

Optimization of treatment protocols and improved risk stratification have enhanced event-free survival rates in pediatric precursor-B acute lymphoblastic leukemia (BCP-ALL) up to 80%–90%.1 Remarkably, these results are obtained without changing the core chemotherapeutic drugs that have been used for decades, including prednisolone, L-asparaginase and vincristine. To cure the remaining 20% of patients and to reduce long-term side-effects in survivors, more personalized targeted therapy is warranted. Optimized high throughput screening has brought tailored therapy a step closer. Recently, high throughput sequencing in 187 high-risk BCP-ALL cases identified a high frequency of recurrent alterations in crucial signaling pathways.2
Novel targeted drugs will most likely serve as adjuvants to current chemotherapy regimens. Prednisolone has been shown to be the most pivotal in treating pediatric BCP-ALL, as in vivo and in vitro response to prednisolone is an important predictor for long-term clinical outcome,3,4 and relapsed leukemic cells gain prednisolone resistance. Hence, to improve clinical outcome, drugs need to be found which reverse resistance to prednisolone. Prednisolone is a glucocorticoid that binds the glucocorticoid receptor, which acts as a transcription factor regulating the expression of numerous genes eventually leading to apoptosis. In this study, we hypothesized that prednisolone resistance in BCP-ALL may be caused by dysregulation of prednisolone responsive survival proteins.
To study our hypothesis, we analyzed phosphorylation levels of 18 key-signaling proteins in leukemic cells obtained from in vitro prednisolone-sensitive and prednisolone-resistant ALL patients (Online Supplementary Table S1a, column 2) using reverse-phase protein arrays. We observed that there was no difference in basal phosphorylation levels of these 18 proteins between prednisolone-resistant and prednisolone-sensitive patients’ cells (Online Supplementary Figure S1). Exposure to prednisolone for 48 h induced discrepant (phosphorylation) changes in 7 out of 18 proteins between prednisolone-resistant and prednisolone-sensitive cases (Figure 1). Remarkably, exposure to prednisolone down-regulated phosphorylation of the RAS-RAF-MEK pathway in prednisolone-sensitive patients cells but not in prednisolone-resistant patients (Figure 1A–E). Levels of RAS, pRAS-GRF1, pARAF, pBRAF, pMEK1/2 and also pSTAT6 were, respectively, 1.5-fold, 2.5-fold, 8.1-fold, 2.3-fold, 2.9-fold and 3.1-fold (P<0.01) higher in prednisolone-resistant compared to prednisolone-sensitive patients’ cells after prednisolone exposure (Figure 1A–F). In contrast, a 3.8-fold induction of cMET phosphorylation was observed in resistant patients’ cells after prednisolone exposure, whereas no change was observed in sensitive cells (Figure 1G).
Figure 1.
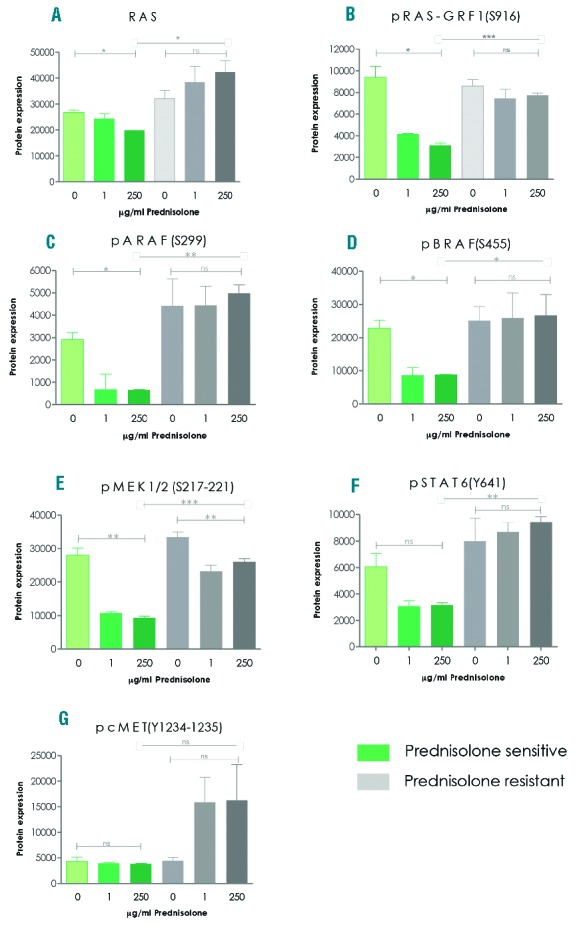
Prednisolone-mediated regulation of pivotal tyrosine-kinase pathway proteins is impaired in prednisolone-resistant primary patient cells. (A–G) Protein phosphorylation was analyzed by means of reverse phase protein array in 3 pediatric BCP-ALL patient cases with in vitro prednisolone-sensitive and 3 cases with in vitro prednisolone-resistant leukemic cells. Samples were exposed for 48 h to 0 mg/mL, 1 mg/mL prednisolone or 250 mg/mL prednisolone. Data are presented as mean plus SEM of 3 independent patients’ samples. (A Kruskal-Wallis test was used to compare 0, 1, 250 mg/mL data points indicated by ⊡-⊡ and a t-test was used to compare data between sensitive and resistant patients indicated by ⊢– ⊣. *P<0.05, **P<0.01, ***P<0.001. ns: not significant.
The finding that prednisolone is incapable of down-regulating the activity of RAS-RAF-MEK pathway in prednisolone resistant patients, prompted us to investigate the presence of RAS-pathway activating mutations. These mutations are known to induce constitutive RAS signaling which results in a survival advantage of cancer cells. We investigated hotspot regions of mutations in the protein coding domains of BRAF, NRAS, HRAS, KRAS, PTPN11 and FLT3 as documented in the COSMIC database (Online Supplementary Table S2) with Ion Torrent deep sequencing (approx. 1000x read depth) in 26 pediatric BCP-ALL patients with known genetic subtype and in vitro prednisolone response (Online Supplementary Table S1a). In addition, we examined in previously described hotspot regions the presence of mutations in cMET (Online Supplementary Table S2) but no cMET mutations were found. Eleven out of 26 patients (42%) (Online Supplementary Table S1a–b) carried activating NRAS and/or KRAS mutations or additional mutations in the FLT3 receptor or regulatory PTPN11 (SHP2) known to trigger RAS-MEK pathways in hematopoietic cells.5 Single heterozygous NRAS/KRAS-mutations were found in 5 cases with one dominant clone, representing 50%–100% of cells (Online Supplementary Table S1; Patients 3, 5, 6, 7 and 9). Two or more RAS-pathway mutations were found in 6 cases (Online Supplementary Table S1; Patients 1, 2, 4, 8, 10 and 11). Of interest, these mutations were often found in subclones as small as 5%. We identified 10 activating point mutations in NRAS and 8 in KRAS, 1 in PTPN11 and 1 in FLT3. We did not find mutations in BRAF and HRAS. Interestingly, we more often detected RAS activating mutations in prednisolone-resistant compared to prednisolone-sensitive patients (62% vs. 23%; P<0.001) (Online Supplementary Table S3). Furthermore, the mutations did not present in one specific genetic subtype (Online Supplementary Table S3) nor did the 2 patients that relapsed in this cohort harbor a RAS-mutation (Online Supplementary Table S1a).
Based on these findings, we examined the effects of inhibitors directed against MEK1/2 (trametinib), BRAF (sorafenib), STAT6 (AS1517499) and cMET (crizotinib) (Online Supplementary Table S4) on leukemic cell survival using an MTT assay. Of the 26 patient samples in this cohort (Online Supplementary Table S1), only 7 survived 4 days in vitro culture, i.e. 5 in vitro prednisolone-resistant and 2 in vitro prednisolone-sensitive BCP-ALL patients’ samples of which 3 are RAS-mutant and 4 are RAS-wild type. All patient leukemic cells expressed high levels of the targeted proteins in leukemic cells compared to normal mononuclear bone marrow cells (Figure 2A). There was no significant difference in protein phosphorylation between RAS-mutant and RAS-wt patient leukemic cells (Figure 2A). Single agent exposure of trametinib, sorafenib, AS1517499 or crizotinib did not induce substantial cell death in any of the leukemic patients’ cell samples (Figure 2B–2E). Western blot analysis verified that trametinib decreased phospho-ERK, sorafenib decreased phospho-BRAF, AS1517499 decreased phospho-AKT and crizotinib decreased phospho-MEK1/2 and phospho-AKT, which are known downstream targets of STAT6 and cMET, respectively (Figure 2F). In conclusion, although these patient cells have high target phosphorylation, MEK1/2, BRAF, STAT6, cMET single agent exposure did not reduce viability of patients’ leukemic cells independent of RAS-mutant status.
Figure 2.

Inhibitors against MEK, BRAF, STAT6, cMET do not reduce viability of patients’ leukemic cells. (A) Protein phosphorylation levels of MEK1/2, BRAF, STAT6 and cMET were analyzed in leukemic cells of 7 BCP-ALL samples by reverse phase protein array. Phosphorylated protein levels in patients were compared to the levels observed in normal bone marrow mononuclear cells (dashed line set at 1). (B–E) Seven BCP-ALL patient cell samples were exposed for four days with the indicated inhibitor in a concentration range between 0.0002–20 μM, after which leukemic cell survival was analyzed by MTT-assay and corrected for effects caused by vehicle itself. (F) MEK1/2(S217–221), ERK1/2(Thr202/Tyr204), AKT(S473) and BRAF(S455) phosphorylation of patient D treated for four days with 5 mM of the indicated inhibitors or 5 mM of the vehicle DMSO was analyzed by western blot. β-actin expression was used as a loading control. Data are presented as mean+SEM.
We next evaluated the prednisolone sensitizing properties of these inhibitors in the same 7 primary BCP-ALL patients’ samples (Figure 3, Online Supplementary Table S1 and Online Supplementary Figure S2). Remarkably, we observed a complete sensitization to prednisolone after trametinib treatment in Patients B and C (P<0.01) (Figure 3B1–C2). This amount of prednisolone sensitization in BCP-ALL patient cells has never been seen before for any other drug/inhibitor/shRNA combination. Patient B harbored a KRAS mutation and C harbored a KRAS and NRAS mutation (Online Supplementary Table S1). Trametinib was not effective in Patient A who harbored a KRAS and NRAS activating mutation (Figure 3A1 and Online Supplementary Table S1). Interestingly, this patient was the only one in whom sorafenib sensitized cells to prednisolone up to 61% compared to prednisolone and vehicle control (Figure A1–A2; P=0.001). These highly sensitizing effects of prednisolone were not seen for AS1517499 (STAT6 inhibitor) or crizotinib (cMET inhibitor). Trametinib and sorafenib had no substantial prednisolone sensitizing effects in the RAS-wild-type patients (Figure 3D1–G2 and Online Supplementary Figure S2). In one RAS-wild-type patient (Figure 3G2), trametinib had an antagonistic effect on prednisolone sensitivity. This finding warns against a broad use of MEK inhibitors, and supports our conclusion that implementation of these inhibitors needs to be guided by relevant biomarkers (e.g. RAS mutation) and pre-clinical in vitro evidence.
Figure 3.

Trametinib (MEK inhibitor) and sorafenib (BRAF inhibitor) restored prednisolone sensitivity in RAS-mutant patients. (A1–G1) Sensitivity to 3.9 mg/mL prednisolone of 7 distinct pediatric BCP-ALL patients’ cell samples co-incubated with 2.5 mM trametinib (MEK1/2 inhibitor), sorafenib (BRAF inhibitor), AS1517499 (STAT6 inhibitor), Crizotinib (cMET inhibitor) or vehicle (DMSO) control measured in a 4-day MTT assay. Data are presented as mean+SEM of a duplicate experiment (t-test *P<0.05, **P<0.01, ***P<0.001). (A2–G2) Dose-response curve of prednisolone combined with the inhibitor giving the most prominent effect in A1–G1. Graphs represent 2.5 or 5.0 mM sorafenib (BRAF inhibitor), trametinib (MEK inhibitor) or vehicle control (DMSO). Data are presented as mean+SEM of a duplicate experiment (repeated measurement two-way ANOVA, interaction inhibitor, *prednisolone *P<0.05, **P<0.01, ***P<0.001). Sensitivity was corrected for cell death induced by the inhibitor/vehicle itself in the absence of prednisolone to facilitate assessment of pure prednisolone sensitizing effects. The upper 3 patients have RAS-mutations; the lower 4 patients are RAS-wild type.
The synergy between prednisolone and MEK/BRAF inhibitors in RAS-mutant patients might be explained by three mechanisms.
MEK/BRAF inhibitors mostly target RAS-mutant signaling.6 Therefore, the limited effect of MEK/BRAF inhibitors as single agents on viability of BCP-ALL cells might be explained by recent findings that targeted RAS-mutant signaling re-sensitizes to tyrosine kinase signaling via wild-type RAS.6 In contrast, prednisolone presumably targets wild-type RAS as is anticipated by the presence of a glucocorticoid response element in RAS,7 and our finding that prednisolone decreases RAS phosphorylation levels in prednisolone sensitive cells, either directly/indirectly. The synergy between MEK/BRAF inhibitors and prednisolone might, therefore, be explained by a prednisolone-mediated decrease in RAS-wild-type signaling and MEK/BRAF targeted decrease in RAS-mutant signaling.
RAS mutations might inhibit glucocorticoid-dependent inhibition of the survival protein AP1.8 Targeting the mutated RAS-pathway genes may enable prednisolone to inhibit AP1, thereby reducing cell viability.
It is known that RAS-activating mutations generate an anti-apoptotic environment by decreasing pro-apoptotic BIM levels,9 increasing anti-apoptotic BCL2 and MCL1 levels.10 We previously showed that prednisolone down-regulates MCL1 in prednisolone-sensitive but not in prednisolone-resistant BCP-ALL cells.11 Targeting MEK/BRAF may, therefore, act synergistically with prednisolone on the balance between pro- and anti-apoptotic proteins.
In this study, we observed a relative high frequency (42%) of RAS-pathway mutant patients, and these mutations seem to be significantly over-represented in prednisolone-resistant compared to prednisolone-sensitive patients. RAS-pathway mutations did not present in all prednisolone-resistant patients, which indicates that resistance to prednisolone is not solely driven by RAS-pathway mutations but may also be driven by other (epi)genetic lesions. Prednisolone resistance has recently been associated with RAS mutations in major clones of infant ALL.12 Remarkably, deep sequencing of BCP-ALL cases in the present study revealed multiple minor clones in 55% of mutant cases. In contrast, most large studies hardly detected concomitant mutations,12–16 and only one study detected multiple mutations in 14% of patients.2 Our finding that RAS-mutations are not mutually exclusive in most leukemic patients not only encourages the need for future mutation analysis by means of deep-sequencing instead of sanger-sequencing, but also signifies the importance of these mutations in leukemia. This is further supported by the finding that RAS mutations predominating at relapse could be retrospectively demonstrated in minor clones at initial diagnosis of the corresponding patients.15,16
In conclusion, targeting RAS-signaling in RAS-mutant BCP-ALL cells with trametinib and sorafenib could serve as a novel therapeutic option to modulate prednisolone resistance and may provide a very promising way to further improve clinical outcome of childhood BCP-ALL.
Acknowledgments
We acknowledge M.W.J. Luijendijk (Erasmus MC) and E.F. Petricoin (George Mason University-Manassas USA) for RPPA data generation. We would like to thank Nicolle Besselink and Stef van Lieshout (University Medical Center Utrecht) for next-generation sequencing data generation and analysis. This work was supported by the Dutch Cancer Society (MLDB, RP, EMCR 2005-3313 and AMC 2008-4265) and by the Center for Personalized Cancer Treatment, which is a collaboration between all University Medical Cancer Centers and the Netherlands Cancer Institute Amsterdam and is supported by grants from KWF/Alpe d’Huzes and Nuts/Ohra. The funding source had no role in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Footnotes
The online version of this article has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Pui C-H, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120(6):1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are fre quently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118(11):3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaspers GJ, Pieters R, Van Zantwijk CH, et al. Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood. 1998;92(1):259–266. [PubMed] [Google Scholar]
- 4.Styczynski J, Piatkowska M, Jaworska-Posadzy A, et al. Comparison of prognostic value of in vitro drug resistance and bone marrow residual disease on day 15 of therapy in childhood acute lymphoblastic leukemia. Anticancer Res. 2012;32(12):5495–5499. [PubMed] [Google Scholar]
- 5.Rosen DB, Minden MD, Kornblau SM, et al. Functional characterization of FLT3 receptor signaling deregulation in acute myeloid leukemia by single cell network profiling (SCNP). PLoS One. 2010;5(10):e13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayes TK, Der CJ. Mutant and wild-type Ras: co-conspirators in cancer. Cancer Discov. 2013;3(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zachos G, Zoumpourlis V, Sekeris C, Spandidos D. Binding of the glucocorticoid and estrogen-receptors to the human h-ras oncogene sequences. Int J Oncol. 1995;6(3):595–600. [DOI] [PubMed] [Google Scholar]
- 8.Johnson TA, Li J, Beer DG. Influence of the glucocorticoid receptor on c-fos inducibility in activated ras-containing mouse lung cells. Mol Carcinog. 1996;17(2):70–77. [DOI] [PubMed] [Google Scholar]
- 9.Weston CR, Balmanno K, Chalmers C, et al. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene. 2003;22(9):1281–1293. [DOI] [PubMed] [Google Scholar]
- 10.Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 2009;7(4):549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ariës IM, Hansen BR, Koch T, et al. The synergism of MCL1 and glycolysis on pediatric acute lymphoblastic leukemia cell survival and prednisolone resistance. Haematologica. 2013;98(12):1905–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Driessen EMC, van Roon EHJ, Spijkers-Hagelstein JAP, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013;98(6):937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tartaglia M, Martinelli S, Cazzaniga G, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104(2):307–313. [DOI] [PubMed] [Google Scholar]
- 14.Paulsson K, Horvat A, Strömbeck B, et al. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes. Chromosomes Cancer. 2008;47(1):26–33. [DOI] [PubMed] [Google Scholar]
- 15.Case M, Matheson E, Minto L, et al. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res. 2008;68(16):6803–6809. [DOI] [PubMed] [Google Scholar]
- 16.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471(7337): 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]