

Abstract
We report the Pd-catalyzed arylation of very hindered α,α,α-trisubstituted primary amines. Kinetics-based mechanistic analysis and rational design have led to the development of two biarylphosphine ligands that allow the transformation to proceed with excellent efficiency. The process was effective in coupling a wide range of functionalized aryl and heteroaryl halides under mild conditions.
Introduction
Organic molecules containing bulky alkyl groups have shown great potential in drug discovery and medicinal chemistry.1a Sterically demanding alkyl substituents such as adamantyl or tert-butyl are often introduced into pharmaceuticals to enhance lipophilicity and/or improve the drug’s metabolic stability by shielding adjacent functional groups or reactive sites from enzymatic degradation.1b−1f Aminoadamantanes themselves have been examined and used as antiviral drugs;1a,1g−1j however, aryl aminoadamantane derivatives and other anilines based on hindered amines such as 3–5 (Figure 1) remain largely unexplored, presumably due to difficulty in their preparation.
Figure 1.

Mechanism of Pd-catalyzed arylation of primary amines and examples of bulky α,α,α-trisubstituted primary amines.
Successful strategies that have previously been used to synthesize these bulky anilines employ an electrophilic amination approach. Amines 1 and 3 have been arylated through a titanium-mediated coupling of the corresponding N-chloroamines with Grignard reagents.2 Additionally, there are examples of transition-metal-free amination of arylboroxines and copper-catalyzed amination of organozinc reagents using 3.3 Recently, Lalic reported an elegant synthesis of hindered tertiary anilines through the copper-catalyzed coupling of aryl boronic esters with O-benzoyl hydroxylamines.4 While these methods are efficient, the electrophilic amine must be separately prepared, and many of the nucleophiles that are employed are moisture-sensitive. A useful alternative is the palladium-catalyzed C–N cross-coupling—an operationally simple and widely used reaction in both industrial and academic settings.5a−5i Although advances in ligand design have overcome many challenges,6 only a few examples of the N-arylation of hindered primary amines have been reported. Amines 1 and 2 have been previously cross-coupled with catalysts with either phosphines or N-heterocyclic carbenes as supporting ligands.7a−7e However, most of these reactions require moderate catalyst loadings (1–5 mol %) and elevated temperatures (90–135 °C) and, most importantly, are limited with regard to the substrate scope. In addition, there are no examples using more hindered and challenging amine substrates such as 3–5. The availability of a general method to obtain a broad range of hindered anilines by a Pd-catalyzed C–N cross-coupling process is desirable. Herein, we describe the development of two related catalyst systems that demonstrate high activity for the coupling of α,α,α-trisubstituted primary amines 1–5 with a variety of (hetero)aryl halides. These complementary systems were developed through an initial ligand screen followed by a rational ligand design based on the results of the kinetic analysis of the reaction (Figure 1).
Initial Ligand Evaluation
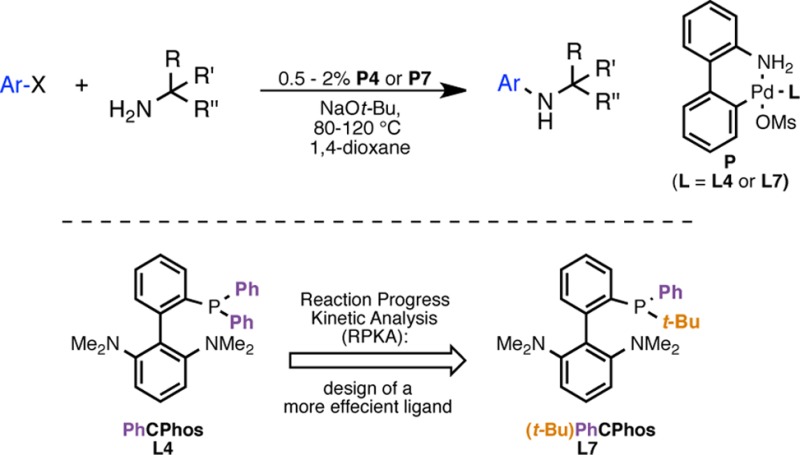
In 2008 we reported BrettPhos (L1; Table 1), a biaryl(dialkyl)phosphine ligand for the Pd-catalyzed arylation of primary amines.8a BrettPhos-based catalyst systems often promote amination reactions at low catalyst loadings and exhibit an exceptionally broad scope for both the aryl halide and the primary amine.8b However, when we evaluated the (2-aminobiphenyl)palladium methanesulfonate precatalyst of L1 (P1),9 for the coupling of 3,5-dimethoxychlorobenzene (6) with tert-octylamine (3), the product 7 was formed in low yield (Table 1, entry 1). Given the size of both L1 and 3, we felt that either amine binding (III, Figure 1) or amine deprotonation (III–IV, Figure 1) could be problematic.10 Therefore, we hypothesized that the use of a smaller ligand would allow for more effective coordination to the Pd(II) center and/or facilitate the approach of the base. Indeed, the yield was significantly improved with the less sterically demanding11 and less electron-rich PhBrettPhos, L2 (Table 1, entry 2). Furthermore, the use of L3, which lacks methoxy groups on the phosphine-containing arene ring, was even more effective (Table 1, entry 3). The use of a ligand in which the distal arene of L3 had been modified by replacing the triisopropyl substituents with two dimethylamino groups (L4) also gave the desired product in good yield (Table 1, entry 4). The catalyst derived from the corresponding dicyclohexylphosphino analogue, CPhos (L5), gave no product, suggesting that the relatively smaller and less electron-donating phenyl groups on the phosphine atom are required to promote the desired transformation.
Table 1. Ligand Evaluation for the Cross-Coupling of 6 and 3a.
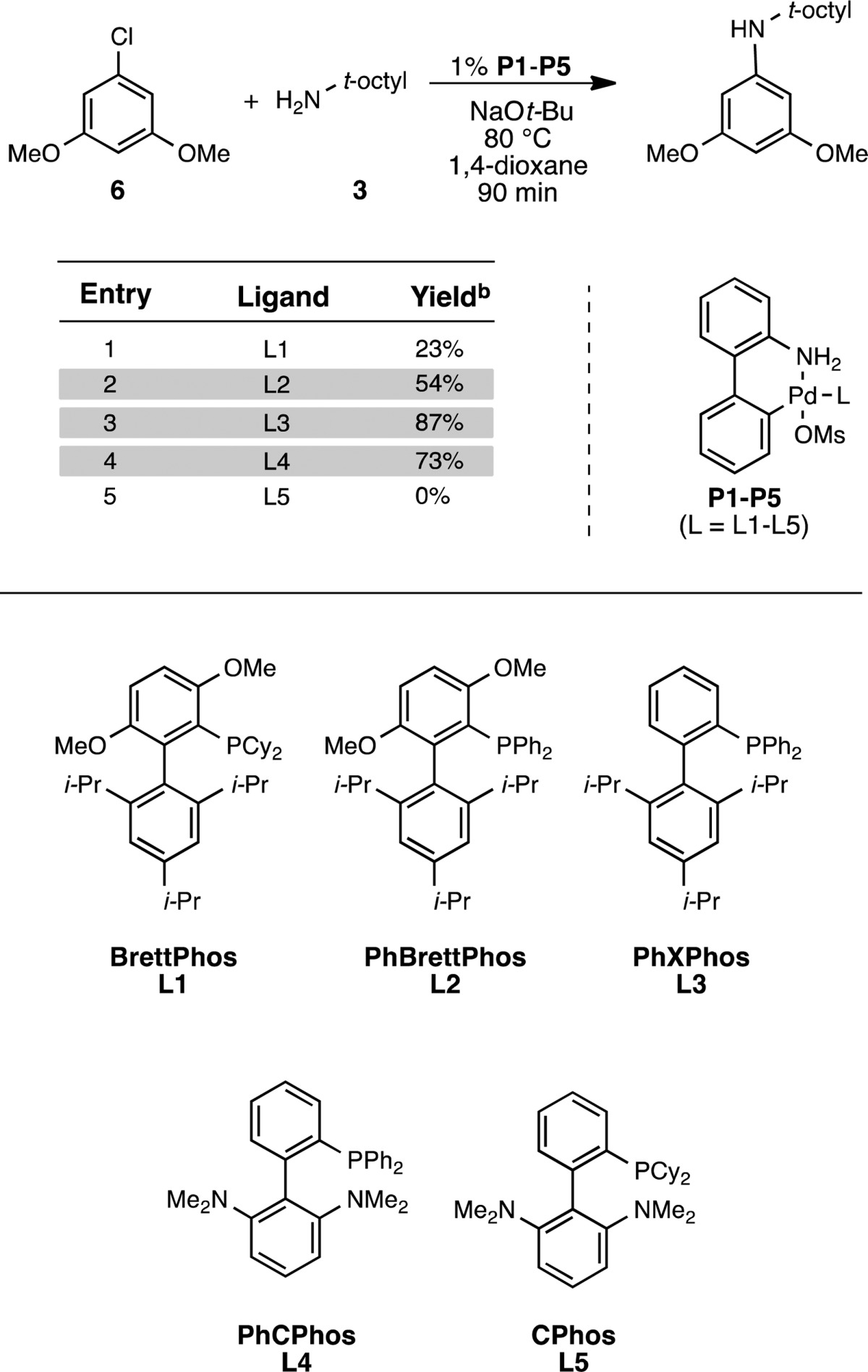
Reaction conditions: aryl halide (0.5 mmol), amine (0.6 mmol), NaO-t-Bu (0.6 mmol), 1,4-dioxane (0.5 mL).
Corrected GC yields.
The most effective precatalysts, P3 and P4, were further evaluated in reactions between 3 and more complex aryl halides, and the use of P4 initially gave the best results (Table 2).
Table 2. Comparison between P3 and P4 and Selected Examples of the Initial Reaction Scope Using P4a.
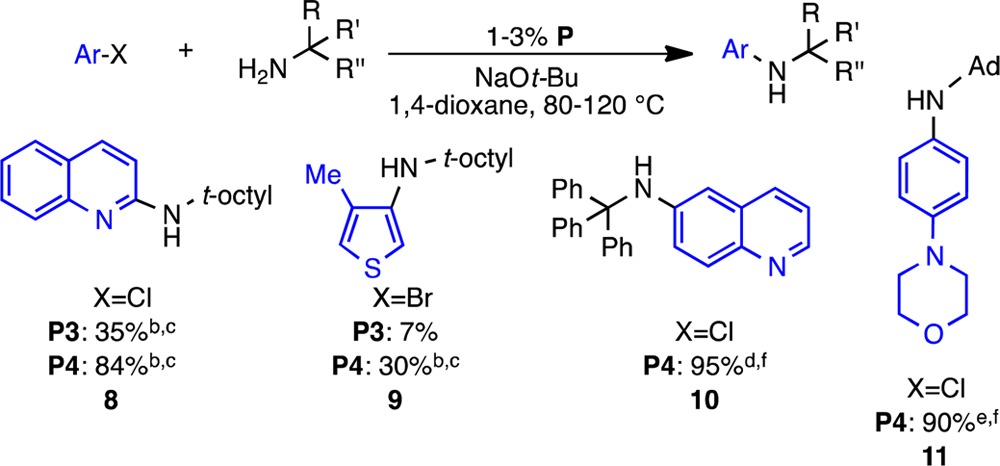
Reaction conditions: aryl halide (0.5 mmol), amine (0.6 mmol), NaO-t-Bu (0.6 mmol), 1,4-dioxane (0.5 mL). Ad = adamantyl
Reaction conditions: 1% P, 80 °C, 90 min.
GC yield.
Reaction conditions: 2% P4, 120 °C, 24 h.
Reaction conditions: 1% P4, 100 °C, 12 h.
Isolated yield.
Reaction Kinetics Analysis
While the results with P4 were promising, relatively high temperatures (100–120 °C) and catalyst loadings (1–3 mol %) were required for the reaction to reach completion. To design an improved catalyst system, we set out to qualitatively explore the reaction rate dependence on each substrate using reaction progress kinetic analysis (RPKA). As described by Blackmond,12a,12b RPKA is a simple, systematic method to obtain a complete picture of a reaction’s kinetic profile from a limited number of experiments performed under synthetically relevant conditions. This method has been successfully used in a number of laboratories12c−12f to elucidate the reaction mechanism of various catalytic processes. The key parameter utilized in RPKA is “excess”, which refers to the difference between the initial concentrations of the two reactants (eq 1), and the kinetic information is obtained from reactions run under “different excess” conditions.
| 1 |
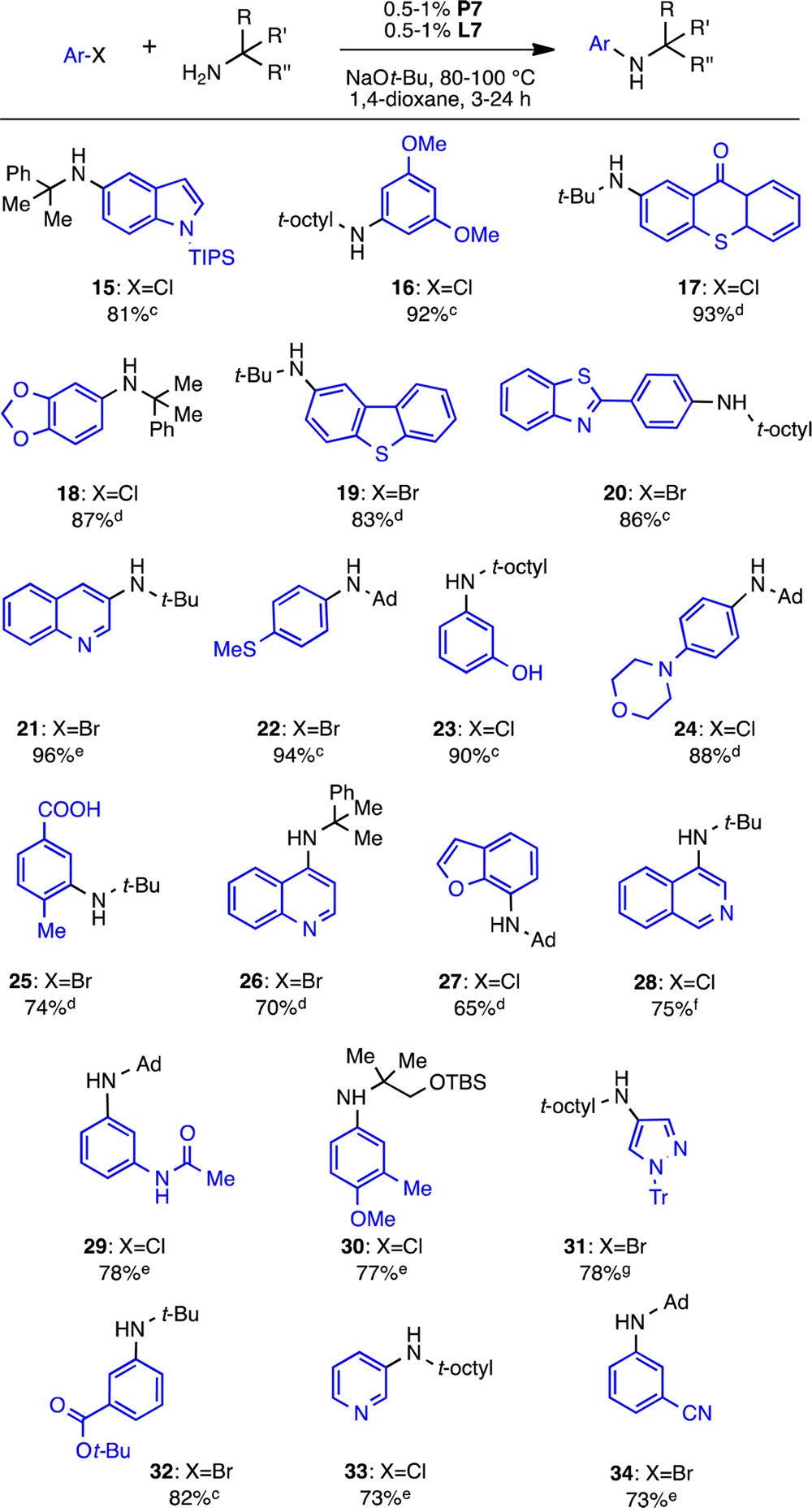
We chose the model reaction between aryl chloride 6 and amine 3 for our kinetic analysis using precatalyst P4. To broaden the study, we also explored the reaction with the corresponding aryl bromide 12. The reactions were monitored in situ by reaction calorimetry along with GC analysis to support the calorimetry results13 (see the Supporting Information). The reaction rate progress over time profiles for both aryl halides are shown together in Figure 2. It is immediately apparent that the shape of the curves is different for each aryl halide and that the reaction for ArBr is notably faster than that for ArCl. These observations suggest that the nature of the aryl halide plays a key role in the kinetics of the reaction.14 Following the RPKA method, the data may be replotted as rate vs [substrate] to determine the rate orders of each substrate (only the most informative graphs are shown for each case; all graphs are included in the Supporting Information).
Figure 2.
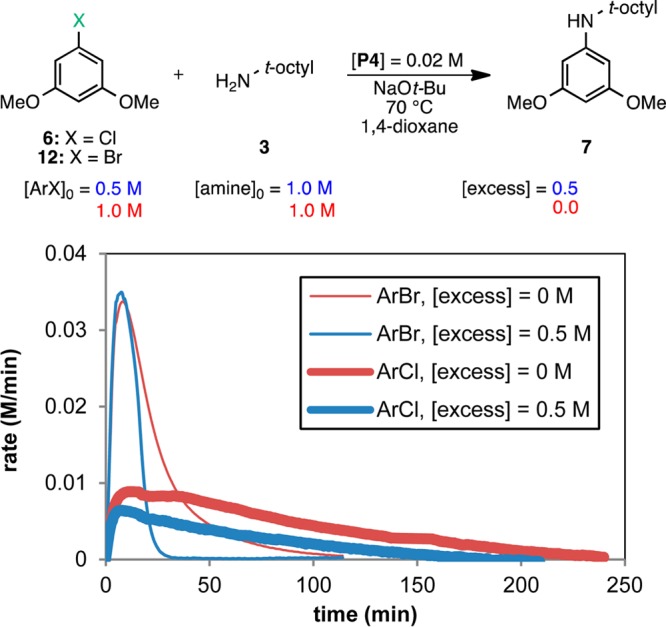
Temporal reaction rate profiles for the reaction of amine 3 (1.0 M) and ArCl 6 or ArBr 12 (concentrations defined by [excess] given in legend; see eq 1) catalyzed by P4 (0.02 M). Reaction at 70 °C in 1,4-dioxane.
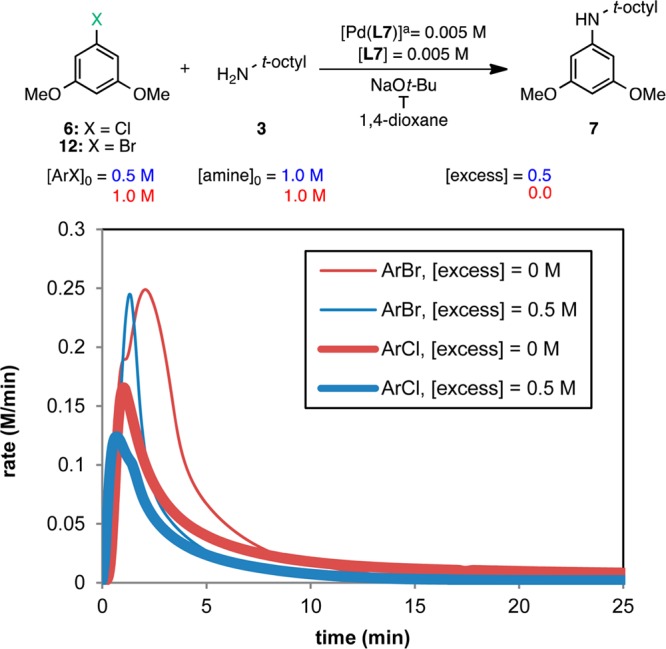
Figure 3 shows the rate vs [ArCl] plot for the two different excess experiments shown in Figure 2 over the range of ArCl concentrations common to both reactions. At any given value of [ArCl], the concentration of amine is different for the two kinetic profiles, as illustrated by the dashed line (when [ArCl] = 0.3 M, [amine] = 0.8 and 0.3 M for the blue and red curves, respectively). An overlay of the curves at different amine concentrations indicates that the rate is independent of the concentration of amine for this range of concentrations. This behavior was unexpected given the steric encumbrance of 3, which we initially predicted to bind to the Pd(II) center with difficulty10 and therefore be involved in the rate-determining step. However, the linear decay of the curves indicates that the reaction has a positive order in [ArCl] and that oxidative addition is (at least partially) rate-determining.15 The fact that the reactions reached different maximum rates when starting at different ArCl concentrations (Figure 2) provides additional evidence for a positive order in aryl halide (since the maximum rates do not differ by a factor of 2, the order in ArCl is fractional). The use of a ligand (L4) with phenyl groups as the phosphine substituents could explain the relatively slow rate of oxidative addition (computational evidence suggests that L2 has a higher energy barrier than its alkyl analogue L1 for this step).16
Figure 3.
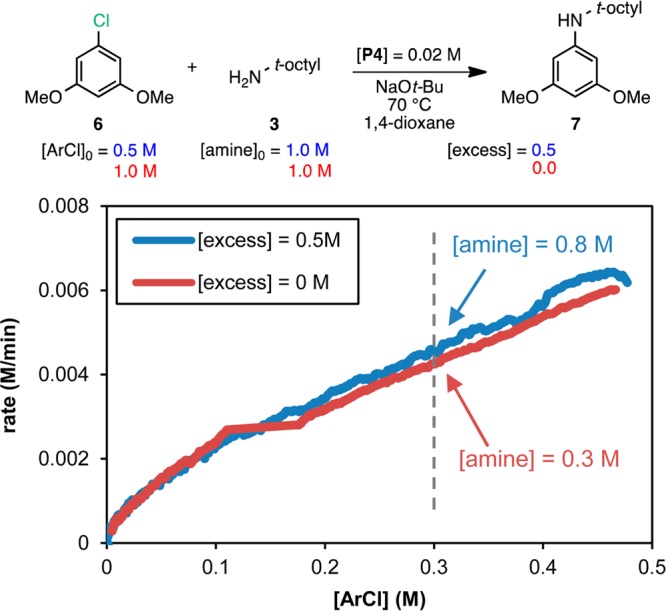
Reaction progress kinetic profiles for the reaction of ArCl 6 with amine 3 plotted as rate vs [ArCl] over the range of concentrations common to the two reactions. The concentration of amine 3 at the point in the reaction progress marked by the dashed line is shown for each reaction.
Further probing of the reaction mechanism is possible by exploring the behavior of aryl bromide 12. Since the rate of oxidative addition is determined by the strength of the C–X bond,17 it was not surprising that the reaction of 12 was ca. 4 times faster than that of the aryl chloride 6 (t1/2 =10 min vs 46 min for [ArX]0 = 0.5 M). In this case, plotting the data of the different excess experiments as rate vs [amine] for the concentrations common to the two reactions provides insights into the aryl bromide reaction kinetics (Figure 4). First, the overlay between the curves reveals identical rates at different ArBr concentrations (as illustrated by the dashed line: when [amine] = 0.75 M, [ArBr] = 0.25 and 0.75 M for the blue and red curves, respectively), indicating zeroth-order kinetics in aryl halide, in contrast to the ArCl case. This observation is confirmed by the fact that the reactions achieve identical maximum rates (∼0.03 M/min) for different initial concentrations of aryl bromide (Figure 2). Second, the rate appears to be relatively insensitive to the concentration of amine. The nearly horizontal curves show the rate slightly decreasing over the range of amine concentrations from 0.9 to 0.5 M, suggesting zeroth-order kinetics in [amine]. Since oxidative addition of aryl bromides is faster, the rate-determining step of the reaction has shifted to a subsequent step in the catalytic cycle, which is independent of both [ArBr] and [amine].18 Therefore, for the reaction of aryl bromide 12 and amine 3 with precatalyst P4, reductive elimination is most likely the rate-determining step.
Figure 4.
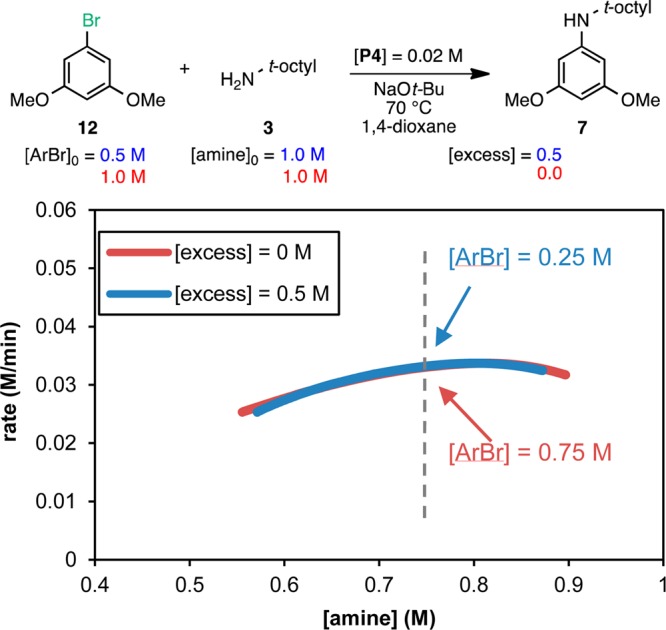
Reaction progress kinetic profiles for the reaction of ArBr 12 with amine 3 plotted as rate vs [amine] over the range of concentrations common to the two reactions. The concentration of ArBr 12 at the point in the reaction progress marked by the dashed line is shown for each reaction.
At approximately 40% conversion, the initial zeroth-order regime shown in Figure 4 gives way to a slow linear decay in reaction rate for the [excess] = 0 M reaction (equal amounts of aryl bromide 12 and amine 3; see the Supporting Information for the full graph). This behavior corresponds to initial saturation kinetics in [amine].12a As the concentration of amine 3 decreases, amine binding to the Pd(II) center becomes slower than reductive elimination and, therefore, rate-determining.
The kinetic analysis of the reactions with two different aryl halides suggests that, to improve the catalyst system, oxidative addition and reductive elimination need to be accelerated. Modifying the ligand to make it both more electron-rich (faster oxidative addition) and larger (faster reductive elimination) might result in faster rates for each step.19 We reasoned that switching from a biaryl(diaryl)phosphine-type ligand to the (dialkyl)phosphino analogue (Figure 5) would accomplish both requirements. However, our initial ligand screen (Table 1) showed that CPhos (L5) gave no product, suggesting this ligand was too large and/or too electron-rich. We thus prepared ligands with one phenyl group and one alkyl group (Figure 5). By incorporating one large alkyl group, we could increase the electron density and size of the ligand, while keeping the smaller aryl group should still allow amine binding and deprotonation to proceed at a rapid rate.
Figure 5.
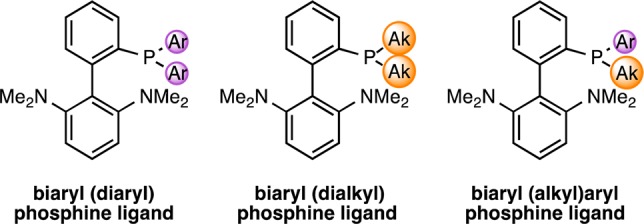
Ligand modifications at the phosphine atom.
We synthesized two biaryl(alkyl)phenyl ligands in which one of the phenyl groups on the phosphine of L4 is replaced with either a cyclohexyl (L6) or a tert-butyl (L7) group (Table 3; see the Supporting Information for details on the preparation of the ligands). The corresponding precatalysts P6 and P7 were prepared and examined under identical conditions used in our previous ligand assessment (Table 1). As hypothesized, the hybrid (alkyl)phenyl phosphines L6 and L7 provided superior catalysts. A system based on ligand L7 proved optimal, giving full conversion of 6 in less than 30 min at 70 °C (Table 3, entry 3).
Table 3. Evaluation of P6 and P7 for the C–N Cross-Coupling Reaction between 6 and 3a.
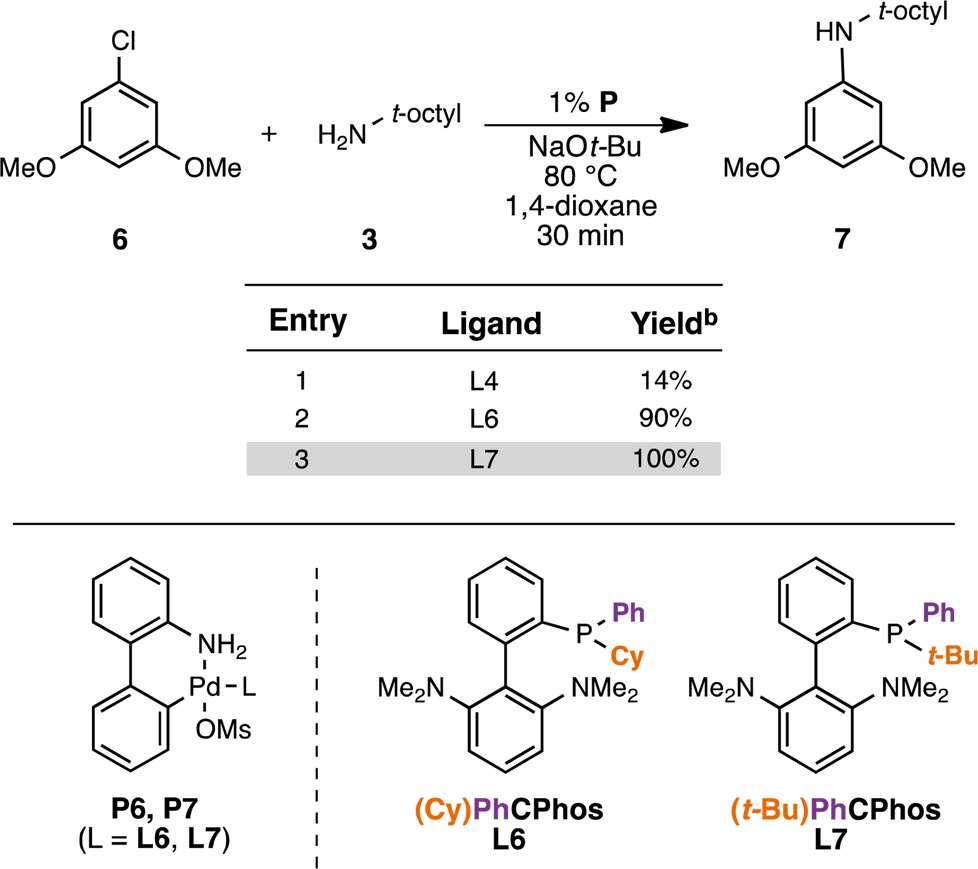
Reaction conditions: aryl halide (0.5 mmol), amine (0.6 mmol), NaO-t-Bu (0.6 mmol), 1,4-dioxane (0.5 mL).
Corrected GC yields.
To understand the difference between L4 and L7, we investigated the kinetics of the reactions between aryl halides 6 and 12 and amine 3 using a catalyst based on L7 under the same reaction conditions used for P4 (2% Pd, 70 °C). The use of ligand L7 dramatically accelerated the rate of the reaction: t1/2 = 2.6 and 1.9 min for 6 and 12, respectively, whereas the half-lives with P4 were 46 and 10 min for 6 and 12 ([ArX]0 = 0.5 M; see the Supporting Information for details). To perform accurate calorimetric kinetic studies of the improved system, we adjusted the reaction conditions by reducing the catalyst loading (from 2 to 0.5 mol %) and the temperature for the aryl bromide (from 70 to 60 °C).20 More consistent results were observed at lower catalyst loadings through in situ generation of an oxidative addition complex from (COD)Pd(CH2TMS)2, ligand L7, and aryl halide than through the use of precatalyst P7 as the Pd source.21 In addition, it was found that better results were obtained in the presence of an extra equivalent of L7 (with respect to the palladium catalyst), which was extended to the general reaction conditions (Table 4).
Table 4. Substrate Scope Using Precatalyst P7a,b.
Reaction conditions: aryl halide (1 mmol), amine (1.2 mmol), NaO-t-Bu (1.2 mmol), 1,4-dioxane (1 mL).
Isolated yields (average of two runs).
Reaction conditions: 0.5% P7, 0.5% L7, 80 °C.
Reaction conditions: 1% P7, 1% L7, 80 °C.
Reaction conditions: 1% P7, 1% L7, 100 °C.
Reaction conditions: 2% P7, 2% L7, 110 °C.
Reaction conditions: 3% P7, 3% L7, 100 °C.
The temporal rate profiles for these experiments are shown in Figure 6. Even with the sharply reduced catalyst concentration (and reduced temperature for the ArBr substrate), the rate of these reactions was ca. 1 order of magnitude greater than that of the reactions with precatalyst P4 (Figures 2 and 6)
Figure 6.
Temporal reaction rate profiles for the reaction of amine 3 (1.0 M) and ArCl 6 or ArBr 12 (concentrations defined by [excess] given in legend; see eq 1). Reaction at 70 °C for ArCl and 60 °C for ArBr in 1,4-dioxane. Note a: Pd(L7) is an oxidative addition complex generated in situ from (COD)Pd(CH2TMS)2, L7, and 12 (see the Supporting Information for details).
Figure 7 shows the data from the different
excess experiments for aryl chloride 6 as rate vs [ArCl]
over the range of ArCl concentrations common to both reactions. Although
the reaction has a more complex profile than with P4,
the nonhorizontal shape of the curves and the different maximum rates
(0.124 and 0.02 M/min for the blue and the red curves, respectively)
imply that there is a positive order in aryl chloride and thus that
oxidative addition remains, at least partially, the rate-determining
step (the reaction has a fractional order in [ArCl]). In contrast
to the ArCl reactions with P4, the two curves are not
overlaid, which could indicate a positive reaction order in [amine]
(the reactions have different rates at different amine concentrations).
However, the convex shape of the rate curves (particularly explicit
for the red curve) is typically a hallmark of catalyst deactivation.22 The likelihood of rate dependence on [amine]
vs catalyst deactivation can be deconvoluted with a quantitative assessment.
We can consider relative rates of the two reactions at any given concentration
of ArCl and amine, with “x” and “y” as the reaction orders in [ArCl] and [amine],
respectively, and kobs as the observed
rate constant, which contains the catalyst concentration (eq 2). Choosing
[ArCl] = 0.5 M, we see from Figure 7 that [amine]
= 1.0 M for the blue curve (the beginning of the reaction) and 0.5
M for the reaction in red (50% conversion) and that their relative
rates differ by a factor of 5 (eq 3). If kobs is identical for the two runs, y is calculated
to be greater than 2 (eq 4a), which is not mechanistically reasonable.
If the reaction is in fact zeroth-order in [amine] (y = 0), eq 4b indicates that kobs for
the red curve has been deactivated by a factor of 5 at 50% conversion.
Although this mathematical argument cannot entirely exclude rate dependence
on [amine], catalyst deactivation is a more likely explanation on
the basis of the shape of the curves and the fact that different maximum
rates are obtained for different ArCl concentrations.
Figure 7.
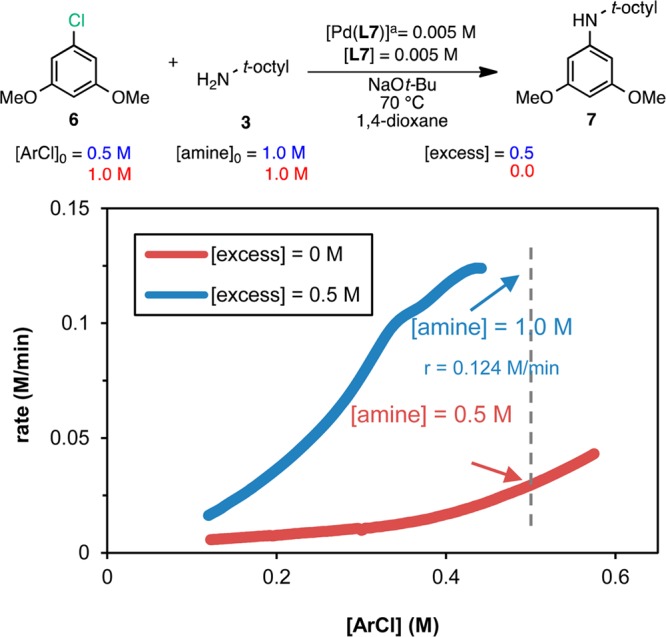
Reaction progress kinetic profiles for the reactions of ArCl 6 with amine 3 plotted as rate vs [ArCl] over the range of concentrations common to the two reactions. The concentration of amine 3 at the point in the reaction progress marked by the dashed line is shown for each reaction. Note a: Pd(L7) is an oxidative addition complex generated in situ from (COD)Pd(CH2TMS)2, L7, and 12 (see the Supporting Information for details).
Lastly, the data for the cross-coupling between aryl bromide 12 and amine 3 with a catalyst based on L7, shown in Figure 6, reveal the fastest reactions run in this work (even at 60 °C). The curves show an uncommon shape in which the rates reach their maximum at ca. 25–35% conversion, which suggests a delay in the catalyst entering the catalytic cycle. In addition, as in the reaction with aryl chloride 6, unusual behavior is observed at the end of the reactions: the curves decay, displaying a convex shape (see the Supporting Information for the full graph), presumably due to catalyst decomposition. Thus, it seems that the catalyst based on L7 has a transient nature in such a fast reaction and does not establish a persistent steady-state kinetics behavior to allow further RPKA analysis. However, since both different excess reactions with aryl bromide 12 reach the same maximum rate independent of their initial concentration, it can be inferred that the reaction is likely zeroth-order in ArBr as with precatalyst P4.23
X-ray Structures of Oxidative Addition Complexes Bearing L4 and L7
To gain a deeper insight into the differences between L4 and L7, we prepared oxidative addition complexes [L4·Pd(4-CNPh)Br] (13) and [L7·Pd(4-CNPh)Br] (14)24 as air-stable yellow solids and analyzed them by single-crystal X-ray crystallography (Figure 8). Similar to what has been previously observed with biaryl(dialkyl)phosphine ligands, the complexes possess a slightly distorted square planar geometry at Pd. As has been observed with L5,25 the dimethylamino substituents were not coplanar with the aryl ring to which they are attached, indicating that no conjugation of the nitrogen lone pairs with the aromatic π-system is occurring. Thus, the dimethylamino groups should be considered as overall electron-withdrawing by induction rather than electron-donating. This could assist the binding of bulky and electron-rich amines by rendering the Pd center more electrophilic.16,26
Figure 8.
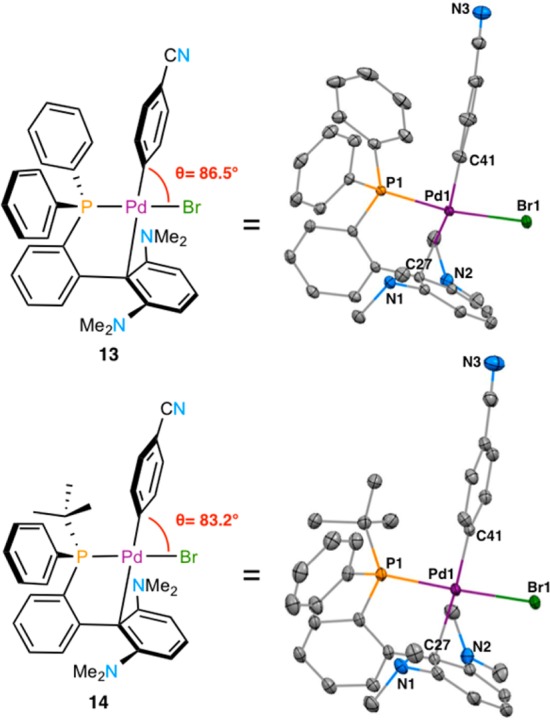
X-ray structures of oxidative addition complexes 13 and 14 and relevant bond lengths. Thermal ellipsoids plotted at 50% probability. Hydrogen atoms omitted for clarity.
The most important distinction between 13 and 14 is the C41–Pd–Br angle (θ), which decreased from 86.5° to 83.2°. We infer that this geometric difference also applies to the amido complexes IV (Figure 1), bringing the two groups to be connected closer in 14 than in 13 and therefore facilitating reductive elimination.19
Reaction Scope
We next set out to examine the substrate scope of the reaction with P7. We were able to reduce catalyst loadings from 1–2% P4 to 0.5 mol % P7 and the temperature from 100–120 to 80 °C for many substrates (Table 4). More challenging substrates required 1 mol % Pd and 100 °C (e.g., with 30 or 33). A variety of electron-deficient, electron-rich, and heteroaryl chlorides and bromides were successfully coupled using this catalyst system. Esters and nitriles (32, 34) were tolerated under the reaction conditions, although free N–H heterocycles and enolizable ketones were not compatible. Protic substrates such as phenols, carboxylic acids, and amides were also successfully coupled by using 2 equiv of base (23, 25, and 29). As previously reported, nitrogen-containing five-membered heterocycles are challenging, albeit important, substrates.8b,27 The reaction with 1-methyl-4-bromopyrazole formed several byproducts in addition to the desired product. However, replacing the methyl group with a trityl protecting group led to the clean formation of 31.
Reactions using ortho-substituted substrates and/or tritylamine 5 were sluggish with P7, and the use of P4 provided better results in these cases (Table 5). This system allowed for the preparation of extremely hindered secondary anilines as 2,6-disubstituted aryl halides (35) and substrates bearing large ortho substituents (36) were successfully coupled in high yields. Additionally, certain heteroaryl halides such as 5-chloro-2,1,3-benzothiadiazole and 4-methyl-3-bromothiophene gave better results using P4 (40, 42; 10–15% yields were obtained using P7). For activated 2-halopyridines and pyrazine (41, 44, 45) the corresponding ArO-t-Bu byproduct was also observed28 but was easily separated from the desired compound. Weaker inorganic bases (Cs2CO3, K3PO4, or K2CO3) that could circumvent this issue were found to be ineffective in promoting the desired transformation. Nonetheless, a wide variety of substrates, including aryl halides containing base-sensitive functional groups, were coupled with deliberate choice of the supporting ligand (L7 or L4)
Table 5. Substrate Scope Using Precatalyst P4a,b.
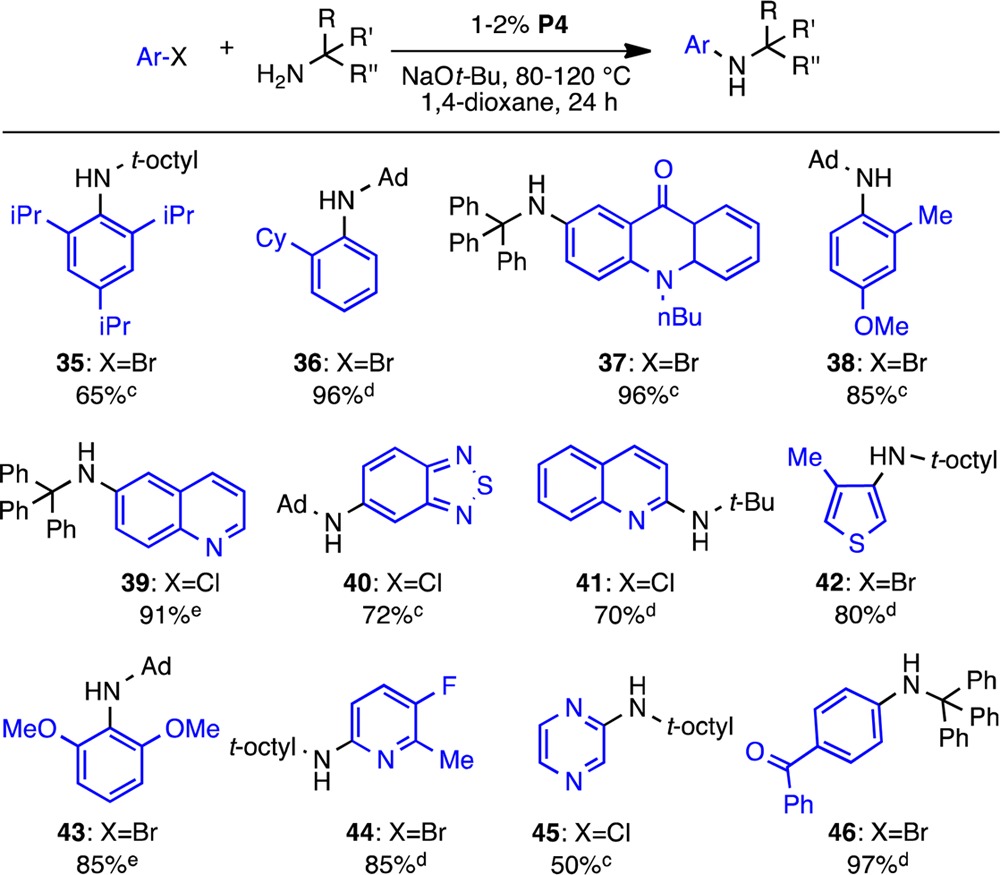
Reaction conditions: aryl halide (1 mmol), amine (1.2 mmol), NaO-t-Bu (1.2 mmol), 1,4-dioxane (1 mL).
Isolated yields (average of two runs).
Reaction conditions: 2% P4, 120 °C.
Reaction conditions: 1% P4, 80 °C.
Reaction conditions: 2% P4, 100 °C.
Conclusions
In summary, we have developed a general method for the cross-coupling of hindered primary amines with a range of (hetero)aryl chlorides and bromides. This transformation occurs under mild conditions and provides the desired products in high yields utilizing two catalyst systems. Ligand L7 was designed after analysis of the reaction mechanism using the RPKA method. Rather than amine binding, it was found that oxidative addition for aryl chlorides and reductive elimination for aryl bromides with the initial catalyst based on ligand L4 were rate-determining. These observations lead to the development of L7, a biaryl(alkyl)arylphosphine ligand that greatly accelerated the reaction with both aryl halides. L7 is the first example of a hybrid (alkyl)aryl ligand that provides superior results as compared to its analogues with identical alkyl- or arylphosphorus substituents,19d offering another tunable site for better control of catalyst activity. We anticipate that this methodology will find applications in both academic and pharmaceutical settings, and we expect the new class of biarylphosphine ligands reported here to influence our future design of ligands.29
Acknowledgments
Research reported in this paper was supported by the National Institutes of Health under Award Number GM58160. P.R.-C. acknowledges the “la Caixa” Fellowship for a predoctorate fellowship. We thank Dr. Peter Müller (Massachusetts Institute of Technology, MIT) for X-ray structural analysis. We thank Nootaree Niljianskul for providing L4 and Dr. Naoyuki Hoshiya for helpful discussions and preliminary studies. We thank Nicholas C. Bruno (MIT), Phillip J. Milner (MIT), Dr. Aaron C. Sather (MIT), and Dr. Michael Pirnot for assistance in the preparation of this manuscript.
Supporting Information Available
Experimental procedures, experimental and spectroscopic data for the new compounds, kinetic graphs, and X-ray crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): MIT has patents on some of the ligands and precatalysts described in this work from which S.L.B. as well as former or current co-workers receive royalty payments.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Wanka L.; Iqbal K.; Schreiner P. R. Chem. Rev. 2013, 113, 3516. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu J.; Obando D.; Liao V.; Lifa T.; Codd R. Eur. J. Med. Chem. 2011, 46, 1949. [DOI] [PubMed] [Google Scholar]; c Lamoureux G.; Artavia G. Curr. Med. Chem. 2010, 17, 2967. [DOI] [PubMed] [Google Scholar]; d Gerzon K.; Kau D. J. Med. Chem. 1967, 10, 189. [DOI] [PubMed] [Google Scholar]; e Meenakshi J.; Vangapandu S.; Sachdeva S.; Singh S.; Singh P. P.; Jena G. B.; Tikoo K.; Ramarao P.; Kaul C. L.; Jain R. J. Med. Chem. 2004, 47, 285. [DOI] [PubMed] [Google Scholar]; f Rapala R. T.; Kraay R. J.; Gerzon K. J. Med. Chem. 1965, 8, 580. [DOI] [PubMed] [Google Scholar]; g Spasov A. A.; Khamidova T. V.; Bugaeva L. I.; Morozov I. S. Pharm. Chem. J. 2000, 34, 1. [PubMed] [Google Scholar]; h Morozov I. S.; Ivanova I. A.; Lukicheva T. A. Pharm. Chem. J. 2001, 35, 5. [Google Scholar]; i Shokova E. A.; Kovalev V. V. Russ. J. Org. Chem. 2012, 48, 1007. [Google Scholar]; j Valente S.; Conte M.; Tardugno M.; Massa S.; Nebbioso A.; Altucci Y.; Mai A. ChemMedChem 2009, 4, 1411. [DOI] [PubMed] [Google Scholar]
- Barker T. J.; Jarvo E. J. Angew. Chem., Int. Ed. 2011, 50, 8325. [DOI] [PubMed] [Google Scholar]
- a Xiao Q.; Tian L.; Tan R.; Xia Y.; Qiu D.; Zhang Y.; Wang J. Org. Lett. 2012, 14, 4230. [DOI] [PubMed] [Google Scholar]; b Berman A. M.; Johnson J. S. J. Org. Chem. 2006, 71, 219. [DOI] [PubMed] [Google Scholar]
- Rucker R. P.; Whittaker A. M.; Dang H.; Lalic G. Angew. Chem., Int. Ed. 2012, 51, 3953. [DOI] [PubMed] [Google Scholar]
- a Schlummer B.; Scholz U. Adv. Synth. Catal. 2004, 346, 1599. [Google Scholar]; b Hartiwg J. F. Nature 2008, 455, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tasler S.; Mies J.; Lang M. Adv. Synth. Catal. 2007, 349, 2286. [Google Scholar]; d Corbet J. P.; Mignani G. Chem. Rev. 2006, 106, 2651. [DOI] [PubMed] [Google Scholar]; e Carey J. S.; Laffan D.; Thomson C.; Williams M. T. Org. Biomol. Chem. 2006, 4, 2337. [DOI] [PubMed] [Google Scholar]; f Dugger R. W.; Ragan J. A.; Brown Ripin D. H. Org. Process Res. Dev. 2005, 9, 253. [Google Scholar]; g Robinson G. E.; Cunningham O. R.; Dekhane M.; McManus J. C.; O’Kearney-McMullan A.; Mirajkar A. M.; Mishra V.; Norton A. K.; Venugopalan B.; Williams E. G. Org. Process Res. Dev. 2004, 8, 925. [Google Scholar]; h Hong J. B.; Davidson J. P.; Jin Q.; Lee G. R.; Matchett M.; O’Brien E.; Welch M.; Bingenheimer B.; Sarma K. Org. Process Res. Dev. 2014, 18, 228. [Google Scholar]; i Liu Y.; Prashad M.; Shieh W.-C. Org. Process Res. Dev. 2014, 18, 239. [Google Scholar]
- a Hartwig J. F. Acc. Chem. Res. 2008, 41, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Surry D. S.; Buchwald S. L. Angew. Chem., Int. Ed. 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Marion N.; Navarro O.; Mei J.; Stevens M. D.; Scott N. M.; Nolan S. P. J. Am. Chem. Soc. 2006, 128, 4101. [DOI] [PubMed] [Google Scholar]; d Abdel-Hadi M.; Avola S.; Dubovyk I.; Hadei N.; Kantchev E. A. B.; O’Brien C. J.; Sayah M.; Valente C.; Organ M. G. Chem.—Eur. J. 2008, 14, 2443. [DOI] [PubMed] [Google Scholar]; e Raders S. M.; Moore J. N.; Parks J. K.; Miller A. D.; Leißing T. M.; Kelley S. P.; Rogers R. D.; Shaughnessy K. H. J. Org. Chem. 2013, 4649. [DOI] [PubMed] [Google Scholar]
- a Wolfe J. P.; Wagaw S.; Marcoux J. F.; Buchwald S. L. Acc. Chem. Res. 1998, 805. [Google Scholar]; b Wolfe J. P.; Buchwald S. L. J. Org. Chem. 2000, 65, 1144. [DOI] [PubMed] [Google Scholar]; c Shen Q.; Ogata T.; Hartwig J. F. J. Am. Chem. Soc. 2008, 130, 6586. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Broggi J.; Clavier H.; Nolan S. P. Organometallics 2008, 27, 5525. [Google Scholar]; e Dumrath A.; Lübbe C.; Neumann H.; Jackstell R.; Beller M. Chem.—Eur. J. 2011, 17, 9599. [DOI] [PubMed] [Google Scholar]; f Tewari A.; Hein M.; Zapf A.; Beller M. Tetrahedron 2005, 61, 9705. [Google Scholar]; g Ehrentraut A.; Zapf A.; Beller M. J. Mol. Catal. A 2002, 182, 5154. [Google Scholar]; h Samblanet D. C.; Schmidt J. A. R. J. Organomet. Chem. 2012, 720, 7. [Google Scholar]; i Park S.-E.; Kang S. B.; Jung K.-J.; Won J.-E.; Lee S.-G.; Yoon Y.-J. Synthesis 2009, 5, 815. [Google Scholar]; j Wheaton C. A.; Bow J-P. J.; Stradiotto M. Organometallics 2013, 32, 6148. [Google Scholar]; k Lundgren R. J.; Sappong-Kumankumah A.; Stradiotto M. Chem.—Eur. J. 2010, 16, 1983. [DOI] [PubMed] [Google Scholar]; l Larsen S. B.; Bang-Andersen B.; Johansen T. N.; Jørgensen M. Tetrahedron 2008, 64, 2938. [Google Scholar]; m Mathes C.; Lehmann C. W.; Furstner A. Chem.—Eur. J. 2001, 7, 5299. [DOI] [PubMed] [Google Scholar]; n Chianese A. R.; Shaner S. E.; Tendler J. A.; Pudalov D. M.; Shopov D. Y.; Kim D.; Rogers S. L.; Mo A. Organometallics 2012, 31, 7359. [Google Scholar]
- a Fors B. P.; Watson D. A.; Biscoe M. R.; Buchwald S. L. J. Am. Chem. Soc. 2008, 130, 13552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Maiti D.; Fors B. P.; Henderson J. L.; Buchwald S. L. Chem. Sci. 2011, 2, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno N. C.; Tudge M. T.; Buchwald S. L. Chem. Sci. 2013, 4, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Biscoe M. R.; Barder T. E.; Buchwald S. L. Angew. Chem., Int. Ed. 2007, 46, 7232. [DOI] [PubMed] [Google Scholar]
- a Tolman C. A. Chem. Rev. 1977, 77, 313. [Google Scholar]; b Brink A.; Roodt A.; Steyl G.; Visser H. G. Dalton Trans. 2010, 39, 5572. [DOI] [PubMed] [Google Scholar]
- a Blackmond D. G. Angew. Chem., Int. Ed. 2005, 44, 4302. [DOI] [PubMed] [Google Scholar]; b Mathew J. S.; Klussmann M.; Iwamura H.; Valera F.; Futran A.; Emanuelsson E. A. C.; Blackmond D. G. J. Org. Chem. 2006, 71, 4711. [DOI] [PubMed] [Google Scholar]; c Zuend S. J.; Jacobsen E. N. J. Am. Chem. Soc. 2009, 131, 15358. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Devery J. J. III; Conrad J. C.; MacMillan D. W. C; Flowers R. A. II. Angew. Chem., Int. Ed. 2010, 49, 6106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Shekhar S.; Ryberg P.; Hartwig J. F.; Mathew J. S.; Blackmond D. G.; Strieter E. R.; Buchwald S. L. J. Am. Chem. Soc. 2006, 128, 3584. [DOI] [PubMed] [Google Scholar]; f Strieter E. R.; Blackmond D. G.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 4120. [DOI] [PubMed] [Google Scholar]
- Due to the limited solubility of precatalyst P4, amine 3 was injected into the reaction vial to initiate the reaction. The reaction mixture was prepared with addition of P4, aryl halide 6, base, and solvent. Before 3 was added, precatalyst P4 was activated in the presence of the base and converted into the respective oxidative addition complex, which enabled precatalyst activation to be excluded from the calorimetry profile.
- a Barrios-Landeros F.; Hartwig J. F. J. Am. Chem. Soc. 2005, 127, 6944. [DOI] [PubMed] [Google Scholar]; b Barrios-Landeros F.; Carrow B. P.; Hartwig J. F. J. Am. Chem. Soc. 2009, 131, 8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attempts to observe the resting state of the catalyst by 31P NMR were unsuccessful; the hypothesis that oxidative addition is the rate-determining step is the most consistent with the available data.
- Calculated reaction barriers for the oxidative addition of PhCl with LPd: L1, ΔG⧧ = 10.8 kcal/mol; L2, ΔG⧧ = 16.5 kcal/mol.Hicks J. D.; Hyde A. M.; Martinez Cuezva A.; Buchwald S. L. J. Am. Chem. Soc. 2009, 131, 16720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The reactivity trend toward oxidative addition follows the order ArI > ArBr ≈ ArOTf > ArCl:; a Crabtree R. H.The Organometallic Chemistry of the Transition Metals, 5th ed.; John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]; b Stille J. K.; Lau K. S. Y. Acc. Chem. Res. 1977, 10, 434. [Google Scholar]; c Alcazar-Roman L. M.; Hartwig J. F. Organometallics 2002, 21, 491. [Google Scholar]
- Addition of different equivalents of base had no effect on the reaction. However, it was observed that the base was not totally soluble in the reaction solvent; therefore, the possibility of deprotonation being the rate-determining step cannot be completely disregarded.
- a Spessard G. O.; Meissler G. L.. Organometallic Chemistry, 2nd ed.; Oxford University Press: New York, 2010. [Google Scholar]; b Yamashita M.; Hartwig J. F. J. Am. Chem. Soc. 2004, 126, 5344. [DOI] [PubMed] [Google Scholar]; c Mann G.; Shelby Q.; Roy A. H.; Hartwig J. F. Organometallics 2003, 22, 2775. [Google Scholar]; d Su M.; Buchwald S. L. Angew. Chem., Int. Ed. 2012, 51, 4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To obtain accurate calorimetry data, the reaction time should be at least 15–20 min.
- These conditions are comparable to those of the reaction setup with precatalyst P4, in which the respective oxidative addition complex was the palladium source as well, thus excluding precatalyst activation from the calorimetry profile.
- Blackmond D. G.; Schultz T.; Mathew J. S.; Loew C.; Rosner T.; Pfaltz A. Synlett. 2006, 18, 3135. [Google Scholar]
- Although the scenario in which the amine deprotonation step is rate-determining could not be completely disregarded, the great acceleration caused by the more hindered ligand, L7, suggests that the rate-determining step is in fact reductive elimination.
- 4-Bromobenzonitrile was used to prepare oxidative addition complexes due to poor crystallinity of oxidative addition complexes of aryl halide 6 or 12.
- Yang Y.; Niedermann K.; Han C.; Buchwald S. L. Org. Lett. 2014, 16, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeev A. G.; Artamkina G. A.; Beletskaya I. P. Tetrahedron Lett. 2003, 44, 4719. [Google Scholar]
- a Baumann M.; Baxendale I. R.; Ley S. V.; Nikbin N. J. Org. Chem. 2011, 7, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhan P.; Li D.; Chen X.; Liu X.; Clercq E. D. Curr. Med. Chem. 2011, 18, 29. [DOI] [PubMed] [Google Scholar]; c Su M.; Hoshiya N.; Buchwald S. L. Org. Lett. 2014, 53, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In the absence of Pd, the ArO-t-Bu product was also observed. It is unclear if its formation in certain cases occurs through an SNAr mechanism or through a cross-coupling pathway.
- Gladysz J. A.; Bedford R. B.; Fujita M.; Gabbai F. P.; Goldberg K. I.; Holland P. L.; Kiplinger J. L.; Krische M. J.; Louie J.; Lu C. C.; Norton J. R.; Petrukhina M. A.; Ren T.; Stahl S. S.; Tilley T. D.; Webster C. E.; White M. C.; Whiteker G. T. Organometallics 2014, 33, 1505. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.