

Abstract
Ebolaviruses are highly lethal filoviruses that cause hemorrhagic fever in humans and nonhuman primates. With no approved treatments or preventatives, the development of an anti-ebolavirus therapy to protect against natural infections and potential weaponization is an urgent global health need. Here, we describe the design, biophysical characterization, and validation of peptide mimics of the ebolavirus N-trimer, a highly conserved region of the GP2 fusion protein, to be used as targets to develop broad-spectrum inhibitors of ebolavirus entry. The N-trimer region of GP2 is 90% identical across all ebolavirus species and forms a critical part of the prehairpin intermediate that is exposed during viral entry. Specifically, we fused designed coiled coils to the N-trimer to present it as a soluble trimeric coiled coil as it appears during membrane fusion. Circular dichroism, sedimentation equilibrium, and X-ray crystallography analyses reveal the helical, trimeric structure of the designed N-trimer mimic targets. Surface plasmon resonance studies validate that the N-trimer mimic binds its native ligand, the C-peptide region of GP2. The longest N-trimer mimic also inhibits virus entry, thereby confirming binding of the C-peptide region during viral entry and the presence of a vulnerable prehairpin intermediate. Using phage display as a model system, we validate the suitability of the N-trimer mimics as drug screening targets. Finally, we describe the foundational work to use the N-trimer mimics as targets in mirror-image phage display, which will be used to identify d-peptide inhibitors of ebolavirus entry.
Keywords: ebolavirus, filovirus entry, ebolavirus GP2, prehairpin intermediate, designed coiled coil, N-trimer, phage display, mirror-image phage display
Introduction
Ebolaviruses are enveloped, negative-strand RNA viruses that cause severe hemorrhagic fever.1 Since its identification in 1976, there have been over 20 reported natural ebolavirus outbreaks, the majority since 2000, and several accidental laboratory exposures with an overall mortality rate >60%.2 Alarmingly, in 2014 the largest known outbreak is occurring in western Africa3 and has crossed international borders. Currently, no vaccines or therapeutics are FDA approved. Because of ease of transmission, high mortality, and potential for a severe impact on public health, the CDC places ebolaviruses in its highest category of potential agents of bioterrorism.4 There is a vital need for preventatives and/or therapeutics to protect against future natural, accidental, or deliberate outbreaks.
Ebolavirus entry into host cells, a critical step to infection, is mediated by the viral surface glycoprotein (GP), a class I fusion protein.5 GP comprises two disulfide-linked subunits, one surface exposed (GP1) and one embedded in the viral membrane (GP2).6,7 Following binding to host cells via cell surface attachment factors, the virus is endocytosed. Endosomal cysteine proteases, cathepsins B and L, cleave off much of GP1, exposing the binding site for the receptor, endosomal NPC1.8–12 At this point, the fusion mechanism is thought to mimic that of other well characterized viral class I fusion proteins, such as HIV-1 and influenza13–15 (Fig. 1). GP2 forms a transient conformation (“prehairpin intermediate”) embedded in both the virus (via the transmembrane domain) and host cell (via the fusion loop) membranes. This prehairpin intermediate exposes a trimeric coiled coil, formed by the N-terminal region (N-trimer), and the C-terminal region (C-peptide). Slow collapse of the intermediate into a very stable trimer-of-hairpins structure, with the C-peptide binding into the grooves on the N-trimer, juxtaposes the virus and cell membranes, leading to membrane fusion. In ebolavirus entry, the low pH of the endosome contributes to the stability of the trimer-of-hairpins.16
Figure 1.
Model for membrane fusion mediated by enveloped virus surface glycoproteins. The HIV-1 and ebolavirus entry events are predicted to be similar. First, the surface glycoprotein (Env for HIV-1, GP for ebolavirus) facilitates viral attachment to the cell and, for ebolavirus, the virus is endocytosed and then cleaved by endosomal proteases. Engagement of the virus receptors (CD4 and a chemokine receptor for HIV-1, NPC1 for ebolavirus) is followed by a conformational change in Env/GP, and insertion of the fusion peptide/loop (brown) into the host cell membrane. At this stage, the virus is in a transient state that bridges both membranes, termed the “prehairpin intermediate,” which is vulnerable to inhibition. In the absence of an inhibitor, the Env/GP structure slowly resolves into the highly stable trimer-of-hairpins structure, juxtaposing the two membranes, and leading to membrane fusion. The inset shows the high resolution structure of the ebolavirus trimer-of-hairpins (PDB: 2EBO).27 In HIV, it has been shown that inhibitors that bind to either the N-trimer (blue) or C-peptide (yellow) regions are capable of inhibiting entry [as reviewed in Ref.17].
In the case of HIV-1, the prehairpin intermediate has been exploited to develop highly potent viral entry inhibitors (Fig. 1). Peptides and proteins that bind with high affinity to either the N-trimer or C-peptide regions prevent formation of the trimer-of-hairpins, thereby halting viral entry [as reviewed in Ref.17]. The most potent of the HIV entry inhibitors, chol-PIE12-trimer, binds to the conserved hydrophobic pockets of the HIV N-trimer and inhibits HIV entry with low picomolar potency.18 Since filoviruses share a similar mechanism of entry as HIV-1, they are likely vulnerable to inhibitors that similarly target the prehairpin intermediate.
There are five known species of ebolavirus, four of which are pathogenic to humans. The vast majority of promising preventative and therapeutic candidates with efficacy against ebolavirus in animal models, such as vaccines, antibodies, and antisense compounds (e.g.,19–23), are species-specific, resulting in limited breadth and difficulty in combating emerging species. The N-trimer of the prehairpin intermediate provides a highly conserved target for potential broad-spectrum inhibitors. Indeed, although the overall sequence identity of GP across all known ebolavirus species is only 42%, the N-peptide region is 90% identical, and all changes are conservative [Fig. 2(A)].
Figure 2.
Conservation of the ebolavirus GP N-trimer and design of peptide N-trimer mimics. (A) Schematic of the primary structure of ebolavirus GP2, indicating the fusion loop (brown), N-trimer (blue), C-peptide (yellow), and transmembrane domain (TM, purple), all shown approximately to scale. The sequences of the N-trimer region (residues 558–596) and the C-peptide region (residues 597–633) (Zaire ebolavirus species, representative Mayinga strain isolated in Zaire in 197660) contained in the peptides described in this study are indicated. In the C-peptide, cysteine 609, which is proposed to disulfide bond with GP1,76 is mutated to alanine in our constructs (red). Below the Zaire N-trimer is an alignment of the sequences from the 4 additional ebolavirus species plus Marburgvirus and Cuevavirus filoviruses. Genbank accession codes are indicated (right). Conserved changes (score of 0 or higher in BLOSUM62 matrix77) are highlighted in gray, nonconserved in cyan. Notably, 3/5 and 5/5 ebolavirus species are 100% identical in the N39 and N21 regions, respectively. The 2014 epidemic is caused by the Zaire ebolavirus species and is 100% identical in this region.3 *Reston and likely Cuevavirus (Lloviu virus) are not pathogenic to humans. (B) Schematics and sequences of the N-trimer mimics and their corresponding binding site mutants [eboIZN21 and eboIZN21(D2); eboIZN39IQ and eboIZN39IQ(D3)]. The designed coiled coils, IZm and IQ, are shown in gray, while the ebolavirus N-trimer is shown in blue. The a and d positions of the coiled-coil heptad repeats are indicated by a larger bolded font, including a stutter at the N-terminal end of the ebolavirus N-trimer as seen in the crystal structures,27,28 where the coil is underwound, leading to an atypical 3-4-4-3 pattern (instead of the standard 3-4, or a-g, periodicity of a heptad repeat). The alanine residues along the C-peptide binding groove that are mutated to aspartate in the binding site mutants are shown (orange).
Here, we describe the development of ebolavirus N-trimer mimics that will be useful in a variety of drug discovery platforms to screen small molecule, antibody, and peptide libraries for entry inhibitors that target this conserved region. Specifically, we have designed and characterized peptide mimics of ebolavirus N-trimers, validated their use as drug discovery tools, and explored conditions that can be applied directly to phage display drug discovery endeavors. In addition, using one of our N-trimers as an inhibitor itself (to target the C-peptide region of the prehairpin intermediate), we have confirmed the vulnerability of the ebolavirus GP prehairpin intermediate to entry inhibition.
Results and Discussion
N-trimer mimic design
Based on our previous HIV-1 work,24,25 we set out to design soluble peptide mimics of the N-trimer region of the ebolavirus GP prehairpin intermediate by fusing stable, soluble, designed trimeric coiled coils to the N-trimer sequence [Fig. 2(B)]. As with HIV-1, the ebolavirus N-trimer aggregates when produced in isolation. We were interested in presenting the entire N-trimer groove as well as a smaller, more conserved region of the N-trimer to provide flexibility in drug screening. Our initial designs, in which we fused the coiled coil IZm(24) to the N-terminus of N-trimer segments of 29 and 39 amino acids, were aggregated as determined by analytical ultracentrifugation (AUC) sedimentation equilibrium experiments (data not shown). To overcome this problem, we fused an additional trimeric coiled coil, GCN4-pIQI’ (IQ)26 to the C-terminus of the ebolavirus N-trimer segment. The resulting peptide, eboIZN39IQ presents the full ebolavirus N-trimer (determined from available trimer-of-hairpins crystal structures27,28) as a trimeric coiled coil, as shown by circular dichroism (CD) [Fig. 3(A)] and AUC [Fig. 3(C) and Table I]. eboIZN39IQ is very stable, as indicated by similar CD spectra at 25, 37, and 50°C (Table I). The ultimate goal for an ebolavirus N-trimer mimic is to use it as a target in drug screening to identify inhibitors of ebolavirus entry. Since these inhibitors will bind to the virus in the endosome, all biophysical analyses were performed at pH 5.8 to mimic endosomal pH.
Figure 3.
Biophysical analyses of ebolavirus N-trimer mimics. (A) CD spectra of 11.4 µM eboIZN39IQ and 11.1 µM eboIZN39IQ(D3) at 25°C. Both spectra indicate a highly helical conformation with 81 and 83% helicity, respectively. (B) CD spectra of 18.0 µM eboIZN21 and 25.3 µM eboIZN21(D2) at 25°C, also indicate a highly helical conformation with 73 and 71% helicity, respectively. (C) Analytical ultracentrifugation (AUC) sedimentation equilibrium analysis of eboIZN39IQ, shown as representative AUC data. 10, 5, and 2.5 µM peptide solutions were centrifuged at 18,000, 21,000, and 24,000 rpm at 4°C on a Beckman XLA. All data were globally fit to a single ideal species, and an observed molecular weight of 39,762 Da was determined for an Mobs/Mcalc of 3.31. The data (open symbols) and fit (solid lines) are shown for the lowest speed.
Table I.
Biophysical Analyses of N-Trimer Mimics Via CD and AUC
| Peptide | [θ222 nm] (deg cm2 dmol−1) 25°C | [θ222 nm] (deg cm2 dmol−1) 37°C | [θ222 nm] (deg cm2 dmol−1) 50°C | Mobs/Mcalc 4°C |
|---|---|---|---|---|
| eboIZN39IQ | −29,400 | −27,900 | −27,100 | 3.24 |
| eboIZN39IQ(D3) | −30,400 | −29,300 | −28,400 | 3.22 |
| eboIZN21 | −25,500 | −24,000 | −22,900 | 3.54 |
| eboIZN21(D2) | −24,800 | −22,800 | −21,800 | 3.15 |
CD scans were performed on the same samples of 11.4 µM eboIZN39IQ, 11.1 µM eboIZN39IQ(D3), 18.0 µM eboIZN21 and 25.3 µM eboIZN21(D2) in 50 mM sodium phosphate, pH 5.8, 150 mM NaCl at 25, 37, and 50°C. The peptides were allowed to equilibrate at each temperature for 10 min, after which no change in signal was seen over time. Sedimentation equilibrium analysis was performed on each peptide at three concentrations each (a starting concentration and two twofold dilutions, with typical starting concentrations between 10 and 30 µM) and a minimum of two speeds, but typically three speeds (18,000, 21,000, and 24,000 rpm). Each data set was globally fit to a single ideal species. Each sedimentation equilibrium analysis was performed 2–4 times and averaged for the above table.
To produce a smaller target that presents a 100% identical region of the N-trimer (across all ebolavirus species), IZm was fused to the N-terminal 21 amino acids of the N-trimer, resulting in eboIZN21 (Fig. 2). Circular dichroism indicates that eboIZN21 is highly helical [Fig. 3(B)], and AUC and gel filtration studies show that it is largely trimeric with a slight tendency to form higher order aggregates (Table I and Supporting Information Fig. S1). X-ray crystallography studies confirm the trimeric coiled-coil structure of eboIZN21 (below). As seen with eboIZN39IQ, eboIZN21 is very stable, showing similar CD spectra at 25, 37, and 50°C (Table I). We also attempted to produce mimics presenting the C-terminal portion of the N-trimer, but they were not soluble (data not shown) and were not studied further.
As negative controls for binding studies and drug discovery efforts, we produced mutant N-trimer mimics aimed at abolishing the C-peptide binding site. Specifically, alanines found along the C-peptide binding groove were mutated to aspartate, introducing binding-disruptive charges along the groove (Fig. 2). The resulting peptides are termed eboIZN39IQ(D3) and eboIZN21(D2). Using CD and AUC, we confirmed that these mutants maintained the stable coiled-coil structure and trimeric nature of their wild-type counterparts (Fig. 3 and Table I).
C-peptide binding characterization
To validate that eboIZN39IQ presents the native conformation of the N-trimer found in the prehairpin intermediate, we characterized binding to its native C-peptide ligand (Fig. 2), which binds along the entire groove of the N-trimer in the postfusion trimer-of-hairpins conformation. Surface plasmon resonance (SPR) analysis (ProteOn XPR36, Bio-Rad) of the interaction of the full-length C-peptide, eboC37, with eboIZN39IQ showed a dissociation constant of 14 nM (Fig. 4), with no binding to the D3 negative control. This tight binding affinity is of the same magnitude as the HIV-1 N-trimer/C-peptide interaction29 and indicates that eboIZN39IQ presents a native N-trimer. A shortened C-peptide (eboC24), missing the 13 N-terminal residues of eboC37, bound to eboIZN39IQ with a dissociation constant of ∼300 nM and did not bind to the D3 negative control (Supporting Information Fig. S2). eboIZN21 was less well behaved on an SPR surface, and we were unable to obtain reproducible data using this target.
Figure 4.
Binding of the ebolavirus C-peptide to the N-trimer mimic. Sensorgram of eboC37 flowed over eboIZN39IQ in a triplicate twofold dilution series starting at 60 nM, plotted with second-order second-neighbor-smoothing with a Savitzky–Golay filter (Prism 6, GraphPad Software). Each replicate dilution series is shown as a distinct color. The kinetic fit of the raw data is shown and yields ka = 9.6 × 105 M/s1, kd = 0.014/s1, and a KD of 14 nM. Inset: The same eboC37 dilutions flowed over an eboIZN39IQ(D3) surface. No binding was observed.
Crystal structure of eboIZN21
To visualize how the N-trimer is presented in the absence of the native C-peptide ligand, as in our mimics, we determined the X-ray crystal structure of eboIZN21 to 2.15 Å. eboIZN21 crystallized as a symmetrical trimer in space group P321 with one monomer in the asymmetric unit. The structure reveals that eboIZN21 is a continuous trimeric coiled coil, as designed [Fig. 5(A)]. Comparing our structure with the two previously reported structures of the ebolavirus 6-helix bundle (PDB IDs: 1EBO28 and 2EBO27) revealed good overall agreement between the N21 residues of our unliganded structure and the C-peptide-bound structures, as indicated by root mean square deviations (rmsd) of 1.2 Å (across 63 atoms, 1EBO) and 1.4 Å (across 61 atoms, 2EBO) when trimers are aligned on Cα residues [Fig. 5(B)]. However, surface renderings show that a hydrophobic pocket in the N21 region of the 6-helix bundle structures, which accommodates residues 619–626 of the bound C-peptide, is collapsed in the isolated eboIZN21 structure [Fig. 5(C–E)].
Figure 5.
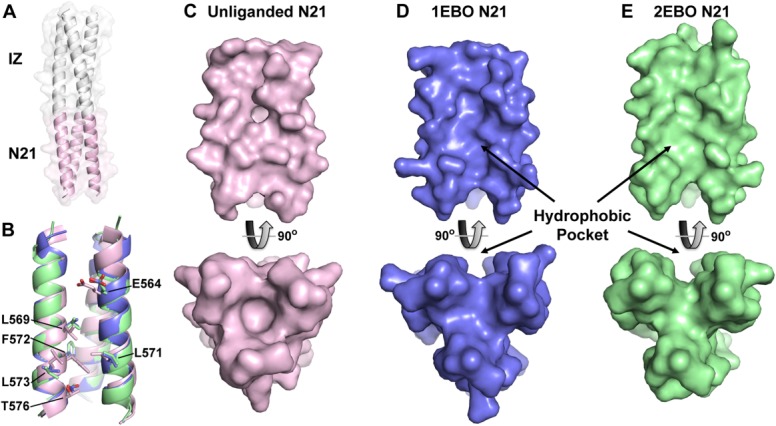
Crystal structure of eboIZN21. (A) Cartoon rendering with a semitransparent surface of the unliganded eboIZN21 structure. The IZ trimerization domain (white) and N21 region (pink) are indicated. The N21 region of available ebolavirus N-trimer structures is shown in isolation in panels B–E. (B) Overlay of the N21 region of the unliganded structure with the N21 region of the two previously solved ebolavirus GP2 core structures containing C-peptide (PDB IDs: 1EBO and 2EBO shown as blue and green, respectively. This color scheme is maintained in panels C–E). Residues that line the N21 groove and have significantly different rotamer conformations in the unliganded structure are shown as sticks and labeled. These residues occupy some of the equivalent space occupied by C-peptide (not shown) in the liganded structures resulting in a less prominent hydrophobic pocket when viewed (in subsequent panels) as a surface. (C) Surface representation of the unliganded N21 region. The bottom panel is the view of N21 from the bottom along its threefold axis and is rotated approximately 90° as compared to the top panel. (D, E) Similar views to (C) of the N21 region from the structures containing C-peptide. The prominent hydrophobic pocket in the 1EBO and 2EBO structures appears to be induced by ligand binding since the pocket is nearly absent in the unliganded structure.
The collapse of this pocket in the unliganded structure results from the side-chain conformations of several residues that fill the pocket. Specifically, in the absence of C-peptide, residues L569, L571, F572, L573, and T576 adopt alternate rotamers to pack together via hydrophobic interactions and thus alter the surface contours of the ligand binding pocket (Supporting Information Fig. S3). The side chain of E564 also adopts an alternate conformation to occupy a distinct portion of the pocket (toward the top of the pocket in Fig. 5). Therefore, as seen with the analogous hydrophobic ligand-binding pocket in the HIV gp41 N-trimer [comparing structures in Refs. (29–32), e.g., Supporting Information Fig. S4], our eboIZN21 unliganded structure indicates that the ebolavirus GP N21 pocket is induced by ligand binding and can likely adopt various conformations depending on the specific ligand.
We used the MONSTER protein interaction server33 to calculate the solvent accessible surface area (SASA) buried at the interface of the ebolavirus and HIV hydrophobic pockets with their C-peptides. The crystal structures of the ebolavirus27,28 and HIV30,31 6-helix bundles reveal that in each case the pocket interacts with 8 C-peptide residues (ITDKIDQI for ebolavirus and WMEWDREI for HIV) [Supporting Information Fig. S4(A,C)]. The buried SASA at the N21 pocket/8-mer C-peptide interface is similar in the 1EBO and 2EBO structures at 393/348 and 387/325 Å2, respectively. These values are comparable to SASA buried at the HIV gp41 pocket/8-mer C-peptide interface (349/310 Å2).30 Finally, a similar analysis between the HIV pocket and our D-peptide entry inhibitor, PIE12 [Supporting Information Fig. S4(B)], reveals that 416/391 A2 of SASA is buried at that interface.32 Given the comparable size of the pocket/C-peptide interface in the ebolavirus and HIV 6-helix bundle structures, combined with the high anti-HIV potency of PIE12-trimer (a trimerized version of PIE12, designed to bind all three pockets of the prehairpin intermediate),32 it is reasonable to expect that potent pocket-specific peptide inhibitors of ebolavirus can also be discovered.
Phage display target validation
Phage display is a powerful screening technology that is used to screen billions of peptides or antibodies against a target of interest to identify specific inhibitors of protein/protein interactions. Indeed, HIV N-trimer mimics were successfully used in phage display screens of both scFv antibodies and D-peptides (see “Mirror-Image Phage Display” below) to identify potent, broadly neutralizing HIV entry inhibitors.25,29,32,34,35 To validate the ebolavirus N-trimer mimics as discovery targets in the context of phage display, we produced phage clones expressing the native binding partners eboC37 and eboC24 [Fig. 2(A)] and assayed their ability to bind to our N-trimer mimics in phage clone binding assays. These experiments were designed to verify ligand binding and to define the best conditions for future phage display discovery efforts.
Phage display selections can be conducted in two formats: solid- and solution-phase. In solid-phase selections, the target is bound to a solid support (here, biotinylated ebolavirus N-trimer mimic is attached to streptavidin-coated magnetic beads), and then the phage are incubated with the immobilized target. Since common phage display libraries are multivalent (multiple copies of the library molecule are expressed on the surface of the phage, due to fusion to multicopy coat proteins), avidity effects improve the apparent binding constant of the library clones. This avidity-induced affinity boost is beneficial when screening naïve phage libraries, where initial binders typically have low target affinities. In solution phase, where both target and phage are incubated in solution, avidity effects are reduced. Following incubation, the bound complexes are captured through a brief interaction with a solid support (again, in this case, through a biotinylated target and streptavidin beads). At equivalent target concentrations, solution-phase selection is more stringent than solid-phase selection. The higher stringency of solution-phase is useful when screening second-generation libraries for affinity maturation (e.g., peptide binding consensus libraries or antibody variable loop mutagenesis libraries), where tight binders must be distinguished from a background of moderate binders.
Both eboC37 and eboC24 clonal phage bound to eboIZN39IQ target significantly over background (both empty beads and negative control eboIZN39IQ(D3) beads) using solution-phase clonal phage binding assays carried out at pH 5.8 to mimic the endosomal environment (Fig. 6). Also, binding of M13KE empty phage to both eboIZN39IQ and eboIZN39IQ(D3) was minimal. These data validate eboIZN39IQ as a phage display target. In addition, these data demonstrate that eboIZN39IQ(D3) serves as an effective negative control, as its clonal C-peptide phage binding is comparable to that of blank beads. In this format, both C-peptide clones bound at similar levels to eboIZN39IQ, although eboC37 had greater background binding to both negative controls.
Figure 6.
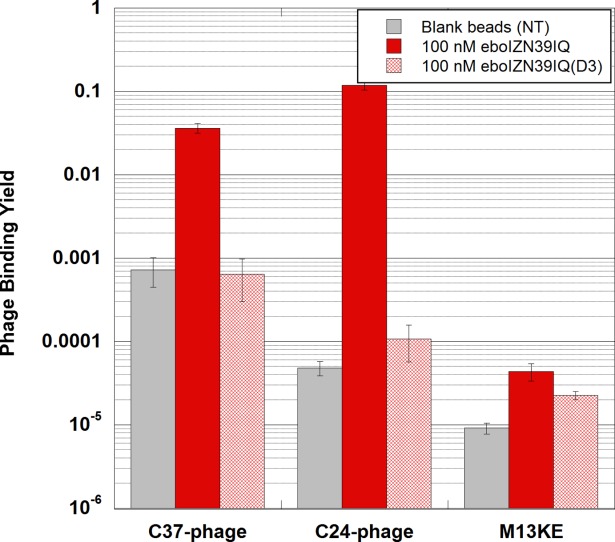
Validation of eboIZN39IQ as a phage display target. Clonal phage expressing ebolavirus C-peptides (eboC37 or eboC24) were incubated with biotinylated eboIZN39IQ in solution followed by capture via magnetic streptavidin beads. Negative target controls include the binding site mutant, eboIZN39IQ(D3), and magnetic beads with no target (NT). Binding of M13KE (phage with no peptide clone) to all targets was also assayed. The fraction of phage bound is reported. Error bars represent standard error across triplicate experiments.
Specific binding (over two orders of magnitude over background) was seen when using eboIZN21 as a target in a solid-phase eboC24 clonal phage binding assay, also validating eboIZN21 as a phage display target (Fig. 7). With the low level of eboC24 phage binding to eboIZN21(D2) (similar to eboC24 binding to blank beads), the binding site mutant is also verified as a negative control. In addition, only a very low level of M13KE empty phage binding to eboIZN21 and eboIZN21(D2) was observed.
Figure 7.
Validation of eboIZN21 as a phage display target. Clonal phage expressing an ebolavirus C-peptide (eboC24) were incubated with biotinylated eboIZN21 bound to streptavidin magnetic beads (solid-phase conditions). Negative target controls include the binding site mutant, eboIZN21(D2), and magnetic beads with no target (NT). Binding of M13KE (phage with no peptide clone) to all targets was also assayed. The fraction of phage bound is reported. Error bars represent standard error for triplicate experiments.
Low phage background binding to targets is required in order to discern specific binding during phage panning rounds. To evaluate this property for our two N-trimer mimics, we analyzed empty M13KE phage binding to both targets under varying conditions (Fig. 8). In both solid- and solution-phase formats, M13KE phage showed significantly higher binding to eboIZN39IQ beads than to blank beads. For the eboIZN21 target, phage background binding was drastically reduced in comparison to eboIZN39IQ, and binding of M13KE phage was similar to both target and blank beads in solid and solution phase. Under stringent conditions (solution phase, 100 nM target) the M13KE background binding to eboIZN39IQ was minimized, and a large affinity difference for eboC24 binding to eboIZN39IQ versus eboIZN21 could be seen. This affinity difference is likely to be biologically relevant because the trimer-of-hairpins structures27,28 show that the binding site of eboC24 extends past the C-terminus of N21. These data indicate the importance of choosing proper stringency conditions in performing phage display selections.
Figure 8.
Comparing the two ebolavirus N-trimer mimics as phage display targets. (A) Phage background binding is greater to eboIZN39IQ than to eboIZN21. Phage binding assay showing M13KE control phage binding to biotinylated eboIZN39IQ and eboIZN21 under both solid-phase (left) and solution-phase (right) conditions. Magnetic beads with no target (NT) were used as a negative control. The fraction of phage bound is reported. Error bars represent the range for duplicate experiments (solid phase) and standard error for four or more replicates (solution phase). (B) High stringency solution-phase binding shows an affinity difference for the specific binding of eboC24 to the two N-trimer mimics. Clonal phage expressing eboC24 were incubated with biotinylated N-trimer in solution followed by capture via magnetic streptavidin beads. Negative target controls include the binding site mutants and magnetic beads with no target (NT). For NT, error bars represent standard error across triplicate experiments. The remaining error bars represent the range for duplicate experiments.
The first step of a phage display discovery process is to screen a naïve phage display library for binding to the desired target. In such a first selection, where the library diversity only partially samples the large potential sequence space (e.g., a naïve peptide 12-mer library has 2012 (>1015) possible sequences, whereas the typical diversity of a phage display library is <1010), the best binders identified are usually modest, with low- to mid-micromolar affinities. Therefore, the selection pressure applied during phage panning must also be modest. Standard naïve phage display starting conditions are 10 µM target presented on solid-phase (i.e., 30 µL of 10 µM target immobilized onto magnetic beads).25 As illustrated in Figure 8, M13KE binding to eboIZN39IQ is nearly saturated at this condition, and therefore it would not be possible to identify binding over background. 10% phage binding is considered saturating, as binding yields of even strong binders do not generally exceed this level (likely due to proteolysis of displayed peptides). Under the same conditions, the eboIZN21 background binding is >600-fold lower and similar to blank bead binding, ideal starting conditions for naïve phage display. Therefore, eboIZN21 is an optimized target for phage display discovery efforts. Additionally, the eboC24-phage can serve as an important positive control during naïve phage display to validate the conditions used to capture weak, but specific binders. Notably, in addition to having ideal behavior in phage display, the N21 region is also identical across all ebolavirus species and highly conserved among filoviruses (95% conserved) [Fig. 2(A)]. eboIZN39IQ is an ideal target for higher stringency solution-phase phage display and could be used to screen secondary libraries for affinity optimization of ligands identified from the naïve library. This could be especially useful for extending the binding interface of the ligands further along the N-trimer groove.
Mirror-image phage display
Mirror-image phage display is an innovative adaptation of standard peptide phage display and is used to identify D-peptides that bind to a target of interest25,36 (Supporting Information Fig. S5). D-peptides are composed of D-amino acids and are the mirror-image of naturally occurring L-peptides. D-peptides have several important potential advantages as drug candidates (as reviewed37). As peptides, they are capable of blocking large protein/protein interactions, which is generally not possible for small molecules. In addition, because they are resistant to protease degradation,38 D-peptides should possess a longer in vivo half life and reduced immunogenicity.39 In our HIV-1 entry inhibitor discovery program,18,25,29,32 we used mirror-image phage display and protein design to develop the highly potent and broadly acting D-peptide entry inhibitor, chol-PIE12-trimer, which is now in advanced preclinical studies. Their resistance to endosomal proteases makes D-peptides especially attractive as ebolavirus entry inhibitors. Our future studies will therefore use the N-trimer mimics to discover D-peptide inhibitors of ebolavirus entry by mirror-image phage display.
In traditional peptide phage display, a library of phage, each with a unique peptide displayed on its surface, is screened against a target.40 In mirror-image phage display, the target is chemically synthesized from D-amino acids and therefore forms the mirror-image structure of the natural L-target (Supporting Information Fig. S5). Phage display using the D-target is performed, and identified L-peptides that bind the D-target are then chemically synthesized using D-amino acids. By the law of symmetry, these D-peptides bind the natural L-target. Unlike with traditional phage display, mirror-image phage display targets are limited in size to those that can be chemically synthesized (although this size limit is continually expanding with modern chemical protein synthesis advances41.
In order to prepare ebolavirus N-trimers as mirror-image phage display targets, we synthesized them as D-peptides. At 48 amino acids each, D-eboIZN21 and D-eboIZN21(D2) were synthesized through standard solid-phase peptide synthesis (SPPS) techniques. Importantly, even though the 101-residue length of eboIZN39IQ is beyond the scope of standard SPPS, modern chemoselective ligation techniques42 allow for its assembly from multiple peptide segments. D-eboIZN39IQ, was assembled using native chemical ligation43 and metal-free desulfurization,44 in which cysteine residues are introduced at native alanine sites to facilitate ligation and then converted back to alanine through desulfurization [Supporting Information Fig. S6(A)]. D-eboIZN39IQ was assembled from three synthetic segments of 27–41 residues. The production of D-eboIZN39IQ(D3) required a different assembly strategy, as one of the alanines used in D-eboIZN39IQ as a ligation junction is mutated to aspartate. Therefore, D-eboIZN39IQ(D3) was assembled from two synthetic segments of 33 and 68 amino acids. The final peptide products were confirmed by LC/MS [e.g., Supporting Information Fig. S6(B,C)].
The law of symmetry dictates that D-peptides will adopt the mirror-image structure of their L-counterparts. CD analysis of D-eboIZN21 confirms it possesses mirror-image helical structure compared to its L-peptide counterpart [Supporting Information Fig. S7(A)]. SPR analysis of D-eboC37 binding to D-eboIZN39IQ shows a similar binding affinity (6 nM) to the l-peptide interaction and validates the functionality of D-eboIZN39IQ [Supporting Information Fig. S7(B)]. Preliminary phage display experiments with these D-targets demonstrate the same M13KE binding properties as the L-versions (data not shown), verifying the strategy of screening naïve libraries with the D-eboIZN21 target and employing D-eboIZN39IQ for subsequent affinity optimization efforts when higher stringency is appropriate.
Vulnerability of the Ebolavirus prehairpin intermediate to a high potency inhibitor
A prerequisite for the success of drug discovery efforts targeting the ebolavirus N-trimer mimics is the exposure of a vulnerable prehairpin intermediate during viral entry. Exogenous C-peptides derived from the transmembrane subunit of the envelope glycoprotein have been used to validate this vulnerable prehairpin intermediate in a variety of viruses (e.g., HIV, SARS, and many paramyxoviruses45–47). For ebolavirus, an early report showed C-peptide inhibition activity at mM concentrations,48 and more recent reports describe improved inhibitory activity (mid µM) of C-peptides with an endosomal localization tag.49,50 Our ebolavirus N-trimer, eboIZN39IQ, provides an additional tool with which to explore the vulnerability of the prehairpin intermediate. In support of this strategy, peptide mimics of the HIV-1 N-trimer inhibit HIV entry at mid nM concentrations by binding to the C-peptide region of the exposed intermediate.24
Indeed, eboIZN39IQ inhibited entry in our pseudovirus system in which ebolavirus GP (representative species, Zaire ebolavirus) is expressed on the surface of an HIV particle [Fig. 9(A)], with an average IC50 of 320 nM. Importantly, the anti-ebolavirus activity of our negative control, eboIZN39IQ(D3), is ∼30-fold diminished, with an IC50 of 11 µM. It is difficult to determine the exact nature of the modest eboIZN39IQ(D3) activity, as it is not seen against a vesicular stomatitis virus glycoprotein pseudotype (VSV), and no morphological changes (indicative of toxicity) were observed. It is possible the modest eboIZN39IQ(D3) activity could be due to residual prehairpin intermediate binding activity. eboIZN39IQ demonstrated modest activity against marburgvirus pseudovirions (another member of the filovirus family), at an IC50 of 5.7 µM, although this was only ∼2-fold better than the eboIZN39IQ(D3) anti-marburgvirus activity.
Figure 9.
Inhibition of filovirus entry by eboIZN39IQ. (A) A representative pseudovirion assay looking at the inhibitory activity of eboIZN39IQ and the negative control, eboIZN39IQ(D3) against ebolavirus (EboV), marburgvirus (MarV), and VSV retroviral pseudotypes. Each point represents the average of quadruplicate measurements normalized to uninhibited control. Error bars represent normalized standard errors. For this particular assay, eboIZN39IQ IC50s are 260 nM against ebolavirus and 5.4 µM against marburgvirus. The eboIZN39IQ(D3) IC50s are 8.9 µM against ebolavirus and 11 µM against marburgvirus. (B) Data for the authentic filovirus immunofluorescence inhibition assay. Each point represents the average of quadruplicate measurements normalized to vehicle control. Strong inhibition of ebolavirus is seen at 10 µM eboIZN39IQ, with an average 33% (±4%) of infected cells compared to vehicle control.
The ability of eboIZN39IQ to inhibit the entry of wild-type ebolavirus and marburgvirus was also assessed using a filovirus immunofluorescence assay under BSL4 conditions [Fig. 9(B)]. Although eboIZN39IQ was significantly less potent in this assay, there is 67% inhibition of entry at the highest concentration tested (10 µM) and no inhibition by our negative D3 control. Also, no activity was seen against marburgvirus. Potency differentials between pseudovirus systems and authentic filoviruses have been seen for other fusion inhibitors [for example in Ref.51]. Taken together, these data validate the presence of a vulnerable prehairpin intermediate during the ebolavirus entry process.
Unlike HIV-1, ebolavirus enters cells via endocytosis and initiates membrane fusion late in the endosomal pathway. Therefore, ebolavirus entry inhibitors will have to enter into and be active in endosomes. Although eboIZN39IQ does not possess a specific tag to localize it to endosomes, it is highly charged on its surface (with both positive and negatively charged side chains), and, interestingly, the inhibitory activity we observed in both the pseudovirus and authentic ebolavirus systems was dependent on the presence of the standard viral assay additive DEAE-dextran. It seems likely that the highly charged N-trimer mimic associates with the anionic cell membrane, especially in the presence of the cationic DEAE-dextran that would reduce electrostatic repulsion between the negative charges of eboIZN39IQ and the membrane, allowing it to access the endosome more efficiently than C-peptides.50 As a structured peptide, eboIZN39IQ would also likely resist proteolysis longer than unstructured C-peptides.
Conclusion
In summary, we have designed and characterized two mimics of the highly conserved ebolavirus GP N-trimer region as it appears in the prehairpin intermediate during viral entry. In addition, through our clonal phage display experiments, we have functionally validated eboIZN39IQ and eboIZN21 as drug discovery targets, especially for phage display screens. Finally, with the characterization of the inhibitory activity of eboIZN39IQ, we have further validated the vulnerability of the ebolavirus prehairpin intermediate by demonstrating potent inhibition. These N-trimer mimics should be valuable for the discovery of small molecules, antibodies and/or peptides that inhibit ebolavirus entry. Specifically, our group is interested in the discovery of D-peptide inhibitors of ebolavirus using mirror-image phage display, and with the two targets and their binding site mutants synthesized in the D configuration, we are now poised for those selections.
It is noteworthy that in addition to the remarkable conservation of the N-trimer region across all ebolavirus species, it is also highly conserved across the filovirus family [see Fig. 2(A)]. Therefore, discovery efforts will likely identify inhibitors with broad filovirus activity. If such activity is suboptimal, it should be possible to design analogous mimics of the marburgvirus N-trimer region and use them in concert with the ebolavirus targets. For example in phage display, rounds of panning could alternate between the ebolavirus and marburgvirus targets, selecting specifically for an inhibitor of both viruses. Although the vast majority of natural filovirus outbreaks have been caused by ebolavirus, marburgvirus still poses a risk both as a natural pathogen (with three outbreaks in the last 10 years) and as a bioterror agent,52 making the discovery of a broad-spectrum inhibitor desirable.
In addition to serving as drug targets, the ebolavirus N-trimer mimics should be useful as cell biological tools. For example, fluorescently labeled N-trimers could be used in cell culture experiments to track the appearance of the prehairpin intermediate during the viral entry event. Such studies would advance insight into filovirus entry dynamics.
Materials and Methods
Reagents
Plasmids and cells were obtained from the indicated sources: pEBB-HXB2 (gift from B. Chen)53, SV-ZeboGPΔmuc and SVMarVGP (gift from M. Farzan),54 BLR(DE3)pLysS E. coli (EMD Millipore, Billerica, MA), BL21-Gold(DE3)pLysS E. coli and XL-1 Blue E. coli (Agilent Technologies, Santa Clara, CA). pNL4–3.Luc.R-E- (N. Landau)55,56 and HOS-CD4-fusin (N. Landau)57,58 were obtained from the NIH AIDS Research and Reference Program. Mammalian cells were propagated in standard tissue culture medium, Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum and l-glutamate (Life Technologies, Grand Island, NY).
Recombinant peptide production and purification
The DNA encoding eboIZN39IQ and eboIZN39IQ(D3) was produced via PCR gene synthesis. The IZm and IQ fragments were PCR amplified from plasmids encoding HIV-1 N-trimer mimics [e.g. in Ref.59]. An NdeI site was included in the 5′ PCR primer for IZm, and a BamHI site was included in the 3′ PCR primer for IQ. The ebolavirus N39 sequence from the species Zaire ebolavirus60 was synthesized in two overlapping oligos with optimized codons and companion primers. All internal primers contained complementary sequences so the three separate components, IZm, N39, and IQ could be annealed and amplified together. The resulting DNA fragment was cloned into the NdeI/BamHI cloning sites of pKA8, validated by sequencing and expressed in BLR(DE3)pLysS cells using an autoinduction protocol. Specifically, cultures were inoculated from a single colony and grown overnight at 37°C in autoinduction media.61 The resultant peptide has an N-terminal His tag (His8) followed by a TEV cleavage site (ENLYFQG). A single tyrosine was placed at the end of the sequence to facilitate concentration determination via absorbance at 280 nm. The peptides were resuspended from inclusion bodies using Ni-binding buffer (20 nM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM imidazole) + 6M GuHCl, and purified via gravity flow Ni affinity chromatography (HIS-Select Nickel Affinity Gel, Sigma Aldrich, St. Louis, MO). The purified peptides were dialyzed into 5% acetic acid and further purified by reverse phase HPLC on a C18 column (Vydac, Grace, Columbia, MD) and lyophilized. Peptide powder was resuspended in water and diluted to 0.2 mg/mL in 50 mM sodium phosphate pH 6.5, 0.5 mM EDTA, 1 mM DTT and digested with a solubility-enhanced tobacco etch virus NIa protease (TEVse, based on published modifications62,63) overnight at 30°C. The digested peptide was dialyzed into 5% acetic acid and then HPLC purified and lyophilized. The final peptide sequences are: GHMDIKKEIEAIKKEQEAIKKKIEAIEKELRQLANETTQ(A/D)LQLFLR(A/D)TTELRTFSILNRK(A/D)IDFLLQRMKQIEDKIEEIESKQKKIENEIARIKKLIGERY, with IZm and IQ shown in bold, the ebolavirus N-trimer in italics, and the three alanine positions that are changed to aspartate in the D3 mutant in parentheses.
Biotinylated eboIZN39IQ and eboIZN39IQ(D3) for SPR analysis and phage display were expressed from plasmids that are modified from those described above. Using PCR, a CGG sequence was added N-terminal to IZ (GHMCGGDIKK…). Expression and purification were as described above with additional reduction steps included to keep the cysteine reduced during purification (100 mM DTT treatment after Ni++ affinity chromatography and 50 mM TCEP treatment after TEV digestion). The purified protein was biotinylated with EZ-link Maleimide–PEG2–biotin (Thermo Scientific, Waltham, MA). The purified lyophilized powder was resuspended at 1 mM in freshly prepared reaction buffer (6 M GuHCl, 150 mM NaCl, 100 mM Na2HPO4, 5 mM TCEP) and the biotinylation reagent was added at 5 mM and allowed to react for 4 h at RT. The biotinylated peptides were purified by reverse phase HPLC on a C18 column (Waters) and lyophilized. The mass of the peptide was confirmed by LC/MS (AB Sciex API 3000 LC/MS/MS system, Framingham, MA).
Peptide synthesis
eboIZN21, eboIZN21(D2), eboC37 and eboC24 were chemically synthesized using solid-phase peptide synthesis (SPPS) with Fmoc-amino acids (AAPPTec, Louisville, KY, and CBL Biopharma, Boulder, CO) on a Prelude peptide synthesizer [Protein Technologies (PTI), Tucson, AZ]. A single tyrosine was placed at the N-terminus of both eboIZN21 and eboIZN21(D2) to facilitate concentration determination via absorbance at 280 nm. The peptides were synthesized on TentaGel R RAM resin (Rapp Polymere, Germany) to yield C-terminal amide peptides. Standard synthesis scales were 25–32 µmol per peptide. Standard amino acid coupling was as follows: 3 × 3 min deprotection with 20% piperidine in DMF followed by 25 min couplings with 72.2 mM amino acid (200 mM stocks in NMP), 71.5 mM HATU (198 mM stock in DMF), and 166.7 mM NMM (600 mM stock in DMF). Biotinylation was achieved with N-Biotinyl-NH-(PEG)2-COOH DIPEA (Novabiochem, EMD Millipore) coupling for 2 h. N-terminal capping was accomplished in 30 min with 2 mL acetic anhydride and 2 mL 0.6M NMM. Peptide cleavage from resin was accomplished offline with 92.5% TFA, 2.5% EDT, 2.5% TIS, 2.5% H2O when the peptide contained Met or Cys residue(s) or with 95% TFA, 2.5% TIS, 2.5% H2O in the absence of any Met/Cys residues followed by precipitation/washing with diethylether. All peptides were purified by reverse-phase HPLC on a Waters (Milford, MA) BEH X-Bridge C18 column (10 μm, 300 Å, 19 × 250 mm) with a water/ACN gradient in 0.1% TFA. All peptides were lyophilized and their molecular weight verified by LC/MS.
D-eboIZN39IQ was assembled from three synthetic peptide segments via native chemical ligation/metal-free desulfurization (Supporting Information Fig. S6). D-peptides were synthesized via Fmoc-SPPS on a PTI PS3 peptide synthesizer at 100 µmol scale. The C-terminal peptide was synthesized on Rink Amide AM resin LL (Novabiochem) and the other two segments were synthesized on Dawson Dbz AM resin (Novabiochem). The C-terminal segment contained an N-terminal cysteine residue in the place of a native alanine for use in native chemical ligation (CIDFLLQRMKQIEDKIEEIESKQKKIENEIARIKKLIGERY). For the same reason, the middle segment contained an N-terminal Boc-L-thiazolidine-4-carboxylic acid (Boc-Thz-OH, Bachem, Torrance, CA) as its N-terminal residue in place of the native alanine at that position((Thz)-NETTQALQLFLRATTELRTFSILNRK). The N-terminus of the N-terminal peptide (GHMDIKKEIEAIKKEQEAIKKKIEAIEKELRQL) was biotinylated with N-Biotinyl-NH-(PEG)2-COOH DIPEA (Novabiochem). For peptides synthesized on Dawson Dbz AM resin, the C-terminal linker was converted to the resin bound N-acyl-benzimidazolinone (Nbz) according to manufacturer instructions. Cleavage of all peptides was performed according to standard procedures. Peptides were purified by reverse-phase HPLC on a Waters BEH X-Bridge C18 column (10 μm, 300 Å, 19 × 250 mm) with a water/acetonitrile gradient in 0.1% TFA. Ligations were performed according to Ref.64 with peptide concentrations ∼2 mM. Following ligation between the C-terminal and middle segments, the N-terminal Thz was converted to cysteine by dissolving the purified ligation product in 6M GuHCl, 200 mM sodium phosphate, 200 mM methoxyamine HCl, pH 4. After Thz to Cys conversion was achieved, the buffer was brought to 200 mM MPAA and 20 mM TCEP, the pH was adjusted to 7, and the N-terminal peptide was added to the solution for the final ligation. Following purification of the ligation product by reverse-phase HPLC, the cysteine residues at the ligation junctions were converted to the native alanine residues via a metal-free, radical-mediated desulfurization strategy essentially as described in Ref.44 except that t-butylthiol was replaced with glutathione, and desulfurizations were performed at 37°C. eboIZN39IQ(D3) was synthesized in an analogous (though simplified) manner using two peptide segments.
Preparation of peptide samples for biophysical analysis
For biophysical analyses, peptide stocks were prepared in water from lyophilized peptide at concentrations of 400 µM or greater for a minimum absorbance at 280 nm of 0.1 in a 1 cm pathlength cuvette. Stocks were centrifuged at 18,000g for 10 min to remove aggregates. Absorbance at 280 nm (using ε280 of 1408 M/cm1 for tyrosine) was used to determine stock concentrations.65 For eboIZN39IQ and eboIZN39IQ(D3), both recombinant and synthetic, UV absorbance consistently overestimated the concentration of the stocks (as evidenced by an unusually high 260/280 ratio as well as CD traces whose shape depicted ideal coiled coils but whose signal had a lower than expected absolute value). This overestimation is likely due to peptide bond absorbance contributing to the signal at 280 nm (101 amino acids with only one near-UV absorbing residue, a tyrosine). Therefore, the concentrations of these stocks were determined via quantitative amino acid analysis, which was performed using a Hitachi L-8800 Amino Acid Analyzer (Hitachi High-Technologies Corporation, Tokyo, Japan). Peptides were hydrolyzed in 5.7N HCl overnight at 105°C in sealed ampoules, and then analyzed via ion-exchange chromatography and postcolumn derivatization with ninhydrin using the Hitachi Analyzer. The peptides were then diluted to the desired concentration in 50 mM sodium phosphate pH 5.8, 150 mM NaCl. For eboIZN21 and eboIZN21(D2), all experiments described in this paper were performed with biotinylated peptide. For eboIZN39IQ and eboIZN39IQ(D3), CD, AUC and viral infectivity were performed with nonbiotinylated material, whereas SPR and phage display used biotinylated material.
Circular dichrosim
Circular dichroism (CD) data were obtained using an AVIV Model 410 spectrophotometer (AVIV, Lakewood, NJ). Samples were analyzed in a 1 mm pathlength quartz cuvette at 25, 37, and 50°C. Prior to CD analysis, prepared samples (in 50 mM sodium phosphate pH 5.8, 150 mM NaCl) were centrifuged at 18,000g for 10 min to remove aggregates. CD data were scanned in triplicate and buffer subtracted. Final CD data were presented according to mean residue ellipticity equation [θ] = 100 × θ/[(n − 1) × (ℓ) × (c)], where θ is the observed ellipticity, n − 1 is the number of peptide bonds, ℓ is the pathlength in cm, and c is the peptide concentration in mM. Percent helicity was calculated from the ellipticity at 222 nm according to the Lifson–Roig-based helix-coil model which defines the dependence on chain length and temperature as described.66 Due to aggregation observed with eboIZN39IQ and eboIZN39IQ(D3) upon initial dilution in the CD buffer, their final concentrations were corrected from the original amino acid analysis values based on the ratio of ellipticity at 222 nm post- and precentrifugation (θ222-postspun/θ222-prespun).
Analytical ultracentrifugation sedimentation equilibrium
Using an Optima XL-A Analytical Ultracentrifuge (Beckman Coulter, Brea, CA), sedimentation equilibrium analysis was performed on each peptide at three concentrations (a starting concentration and two 2-fold dilutions, with typical starting concentrations between 10 and 30 µM). Dilutions were prepared in matching buffer (50 mM sodium phosphate, 150 mM NaCl, pH 5.8), and the same buffer was used for blanks. Each sample was spun until equilibrium, typically ∼24 h, at a minimum of two speeds, but typically three speeds (18,000, 21,000 and 24,000 rpm). Each data set was globally fit to a floating molecular weight single ideal species with a nonlinear least squares algorithm as implemented in HETEROANALYSIS.67 Fits are reported as the observed (i.e., fit) molecular weight divided by the calculated molecular weight of a monomer (Mobs/Mcalc). Buffer densities and protein partial specific volumes were calculated with SEDNTERP (version 1.09).68 For the biotinylated peptides, partial specific volumes were adjusted based on reported values for PEG.69
Surface plasmon resonance
SPR analysis was conducted on the Bio-Rad (Hercules, CA) ProteOn XPR36 instrument in PBS* running buffer (50 mM sodium phosphate, 150 mM NaCl, pH 5.8) + 0.1 mg/mL BSA and 0.01% Tween 20. Approximately 600 RUs of biotin-eboIZN39IQ (in the presence of eboC37) and biotin-eboIZN39IQ(D3) targets (200 nM stocks ultracentrifuged for 30 min at 45,000 rpm) were loaded at 67 nM on to the NLC neutravidin-coated chip (Bio-Rad), followed by blocking with 450 μM biotin. Using the one-shot kinetics method, a twofold dilution series was performed in triplicate at RT starting at 60 nM for eboC37 and a threefold dilution series in triplicate at RT starting at 800 nM for eboC24. 10 min dissociation time for eboC37 and 5 min dissociation time for eboC24 were used to ensure the response fully recovered to baseline prior to the next injection. Data were corrected by subtracting blank surface and blank buffer reference injections, and the kinetics (for eboC37) and equilibrium (for eboC24) were globally fit to the Langmuir model for 1:1 binding70 using ProteOn Manager software (Bio-Rad).
Crystallization
eboIZN21 was dissolved in ddH20 to a concentration of ∼10 mg/mL and centrifuged at 18,000g for 10 min. Sitting-drop vapor-diffusion crystal trials were set up using a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, CA). Crystals grew at 4°C in drops containing a 2:1 ratio of peptide to well solution, which consisted of 30% (v/v) 1,2-propanediol, 100 mM HEPES pH 7.5, 20% (v/v) PEG-400. The crystals were flash frozen in liquid nitrogen without the need for additional cryoprotection and determined to be in space group P321 with unit cell dimensions a = b = 38.51 Å, c = 72.59 Å.
Data collection, structure determination, and refinement
A native dataset was collected at beam line 7-1 of the Stanford Synchrotron Radiation Lightsource. Data were integrated and scaled to 2.15 Å resolution using HKL2000.71 In order to rule out the possibility of twinning, data were initially scaled in space group P3 and analyzed with the program Xtriage,71 which indicated that the data are untwinned and that the correct space group is P321. A model that consisted of a canonical helix appropriate to the size and sequence of the IZ domain and the N21 region of ebolavirus GP (PDB ID: 1EBO) was used for molecular replacement using the program Phaser.72 A single eboIZN21 monomer was found in the asymmetric unit with the trimeric structure generated by the crystallographic threefold. Subsequent model building, structure refinement, and validation were performed with Coot,73 PHENIX Refine,74 and MolProbity75 software, respectively. The final model was refined to crystallographic R/Rfree values of 0.272/0.294 with good geometry (Supporting Information Table S1). Additional refinements were carried out in space group P3 allowing all possible twin laws, to further verify (by monitoring Rfree) that the correct space group is P321 with a monomer in the asymmetric unit. A composite omit map agreed well with the final model, indicating good sidechain density throughout. The atomic coordinates and structure factors have been deposited in the Protein Data Bank, http://www.pdb.org (PDB ID: 4R0R).
Clonal phage production
Forward and reverse sandwich oligonucleotides encoding the C-peptide clones were designed based on the primary sequence of each clone. The forward and reverse eboC37 oligonucleotides were: ATGCGGTACCTTTCTATTCTCATTCTTGGGGCGGCACCTGCCATATTCTGGGCCCGGATTGCGCGATTGAACCGCATGATTGGACCAAAA and CCTTTTCGGCCGAACCCCCACCTTTATCCACAAAATCATGAATAATCTGATCAATTTTATCGGTAATGTTTTTGGTCCAATCATGCGGTT. The forward and reverse eboC24 oligonucleotides were: ATGCGGTACCTTTCTATTCTCATTCTATTGAACCGCATGATTGGACCAAAAACATTACCG and CCTTTTCGGCCGAACCCCCACCTTTATCCACAAAATCATGAATAATCTGATCAATTTTATCGGTAATGTTTTTGGTCCAATCATGCGGTT. The oligonucleotide sandwich was annealed with 5 µg of each primer in 50 µL total volume in ddH2O by heating to 95°C and slow cooling and then extended with Klenow Fragment [New England Biolabs (NEB), Ipswich, MA] according to the manufacturer's protocol. The inserts and M13KE cloning vector backbone (NEB) were digested with Acc65I and EagI-HF. The insert DNA was EtOH precipitated, gel purified from a 6% TBE acrylamide gel, extracted from the gel by incubating gel slices in a minimal volume of extraction buffer (100 mM NaOAc pH 4.5, 1 mM EDTA, 0.1% SDS) for 16 h at 37°C, and ethanol precipitated. The inserts and plasmid backbone were ligated and transformed into SS320 electrocompetent cells and plated on LB/IPTG/X-gal plates (LB agar, 25 µg/mL tetracycline, 1 mM IPTG, 0.1 mg/mL X-gal). The DNA from specific phage plaques was PCR amplified and Sanger sequenced (Eton Bioscience, Durham, NC), and those containing the correct DNA were subsequently amplified from a single plaque.
Phage amplification
A single plaque was added to XL-1 Blue cells (OD600 0.5–1), diluted to 40 mL of OD600 0.05 in LB +25 µg/mL tetracycline, and shaken at 220 rpm at 37°C for 4.5–5 h. Cells were pelleted by centrifugation, and the phage supernatant was sterile filtered. Phage were precipitated by adding 1/6th volume of PEG-NaCl [20% w/v polyethylene glycol-8000 (Fisher Scientific, Pittsburgh, PA), 2.5M NaCl] and incubating overnight at 4°C. Precipitated phage were then pelleted via centrifugation and resuspended in TBS (50 mM Tris–HCl, 150 mM NaCl, pH 7.4). They were PEG-precipitated again (∼1 h on ice), centrifuged and resuspended in 200 µL TBS. Aliquots were flash frozen and stored at −20°C with a working stock left at 4°C if imminent experiments were planned.
Clonal phage binding assay
For each phage binding reaction, 30 µL streptavidin-coated magnetic beads (Life Technologies, Dynabeads MyOne, Streptavidin T1) at 10 mg/mL were magnetically pelleted and washed with 3.3× bead volume TBS. The beads were then blocked in 3.3× bead volume 100% SB (Thermo Scientific, SuperBlock Blocking Buffer in TBS, pH 7.4) for 10 min at RT and rinsed with equal volume of 100% SB* (SB adjusted to pH 5.8 with HCl). Solution-phase beads were then resuspended in 3.3× bead volume of 100% SB* and stored at 4°C for up to 24 h. Solid-phase beads were resuspended in 3.3× bead volume PBS* + 10% SB*. To load target on to solid-phase beads, 1× bead volume of an appropriate target concentration (e.g., 10 µL of 10 µM target for 10 µL beads) was added and incubated for 10 min followed by addition of 3.3× bead volume 5 mM D-Biotin (in PBS* + 10% SB*) and incubation for an additional 5 min. For blank (no target) beads, 3.3× bead volume of 5 mM D-Biotin was added and incubated for 5 min. All beads were then magnetically pelleted, washed in PBS*, and resuspended in 1× bead volume PBS* + 10% SB*.
Solid-phase binding reactions were incubated in 96-well format (Costar, sterile polystyrene, V-bottom, nontreated, Corning, Corning, NY) with shaking at 700 rpm for 2 h at RT either as 30 or 100 µL reactions in 1× PBST* (PBS* + 0.01% Tween 20) + 10% SB* and 1010 plaque-forming units (pfu) of the phage clone. All washes and elution were done on the KingFisher Duo magnetic particle processor (Thermo Scientific). The binding reaction was mixed on the KingFisher for 1 min at medium speed and the beads collected by 5 s dips of the magnet through the sample, repeated five times (5 × 5 s). All washes were done with PBST* (wash 1: 700 µL; wash 2: 800 µL; wash 3: 900 µL; washes 4–7: 1000 µL), mixed at slow speed for 1.5 min, and beads collected 3 × 3 s. Bound phage were eluted with 50 µL EB (0.2 M glycine, pH 2.2) for 10 min, beads collected 5 × 5 s, and neutralized with 7.5 µL NB (1 M Tris, pH 9.1). Dilutions of eluted phage were used to infect XL-1 Blue cells and then plated in top agar (40% LB agar/60% LB) on LB/IPTG/X-gal plates. Blue plaques were then counted to determine phage titers.
Solution-phase binding reactions were performed similarly to solid-phase 30 µL reactions. Instead of adding target-loaded beads to the binding reaction, an appropriate volume of 10× soluble target was added to the reaction (final 1× soluble target) just before phage were added. Additionally, on the Kingfisher Duo, target and bound phage were pulled down in a rapid 1 min magnetic pelleting step (1 min slow mixing, 5 × 5 s bead collect). All washes were done with PBST* except wash 1 which contained 5 mM D-Biotin to block unoccupied streptavidin sites (wash 1: 150 µL; wash 2: 700 µL; wash 3: 800 µL; wash 4: 900 µL; wash 5: 1000 µL), mixed at slow speed for 25 s, and beads collected 3 × 3 s.
Pseudovirus infectivity assays
Single-cycle pseudovirions were produced with a pNL4-3 HIV-1 genome (with firefly luciferase inserted into the nef gene and frameshift mutations in both Env and Vpr) and filovirus GP on their surface (or VSV for a specificity control). These pseudovirions were produced by co-transfecting 293T human embryonic kidney cells with the described HIV-1 genome (pNL4-3.Luc.R-E-) and a plasmid encoding the desired virus glycoprotein (SV-ZeboGPΔmuc for Zaire ebolavirus GP lacking the mucin domain, SV-MarVGP for marburgvirus (Musoke strain) GP and pMDG VSV-G for VSV) in the presence of polyethylenimine (PEI) transfection reagent (Polysciences, Inc., Warrington, PA). Pseudovirus-containing supernatant was collected and filtered through a 0.45 µM filter 38–43 h post-transfection. For ebolavirus and marburgvirus, pseudovirions were concentrated by centrifuging through a 20% sucrose/TNE (10 mM Tris pH 7.6, 100 mM NaCl, 1 mM EDTA) cushion (26,000 rpm, 2 h), and the pellet resuspended in TNE, aliquoted and stored at −80°C.
To measure inhibition of infectivity, 90 µL of each inhibitor dilution and 8.9 µg/mL DEAE–dextran were added to HOS–CD4–fusion cells in a 96-well format. For each assay, a total of six inhibitor dilutions were tested, each in quadruplicate. The plates were transferred to BSL3, and 10 µL of pseudovirus diluted in media was added to each well 30–60 min after the inhibitor addition (final DEAE–dextran concentration of 8 µg/mL). Virus was diluted in order to yield a robust luciferase signal. 24 h later, all wells were inspected under a light microscope to check for gross morphological changes. Virus and inhibitor were removed via aspiration, and fresh media was replenished. 20–24 h later, the cells were lysed, and the luciferase activity was measured (Bright-Glo luciferase assay system, Promega, Madison, WI). To determine IC50 values, the data from each inhibitor concentration series were normalized to the noninhibitor control signal and fit to a Langmuir equation: y = 1/(1+[inhibitor]/IC50) (Kaleidagraph, Synergy Software, Reading, PA). The curve fit was weighted by the normalized standard error of each concentration point (with a minimum error allowed of 1%). Provided IC50s are averages of two to four replicate experiments.
Filovirus immunofluorescence assays
Vero cells were seeded in 96 well black plates. Peptides and vehicle control were diluted to 1.1X final concentration in culture media [MEM, 5% FBS, gentamicin (10 µg/mL)] and 90 µL/well were incubated on the plate for 1 h at 37°C. In the BSL4 10 µL of virus diluted in media and DEAE–dextran was added to each well (final 8 µg/mL DEAE–dextran). Ebola virus (Kikwit, a representative strain of the Zaire ebolavirus species) and Marburg virus (Ci67) were diluted in culture media to yield a robust signal in the assay (∼20% infected cells). After 1 h at 37°C, the virus and peptides were removed, the wells were washed with PBS and media with peptide was used to replenish the wells. At 24 h postinfection, each well was visualized via light microscope to look for any gross morphological abnormalities. At 48 h postinfection, the wells were washed with PBS and then the cells fixed with 10% formalin. After blocking, the fixed cells were incubated with GP-specific mAb (9G4 for Marburg virus, KZ52 for Ebola virus) followed by incubation with FITC-labeled secondary antibody (goat antimouse or antihuman, respectively). Nuclei were stained with Hoechst solution. Cells were imaged using an Operetta high content device (PerkinElmer, Waltham, MA) and images were analyzed using Harmony software to determine percent of infected cells in a given well. Data were plotted normalized to the vehicle control.
Acknowledgments
We thank Hyung Kim and Dennis Winge for amino acid analysis. We also thank Yu-Chan Chen, Andrew Steiner, Ruei-Lin Hsu, and Niladri Sinha for help with molecular biology, protein purification, and preliminary characterization of the N-trimer mimics and C-peptides. For help with peptide synthesis and purification, we thank Maritza Quintero and Dasha Pruss, and we thank Matthew Movsesian for assistance and advice. This research was funded by a University of Utah Funding Incentive Seed grant to D.M.E. and M.S.K., NIH grant AI102347 to B.D.W. and M.S.K, and NIH grant GM82545 to D.M.E. and C.P.H.. We thank the U.S. Air Force for support of T.R.C.. Portions of this research were carried out at the Stanford Synchrotron Radiation Light Source, (SSRL), which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, NIGMS. Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army, U.S. Air Force, the NIH or NIGMS. The mention of trade names or commercial products does not constitute endorsement or recommendation for use by the Department of the Army, the Department of Defense, or the U.S. Air Force.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
REFERENCES
- 1.Sanchez A, Kahn AS, Zaki SR, Nabel GJ, Ksiazek TG, Peters CJ. Fields virology. Philadelphia: Lippincott, Williams and Wilkins; 2001. [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2014. Chronology of Ebola hemorrhagic fever outbreaks. http://www.cdc.gov/vhf/ebola/resources/outbreak-table.html.
- 3.Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N, Soropogui B, Sow MS, Keita S, De Clerck H, Tiffany A, Dominguez G, Loua M, Traore A, Kolie M, Malano ER, Heleze E, Bocquin A, Mely S, Raoul H, Caro V, Cadar D, Gabriel M, Pahlmann M, Tappe D, Schmidt-Chanasit J, Impouma B, Diallo AK, Formenty P, Van Herp M, Gunther S. Emergence of Zaire Ebola virus disease in Guinea—Preliminary report. New Engl J Med. 2014;371:1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 4.Bossi P, Garin D, Guihot A, Gay F, Crance JM, Debord T, Autran B, Bricaire F. Bioterrorism: Management of major biological agents. Cell Mol Life Sci. 2006;63:2196–2212. doi: 10.1007/s00018-006-6308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volchkov VE, Feldmann H, Volchkova VA, Klenk HD. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc Natl Acad Sci USA. 1998;95:5762–5767. doi: 10.1073/pnas.95.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volchkov VE, Volchkova VA, Stroher U, Becker S, Dolnik O, Cieplik M, Garten W, Klenk HD, Feldmann H. Proteolytic processing of Marburg virus glycoprotein. Virology. 2000;268:1–6. doi: 10.1006/viro.1999.0110. [DOI] [PubMed] [Google Scholar]
- 8.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misasi J, Chandran K, Yang JY, Considine B, Filone CM, Cote M, Sullivan N, Fabozzi G, Hensley L, Cunningham J. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J Virol. 2012;86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 14.Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621–635. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White JM, Schornberg KL. A new player in the puzzle of filovirus entry. Nat Rev Microbiol. 2012;10:317–322. doi: 10.1038/nrmicro2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison JS, Higgins CD, Chandran K, Lai JR. Designed protein mimics of the Ebola virus glycoprotein GP2 alpha-helical bundle: Stability and pH effects. Protein Sci. 2011;20:1587–1596. doi: 10.1002/pro.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Root MJ, Steger HK. HIV-1 gp41 as a target for viral entry inhibition. Curr Pharm Des. 2004;10:1805–1825. doi: 10.2174/1381612043384448. [DOI] [PubMed] [Google Scholar]
- 18.Francis JN, Redman JS, Eckert DM, Kay MS. Design of a modular tetrameric scaffold for the synthesis of membrane-localized d-peptide inhibitors of HIV-1 entry. Bioconjug Chem. 2012;23:1252–1258. doi: 10.1021/bc300076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dye JM, Herbert AS, Kuehne AI, Barth JF, Muhammad MA, Zak SE, Ortiz RA, Prugar LI, Pratt WD. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci USA. 2012;109:5034–5039. doi: 10.1073/pnas.1200409109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geisbert TW, Lee AC, Robbins M, Geisbert JB, Honko AN, Sood V, Johnson JC, de Jong S, Tavakoli I, Judge A, Hensley LE, Maclachlan I. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: A proof-of-concept study. Lancet. 2010;375:1896–1905. doi: 10.1016/S0140-6736(10)60357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marzi A, Feldmann H, Geisbert TW, Falzarano D. Vesicular stomatitis virus-based vaccines for prophylaxis and treatment of filovirus infections. J Bioterror Biodef S1. 2011 doi: 10.4172/2157-2526.S1-004. (4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, Strong JE, Plummer F, Corbett CR, Alimonti JB, Kobinger GP. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci Transl Med. 2012;4:138ra181. doi: 10.1126/scitranslmed.3003876. [DOI] [PubMed] [Google Scholar]
- 23.Warren TK, Warfield KL, Wells J, Swenson DL, Donner KS, Van Tongeren SA, Garza NL, Dong L, Mourich DV, Crumley S, Nichols DK, Iversen PL, Bavari S. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med. 2010;16:991–994. doi: 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- 24.Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci USA. 2001;98:11187–11192. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 26.Eckert DM, Malashkevich VN, Kim PS. Crystal structure of GCN4-pIQI, a trimeric coiled coil with buried polar residues. J Mol Biol. 1998;284:859–865. doi: 10.1006/jmbi.1998.2214. [DOI] [PubMed] [Google Scholar]
- 27.Malashkevich VN, Schneider BJ, McNally ML, Milhollen MA, Pang JX, Kim PS. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc Natl Acad Sci USA. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 29.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-Peptide Inhibitors of HIV-1 Entry. Proc Natl Acad Sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 31.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 32.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. Design of a potent d-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84:11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salerno WJ, Seaver SM, Armstrong BR, Radhakrishnan I. MONSTER: Inferring non-covalent interactions in macromolecular structures from atomic coordinate data. Nucleic Acids Res. 2004;32:W566–W568. doi: 10.1093/nar/gkh434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller MD, Geleziunas R, Bianchi E, Lennard S, Hrin R, Zhang H, Lu M, An Z, Ingallinella P, Finotto M, Mattu M, Finnefrock AC, Bramhill D, Cook J, Eckert DM, Hampton R, Patel M, Jarantow S, Joyce J, Ciliberto G, Cortese R, Lu P, Strohl W, Schleif W, McElhaugh M, Lane S, Lloyd C, Lowe D, Osbourn J, Vaughan T, Emini E, Barbato G, Kim PS, Hazuda DJ, Shiver JW, Pessi A. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc Natl Acad Sci USA. 2005;102:14759–14764. doi: 10.1073/pnas.0506927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montgomery DL, Wang YJ, Hrin R, Luftig M, Su B, Miller MD, Wang F, Haytko P, Huang L, Vitelli S, Condra J, Liu X, Hampton R, Carfi A, Pessi A, Bianchi E, Joyce J, Lloyd C, Geleziunas R, Bramhill D, King VM, Finnefrock AC, Strohl W, An Z. Affinity maturation and characterization of a human monoclonal antibody against HIV-1 gp41. MAbs. 2009;1:462–474. doi: 10.4161/mabs.1.5.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schumacher TN, Mayr LM, Minor DL, Jr, Milhollen MA, Burgess MW, Kim PS. Identification of d-peptide ligands through mirror-image phage display. Science. 1996;271:1854–1857. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- 37.Weinstock MT, Francis JN, Redman JS, Kay MS. Protease-resistant peptide design-empowering nature's fragile warriors against HIV. Biopolymers. 2012;98:431–442. doi: 10.1002/bip.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zawadzke LE, Berg JM. A racemic protein. J Am Chem Soc. 1992;114:4002–4003. [Google Scholar]
- 39.Dintzis HM, Symer DE, Dintzis RZ, Zawadzke LE, Berg JM. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins. 1993;16:306–308. doi: 10.1002/prot.340160309. [DOI] [PubMed] [Google Scholar]
- 40.Noren KA, Noren CJ. Construction of high-complexity combinatorial phage display peptide libraries. Methods. 2001;23:169–178. doi: 10.1006/meth.2000.1118. [DOI] [PubMed] [Google Scholar]
- 41.Weinstock MT, Jacobsen MT, Kay MS. Synthesis and folding of a mirror-image enzyme reveals ambidextrous chaperone activity. Proc Natl Acad Sci USA. 2014;111:11679–11684. doi: 10.1073/pnas.1410900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hackenberger CP, Schwarzer D. Chemoselective ligation and modification strategies for peptides and proteins. Angew Chem Int Ed Engl. 2008;47:10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- 43.Blanco-Canosa JB, Dawson PE. An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew Chem Int Edit. 2008;47:6851–6855. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan Q, Danishefsky SJ. Free-radical-based, specific desulfurization of cysteine: A powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew Chem Int Ed Engl. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 45.Joshi SB, Dutch RE, Lamb RA. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- 46.Liu IJ, Kao CL, Hsieh SC, Wey MT, Kan LS, Wang WK. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antiviral Res. 2009;81:82–87. doi: 10.1016/j.antiviral.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell–cell fusion. AIDS Res Hum Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe S, Takada A, Watanabe T, Ito H, Kida H, Kawaoka Y. Functional importance of the coiled-coil of the Ebola virus glycoprotein. J Virol. 2000;74:10194–10201. doi: 10.1128/jvi.74.21.10194-10201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higgins CD, Koellhoffer JF, Chandran K, Lai JR. C-peptide inhibitors of Ebola virus glycoprotein-mediated cell entry: Effects of conjugation to cholesterol and side chain-side chain crosslinking. Bioorgan Med Chem Lett. 2013;23:5356–5360. doi: 10.1016/j.bmcl.2013.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller EH, Harrison JS, Radoshitzky SR, Higgins CD, Chi X, Dong L, Kuhn JH, Bavari S, Lai JR, Chandran K. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J Biol Chem. 2011;286:15854–15861. doi: 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, Butler MM, Peet NP, Majgier-Baranowska H, Williams JD, Patel I, Moir DT, Bavari S, Ray R, Farzan MR, Rong L, Bowlin TL. Identification of a small-molecule entry inhibitor for filoviruses. J Virol. 2011;85:3106–3119. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roffey R, Tegnell A, Elgh F. Biological warfare in a historical perspective. Clin Microbiol Infect. 2002;8:450–454. doi: 10.1046/j.1469-0691.2002.00501.x. [DOI] [PubMed] [Google Scholar]
- 53.Chen BK, Saksela K, Andino R, Baltimore D. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994;68:654–660. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuhn JH, Radoshitzky SR, Guth AC, Warfield KL, Li W, Vincent MJ, Towner JS, Nichol ST, Bavari S, Choe H, Aman MJ, Farzan M. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J Biol Chem. 2006;281:15951–15958. doi: 10.1074/jbc.M601796200. [DOI] [PubMed] [Google Scholar]
- 55.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 56.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 58.Landau NR, Littman DR. Packaging system for rapid production of murine leukemia virus vectors with variable tropism. J Virol. 1992;66:5110–5113. doi: 10.1128/jvi.66.8.5110-5113.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamburger AE, Kim S, Welch BD, Kay MS. Steric accessibility of the HIV-1 gp41 N-trimer region. J Biol Chem. 2005;280:12567–12572. doi: 10.1074/jbc.M412770200. [DOI] [PubMed] [Google Scholar]
- 60.Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc Natl Acad Sci USA. 1996;93:3602–3607. doi: 10.1073/pnas.93.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 62.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van den Berg S, Lofdahl PA, Hard T, Berglund H. Improved solubility of TEV protease by directed evolution. J Biotechnol. 2006;121:291–298. doi: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 64.Blanco-Canosa JB, Dawson PE. An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew Chem Int Ed Engl. 2008;47:6851–6855. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 66.Rohl CA, Baldwin RL. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of the helix-coil transition in peptides. Biochemistry. 1997;36:8435–8442. doi: 10.1021/bi9706677. [DOI] [PubMed] [Google Scholar]
- 67.Cole JL. Analysis of heterogeneous interactions. Methods Enzymol. 2004;384:212–232. doi: 10.1016/S0076-6879(04)84013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laue T, Shah B, Ridgeway T, Pelletier S. Analytical ultrancentrifugation in biochemistry and polymer science. Roy Soc Chem. 1992:90–125. Computer-aided interpretation of analytical sedimentation data for proteins. [Google Scholar]
- 69.Wohlfarth C. Partial specific volume of poly(ethylene glycol) In: Lechner MD, Arndt KF, editors. Polymer solutions. Berlin: Springer; 2010. [Google Scholar]
- 70.O'Shannessy DJ, Brigham-Burke M, Soneson KK, Hensley P, Brooks I. Determination of rate and equilibrium binding constants for macromolecular interactions using surface plasmon resonance: Use of nonlinear least squares analysis methods. Anal Biochem. 1993;212:457–468. doi: 10.1006/abio.1993.1355. [DOI] [PubMed] [Google Scholar]
- 71.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter JCW, Sweet RM, editors. Methods in enzymology. San Diego: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 72.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 74.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jeffers SA, Sanders DA, Sanchez A. Covalent modifications of the ebola virus glycoprotein. J Virol. 2002;76:12463–12472. doi: 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information