

Abstract
Population genomic studies have the potential to address many unresolved questions about microbial pathogens by facilitating the identification of genes underlying ecologically important traits such as novel virulence factors and adaptations to humans or other host species. Additionally, this framework improves estimations of population demography and evolutionary history to accurately reconstruct recent epidemics and identify the molecular and environmental factors that resulted in the outbreak. The Lyme disease bacterium, Borrelia burgdorferi, exemplifies the power and promise of the application of population genomics to microbial pathogens. We discuss here the future of evolutionary studies in B.burgdorferi - focusing on the primary evolutionary forces of horizontal gene transfer, natural selection, and migration - as investigations transition from analyses of single genes to genomes.
Keywords: Population genomics, horizontal gene transfer, natural selection, demography, microbial pathogens, vector-borne pathogens
Population genomics of microbial pathogens
The central purpose of population genetics (see Glossary) is to identify environmental, historical, and evolutionary processes that shape naturally occurring genetic variation. The power to detect and describe these processes from natural populations has increased in parallel with advances in sequencing technologies, which provide ever increasing amounts of genetic information. This trend has resulted in dramatic improvements in both the accuracy and resolution of evolutionary inferences and has expanded the types of hypotheses that are addressable [1–3]. The application of population genetic theory to molecular data from DNA sequences, which describes the relatedness among alleles, reveals the history of past evolutionary processes
Population genetic analyses of individual loci have resulted in outstanding progress in the study of many microbial pathogens [4]. However, analyses of a single locus or multilocus datasets are often inadequate to study evolutionary processes on fine spatial and temporal scales, precisely the scales at which evolutionary and mechanistic processes can be detected and described [5, 6]. By contrast, genomic datasets from populations of individuals are likely to contain sufficient information to precisely infer demographic and evolutionary processes at very fine scales by applying classical population genetic theory across all genetic loci [7]. Simultaneous analysis of all genomic loci is more powerful than the sum of analyses of individual loci because loci with distinct patterns of variation, caused by a specific evolutionary process, can be identified against the backdrop of the genome-wide allele frequency distributions which are affected by neutral and demographic processes [8–10].
Investigations of microbial pathogens are particularly poised to take advantage of the power of population genomics due to the minimal cost of sequencing small genomes [11–16]. Full genome datasets can be acquired from every sampled individual such that all novel variations from multiple environmental conditions and spatial scales can be detected [17–19]. The developing single-cell and selective whole genome amplification technologies circumvent the need for laboratory culture and the biases associated with culturing, providing cost-effective and practical methods to obtain sufficiently pure genomic DNA for high-throughput sequencing [11–13, 15].
Remarkable advances in the evolutionary biology of microbial pathogens, such as the identification of host-associated and virulence mutations [17, 18], the identification of selective pressures within and among environments [20], the discovery of genetic variation in pathogen populations previously thought to be clonal [21–23], and the reconstruction of recent epidemics [24–30] have already been realized in population genomic studies. Population genomic studies can also directly impact public health [31] as demonstrated by a recent study of Staphylococcus aureus in hospital patients that found no evidence of inter-patient transmission, in direct contrast to previous conclusions based on low-resolution typing methods [32]. The results of this study can guide hospital infection-control policies and may affect health insurance reimbursement policies [32].
The power and promise of population genomics for bacterial pathogens is well illustrated by the bacterial spirochete, Borrelia burgdorferi sensu stricto, a member of the B. burgdorferi sensu lato species complex [33–37]. B. burgdorferi is an obligate vector-borne pathogen, transmitted among vertebrate wildlife species by ticks in the genus Ixodes [38, 39]. Infected ticks can bite and transmit B. burgdorferi to humans resulting in Lyme borreliosis, the most common vector-borne disease in North America [40]. Population genomic analyses are ideal for identifying genes that are locally adapted to host species or to different geographic and climatic conditions [17, 41]. Additionally, population genomic analyses can infer historical and recent range expansions at fine spatial and temporal scales with the potential to identify causative mechanisms [25, 30, 39, 41]. Genomic studies of the currently available B. burgdorferi genomes have been comprehensively reviewed elsewhere [39, 42, 43]. Here we discuss the future of B. burgdorferi population biology as investigations transition from analyses of single genes to multiple genes to genomes.
Genomic architecture of B. burgdorferi
The genomic architecture of Borrelia is unique among bacteria, consisting of a linear chromosome (~1Mb) containing primarily housekeeping genes and numerous linear and circular plasmids (collectively ~0.6Mb) that contain the majority of genes necessary for infection of vertebrate hosts and tick vectors [42, 44, 45]. The order of genes on the plasmids is highly variable among strains which impedes efficient genome assembly but has little effect on many population genomic analyses [23]. The variation in gene content on plasmids, however, can impact the interpretation of investigations of horizontal gene transfer, natural selection, and phylogeography. In the following sections we explore some of the potential impacts of the complex genomic architecture of B. burgdorferi on the interpretation of population genomic analyses.
Horizontal gene transfer
The population genetic processes that govern evolutionary change include mutation and recombination, natural selection, genetic drift, and gene flow. Although population genomic analyses are poised to make dramatic advances in each of these areas, B. burgdorferi genomic data have been used most extensively in the study of mutation and recombination. Horizontal gene transfer is an important evolutionary force that can accelerate adaptation by mitigating the effects of clonal interference, increasing the effective strength of natural selection, and combining beneficial mutations in a single genome and purging deleterious mutations [46–49].
Early investigations of horizontal gene transfer focusing on two chromosomal loci and one plasmid-borne locus suggested that B. burgdorferi was perfectly clonal [50, 51]. The strict clonality of B. burgdorferi has been consistently supported by multilocus studies with the notable exceptions of the surface-exposed ospC and ospE/F-related loci [23, 42, 52–55]. These apparent recombination hotspots are perhaps more indicative of natural selection favoring diversity than a proclivity for recombination as horizontally transferred alleles under diversifying selection are more likely to be retained, and thus detected [47]. Among all other loci, only very limited recombination was detected when B. burgdorferi from geographically-isolated regions were analyzed together [56], and no recombination was detected when only one geographic region was analyzed [57]. Simultaneous analysis of eight loci was finally able to detect a strikingly low rate of horizontal gene transfer among isolates from a single geographic region [58].
Despite the conclusion of nearly complete clonality derived from single and multilocus studies, evidence from genome-level investigations suggested that three-quarters of the standing sequence diversity in B. burgdorferi can be attributed to re-assortment of polymorphisms through recombination while only one-quarter is due to point mutations [23]. Although linkage disequilibrium is expected to be inversely correlated with genetic distance, the pattern of linkage disequilibrium in B. burgdorferi found in these studies was much more complex [23, 43, 59]. These studies suggest that horizontal gene transfer among B. burgdorferi lineages is ‘localized,’ leading to the anomalous result of a positive correlation between linkage disequilibrium and genetic distance [23]. Specifically, evidence of recombination was apparent among nucleotides within 500 bases of each other, but not at larger genetic distances, resulting in the conclusion of “pervasive localized recombination but genome-wide clonality” [23]. The authors of this study caution that recombination and point mutation rate estimates are strongly affected by sampling biases which could alter the interpretation of their results [23]. That is, this counter-intuitive result may be explained by an overestimation of recombination rates caused by analyses of a set of genomes which were not sampled randomly but chosen to represent the global diversity of B. burgdorferi [23]. Supporting this interpretation, a recent study of randomly sampled B. burgdorferi genomes estimated a much lower recombination rate [60]. Future studies of horizontal gene transfer in B. burgdorferi using randomly sampled genomes within powerful analytical frameworks [61–63] are likely to resolve outstanding questions regarding the rate of horizontal gene transfer in natural B. burgdorferi populations.
Natural selection
Innumerable population genetic studies in many pathogen species have identified genes under natural selection, including the ospC and vlsE loci in B. burgdorferi. The ospC locus encodes a surface exposed protein that may promote dissemination in vertebrate hosts, although its precise function is still debated [64–67]. Alleles at the ospC locus have an exceptionally long coalescence time and are found in nearly every natural B. burgdorferi population at relatively even frequencies, both of which suggest that balancing selection maintains the variation at ospC [23, 68, 69]. Balancing selection acting on ospC occurs through either multiple niche polymorphism or negative frequency dependent selection, both of which have some empirical or theoretical support. Empirical studies from natural populations and laboratory experiments have demonstrated that there are non-random associations between ospC alleles and host species supporting the multiple niche polymorphism model of balancing selection [65, 70–73] whereas mathematical modeling has demonstrated that negative frequency dependent selection could be sufficient to maintain the ospC polymorphism in the absence of multiple niche polymorphism [23]. Natural selection favoring an increased evolutionary rate at the immune evasion locus, vlsE, has also been described [74–80]. Although greater rates of vlsE sequence evolution provide a selective advantage by enhancing immune evasion capability, empirical data suggests that the rate of sequence evolution is limited by natural selection for molecular functionality such as highly-efficient translation [81].
Despite some success in identifying loci under natural selection and associating those genes with an adaptive trait, there are multiple challenges with the candidate gene approach. First, there are no data-derived expectations of the effects of neutral evolutionary or demographic processes on patterns of sequence variation [82]. Without an empirical neutral model, locus-specific selection cannot be separated from neutral processes or processes that have genome-wide effects. Targets of natural selection are especially difficult to identify in highly clonal species like B. burgdorferi without comparison to many other loci throughout the genome [83]. Second, candidate loci typically are chosen due to their antigenicity or surface exposure, which may not be indicative of an ecologically-relevant gene or even a gene with alleles that vary in their effects on fitness. For example, both the FlaB and OspA proteins are antigenic but have little or no allelic variation within populations and no evidence of positive natural selection has been detected [39, 84–86]. Third and most importantly, the candidate gene approach cannot identify the genes relevant to the phenotype under investigation if they are not included in the a priori chosen set of loci. Thus, many genes that are relevant to the phenotype under investigation will not be identified without whole genome data.
Population genomic analyses can address many of these shortcomings. Not only does population genomics eliminate the need for a priori candidate genes, but these approaches can also distinguish selective, neutral, and demographic processes that shape patterns of allelic variation differentially across loci [8–10]. For example, although selective sweeps and population bottlenecks have similar effects on the coalescence times and genetic diversity of a single locus, population bottlenecks affect all loci whereas the impact of selection is locus-specific (Figure 1). Population genomic analyses provide a framework to simultaneously estimate both genome-wide averages and statistical outliers in order to identify genes under natural selection with minimum a priori assumptions [8, 10]. Genome-wide averages provide a baseline of demographic and neutral processes while outliers indicate an evolutionary process such as natural selection acting on specific loci [8, 10]. Although several commonly used statistical tests are ineffective in highly clonal populations such as B. burgdorferi (Tajima’s D; FST), analyses that assess the rate of fixation of amino acids (dN/dS; Relative Rates tests), especially those that are informed by the neutral phylogeny (Zonal analysis; Convergence tests), can be highly effective in clonal pathogen species [7]. For example, 39 novel targets of positive natural selection for drug resistance were identified in Mycobacterium tuberculosis, a highly-clonal species, using the Phylogenetic Convergence test and a genome-wide phylogeny from 123 strains [87].
Figure 1.

Population genomic analyses reveal selective, neutral, and demographic processes affecting loci across the genome. Population genetic summary statistics, such as πA/πS, can be calculated as continuous variables along the length of the genome. Evolutionary processes such as (I) regions under purifying selection, (II) neutrally evolving regions, (III) regions experiencing positive selection, and (IV) horizontal gene transfer events leave distinct signatures on these summary statistics. The topology of phylogenies, including branch lengths and evolutionary relationships, inferred from different loci along the genome also reflect the particular evolutionary or demographic processes governing the allelic variation along the genome.
Population genomic studies of adaptive molecular variation in B. burgdorferi have the potential to identify genes underlying ecologically-relevant traits and describe the fitness consequences of allelic variation at these loci. Although single-gene analyses have made some advances, many of the major unresolved questions can be readily addressed through population genomic analyses including why only some B. burgdorferi strains are regularly found in human infections. Why are B. burgdorferi strains associated with different symptoms in humans? Are B. burgdorferi locally adapted to environmental conditions or host species? How many genes influence host species specialization and what are their relative effect sizes? To what extent does linkage disequilibrium and genomic architecture limit adaptation? Similar questions have been addressed using a population genomic framework in other bacterial pathogen systems. For example, population genomic analysis of 3,615 genome sequences permitted the description of the timing of mutations, horizontal gene transfer events, and natural selection that led to the “flesh-eating” Streptococcus epidemic [18].
Phylogeography and Historical Demography
Emerging and re-emerging infectious pathogens are a substantial burden on both human health and economics [88–90]. Emergence and spread of pathogens is thought to be driven largely by environmental and ecological factors, many of which are changing rapidly due to human activity [91–93]. The history of migratory events and changes in population sizes in microbial populations which have led to the current species distribution is most readily studied using phylogeographic tools. Phylogeography incorporates geographic, ecological, and environmental correlates onto sequence-based phylogenies to address hypotheses and interpret patterns of present day species distributions in light of historical events [94]. Results from very fine scale phylogeographic investigations can identify mechanistic drivers of demography which can be used to predict future population or range expansions [30, 95]. The resolution and accuracy of phylogeographic studies is correlated with the amount of DNA sequence information analyzed and the number of individuals sampled (Figure 2)[21, 96, 97]. Thus, genome-level datasets with population-level sampling will improve inferences about historical demography and evolutionary history.
Figure 2.
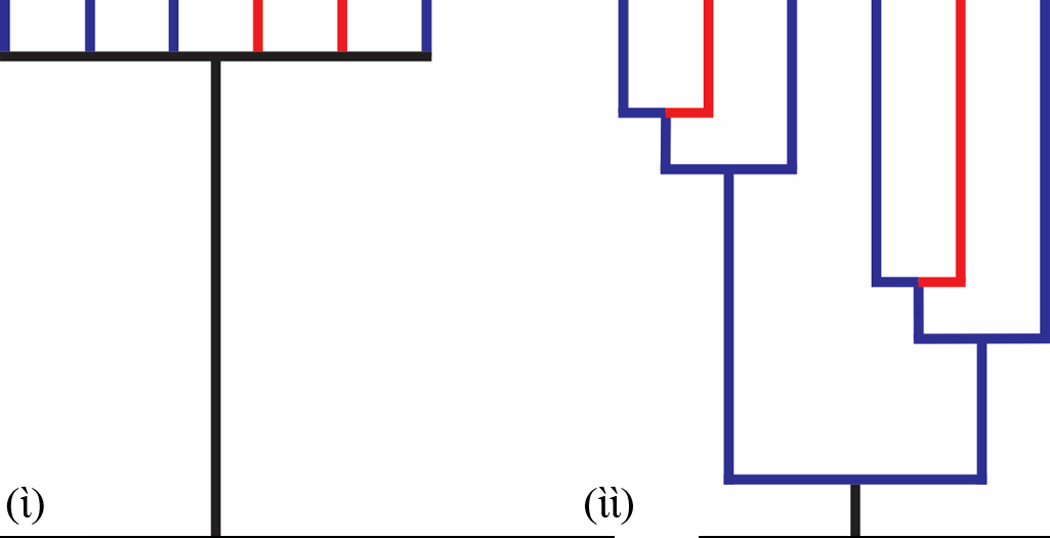
The resolution and accuracy of phylogeographic inferences increases with increasing DNA sequence information. (ὶ) Limited sequence information results in a largely unresolved phylogeny which provides little power to infer historical demography and migratory history. For example, the direction and timing of migratory events between locations (red or blue) cannot be inferred from unresolved trees if the ancestral location of a clade cannot be reconstructed. (ὶὶ) The ancestral location (blue) can be inferred from fully resolved phylogenies permitting the detection of migration events (from the blue location to red location). Further, the relative timing of migration events can be inferred from the branch lengths of the phylogeny. The accuracy of the inferred direction and timing of migration events depends on the statistical confidence in the reconstructed phylogeny [95, 97].
B. burgdorferi populations exist in four major isolated regions: the Northeastern, Midwestern, and far western North America, and Europe. At these coarse spatial scales, sequence information from a single chromosomal locus was sufficient to detect barriers to gene flow between the Midwestern and Northeastern United States [98]. However, the among-region population genetic structure that was detected was driven by the absence of one of the three basal phylogenetic clades from the Midwestern region, potentially due to natural selection [98]. Analyses using only the clades present in both regions is not sufficient to detect population genetic structure even at these coarse scales and the sequence information from a single gene is also insufficient to detect barriers to gene flow within the geographic regions [98].
Phylogeographic analyses of multiple loci from B. burgdorferi sampled across a larger geographic range improved the resolution of the inferred migratory history of B. burgdorferi. Evidence of limited historical gene flow from multi-locus studies suggested that past migration events originated in eastern America and subsequently colonized the American Midwest [58, 99], which agrees with the historical migration of the tick vector [98, 100, 101]. However, phylogenetic analyses of multiple loci have not resolved the debate on the geographic origin of B. burgdorferi, as a European and an American origin both have statistical support [52, 102–104]. Fine and even coarse temporal and spatial scale resolution of the directions, rates, and timing of migration events, as well as the geographic origin of the species, will require analyses of genomes from geographically stratified samples and multiple outgroup genomes.
The rate of human Lyme borreliosis incidence and the geographic range of affected areas have both increased continuously since the disease was described by Dr. Willy Burgdorfer and colleagues in 1981 [105, 106]. Application of coalescent-based phylogeography, as implemented in BEAST and other programs [107], to randomly sampled genomes from natural B. burgdorferi populations will allow fine scale reconstruction of the direction and timing of these recent population and range expansions. These analytical frameworks can also be used to correlate ecological and environmental parameters with the demographic history of B. burgdorferi to identify factors that have led to the recent upsurge in Lyme borreliosis. Coalescent-based phylogeography has been used to reconstruct the spatial epidemic history of many pathogens including Avian influenza and rabies viruses [30, 108]. Additionally, this framework was used to identify environmental factors, such as the construction of transportation networks in Africa, that drove the early population dynamics of HIV-1 which lead to the current global pandemic [109].
Challenges for future B. burgdorferi population genomic studies
The analytical power of population genomics offers great promise, but it also has the potential to lead the field astray. All population genomic analyses have underlying assumptions that, when violated, can result in inaccurate inferences that nonetheless have strong statistical support. It falls upon researchers to determine if their data conform to the assumptions of the analyses as the majority of analytical platforms cannot detect datasets that violate their assumptions. The sets of sequenced genomes that are publically available for many species rarely conform to the assumptions of population genomic analyses; the available B. burgdorferi genomes are no exception. The 42 currently available complete and draft B. burgdorferi genomes were collected from disparate geographic regions at different time points, only one of which was isolated from a known natural vertebrate host, and many were collected from humans, a dead-end species for this pathogen [110]. Although it is tempting to draw population inferences from these genomes, these samples do not constitute random samples from a population that shares an evolutionary history. Future studies that obtain host and environment-associated information in addition to randomly sampled B. burgdorferi genomes from one or multiple populations can properly harness the power of population genomics to accurately infer mutational processes, adaptation to local environments, and population demography.
The association of B. burgdorferi phenotypes with the presence or absence of genes can be challenging as genome assembly is complicated by the complex genomic architecture. Incorrect genome assembly can result in apparent but not actual gene deletions that can conceal or identify associations between genes and phenotypes. Furthermore, B. burgdorferi in laboratory culture quickly lose plasmids, resulting in true gene loss which is unrelated to the phenotype observed in nature [111]. Proper experimental controls as well as long-read sequencing [112, 113] and advances in culture-free sequencing technologies [12, 13, 16] can be used to overcome some of these challenges.
Coalescence-based analyses assume that all loci in a genomic region share a common evolutionary history, an assumption that is violated by horizontal gene transfer. Genomic regions that do not share a common evolutionary history due to horizontal gene transfer can be readily identified in many analyses [61, 62, 63] and unlinked regions must be analyzed independently. An important consideration for multilocus and genomic B. burgdorferi studies is that loci will not share a common evolutionary history if they are derived from different strains in a mixed infection. Loci from different strains in a mixed infection that are incorrectly assembled into a single genome will cause erroneous conclusions from coalescence-based analyses. Genomic regions that do not share a common evolutionary history due incorrect assignment of loci among genomes that were sequenced from a mixed infection can be overcome with deeper sequencing [114] and using computational methods to better assign polymorphic bases [115].
Concluding remarks
Population genomic analyses separate processes that affect specific loci such as natural selection from processes that affect all loci such as genetic drift, migration, and changes in population size. The population demography and evolutionary history of B. burgdorferi can be accurately inferred from genome-wide data while the specific loci can be associated with an ecologically relevant trait. Future population genomic studies of B. burgdorferi should occur in four distinct phases [82]. First, the sample of B. burgdorferi genomes must conform to the assumptions of empirical population genetic data, most notably random sampling from natural populations. Second, each genome sequence must be derived from an individual strain even if the strain was derived from a sample containing multiple strains. Third, genome-wide data should be used to generate null models and estimate the average effects of neutral and demographic processes. Fourth, specific loci that are statistically distinct can be associated with ecologically relevant traits. The final two analytical phases provide a powerful framework to explore the evolutionary history of B. burgdorferi at fine and coarse scales (10’s of kilometers and 1000’s of kilometers, respectively), address many major unresolved questions, and identify candidate loci for future functional investigations.
Population genomics has the potential to unlock the mysteries of adaptive evolution and to refine inferences about evolutionary histories. Genome-wide averages derived from the application of population genetic statistics to randomly sampled full genomes provides a baseline of demographic and neutral processes against which adaptive molecular variation can be discovered at all loci. Identification of the loci that underlie the genetic basis of adaptation for particular traits is perhaps the most exciting application of the population genomics. However, population genomics approaches are equally valuable for improving inferences about the population demography and evolutionary history associated with the recent emergence of Lyme borreliosis.
Highlights.
The complicated ecology and evolutionary history of the Lyme disease pathogen can be resolved using population genomics
Population genomics provides the power to detect fine-scale evolutionary processes including horizontal gene transfer
Population genomic analyses can identify genes underlying ecologically-relevant traits including adaptations to host species
Correlating ecological and environmental parameters with demographic history can elucidate the causes of the recent re-emergence of Lyme disease
Acknowledgements
This work was supported in part by grants from the NIH AI076342, NIH AI097137, NSF DEB-1354184, the Burroughs Wellcome Fund (1012376), the Lyme Research Alliance, the Bay Area Lyme Foundation, and the NSF GRFP DGE-1321851. The authors are very grateful to the anonymous reviewers and the editors who helped to refine and improve the writing of this manuscript.
Glossary
- Population genetics
the study of how natural selection, genetic drift, mutation, and gene flow affect allele frequencies in a population
- Horizontal gene transfer
the transfer of genetic components between organisms of the same generation rather than vertical transfer from parent to offspring
- Linkage disequilibrium
the non-random association of alleles at two or more loci
- Balancing selection
refers to selective processes that favor the maintenance of multiple alleles in a population
- Multiple niche polymorphism
a type of balancing selection in which diversity is maintained when the environment is heterogeneous and no single genotype has the highest fitness in all environments
- Negative frequency dependent selection
a type of balancing selection in which an allele confers a fitness advantage when it is rare in a population
- Historical demography
the quantitative study of historic population parameters such as changes in population size and migration rates
- Phylogeography
the study of the environmental, ecological, and evolutionary processes that are responsible for the current geographic distribution of individuals and species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shapiro BJ, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stukenbrock EH, et al. The making of a new pathogen: insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res. 2011;21:2157–2166. doi: 10.1101/gr.118851.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews KR, Luikart G. Recent novel approaches for population genomics data analysis. Mol. Ecology. 2014;23:1661–1667. doi: 10.1111/mec.12686. [DOI] [PubMed] [Google Scholar]
- 4.Smith JM, et al. Population structure and evolutionary dynamics of pathogenic bacteria. BioEssays. 2000;22:1115–1122. doi: 10.1002/1521-1878(200012)22:12<1115::AID-BIES9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 5.Margos G, et al. Population genetics, taxonomy, phylogeny and evolution of Borrelia burgdorferi sensu lato. Inf., Gen. and Evol. 2011;11:1545–1563. doi: 10.1016/j.meegid.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coletta-Filho HD, et al. Spatial genetic structure of a vector-borne generalist pathogen. App. and Env. Microbiol. 2011;77:2596–2601. doi: 10.1128/AEM.02172-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapiro BJ, et al. Looking for Darwin's footprints in the microbial world. Trends in Microbiol. 2009;17:196–204. doi: 10.1016/j.tim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Hohenlohe PA, et al. Using population genomics to detect selection in natural populations: key concepts and methodological considerations. Int. J. of Plant Sci. 2010;171:1059–1071. doi: 10.1086/656306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright SI, et al. The effects of artificial selection on the maize genome. Science. 2005;308:1310–1314. doi: 10.1126/science.1107891. [DOI] [PubMed] [Google Scholar]
- 10.Black WCI, et al. Population genomics: Genome-wide sampling of insect populations. Annu. Rev. of Ent. 2001;46:441–469. doi: 10.1146/annurev.ento.46.1.441. [DOI] [PubMed] [Google Scholar]
- 11.Lasken RS, McLean JS. Recent advances in genomic DNA sequencing of microbial species from single cells. Nature Rev. Gen. 2014;15:577–584. doi: 10.1038/nrg3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leichty AR, Brisson D. Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics. 2014;198:473–481. doi: 10.1534/genetics.114.165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seth-Smith HMB, et al. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013;23:855–866. doi: 10.1101/gr.150037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson MF, et al. Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Gen. 2012;8:e1003129. doi: 10.1371/journal.pgen.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pain A, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455:799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellegaard KM, et al. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Gen. 2013;9:e1003381. doi: 10.1371/journal.pgen.1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheppard SK, et al. Genome-wide association study identifies vitamin B 5 biosynthesis as a host specificity factor in Campylobacter. Proc. Natl. Acad. Sci. U.S.A. 2013;110:11923–11927. doi: 10.1073/pnas.1305559110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasser W, et al. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1768–E1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pespeni MH, et al. Genome-wide polymorphisms show unexpected targets of natural selection. Proc. R. Soc. B. 2011 doi: 10.1098/rspb.2011.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieberman TD, et al. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nature Gen. 2014;46:82–87. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. of Microbiol. 2008;62:53–70. doi: 10.1146/annurev.micro.62.081307.162832. [DOI] [PubMed] [Google Scholar]
- 22.Namouchi A, et al. After the bottleneck : Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 2012;22:721–734. doi: 10.1101/gr.129544.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haven J, et al. Pervasive recombination and sympatric genome diversification driven by frequency-dependent selection in Borrelia burgdorferi the Lyme disease bacterium. Genetics. 2011;189:951–966. doi: 10.1534/genetics.111.130773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castro-Nallar E, et al. The evolution of HIV: inferences using phylogenetics. Mol. Phylogenetics and Evol. 2012;62:777–792. doi: 10.1016/j.ympev.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morelli G, et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nature Gen. 2010;42:1140–1143. doi: 10.1038/ng.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasko DA, et al. Origins of the E.coli strain causing an outbreak of hemolyticuremic syndrome in Germany. New England J. of Med. 2011:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fitzgerald JR, et al. Evolutionary genomics of Staphylococcus aureus : insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8821–8826. doi: 10.1073/pnas.161098098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broekhuijsen M, et al. Genome-wide DNA microarray analysis of Francisella tularensis strains demonstrates extensive genetic conservation within the species but identifies regions that are unique to the highly virulent F. tularensis subsp. tularensis. J. of Clin. Microbiol. 2003;41:2924–2931. doi: 10.1128/JCM.41.7.2924-2931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruan Y, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. The Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemey P, et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Path. 2014;10:e1003932. doi: 10.1371/journal.ppat.1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Didelot X, et al. Transforming clinical microbiology with bacterial genome sequencing. Nature Rev. Gen. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long SW, et al. Absence of patient-to-patient intrahospital transmission of Staphylococcus aureus as determined by whole-genome sequencing. mBio. 2014;5:e01692–14. doi: 10.1128/mBio.01692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukunaga M, et al. Phylogenetic analysis of Borrelia species based on flagellin gene sequences and its application for molecular typing of Lyme disease borreliae. Int. J. of Sys. and Evol. Microbiol. 1996;46:898–905. doi: 10.1099/00207713-46-4-898. [DOI] [PubMed] [Google Scholar]
- 34.Richter D, et al. Delineation of Borrelia burgdorferi sensu lato species by multilocus sequence analysis and confirmation of the delineation of Borrelia spielmanii sp. nov. Int. J. of Sys. and Evol. Microbiol. 2006;56:873–881. doi: 10.1099/ijs.0.64050-0. [DOI] [PubMed] [Google Scholar]
- 35.Postic D, et al. Multilocus sequence analysis of atypical Borrelia burgdorferi sensu lato isolates--description of Borrelia californiensis sp. nov., genomospecies 1 and 2. Int. J. of Med. Microbiol. 2007;297:263–271. doi: 10.1016/j.ijmm.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Margos G, et al. A new Borrelia species defined by multilocus sequence analysis of housekeeping genes. App. and Env. Microbiol. 2009;75:5410–5416. doi: 10.1128/AEM.00116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morlon H, et al. Explosive radiation of a bacterial species group. Evolution. 2012;66:2577–2586. doi: 10.1111/j.1558-5646.2012.01598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol. Rev. 1986;50:381–400. doi: 10.1128/mr.50.4.381-400.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu WG, Martin CL. Evolutionary genomics of Borrelia burgdorferi sensu lato: Findings, hypotheses, and the rise of hybrids. Inf., Gen. and Evol. 2014;27:576–593. doi: 10.1016/j.meegid.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rizzoli A, et al. Lyme borreliosis in Europe. Eurosurveillance. 2011;16:19906. [PubMed] [Google Scholar]
- 41.Coleman ML, Chisholm SW. Ecosystem specific selection pressures revealed through comparative population genomics. Proc. Natl. Acad. Sci. U.S.A. 2010;17:18634–18639. doi: 10.1073/pnas.1009480107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casjens SR, et al. Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS ONE. 2012;7:e33280. doi: 10.1371/journal.pone.0033280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brisson D, et al. Genetics of Borrelia burgdorferi. Annu. Rev. of Gen. 2012;46:515–536. doi: 10.1146/annurev-genet-011112-112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaconas G, Kobryn K. Structure, function, and evolution of linear replicons in Borrelia. Annu. Rev. of Microbiol. 2010;64:185–202. doi: 10.1146/annurev.micro.112408.134037. [DOI] [PubMed] [Google Scholar]
- 45.Samuels DS. Gene regulation in Borrelia burgdorferi. Annu. Rev. of Microbiol. 2011;65:479–499. doi: 10.1146/annurev.micro.112408.134040. [DOI] [PubMed] [Google Scholar]
- 46.Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011;35:957–976. doi: 10.1111/j.1574-6976.2011.00292.x. [DOI] [PubMed] [Google Scholar]
- 47.Vos M. Why do bacteria engage in homologous recombination? Trends in Microbiol. 2009;17:226–232. doi: 10.1016/j.tim.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Polz MF, et al. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends in Gen. 2013;29:170–175. doi: 10.1016/j.tig.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gyles C, Boerlin P. Horizontally transferred genetic elements and their role in pathogenesis of bacterial disease. Vet. Path. Online. 2013 doi: 10.1177/0300985813511131. 0300985813511131. [DOI] [PubMed] [Google Scholar]
- 50.Dykhuizen DE, et al. Borrelia burgdorferi is clonal : Implications for taxonomy and vaccine development. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10163–10167. doi: 10.1073/pnas.90.21.10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith JM, Smith NH. Detecting recombination from gene trees. Mol. Biol. and Evol. 1998;15:590–599. doi: 10.1093/oxfordjournals.molbev.a025960. [DOI] [PubMed] [Google Scholar]
- 52.Margos G, et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8730–8735. doi: 10.1073/pnas.0800323105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stevenson B, Miller JC. Intra- and interbacterial genetic exchange of Lyme disease spirochete erp genes generates sequence identity amidst diversity. J. of Mol. Evol. 2003;57:309–324. doi: 10.1007/s00239-003-2482-x. [DOI] [PubMed] [Google Scholar]
- 54.Barbour AG, Travinsky B. Evolution and distribution of the ospC gene, a transferable serotype determinant of Borrelia burgdorferi. mBio. 2010;1:e00153–10. doi: 10.1128/mBio.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brisson D, et al. Distribution of cp32 prophages among Lyme disease-causing spirochetes and natural diversity of their lipoprotein-encoding erp loci. App. and Env. Microbiol. 2013;79:4115–4128. doi: 10.1128/AEM.00817-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brisson D, et al. Evolution of northeastern and midwestern Borrelia burgdorferi United States. Em. Inf. Dis. 2010;16:911–917. doi: 10.3201/eid1606.090329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bunikis J, et al. Sequence typing reveals extensive strain diversity of the Lyme borreliosis agents Borrelia burgdorferi in North America and Borrelia afzelii in Europe. Microbiology. 2004;150:1741–1755. doi: 10.1099/mic.0.26944-0. [DOI] [PubMed] [Google Scholar]
- 58.Margos G, et al. Two boundaries separate Borrelia burgdorferi populations in North America. App. and Env. Microbiol. 2012;78:6059–6067. doi: 10.1128/AEM.00231-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mongodin EF, et al. Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation. BMC Genomics. 2013;14:693–693. doi: 10.1186/1471-2164-14-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacquot M, et al. Comparative population genomics of the Borrelia burgdorferi species complex reveals high degree of genetic isolation among species and underscores benefits and constraints to studying intra-specific epidemiological processes. PLoS ONE. 2014;9:e94384. doi: 10.1371/journal.pone.0094384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin DP, et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mailund T, et al. CoaSim: A flexible environment for simulating genetic data under coalescent models. BMC Bioinf. 2005;6:252. doi: 10.1186/1471-2105-6-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hellenthal G, Stephens M. Insights into recombination from population genetic variation. Curr. Opin. in Gen. & Dev. 2006;16:565–572. doi: 10.1016/j.gde.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 64.Önder Ö, et al. OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J. of Biol. Chem. 2012;287:16860–16868. doi: 10.1074/jbc.M111.290775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lagal V, et al. Borrelia burgdorferi sensu stricto invasiveness is correlated with OspC-plasminogen affinity. Microbes and Infection. 2006;8:645–652. doi: 10.1016/j.micinf.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 66.Radolf JD, Caimano MJ. The long strange trip of Borrelia burgdorferi outer-surface protein C. Mol. Microbiol. 2008;69:1–4. doi: 10.1111/j.1365-2958.2008.06226.x. [DOI] [PubMed] [Google Scholar]
- 67.Seemanapalli SV, et al. Outer surface protein C is a dissemination-facilitating factor of Borrelia burgdorferi during mammalian infection. PLoS ONE. 2010:e15830. doi: 10.1371/journal.pone.0015830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang IN, et al. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics. 1999;151:15–30. doi: 10.1093/genetics/151.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oiu WG, et al. A population genetic study of Borrelia burgdorferi sensu stricto from eastern Long Island, New York, suggested frequency-dependent selection, gene flow and host adaptation. Hereditas. 1997;127:203–216. doi: 10.1111/j.1601-5223.1997.00203.x. [DOI] [PubMed] [Google Scholar]
- 70.Dykhuizen DE, et al. The propensity of different Borrelia burgdorferi sensu stricto genotypes to cause disseminated infections in humans. Am. J. of Trop. Med. and Hyg. 2008;78:806–810. [PMC free article] [PubMed] [Google Scholar]
- 71.Vuong HB, et al. Occurrence and transmission efficiencies of Borrelia burgdorferi ospC types in avian and mammalian wildlife. Inf., Gen. and Evol. 2013;27:594–600. doi: 10.1016/j.meegid.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hanincova K, et al. Epidemic spread of Lyme borreliosis, Northeastern United States. Em. Inf. Dis. 2006;12:604–611. doi: 10.3201/eid1204.051016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brisson D, Dykhuizen DE. OspC diversity in Borrelia burgdorferi: different hosts are different niches. Genetics. 2004;168:713–722. doi: 10.1534/genetics.104.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang JR, et al. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–285. doi: 10.1016/s0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 75.Coutte L, et al. Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Path. 2009;5:e1000293. doi: 10.1371/journal.ppat.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Inf. and Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Labandeira-Rey M, et al. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Inf. and Immun. 2003;71:4608–4613. doi: 10.1128/IAI.71.8.4608-4613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lawrenz MB, et al. Effects of VlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Inf. and Immun. 2004;72:6577–6585. doi: 10.1128/IAI.72.11.6577-6585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bankhead T, Chaconas G. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 2007;65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- 80.Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou W, Brisson D. Potentially conflicting selective forces that shape the vls antigenic variation system in Borrelia burgdorferi. Inf., Gen. and Evol. 2014;27:559–565. doi: 10.1016/j.meegid.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luikart G, et al. The power and promise of population genomics: from genotyping to genome typing. Nature Rev. Gen. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- 83.Achtman M. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Phil. Trans. R. Soc. B. 2012;370:860–867. doi: 10.1098/rstb.2011.0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vaz A, et al. Cellular and humoral immune responses to Borrelia burgdorferi antigens in patients with culture-positive early Lyme disease. Inf. and Immun. 2001;69:7437–7444. doi: 10.1128/IAI.69.12.7437-7444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu Y, et al. Profiling the humoral immune response to Borrelia burgdorferi infection with protein microarrays. Microb. Path. 2008;45:403–407. doi: 10.1016/j.micpath.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 86.Baum E, et al. Diversity of antibody responses to Borrelia burgdorferi in experimentally infected beagle dogs. Clin. and Vacc. Imm. 2014;21:838–846. doi: 10.1128/CVI.00018-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farhat MR, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nature Gen. 2013;45:1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fonkwo PN. Pricing infectious disease. EMBO Reports. 2008;9:S13–S17. doi: 10.1038/embor.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang X, et al. Economic impact of Lyme disease. Em. Inf. Dis. 2006;12:653–660. doi: 10.3201/eid1204.050602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cascio A, et al. The socio-ecology of zoonotic infections. Clin. Microbiol. and Inf. 2011;17:336–342. doi: 10.1111/j.1469-0691.2010.03451.x. [DOI] [PubMed] [Google Scholar]
- 91.LoGuidice K, et al. Impact of host community composition on Lyme disease risk. Ecology. 2008;89:2841–2849. doi: 10.1890/07-1047.1. [DOI] [PubMed] [Google Scholar]
- 92.Patz JA, et al. Unhealthy landscapes: Policy recommendations on land use change and infectious disease emergence. Env. Health Persp. 2004;112:1092–1098. doi: 10.1289/ehp.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Daszak P, et al. Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Tropica. 2001;78:103–116. doi: 10.1016/s0001-706x(00)00179-0. [DOI] [PubMed] [Google Scholar]
- 94.Avise JC, et al. The intraspecific phylogeography: mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. of Ecol. and Sys. 2014;18:489–522. [Google Scholar]
- 95.Faria NR, et al. The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014;346:56–61. doi: 10.1126/science.1256739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parks M, et al. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7:84–84. doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Benavides JA, et al. Limitations to estimating bacterial cross-species transmission using genetic and genomic markers: inferences from simulation modeling. Evol. App. 2014;7:774–787. doi: 10.1111/eva.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Humphrey PT, et al. Uncoordinated phylogeography of Borrelia burgdorferi and its tick vector, Ixodes scapularis. Evolution. 2010;64:2653–2663. doi: 10.1111/j.1558-5646.2010.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoen AG, et al. Phylogeography of Borrelia burgdorferi in the eastern United States reflects multiple independent Lyme disease emergence events. Proc. Natl. Acad. Sci. U.S.A. 2009;106:15013–15018. doi: 10.1073/pnas.0903810106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ogden NH, et al. Investigation of genotypes of Borrelia burgdorferi in Ixodes scapularis ticks collected during surveillance in Canada. App. and Env. Microbiol. 2011;77:3244–3254. doi: 10.1128/AEM.02636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khatchikian CE, et al. Geographical and environmental factors driving the increase in the Lyme disease vector Ixodes scapularis. Ecosphere. 2012;3:1–18. doi: 10.1890/ES12-00134.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qiu WG, et al. Wide distribution of a high-virulence Borrelia burgdorferi clone in Europe and North America. Em. Inf. Dis. 2008;14:1097–1104. doi: 10.3201/eid1407.070880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ras NM, et al. Borrelia burgdorferi sensu stricto, a bacterial species “made in the USA”? Int. J. of Sys. and Evol. Microbiol. 1997;47:1112–1117. doi: 10.1099/00207713-47-4-1112. [DOI] [PubMed] [Google Scholar]
- 104.Dykhuizen DE, Baranton G. The implications of a low rate of horizontal transfer in Borrelia. Trends in Microbiol. 2001;9:344–350. doi: 10.1016/s0966-842x(01)02066-2. [DOI] [PubMed] [Google Scholar]
- 105.Steere AC, et al. The emergence of Lyme disease. J. of Clin. Inv. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kurtenbach K, et al. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nature Rev. Microbiol. 2006;4:660–669. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- 107.Drummond AJ, et al. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol. Biol. and Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Faria NR, et al. Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Phil. Trans. R. Soc. B. 2013;368:20120196. doi: 10.1098/rstb.2012.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vrancken B, et al. The genealogical population dynamics of HIV-1 in a large transmission chain: bridging within and among host evolutionary rates. PLoS Comp. Biol. 2014;10:e1003505. doi: 10.1371/journal.pcbi.1003505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wattam AR, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nuc. Ac. Res. 2013;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barbour AG. Plasmid analysis of Borrelia burgdorferi the Lyme disease agent. J. of Clin. Microbiol. 1988;26:475–478. doi: 10.1128/jcm.26.3.475-478.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ashton PM, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nature Biotech. 2014 doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 113.Hoffmann M, et al. Complete genome sequence of a multidrug-resistant Salmonella enterica serovar Typhimurium var. 5- strain isolated from chicken breast. Genome Announc. 2013;1:e01068–13. doi: 10.1128/genomeA.01068-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zagordi O, et al. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinf. 2011;12:119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eyre DW, et al. Detection of mixed infection from bacterial whole genome sequence data allows assessment of its role in Clostridium difficile transmission. PLoS Biol. 2013;9:e1003059. doi: 10.1371/journal.pcbi.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]