 349. [Google Scholar]; b Fujita K.-i.; Nonogawa M.; Yamaguchi R. Chem. Commun. 2004, 1926. [DOI] [PubMed] [Google Scholar]; c Hull K. L.; Sanford M. S. J. Am. Chem. Soc. 2007, 129, 11904. [DOI] [PubMed] [Google Scholar]; d Dwight T. A.; Rue N. R.; Charyk D.; Josselyn R.; Deboef B. Org. Lett. 2007, 9, 3137. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yanagisawa S.; Sudo T.; Noyori R.; Itami K. Tetrahedron 2008, 64, 6073. [Google Scholar]; f Brasche G.; Garcia-Fortanet J.; Buchwald S. L. Org. Lett. 2008, 11, 2207. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Join B.; Yamamoto T.; Itami K. Angew. Chem., Int. Ed. 2009, 48, 3644. [DOI] [PubMed] [Google Scholar]; h Shirakawa E.; Itoh K.-i.; Higashino T.; Hayashi T. J. Am. Chem. Soc. 2010, 132, 15537. [DOI] [PubMed] [Google Scholar]; i Liu W.; Cao H.; Lei A. Angew. Chem., Int. Ed. 2010, 49, 2004. [DOI] [PubMed] [Google Scholar]; j Sun C.-L.; Li H.; Yu D.-G.; Yu M.; Zhou X.; Lu X.-Y.; Huang K.; Zheng S.-F.; Li B.-J.; Shi Z.-J. Nat. Chem. 2010, 2, 1044. [DOI] [PubMed] [Google Scholar]; k Storr T. E.; Greaney M. F. Org. Lett. 2013, 15, 1410. [DOI] [PubMed] [Google Scholar]
- For an example of a selective scandium-catalyzed ortho-alkylation of anisoles, see:Oyamada J.; Hou Z. Angew. Chem., Int. Ed. 2012, 51, 13000. [DOI] [PubMed] [Google Scholar]
- Ricci P.; Krämer K.; Cambeiro X. C.; Larrosa I. J. Am. Chem. Soc. 2013, 135, 13258. [DOI] [PubMed] [Google Scholar]
- Gorelski S. I.; Lapointe D.; Fagnou K. J. Org. Chem. 2012, 77, 658. [DOI] [PubMed] [Google Scholar]
- Interestingly, TMP has recently been shown to undergo Pd-mediated sp3-C–H activation; see:McNally A.; Haffemayer B.; Collins B. S. L.; Gaunt M. J. Nature 2014, 510, 129. [DOI] [PubMed] [Google Scholar]
- Normalized total regioselectivity of arylation events, calculated as ortho = (3aa + 2 × 3aa′ + 3aa″) and para = 3aa″.
- Cr-complexes 1a–t were synthesized from the corresponding arene (1.0 equiv) and Cr(CO)6 (1.3 equiv) in high yields of 75–90%.
- For reviews on late-stage functionalization, see:; a Gutekunst W. R.; Baran P. S. Chem. Soc. Rev. 2011, 40, 1976. [DOI] [PubMed] [Google Scholar]; b McMurray L.; O’Hara F.; Gaunt M. J. Chem. Soc. Rev. 2011, 40, 1885. [DOI] [PubMed] [Google Scholar]; c Wencel-Delord J.; Glorius F. Nat. Chem. 2013, 5, 369. [DOI] [PubMed] [Google Scholar]
- Bansal R.; Acharya P. C. Chem. Rev. 2014, 114, 6986. [DOI] [PubMed] [Google Scholar]
- For selected examples of late-stage functionalization of steroidal compounds, see ref (5b) and:; a Tang P.; Furuya T.; Ritter T. J. Am. Chem. Soc. 2010, 132, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bigi M. A.; White M. C. J. Am. Chem. Soc. 2013, 135, 7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic J. P.; Rose-Munch F.; Rose E.; Dromzee Y. J. Am. Chem. Soc. 1993, 115, 6434. [Google Scholar]
- a McGrew G. I.; Temaismithi J.; Carroll J. P.; Walsh P. J. Angew. Chem., Int. Ed. 2010, 49, 5541. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang J.; Stanciu C.; Wang B.; Hussain M. M.; Da C.-S.; Carroll P. J.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2011, 50, 20552. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McGrew G. I.; Stanciu C.; Zhang J.; Carroll J. P.; Dreher S. D.; Walsh P. J. Angew. Chem., Int. Ed. 2012, 51, 11510. [DOI] [PubMed] [Google Scholar]
- A competition experiment between (anisole)tricarbonylchromium(0) (1b) and (o-fluorotoluene)tricarbonylchromium(0) revealed the latter to be 4.5 times more reactive. See SI for details.
- Arylation of (fluoroanisole)tricarbonylchromium(0) complexes revealed that, while (o-fluoroanisole)tricarbonylchromium(0) can be selectively arylated ortho to the fluorine (see ref (11)), (meta-) and (para-fluoroanisole)tricarbonylchromium(0) complexes led to complicated mono- and bis-arylated regioisomeric mixtures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.