

Abstract
Recent studies have revealed an important role for LTBP-4 in elastogenesis. Its mutational inactivation in humans causes autosomal recessive cutis laxa type 1C (ARCL1C), which is a severe disorder caused by defects of the elastic fiber network. Although the human gene involved in ARCL1C has been discovered based on similar elastic fiber abnormalities exhibited by mice lacking the short Ltbp-4 isoform (Ltbp4S−/−), the murine phenotype does not replicate ARCL1C. We therefore inactivated both Ltbp-4 isoforms in the mouse germline to model ARCL1C. Comparative analysis of Ltbp4S−/− and Ltbp4-null (Ltbp4−/−) mice identified Ltbp-4L as an important factor for elastogenesis and postnatal survival, and showed that it has distinct tissue expression patterns and specific molecular functions. We identified fibulin-4 as a previously unknown interaction partner of both Ltbp-4 isoforms and demonstrated that at least Ltbp-4L expression is essential for incorporation of fibulin-4 into the extracellular matrix (ECM). Overall, our results contribute to the current understanding of elastogenesis and provide an animal model of ARCL1C.
KEY WORDS: Latent transforming growth factor β-binding protein 4, Ltbp-4, Ltbp-4L, Ltbp-4S, Autosomal recessive cutis laxa type 1C, ARCL1C, Elastogenesis, Extracellular matrix, ECM, Fibulin-4, Fibulin-5
INTRODUCTION
The extracellular matrix (ECM) determines and controls the biochemical and mechanical properties of all mammalian tissues including specific ligand-binding properties, elasticity and stiffness (Frantz et al., 2010). As integral ECM components, elastic fibers enable elastic recoil and resilience in all elastic tissues such as arteries, lung, skin and cartilage. Despite being indispensable for survival and normal tissue function (Hornstra et al., 2003; Li et al., 1998), the molecular mechanisms controlling elastic fiber formation have only recently been investigated in more detail (Choudhury et al., 2009; Dabovic et al., 2015; Hirai et al., 2007; Horiguchi et al., 2009; Noda et al., 2013; Sideek et al., 2014). Elastic fibers consist of an internal core of cross-linked monomeric elastin (tropoelastin) surrounded by a network of fibrillin-rich microfibrils (Kielty et al., 2002; Mithieux and Weiss, 2005). Elastic fiber formation in the ECM is a complex process, which involves the formation of fibrillin-rich microfibrillar scaffolds and the deposition of the tropoelastin molecules onto these scaffolds. These processes are followed by the cross-linking of tropoelastin after oxidative deamination of lysine residues by enzymes of the lysyl oxidase family, leading to the formation of covalent cross-links such as desmosines and isodesmosines (Mithieux and Weiss, 2005; Wagenseil and Mecham, 2007; Yanagisawa and Davis, 2010). This process is strongly influenced by the composition of the microfibrillar scaffold. Although the role of several microfibrillar proteins in elastic fiber formation has been studied in more detail (Mithieux and Weiss, 2005; Ramirez et al., 2004; Todorovic and Rifkin, 2012), the impact of the microfibril-associated latent transforming growth factor β-binding protein 4 (LTBP-4) on elastic fiber formation is still poorly understood.
LTBP-4 belongs to a family of four (LTBP-1–LTBP-4) secreted ECM glycoproteins with structural homology to fibrillins (fibrillin-1–fibrillin-3). LTBP-1, LTBP-3 and LTBP-4 covalently bind the small latent complex (SLC) of TGFβ (TGFβ-LAP; LAP, latency-associated peptide) and deposit these complexes into the ECM after secretion (Todorovic and Rifkin, 2012). LTBP-1 and LTBP-3 bind all three TGFβ isoforms, whereas LTBP-4 binds only TGFβ1 (Saharinen et al., 1998). In addition to a TGFβ-related function, LTBPs have structural functions within the ECM (Isogai et al., 2003). For example, LTBP-2, a non TGFβ carrier, interacts with fibulin-5 and thus negatively regulates elastogenesis (Sideek et al., 2014), whereas LTBP-4 facilitates elastogenesis by interacting with fibulin-5 (Dabovic et al., 2015; Noda et al., 2013).
Mammalian cells express two major isoforms of LTBP-4, which in analogy to the long and short isoforms of LTBP-1 discovered earlier are also called long (LTBP-4L; human: NM_001042544.1; murine: NM_175641.2) and short (LTBP-4S; human, NM_001042545.1; murine, NM_001113549.1). These isoforms are encoded by two N-terminal splice variants that are independently expressed using their own promoters (Kantola et al., 2010). In humans LTBP-4L and LTBP-4S are differentially expressed in a tissue-specific manner and show distinct deposition patterns in the ECM (Kantola et al., 2010), presumably due to their different N-termini. For ECM-targeting, the N-terminus of LTBP-4 appears to be particularly important because it enables binding to fibronectin and thereby guides LTBP-4 deposition into the early ECM (Kantola et al., 2008).
TRANSLATIONAL IMPACT.
Clinical issue
Recent studies revealed an important role for latent TGFβ-binding protein-4 (LTBP-4) in elastogenesis (formation of elastic fibers). Its mutational inactivation in humans causes autosomal recessive cutis laxa type 1C (ARCL1C), which is a severe disorder caused by defects of the elastic fiber network. Although the human gene involved in ARCL1C was discovered based on similar elastic fiber abnormalities exhibited by Ltbp4S−/− mice (which lack the short Ltbp-4 isoform), the murine phenotype does not replicate human ARCL1C. The authors therefore hypothesize that inactivation of both the long (Ltbp-4L) and short Ltbp-4 isoforms in the mouse germline might model ARCL1C.
Results
Comparative analysis of Ltbp4S−/− and Ltbp4-null (Ltbp4−/−) mice identify important and partially non-overlapping functions of Ltbp-4L and Ltbp-4S in survival and elastic fiber formation, as well as unique and overlapping expression of the two isoforms depending on the analyzed tissue, with characteristic tissue localization patterns. Furthermore, the authors identify fibulin-4 as a new interaction partner of both Ltbp-4 isoforms and demonstrate that Ltbp-4L is crucial for postnatal survival. Moreover, the authors show that specific N-glycosylation of Ltbp-4S modulates its binding affinity to fibulin-4- and fibulin-5.
Implications and future directions
Overall, the results contribute to the current understanding of elastogenesis and provide an animal model of ARCL1C. The study documents that Ltbp-4L and Ltbp-4S have distinct roles in elastic fiber formation and postnatal survival in mice and identify fibulin-4 as an interaction partner of both Ltbp-4 isoforms. Future experiments involving the generation of Ltbp4L-knockout and tissue-specific-Ltbp4-knockout mouse lines are planned to gain further insights into the isoform-specific functions of Ltbp-4.
We and others have previously reported that mice with an inactivating mutation of Ltbp4S (Ltbp4S−/− mice) are born with alveolar septation defects that deteriorate with age and eventually develop massive pulmonary emphysema associated with dilated cardiomyopathy (Dabovic et al., 2009; Sterner-Kock et al., 2002).
In humans, null mutations in the LTBP4 gene cause autosomal recessive cutis laxa type 1C (ARCL1C; initially called Urban-Rifkin-Davis syndrome), which is a rare congenital connective tissue disorder characterized by severe craniofacial anomalies, lax skin and severe abnormalities in several visceral organs including the lung. However, ARCL1C patients have several lesions that have not been described in Ltbp4S−/− mice (Callewaert et al., 2013; Sterner-Kock et al., 2002; Urban et al., 2009). A possible explanation for this could be that Ltbp-4L expressed in Ltbp4S−/− mice compensates in part for the loss of Ltbp-4S.
To address Ltbp-4 isoform-specific functions, we generated Ltbp4-null (Ltbp4−/−) mice that, like the ARCL1C patients, do not express any Ltbp-4. Here, we show that Ltbp4−/− mice nearly replicate the features of ARCL1C syndrome. We further show that Ltbp-4L and Ltbp-4S have important and partially non-overlapping roles in survival and elastic fiber formation, identify fibulin-4 as a new interacting partner of both Ltbp-4 isoforms and demonstrate that at least Ltbp-4L expression is essential for incorporation of fibulin-4 into the ECM.
RESULTS
Complete inactivation of the Ltbp4 gene in mice results in weight loss, growth reduction and neonatal death
We previously established and characterized Ltbp4S-knockout (Ltbp4S−/−) mice (Sterner-Kock et al., 2002), which show similar elastic fiber defects in the lung to those found in ARCL1C patients; however the mice survived to adulthood without manifestation of major clinical symptoms. To investigate whether the less-severe phenotype of Ltbp4S−/− mice was due to the presence of Ltbp-4L, which might have important functions during development, we generated Ltbp4-null (Ltbp4−/−) mice lacking both Ltbp-4 isoforms (supplementary material Fig. S1). Indeed, unlike Ltbp4S−/− mice, Ltbp4−/− mice showed no expression of Ltbp-4 (Fig. 1A; supplementary material Fig. S2).
Fig. 1.

Clinical and morphological analyses of Ltbp-4 deficient mice. (A) Representative immunoblots of lung, aorta, skin and hearts showing the reduced or absent expression of Ltbp-4 in Ltbp4S−/− and Ltbp4−/− mice compared to WT mice. (B) Kaplan–Meier survival curve revealing the significantly higher neonatal mortality in Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥23; **P<0.01 versus WT). (C) Ltbp4−/− mice showed reduced body size compared to Ltbp4S−/− and WT mice. Scale bar: 0.5 cm. (D) The P4-P12 weight curve showed significantly reduced body weight of Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥8; **P<0.01 versus WT). (E) In Ltbp4S−/− mice, the pulmonary parenchyma showed enlarged alveolar spaces with reduced numbers of alveoli and multifocal areas of atelectasis compared to WT mice. Ltbp4−/− lungs revealed lack of lobular architecture, severely enlarged alveolar spaces and emphysematous areas compared to WT mice. Scale bars: 40 μm. (F,G) Aortas showed marked thickening of the aortic wall in Ltbp4S−/− and Ltbp4−/− mice compared to WT mice (n≥3; *P<0.05, **P<0.01). Scale bars: 40 μm. (H,I) Ltbp4−/− mice showed reduced dermal thickness compared to Ltbp4S−/− and WT mice (n≥5;**P<0.01). Scale bars: 200 μm. (J) The epidermal hydration was increased in Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥9; *P<0.05). (K) The transepidermal water loss tended to be higher in Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥9; not significant). (L) The net-elasticity of the skin of both Ltbp-4-deficient mice tended to be lower compared to WT mice (n≥9; not significant). (M) Representative images showing the increased size of hearts of Ltbp4−/− mice compared to hearts of WT and Ltbp4S−/− mice. Scale bar: 0.1 cm. (N) Heart weight:body weight ratios showing that hearts of Ltbp4−/− mice were significantly heavier than hearts of WT and Ltbp4S−/− mice (n≥7; **P<0.01). (O) Wall thickness of the left ventricle was not changed in Ltbp4S−/− and Ltbp4−/− mice compared to WT mice (n≥6; not significant). (P) Wall thickness of the right ventricle was significantly increased in Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥6; *P<0.05). (Q) Representative short-axis images from MRI analysis revealing a flattened interventricular septum resulting in a more oval shape of the left ventricle in Ltbp4S−/− and Ltbp4−/− mice compared to WT mice.
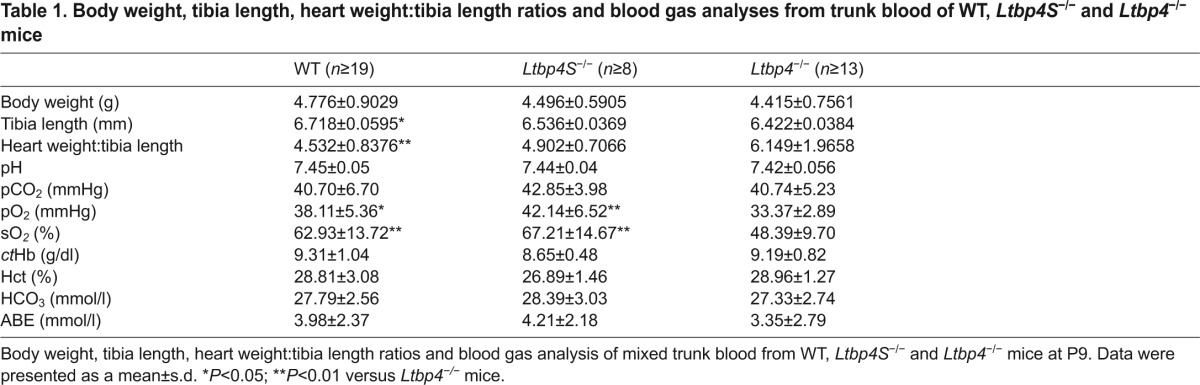
Complete lack of Ltbp-4 expression reduced survival dramatically as most Ltbp4−/−mice died between P8 and P14 (Fig. 1B), whereas Ltbp4S−/− mice survived to adulthood (Sterner-Kock et al., 2002). Furthermore, Ltbp4−/− mice were smaller than Ltbp4S−/− mice and showed decreased body weight as compared with their wild-type (WT) littermates (Fig. 1C,D; Table 1).
Table 1.
Body weight, tibia length, heart weight:tibia length ratios and blood gas analyses from trunk blood of WT, Ltbp4S−/− and Ltbp4−/− mice
Ltbp-4-deficient mice show morphological abnormalities in lung, aorta and skin
Similar to the ARCL1C lung histopathology, the lungs of Ltbp4−/− mice lacked a lobular architecture and showed severely enlarged alveolar spaces (Fig. 1E). Lungs from Ltbp4S−/− mice also exhibited enlarged alveolar spaces with multifocal areas of atelectasis although these were much less pronounced than in the Ltbp4−/− lungs (Fig. 1E). This difference was reflected by a significantly reduced oxygen saturation (sO2) and partial oxygen pressure (pO2) in Ltbp4−/− mice when compared with Ltbp4S−/− and WT mice (Table 1).
Aortas of Ltbp-4-deficient mice were malformed and tortuous (supplementary material Fig. S3) (Noda et al., 2013). Aortic walls were twice as thick in Ltbp4−/− mice as in WT mice (Fig. 1F,G). The aortic wall thickening could not be attributed to extra elastic lamellae (Fig. 1F), increased deposition of glycosaminoglycans in the ECM, increased numbers of smooth muscle cells or increased proliferation (supplementary material Fig. S4).
Ltbp4−/− mice showed reduced dermal thickness compared to Ltbp4S−/− and WT mice (Fig. 1H,I), whereas the thickness of the epidermis was not changed (data not shown). The epidermal hydration was increased (Fig. 1J) and the transepidermal water loss (Fig. 1K) tended to be higher but did not reach statistical significance in Ltbp4−/− mice compared to Ltbp4S−/− and WT mice. The net-elasticity of the skin of both Ltbp-4-deficient mice tended to be lower but did not reach statistical significance compared to WT mice (Fig. 1L).
Ltbp4−/− mice develop cardiac hypertrophy of the right ventricle with flattening of the interventricular septum
Hearts of Ltbp4−/− but not of Ltbp4S−/− mice were larger when compared to WT hearts (Fig. 1M) as indicated by higher heart to body weight (Fig. 1N) and heart weight to tibia length ratios (Table 1). Stereological analysis revealed significantly increased right ventricular wall thickness in Ltbp4−/− hearts as compared to Ltbp4S−/− or WT hearts, but showed no change in the left ventricular walls (Fig. 1O,P). The left ventricular end-diastolic volume (LVEDV), the left ventricular end-systolic volume (LVESV) and the stroke volume (SV) tended to be lower and the ejection fraction (EF) tended to be higher but did not reach statistical significance in Ltbp4−/− mice compared to WT mice (supplementary material Table S1).
Ltbp4−/− mice and to a lesser extent Ltbp4S−/− mice develop flattened interventricular septae resulting in a more oval shape of the left ventricle on short axis views (Fig. 1Q). The ratio between the maximum and minimum diameter of the left ventricle measured on end-diastolic short axis images was 1.5 in Ltbp4−/− and 1.4 in Ltbp4S−/− mice. In contrast, the ratio for WT ventricles was 1.1, consistent with a round ventricle shape (Fig. 1Q).
Overlapping and distinct expression and localization patterns of Ltbp-4L and Ltbp-4S
Because the different phenotypes of Ltbp4S−/− and Ltbp4−/− mice might reflect differences in tissue-specific expression and localization of the Ltbp-4 isoforms, we quantified the amount of Ltbp4L and Ltbp4S transcripts in various tissues of WT, Ltbp4S−/− and Ltbp4−/− mice and related these results to Ltbp-4 protein expression.
WT lungs expressed comparable amounts of Ltbp4L and Ltbp4S mRNA, whereas in lungs from Ltbp4S−/− mice only Ltbp4L was expressed. In the lungs of Ltbp4−/− mice neither Ltbp4L nor Ltbp4S mRNA was expressed (Fig. 2A,B). Ltbp-4 immunoreactivity was found primarily in the bronchial and bronchiolar walls, as well as in the parenchyma and vascular walls, of WT lungs (Fig. 2C). There was no difference in the tissue distribution of Ltbp-4 between WT and Ltbp4S−/− lungs (Fig. 2C), indicating that Ltbp-4 isoform expression is overlapping in the lung. Ltbp-4 immunoreactivity was completely absent in Ltbp4−/− lungs (Fig. 2C).
Fig. 2.

Localization patterns of Ltbp-4L, Ltbp-4S and elastin. (A) Quantitative PCRs of lung, aorta, skin and heart of WT and Ltbp4S−/− mice showing that each tissue had different amounts of Ltbp4L mRNA. There was no mRNA expression of Ltbp4L in lung, aorta, skin and heart of Ltbp4−/− mice (n≥3; §, no expression detectable). (B) Quantitative PCRs of lung, aorta, skin and heart of WT mice showing that each tissue had different amounts of Ltbp4S mRNA. There was no mRNA expression of Ltbp4S in lung, aorta, skin and heart of Ltbp4S−/− and Ltbp4−/− mice (n≥3; §, no expression detectable). (C) Representative images of Ltbp-4 immunoreactivity of lungs from WT, Ltbp4S−/− and Ltbp4−/− mice. Ltbp-4 was localized particularly in bronchial and bronchiolar walls and in vascular walls of WT and Ltbp4S−/− mice. Lungs of Ltbp4−/− mice were negative for Ltbp-4 immunoreactivity. Scale bars: 20 μm. (D) Representative images of Ltbp-4 immunoreactivity of aortas from WT, Ltbp4S−/− and Ltbp4−/− mice. Black arrows point to the aortic luminal side and black arrowheads to the adventitia. Ltbp-4 immunoreactivity was present in the vicinity of aortic elastic lamella throughout the entire aorta (from the endothelial lining to the adventitia) of WT mice and in the vicinity of the internal elastic lamella (IEL) and in the adventitia of Ltbp4S−/− mice. The aortic intramural elastic lamella of Ltbp4S−/− mice and the entire aorta of Ltbp4−/− mice showed no immunoreactivity for Ltbp-4. Scale bars: 20 μm. (E) Representative images of Ltbp-4 immunoreactivity of skin from WT, Ltbp4S−/− and Ltbp4−/− mice. In WT skin, Ltbp-4 immunoreactivity was present in the entire dermis, whereas it was completely absent in the epidermis. There was no difference in the tissue distribution of Ltbp-4 between WT and Ltbp4S−/− skin. The skin of Ltbp4−/− mice expressed no Ltbp-4. Scale bars: 20 μm. (F) Representative images of Ltbp-4 immunoreactivity of hearts from WT, Ltbp4S−/− and Ltbp4−/− mice. Upper panel: black arrows point to the epicardium of the heart. Ltbp-4 immunoreactivity was present within the myocardium and in the epicardium of WT mice. The myocardium and the epicardium of Ltbp4S−/− and Ltbp4−/− mice were negative for Ltbp-4 immunoreactivity. Lower panel: black arrows point to the endocardium. The endocardium of WT and Ltbp4S−/− mice clearly has Ltbp-4 immunoreactivity, whereas Ltbp4−/− mice were negative for Ltbp-4 immunoreactivity. Scale bars: 20 μm. (G) Representative histochemical elastica stainings of lungs (upper panels) and aortas (lower panels) displaying moderate elastic fiber fragmentation with intact and disrupted elastic fibers in Lbp4S−/− mice compared to WT mice and an increased degree of fragmentation of the elastic fibers in Ltbp4−/− mice compared to Lbp4S−/− mice. Scale bars: 20 μm. (H) Representative semi-thin sections of lungs showing elastic fibers with fragmented and intact parts in Lbp4S−/− mice and total disruption of elastic fibers in Ltbp4−/− mice compared to WT mice. Scale bars: 6 μm. (I) Quantitative analysis of disruptions of the IEL showing significantly higher numbers of disruptions in Ltbp4−/− mice compared to WT and Ltbp4S−/− mice and significantly higher numbers of disruptions in Ltbp4S−/− mice compared to WT mice (n=6; n.d., not detectable; *P<0.05, **P<0.01).
Aortas of WT mice expressed comparable amounts of Ltbp4L and Ltbp4S mRNA, whereas the aortas of Ltbp4S−/− mice expressed exclusively Ltbp4L mRNA. Aortas of Ltbp4−/− mice expressed neither Ltbp4 isoform (Fig. 2A,B). In WT aortas, Ltbp-4 immunoreactivity was present in the vicinity of the aortic elastic lamellae throughout the entire aortic wall extending from the endothelial lining to the adventitia (Fig. 2D). However, in Ltbp4S−/− mice, Ltbp-4 was only detectable in the vicinity of the internal elastic lamella (IEL) and in the adventitia. Intramural elastic lamellae of Ltbp4S−/− mice were negative for Ltbp-4 staining, suggesting that Ltbp-4L expression is restricted to the adventitia and the IEL (Fig. 2D). Aortas of Ltbp4−/− mice expressed no Ltbp-4 (Fig. 2D).
In WT skin, Ltbp4S was the major isoform expressed, representing about 98% of the total Ltbp4 transcripts. In Ltbp4S−/− skin, only Ltbp4L mRNA was expressed and in Ltbp4−/− skin neither Ltbp4L nor Ltbp4S mRNA was expressed (Fig. 2A,B). In WT skin, Ltbp-4 immunoreactivity was present in the entire dermis and was completely absent in the epidermis (Fig. 2E). There was no difference in the tissue distribution of Ltbp-4 between WT and Ltbp4S−/− skin (Fig. 2E), indicating that Ltbp-4 isoform expression is overlapping in the skin. The skin of Ltbp4−/− mice expressed no Ltbp-4 (Fig. 2E).
Ltbp4S was the major isoform expressed in WT hearts, representing about 93% of the total Ltbp4 transcripts. Hearts of Ltbp4S−/− mice expressed only Ltbp4L mRNA. Ltbp4−/− hearts expressed neither Ltbp4L nor Ltbp4S mRNA (Fig. 2A,B). In the heart, Ltbp-4 immunoreactivity was detectable in the epicardium, myocardium and endocardium of WT mice (Fig. 2F). In Ltbp4S−/− hearts Ltbp-4 immunoreactivity was only detectable in the endocardium, implying that Ltbp-4L expression is restricted to this region. Ltbp4−/− hearts showed no Ltbp-4 immunoreactivity (Fig. 2F).
Ltbp-4 deficiency in mice results in the failure to form an intact elastic fiber network
We and others have shown that Ltbp4S−/− mice display severe defects in elastic fiber formation (Dabovic et al., 2015; Noda et al., 2013; Sterner-Kock et al., 2002; Urban et al., 2009). Elastic fiber fragmentation was present in aortas and lungs of both Ltbp-4-deficient mice (Fig. 2G). In Ltbp4S−/− mice, elastic fibers, although fragmented, consisted of short intact fibers dispersed between scattered patches of elastin. In contrast, Ltbp4−/− tissues showed only scattered patches of elastin (Fig. 2G). These observations were confirmed on ultrastructural images of semi-thin sections (Fig. 2H, supplementary material Fig. S5A).
Major differences in elastic fiber structure between the Ltbp-4-deficient mice were found in the subluminal region of the aorta, where Ltbp-4L is preferentially expressed (Fig. 2D). Whereas Ltbp4S−/− aortas exhibited only moderate disruptions of the IEL adjacent to the endothelial lining, disruptions were significantly more severe in the aortas of Ltbp4−/− mice (Fig. 2I).
There were no significant differences in tropoelastin mRNA expression (supplementary material Fig. S5B), in the amount of elastin (supplementary material Fig. S5C) or in the ratio of desmosine (DES) and isodesmosine (IDES) to elastin (supplementary material Fig. S5D) between the genotypes, indicating that the elastic fiber defects observed in the Ltbp-4-deficient mice were not due to altered elastin levels or ineffective elastin cross-linking.
Ltbp-4 modulates fibulin-5 expression and matrix deposition
It has been suggested that Ltbp-4S promotes elastic fiber assembly by facilitating tropoelastin deposition onto microfibrils through direct interactions with fibulin-5 (Dabovic et al., 2015; Noda et al., 2013). There was an 80% reduction of fibulin-5 mRNA levels in Ltbp4S−/− and Ltbp4−/− lungs compared to WT lungs (supplementary material Fig. S6A). Moreover, the normal fibrillar structure of fibulin-5 was disrupted in the lungs and aortas of both Ltbp-4-deficient mice and replaced by scattered amorphous patches, suggesting that there is defective deposition of fibulin-5 into the ECM (supplementary material Fig. S6B). Ltbp4S−/− fibroblasts expressed ~50% and Ltbp4−/− ~10% of the normal amount of fibulin-5 mRNA and protein (supplementary material Fig. S6C,D). Moreover, fibulin-5 ECM deposition was defective in both Ltbp4S−/− and Ltbp4−/− lung fibroblasts and similar to the ECM deposition observed in the corresponding Ltbp-4-deficient tissues (supplementary material Fig. S6B,E).
Deposition of fibulin-4 on microfibrils requires Ltbp-4L
Previous observations in elastin-producing cell cultures have shown that fibulin-4 and fibulin-5 are required for tropoelastin deposition into the ECM (Yamauchi et al., 2010). Although the interaction between Ltbp-4S and fibulin-5 has recently been shown to be essential for elastic fiber formation (Noda et al., 2013), functional interactions between Ltbp-4 and fibulin-4 in this process have not yet been described. The expression of fibulin-4 declined in lungs from both Ltbp4S−/− and Ltbp4−/− mice (Fig. 3A). However, fibulin-4 deposition differed significantly between the knockout strains. Whereas fibulin-4 deposition into the ECM of Ltbp4S−/− mice was comparable to that in WT mice, Ltbp4−/− mice exhibited a punctuate deposition pattern (Fig. 3B). We confirmed the decline of fibulin-4 expression in lung fibroblasts from Ltbp-4-deficient mice (Fig. 3C,D), whereas only Ltbp4−/− cells showed a defective fibulin-4 matrix deposition (Fig. 3E). Moreover, to normalize fibulin-4 levels, recombinant full-length fibulin-4 (rfibulin-4) was added to primary lung fibroblasts. Primary lung fibroblasts from Ltbp4S−/− and WT mice cultured with rfibulin-4 revealed fibrillar deposition of the recombinant protein, whereas the deposition of rfibulin-4 appeared scattered and not fibrillar in primary lung fibroblasts from Ltbp4−/− mice (Fig. 3F), suggesting that at least Ltbp-4L is required for fibrillar deposition of fibulin-4.
Fig. 3.

Ltbp-4L is necessary for fibrillar matrix deposition of fibulin-4. (A) Fibulin-4 mRNA expression showed significant downregulation in lungs from Ltbp4S−/− and Ltbp4−/− mice compared to WT mice (n=4; *P<0.05). (B) Representative images showing fibulin-4 immunoreactivity and disruption of the fibrillar structure of fibulin-4 fibers in aortas (upper panel) and lungs (lower panel) from Ltbp4−/− mice compared to WT and Ltbp4S−/− mice. Scale bars: 20 μm (upper panels), 50 μm (lower panels). (C) Fibulin-4 mRNA expression showed significant downregulation in lung fibroblasts isolated from Ltbp4−/− mice compared to Ltbp4S−/− and WT mice (n≥5; **P<0.01). (D) Representative immunoblot of lung fibroblasts (left) and its densitometric analysis (right) revealing significant downregulation of fibulin-4 in Ltbp4S−/− and Ltbp4−/− mice compared to WT mice (n≥4; *P<0.05; **P<0.01). Protein as well as mRNA expression of fibulin-4 of the WT was set to 1. (E) Representative immunofluorescence staining of Ltbp-4 and fibulin-4 revealing reduced expression and disrupted fibrillar structure of fibulin-4 in primary lung fibroblasts from Ltbp4−/− mice compared to Ltbp4S−/− and WT mice. Scale bars: 100 μm. (F) Representative immunofluorescence staining revealing fibrillar deposition of recombinant full-length fibulin-4 (rfibulin-4; 20 nm) in primary lung fibroblasts from Ltbp4S−/− and WT mice, whereas rfibulin-4 appeared scattered and not fibrillar in primary lung fibroblasts from Ltbp4−/− mice. For detection of rfibulin-4, an anti-strep antibody was used. Scale bars: 50 μm.
N-terminal regions of Ltbp-4L and Ltbp-4S interact with fibulin-4
The N-terminal region of Ltbp-4S directly interacts with fibulin-5 (Noda et al., 2013). Based on our data showing that at least Ltbp-4L is required for fibulin-4 deposition (Fig. 3B,E), we performed protein-protein interaction studies using N-terminal Ltbp-4L (Ltbp-4L-2xStrep) and Ltbp-4S (Ltbp-4S-2xStrep) fragments conjugated to a 2xStrep tag (Fig. 4A) and recombinant full-length fibulin-4 and fibulin-5 (rfibulin-4 and rfibulin-5). Surface plasmon resonance analysis revealed that Ltbp-4L-2xStrep and Ltbp-4S-2xStrep interact with rfibulin-4 and rfibulin-5 (Fig. 4B,C). However, the molecular dissociation of Ltbp-4L-2xStrep from both rfibulins was significantly slower than that of Ltbp-4S-2xStrep (supplementary material Table S2), indicating that the 126 N-terminal amino acids specific to Ltbp-4L are responsible for a stronger binding affinity of Ltbp-4L-2xStrep to rfibulin-4 and rfibulin-5.
Fig. 4.

Interaction studies of the Ltbp-4L and Ltbp-4S N-terminal regions with full-length fibulin-4 and fibulin-5. (A) Domain structure of full-length Ltbp-4L and Ltbp-4S and the recombinantly expressed Ltbp-4L (Ltbp-4L-2xStrep) and Ltbp-4S (Ltbp-4S-2xStrep) N-terminal fragments. The full-length proteins consist of 4-cystein (4-cys) repeats (white rhombi), non-Ca2+-binding EGF-like repeats (black rectangles), Ca2+-binding EGF-like repeats (white rectangles), hybrid domains (black ellipses) and 8-cystein (8-cys) repeats (white ellipses). The N-terminal fragments consist of two (Ltbp-4L-2xStrep) or one (Ltbp-4S-2xStrep) unique 4-cys repeats, the common non-Ca2+-binding EGF-like repeat and a C-terminal 2xStrep tag (red ellipses). Binding sites for ECM proteins, putative N-glycosylation sites (green lines) as well as the amino acid (aa) lengths are indicated. (B,C) Sensorgrams from surface-plasmon resonance interaction experiments showed a stronger binding affinity of Ltbp-4L-2xStrep (0–320 nM) ‘flown’ over immobilized recombinant full-length fibulin-5 (B; rfibulin-5) or immobilized recombinant full-length fibulin-4 (C; rfibulin-4) compared to Ltbp-4S-2xStrep (0–80 nM) flown over immobilized rfibulin-5 (B) or rfibulin-4 (C). The results are expressed as resonance units (RUs; n=2). (D) Deglycosylation digest with PNGase F of denatured recombinant full-length human LTBP-4S (rLTBP-4S) showing that there is a shift towards lower molecular mass positions. (E) Upper panel: deglycosylation of Ltbp-4L-2xStrep and Ltbp-4S-2xStrep. Ltbp-4L-2xStrep was unaffected, whereas Ltbp-4S-2xStrep showed a shift towards lower molecular weight positions. Lower panel: Ltbp-4S-2xStrep was digested with PNGase F under native (left lanes) and denaturing (right lanes) conditions. Both conditions resulted in a shift towards lower molecular weight positions. (F,G) After digest under non-denaturing conditions Ltbp-4L-2xStrep and Ltbp-4S-2xStrep were both able to bind to rfibulin-4 and -5 immobilized on a Biacore chip. Ltbp-4L-2xStrep binding was not affected, whereas Ltbp-4S-2xStrep showed an increase in binding of 15% to 20% after deglycosylation. The continuous lines represented the response before and the dashed lines after deglycosylation (n=2).
Ltbp-4 isoform-specific N-glycosylation modulates fibulin binding
In silico analysis revealed one putative N-glycosylation site within the specific N-terminal amino acid sequence of Ltbp-4S whereas the N-terminus of Ltbp-4L was predicted to only be subject to O-linked glycosylation (Fig. 4A). We verified N-linked glycosylation in a PNGase F deglycosylation assay with recombinant full-length human LTBP-4S (Fig. 4D; rLTBP-4S) and also with Ltbp-4S-2xStrep (Fig. 4E). PNGase F digest of Ltbp-4L-2xStrep showed no difference in band retardation (Fig. 4E). In order to test whether N-glycosylation of Ltbp-4L-2xStrep and Ltbp-4S-2xStrep contributes to rfibulin-4 and rfibulin-5 binding, the PNGase F deglycosylation assay was also performed under non-denaturating conditions to enable subsequent surface plasmon resonance analysis (Fig. 4E).
We demonstrated enhanced binding of Ltbp-4S-2xStrep to rfibulin-4 and rfibulin-5 after deglycosylation with a signal increase of 15% to 20% whereas binding between Ltbp-4L-2xStrep and rfibulin-4 or rfibulin-5 was not changed after deglycosylation (Fig. 4F,G).
DISCUSSION
The human gene involved in ARCL1C was discovered based on the ultrastructural similarity of elastic fiber defects exhibited in mice lacking Ltbp-4S (Ltbp4S−/−) and a subcohort of cutis laxa patients (Sterner-Kock et al., 2002; Urban et al., 2009). However, unlike ARCL1C patients, Ltbp4S−/− mice live to adulthood despite severe elastic fiber defects (Sterner-Kock et al., 2002), whereas ARCL1C patients die very early due to extensive developmental abnormalities in multiple visceral organs (Urban et al., 2009). These phenotypic differences are likely due to the inactivation of both LTBP-4 isoforms in ARCL1C patients, which does not occur in Ltbp4S−/− mice. To test this hypothesis we generated Ltbp4-knockout (Ltbp4−/−) mice lacking both Ltbp-4 isoforms.
Ltbp4−/− mice grew significantly slower than their WT littermates, developed dermal and cardiopulmonary abnormalities and died within 2 weeks after birth, most likely due to respiratory failure caused by extensive pulmonary emphysema similar to that observed in ARCL1C patients (Table 2) (Callewaert et al., 2013; Urban et al., 2009).
Table 2.
Clinical and morphological characteristics of individuals with ARCL1C compared to Ltbp4S−/− and Ltbp4−/− mice

The observed lung, aortic, skin and cardiac phenotype of Ltbp4S−/− and Ltbp4−/− mice was more severe in the Ltbp4−/− mice (Table 2), suggesting that Ltbp-4L compensates for some Ltbp-4S functions. Ltbp-4L functions seem particularly important during postnatal development and survival to adulthood (Sterner-Kock et al., 2002). For instance, in the skin, the relatively low Ltbp-4L expression compared with Ltbp-4S seems to be important for dermal thickness and density. The distinct tissue expression patterns of Ltbp-4L in heart and aorta might result from the activation of the two known Ltbp-4 promoters under the control of independent transcription factors (Bultmann et al., 2013; Kantola et al., 2010).
Ltbp4−/− mice displayed right ventricular hypertrophy in the heart, which Ltbp4S−/− mice do not. This could be either a primary consequence caused by the additional loss of Ltbp-4L or secondary to increased pulmonary resistance leading to pulmonary hypertension (Bleeker et al., 2006). The human ARCL1C syndrome includes pulmonary artery stenosis, pulmonary hypertension, atrial septal defects, patent foramen ovale, heart valve defects and a mild right ventricular hypertrophy in one case (Urban et al., 2009). In contrast to older Ltbp4S−/− mice (Sterner-Kock et al., 2002), no cardiomyopathy was found in ARCL1C patients, but these conditions might require a longer time to develop in humans (Urban et al., 2009).
The disruption of the aortic IEL in mice seems to be dependent on the expression of Ltbp-4L because we found that only Ltbp4−/− but not Ltbp4S−/− mice had severely disrupted IEL. Fibulin-2 and fibulin-5 have been reported to function cooperatively to form the IEL during postnatal development (Chapman et al., 2010). Interestingly, Ltbp-4 deficiency leads to upregulation of fibulin-2, especially in the area of the endothelial lining of the vessel walls in the heart (supplementary material Fig. S7). Obviously, this upregulation of fibulin-2 could protect the IEL only in Ltbp4S−/− mice, indicating a possible direct or indirect interaction between Ltbp-4L and fibulin-2. Similar to other models of disturbed elastogenesis, including elastin (Eln−/−) (Li et al., 1998), fibulin-4 (Fibulin-4−/− and Fibulin-4R/R) (Hanada et al., 2007; McLaughlin et al., 2006), and lysyl oxidase (Lox−/−) (Mäki et al., 2005) mutant mice, Ltbp4−/− and to a lesser extent Ltbp4S−/− mice develop thick aortic walls. Ultrastructural analysis of aortas (data not shown) and elastic lung arteries (supplementary material Fig. S5) revealed the presence of amorphous material between elastic fibers in Ltbp4−/− mice, a phenotype that has already been described in Fibulin-4R/R mice (Hanada et al., 2007). However, we were not able to characterize this material in more detail.
Fragmented elastic fibers are the hallmark of LTBP-4 deficiency in both humans and mice (Sterner-Kock et al., 2002; Urban et al., 2009). Ltbp4−/− mice and ARCL1C patients (Callewaert et al., 2013; Urban et al., 2009) show massive elastic fiber fragmentation, which is significantly less severe in Ltbp4S−/− mice. Formation of functional elastic fibers requires cross-linking of tropoelastin monomers after oxidative deamination by lysyl oxidases and organization of the insoluble elastin matrix onto a pre-assembled fibrillin microfibril scaffold (Mithieux and Weiss, 2005; Wagenseil and Mecham, 2007; Yanagisawa and Davis, 2010). In addition to structural proteins and cross-linking enzymes, elastic fiber formation requires multiadhesive adaptor proteins, such as fibulin-4 (Horiguchi et al., 2009; McLaughlin et al., 2006), fibulin-5 (Nakamura et al., 2002; Yanagisawa et al., 2002) and Ltbp-4 (Noda et al., 2013; Sterner-Kock et al., 2002; Urban et al., 2009), all of which are present on microfibrils. Fibulin-4 and fibulin-5 have crucial non-redundant functions in the assembly of elastic fibers (Papke and Yanagisawa, 2014).
Fibulin-5 mediates coacervation of tropoelastin prior to cross-linking (Hirai et al., 2007; Wachi et al., 2008) by binding to Ltbp-4S (Noda et al., 2013), whereas fibulin-4 enhances the binding of tropoelastin to lysyl oxidase and thus facilitates cross-linking (Choudhury et al., 2009; Horiguchi et al., 2009). Fibulin-5−/− mice have severely disorganized elastic fibers throughout the body and develop severe and progressive elastinopathy postnatally, but survive to adulthood (Nakamura et al., 2002; Yanagisawa et al., 2002). Fibulin-4−/− mice fail to form elastic fibers and die perinatally (Horiguchi et al., 2009; McLaughlin et al., 2006), whereas hypomorphic Fibulin-4R/R mice can survive up to 14 weeks (Hanada et al., 2007). This suggests that at least 25% fibulin-4 expression, and probably its proper deposition, but not fibulin-5, is crucial for postnatal survival. The expression of fibulin-5 and to a lesser extent fibulin-4 was repressed in Ltbp4S−/− and Ltbp4−/− mice, suggesting that Ltbp-4L and Ltbp-4S are required for adequate fibulin-4 and fibulin-5 expression. The molecular mechanisms leading to fibulin-4 and fibulin-5 repression in Ltbp-4-deficient mice remain to be investigated. This repression could possibly be mediated by an outside-in signaling cascade triggered by cellular sensing of an altered ECM in Ltbp-4-deficient mice.
The fibrillar deposition of fibulin-5 is destroyed in both Ltbp-4-deficient mouse strains. However, matrix deposition of fibulin-4 appeared to be unaltered in Ltbp4S−/− mice, whereas it was highly defective in Ltbp4−/− mice, suggesting that Ltbp-4L is important for this process. The ECM deposition of other ECM proteins, such as fibronectin and fibrillin-1, was not affected by Ltbp-4 deficiency (supplementary material Fig. S8). We showed that Ltbp-4L and Ltbp-4S bind to fibulin-4 and fibulin-5 through their N-termini with different affinities. The much higher affinity of Ltbp-4L to fibulin-4 and fibulin-5 might reflect its importance during early postnatal development when rapid elastic fiber formation is mandatory, for example, during postnatal lung maturation. A possible mechanism to modify protein-protein binding is the attachment of glycosaminoglycan (GAG) chains (Lowery et al., 2014; Torreno-Pina et al., 2014). We verified the predicted specific N-glycosylation of Ltbp-4S in vitro and in vivo (supplementary material Fig. S9) and showed that deglycosylation enhances binding of Ltbp-4S but not of Ltbp-4L to fibulin-4 and fibulin-5. N-glycosylation patterns can vary between different tissues and thereby possibly modulate Ltbp-4S binding to fibulin-4 and fibulin-5. In situations of high glycosylation of the Ltbp-4S N-terminus, Ltbp-4L-driven binding mechanisms might be favored. However, another possibility is that this specific N-glycosylation site is used to enable Ltbp-4S to bind to additional unknown factors.
Besides their obvious role within the ECM, Ltbp-4L and Ltbp-4S are reported to modulate the activity of TGFβ by facilitating its secretion and deposition into the ECM (Annes et al., 2004; Miyazono and Heldin, 1991). However, TGFβ activity was not affected in lung fibroblasts of the two Ltbp4-knockout strains (supplementary material Fig. S10), suggesting that the observed phenotypes are largely attributable to the loss of Ltbp-4 structural functions.
Our results suggest that Ltbp-4L and Ltbp-4S orchestrate elastin deposition onto the ECM fibrillin microfibrils by binding fibulin-4 and fibulin-5, which in turn both target tropoleastin (Choudhury et al., 2009; Nakamura et al., 2002). In the absence of Ltbp-4S, the tropoelastin–fibulin-4–fibulin-5 complexes are deposited only partially onto microfibrils, resulting in an ECM with scattered fibrillar elastic fibers surrounded by amorphous tropoelastin-fibulin aggregates. In ECMs produced by cells lacking Ltbp-4L and Ltbp-4S, fibrillar fibers are non-existent and only amorphous aggregates are observed, strongly suggesting that both Ltbp-4 isoforms are required for normal elastogenesis (Fig. 5).
Fig. 5.

Proposed model for the role of Ltbp-4L and Ltbp-4S in elastogenesis. In the presence of Ltbp-4L and Ltbp-4S, microaggregation of tropoelastin, which is tethered to fibulin-4 and fibulin-5, deposits linearly onto microfibrils. Subsequent coalescence of tropoelastin takes place on microfibrils, resulting in fibrillar deposition of the tropoelastin–fibulin-4–fibulin-5 complex. In the absence of Ltbp-4S, the tropoelastin–fibulin-4–fibulin-5 complex only partly deposits on microfibrils, resulting in scattered fibrillar elastic fibers as well as amorphous aggregates of tropoelastin–fibulin-4–fibulin-5, which grow to form globular structures. In the absence of Ltbp-4L and Ltbp-4S, all tropoelastin–fibulin-4–fibulin-5 complexes form dysmorphous, non-fibrillar globular structures. Modified from Noda et al. (Noda et al., 2013).
In summary, we characterized a new mouse model for ARCL1C that will help to elucidate the pathogenesis of this severe disease. We further showed that Ltbp-4L and Ltbp-4S have important and partially non-overlapping roles in survival and elastic fiber formation, and identified fibulin-4 as a new interacting partner of both Ltbp-4 isoforms. Moreover, we showed that specific N-glycosylation of Ltbp-4S influences its fibulin-4- and fibulin-5-binding affinity. Future experiments involving the generation of Ltbp4L- and tissue-specific-Ltbp4-knockout mouse lines are planned to gain further insights into the isoform-specific functions of Ltbp-4.
MATERIALS AND METHODS
Animals, breeding and genotyping
The embryonic stem cell clone E301B04 (E14TG2a.4, 129P2) carrying a gene-trap insertion in the fifth intron of the Ltbp4 gene (supplementary material Fig. S1) was purchased from the German Gene Trap Consortium (GGTC, Helmholtz-Zentrum München, Munich, Germany). These cells were used to derive chimeric mice that were mated with C57BL/6N female mice. The offspring was tested for transgene germline transmission and backcrossed to C57BL/6N background for ten generations. Genotyping of Ltbp4-knockout mice was performed using 5mL4_gen, 3mL4_gen and betageo_gen primers (supplementary material Table S3) complementary to the sequences flanking the gene-trap insertion sites. Generation and genotyping of Ltbp4S−/− mice was performed as described previously (Sterner-Kock et al., 2002). Animals were housed in a 12-hour-light-12-hour-dark cycle and were fed a standard rodent diet (ssniff Spezialdiäten, Soest, Germany).
Unless otherwise stated, all animals were sacrificed at postnatal day 9 (P9) by decapitation and autopsies were carried out using standard protocols. For blood gas analyses (BGA), trunk blood was collected in a heparinised 1.5 ml tube and pH, pCO2, pO2, sO2, ctHB, Hct, HCO3 and ABE were analyzed using an ABL800 Flex blood gas analyzer (Radiometer, Willich, Germany). All animal procedures were performed in accordance with the German Laws for Animal Protection and were approved by the Institutional Animal Care and Use Committee: Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (Recklinghausen, Germany).
mRNA expression analysis
Total RNA isolation and real-time PCR were performed as described previously (Bultmann et al., 2013). All primers and probes are listed in supplementary material Table S3. Quantification of mRNA levels for Ltbp4, Ltbp4S and Ltbp4L were calculated using the standard curve method (Bultmann et al., 2013). Relative expression of tropoelastin, fibulin-5 and fibulin-4 was adjusted for total RNA content by normalizing to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) expression. Calculations were performed by a comparative 2−ΔΔCT method (Livak and Schmittgen, 2001).
SDS-PAGE and immunoblotting
Protein expression levels were determined by western blotting, using SDS-PAGE as described previously (Bultmann et al., 2013). All antibodies used are listed in supplementary material Table S4.
Histology and immunohistochemistry
Mice were sedated with ketamine and xylazine. The heart was punctured and perfused with PBS and 4% paraformaldehyde according to standard protocols. Perfusion-fixed aortas, lungs and hearts were dissected and embedded in paraffin. 4-μm sections were stained with haematoxylin-eosin, resorcin-fuchsin (elastin) or alcian blue (glucosaminoglycans). For immunohistochemistry, tissue sections were deparaffinized and antigen retrieval was performed in 10 mM citrate puffer (pH 6.0) for 20 minutes in a microwave (Ltbp4, PCNA) or by using proteaseXXIV (DCS Innovative Diagnostik-Systeme, Hamburg, Germany) for 6 minutes at room temperature (fibulin-4, fibulin-5) followed by treatment with 3% H2O2 for 15 minutes at room temperature. Incubation with primary antibodies (supplementary material Table S4) in dilution buffer was performed for 1 hour at room temperature. For detection, Supervision 2-Single Species HRP Polymer (DCS Innovative Diagnostik-Systeme, Hamburg, Germany) and Permanet AEC-Kit (ZYTOMED Systems, Berlin, Germany) were used according to manufacturers’ protocols.
Analyses of skin parameters
The epidermal and dermal thicknesses were determined in skin sections. Epidermal hydration, transepidermal water loss and net-elasticity of the skin were analyzed using the Multi Probe Adapter (MPA) System (Courage+Khazaka electronic, Cologne, Germany) with the Corneometer CM 825-, Tewameter TM 300- or Cutometer MPA 580-probe, respectively, and freshly sacrificed mice according to the manufacturer’s instructions.
Stereological analyses of the cardiac ventricles
The stereological analyses of the cardiac ventricles were determined in myocardial sections fixed in situ and stained with haematoxylin-eosin as described previously (Hilfiker-Kleiner et al., 2004; Ricke-Hoch et al., 2014).
Echocardiography of neonatal mice
High-resolution mouse echocardiography was performed in WT and Ltbp4−/− mice using a commercially available ultrasound system (Philips iE33, Philips, Hamburg, Germany) equipped with a 15 MHz linear array transducer utilizing harmonic imaging (L15-io7, Philips, Hamburg, Germany). Volumetric analysis of left-ventricular structures was performed to assess left-ventricular morphology and function (Ghanem et al., 2006; Zheng-Fischhöfer et al., 2006).
Magnetic resonance imaging of neonatal mice
Magnetic resonance imaging (MRI) studies were performed in two mice per genotype using a clinical 3 Tesla MR whole-body system (Achieva 2.3, Philips, Best, The Netherlands) as described previously (Bunck et al., 2009). For optimal MR signal detection, a dedicated solenoid receive-only coil was used (Philips, Forschungslaboratorien, Aachen, Germany). After positioning of the mouse in the isocenter of the magnet gradient, echo scout images were acquired in a horizontal orientation for localization of the heart. For cine imaging, we used a retrospectively ECG-triggered, segmented gradient echo sequence. The number of signal averages was set to eight to achieve good signal-to-noise ratios. The reconstructed in-plane spatial resolution was 0.13×0.13 mm; slice thickness was set to 0.8 mm. Per heart cycle, 30 phases were imaged. For left-ventricular assessment, standard imaging planes were obtained according to the recommendations by Schneider et al. (Schneider et al., 2006).
Electron microscopy
For transmission electron microscopy, organ tissues were fixed in 5% glutaraldehyde in 0.4 M PBS buffer (pH 7.2–7.3). The samples were washed three times with 0.1 M cacodylate buffer and stored in this buffer at 4°C until plastic embedding. Afterwards preparations were postfixed with 2% osmium tetraoxide in 0.1 M cacodylate buffer for 2 hours at 4°C. Before embedding in araldite (Novartis Pharma, Nürnberg, Germany) the specimens were dehydrated in a graded series of ethanol. Sections of plastic-embedded specimens were cut with a diamond knife for thin sections and ultrathin sections on an ultra-microtome (Reichert, Buffalo, NY, USA). The 0.5-μm thin slices were stained with Methylene Blue and examined using a Zeiss Axiovert 200 (Zeiss, Oberkochen, Germany). Ultrathin sections (60 nm) were mounted on formvar-coated copper grids, stained with 0.2% uranyl acetate and lead citrate and then examined with a Zeiss EM 902 A electron microscope (Zeiss, Oberkochen, Germany).
Isolation of elastin, and quantification of desmosine and isodesmosine
Pure elastin, free of contaminants by other ECM components, was isolated from biopsies of mouse lung as described previously (Schmelzer et al., 2012). Isolated elastin samples were weighed and then hydrolyzed at a concentration of 1 mg/ml in 6 M HCl at 105°C for 24 hours, respectively, to allow the liberation of DES and IDES. After incubation, the samples were evaporated to dryness at 60°C, and the residues were taken up in acetonitrile:water (1:1, v/v) prior to liquid chromatography mass spectrometry (LC-MS) analysis. LC-MS analysis of DES and IDES was carried out using an Agilent 1100 LC system (Agilent Technologies, Waldbronn, Germany) coupled to a quadrupole ion trap mass spectrometer Finnigan LCQ (Thermo Fisher Scientific, Waltham, MA) by an electrospray interface. Isocratic chromatographic separation was performed over 8 minutes at a flow rate of 0.2 ml/min on a Reprosil-Pur 120 C18 AQ 3 μm column (150×2 mm, Maisch, Ammerbuch-Entringen, Germany). 0.1% formic acid/methanol (97.5:2.5, v/v) was used as the mobile phase and the column temperature was 40°C. Under these chromatographic conditions DES and IDES co-elute. For mass spectrometric analyses, the following parameters were used: positive ion mode, electrospray voltage: 4.5 kV, heated capillary temperature: 220°C.
Isolation of primary lung fibroblasts
Mouse lung fibroblasts were isolated according to described protocols (Seluanov et al., 2010). In brief, tissues were finely chopped, and digested with 0.12 Wünsch units/ml Liberase TM research Grade (Roche Diagnostics, Mannheim, Germany) for 30 minutes at 37°C. Cells were outgrown in DMEM/F12 (Gibco, Life Technologies, Darmstadt, Germany) containing 15% FCS (Biochrom, Berlin, Germany) and antibiotic/antimycotic (Life Technologies, Darmstadt, Germany) at 37°C in 5% CO2. After the first passage, remaining tissue fragments were discarded and cells were incubated in EMEM (Gibco, Life Technologies, Darmstadt, Germany) containing 15% FCS and penicillin-streptomycin (Life Technologies, Darmstadt, Germany).
Expression of recombinant proteins and deglycosylation assay
Full-length rLTBP-4S was purified as previously described (Noda et al., 2013). The first 253 amino acids of mouse Ltbp-4L (Ser25-Asp277) and the first 185 amino acids of mouse Ltbp-4S (Arg27-Asp211) after the signal peptide, as well as mouse full-length fibulin-4 and fibulin-5 after the signal peptide, were cloned into a modified pCEP-Pu vector containing a 3ʹ 2xStrepII tag using the restriction sites NheI and XhoI, and stably expressed in human embryonic kidney (HEK) 293 EBNA cells. The Strep-tagged proteins were purified from conditioned medium. Collected supernatants were supplemented with 1 mM phenylmethylsulfonyl fluoride, filtered and passed over a streptactin-Sepharose column. The recombinant proteins were eluted with elution buffer (100 mM Tris-HCl pH 7.4, 150 mM NaCl) containing 2.5 mM d-desthiobiotin.
Deglycosylation of denatured rLTBP-4S (200 ng) or homogenized WT lung (20 μg) was performed using 250 units recombinant PNGase F (New England Biolabs, Frankfurt am Main, Germany) for 3 hours at 37°C, or of native or denatured Ltbp-4L-2xStrep and Ltbp-4S-2xStrep (10 μg) using 500 units of PNGase F for 48 hours at 37°C according to the manufacturer’s protocol.
Immunofluorescence analysis
Primary lung fibroblasts were seeded on glass coverslips and cultured for 7 days in EMEM, 15% FCS and penicillin-streptomycin with or without recombinant full-length fibulin-4 (20 nm). Cells were fixed in methanol or PFA for 20 minutes at −20°C, and blocked with 5% BSA in PBS for 1 hour at room temperature. Cells were incubated with primary antibodies (supplementary material Table S4) in 1% BSA overnight at 4°C. Secondary antibodies were coupled to Alexa-Fluor-488 donkey anti-rabbit IgG, or Alexa-Fluor-546 donkey anti-goat IgG (Life Technologies, Darmstadt, Germany). Nuclei were counterstained with DAPI (Life Technologies, Darmstadt, Germany). Images were taken using a confocal LSM710 microscope (Zeiss, Oberkochen, Germany).
Surface plasmon resonance
Binding analyses were performed by using a BIAcore2000 (GE Healthcare GmbH, Solingen, Germany). 570 RUs of rfibulin-4, and 390 RUs of rfibulin-5 were covalently coupled to carboxymethyldextran hydrogel 500M sensor chips (XanTec, Düsseldorf, Germany) using the amine coupling kit following the manufacturer’s instructions (GE Healthcare, Solingen, Germany). Interactions between Ltbp-4L-2xStrep or Ltbp-4S-2xStrep and rfibulin-4 and rfibulin-5 were performed as previously described (Sengle et al., 2008). For determination of the kinetic constants, each individual sensorgram was fitted using the BIAevaluation 4.1 software (1:1 Langmuir interaction model) according to the manufacturer’s instructions. The determined pseudo-first order kinetic constants kobs were plotted versus the analyte concentrations and a linear best fit was applied. The average koff equals the intersection with the y-axis. The slope of the fitted straight line is a measure of the kon rate. Apparent equilibrium dissociation constants (Kd) were then calculated as the ratio of koff to kon.
TGFβ activity bioassay
TGFβ levels were assessed in 48-hour conditioned medium of primary lung fibroblasts that were cultured in DMEM (2% FCS, 1% penicillin-streptomycin, 10% BSA). TGFβ activity was determined by using a plasminogen activator inhibitor-1 (PAI-1) promoter luciferase reporter assay (Abe et al., 1994) using mink lung epithelial cells stably expressing the firefly luciferase reporter gene under the control of a TGFβ-specific, truncated PAI-1 promoter.
Active TGFβ was determined after overnight incubation of the reporter cells with conditioned medium or with recombinant human TGFβ1 standards. The reporter cells were lysed and luciferase activity was measured using the Beetle-Juice kit (PJK, Kleinblittersdorf, Germany) and a GloMax-Multi Detection System (Promega, Mannheim, Germany).
Statistical evaluation
Data are presented as a mean±s.d. Differences between groups were analyzed by Mann-Whitney test, log-rank test, Student’s t-test or ANOVA, followed by Bonferroni correction as appropriate. Statistical significance of post-hoc analyses was taken at P<0.05. Calculations were performed using SPSS21 (IBM Deutschland, Ehningen, Germany).
Supplementary Material
Acknowledgments
We thank Reinhild Brinker, Andrea Elbers, Petra Heid, Irmgard Henke, Manuela Lerwe, Christian Meißner and Karolin Scharnetzki for excellent technical assistance. We thank the CECAD Imaging Facility and Christian Jüngst for their support. We are grateful to Takako Sasaki, Oita University, Japan for providing the anti-fibulin-4 antibody.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
I.B.M., A.C., G.S. and A.S.K. designed research; I.B.M., A.C., A.C.M., K.D., A.P.W., T.I., F.T.W., A.C.B., W.B., A.G., A.H., G.S. and A.S.K. performed research; F.W., T.N. and K.K. contributed new reagents and/or analytic tools; I.B.M., A.C., A.P.W., F.T.W., A.C.B., W.B., A.G., A.H., G.S. and A.S.K. analyzed data; I.B.M., A.C., G.S., H.v.M. and A.S.K. wrote the paper.
Funding
I.B.M. was supported by the German Academic Exchange Service DAAD. A.H. was supported by the Deutsche Forschungsgemeinschaft (grant number HE 6190/1–2). H.v.M. was supported by the Deutsche Forschungsgemeinschaft [grant number SFB 815] and the Bundesministerium für Bildung und Forschung [grant number NGFN-plus 01G50858]. G.S. was supported by the Deutsche Forschungsgemeinschaft [grant number SFB829/project B12]. A.S.K. was supported by Hochhaus-, Maria Pesch and Imhoff-Stiftung and by UNDP [grant number EU3631500121]. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supplementary material
Supplementary material available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.018960/-/DC1
References
- Abe M., Harpel J. G., Metz C. N., Nunes I., Loskutoff D. J., Rifkin D. B. (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276–284. [DOI] [PubMed] [Google Scholar]
- Annes J., Vassallo M., Munger J. S., Rifkin D. B. (2004). A genetic screen to identify latent transforming growth factor beta activators. Anal. Biochem. 327, 45–54. [DOI] [PubMed] [Google Scholar]
- Bleeker G. B., Steendijk P., Holman E. R., Yu C. M., Breithardt O. A., Kaandorp T. A., Schalij M. J., van der Wall E. E., Bax J. J., Nihoyannopoulos P. (2006). Acquired right ventricular dysfunction. Heart 92 Suppl. 1, i14–i18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultmann I., Conradi A., Kretschmer C., Sterner-Kock A. (2013). Latent transforming growth factor β-binding protein 4 is downregulated in esophageal cancer via promoter methylation. PLoS ONE 8, e65614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunck A. C., Engelen M. A., Schnackenburg B., Furkert J., Bremer C., Heindel W., Stypmann J., Maintz D. (2009). Feasibility of functional cardiac MR imaging in mice using a clinical 3 Tesla whole body scanner. Invest. Radiol. 44, 749–756. [DOI] [PubMed] [Google Scholar]
- Callewaert B., Su C. T., Van Damme T., Vlummens P., Malfait F., Vanakker O., Schulz B., Mac Neal M., Davis E. C., Lee J. G., et al. (2013). Comprehensive clinical and molecular analysis of 12 families with type 1 recessive cutis laxa. Hum. Mutat. 34, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman S. L., Sicot F. X., Davis E. C., Huang J., Sasaki T., Chu M. L., Yanagisawa H. (2010). Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler. Thromb. Vasc. Biol. 30, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R., McGovern A., Ridley C., Cain S. A., Baldwin A., Wang M. C., Guo C., Mironov A., Jr, Drymoussi Z., Trump D., et al. (2009). Differential regulation of elastic fiber formation by fibulin-4 and -5. J. Biol. Chem. 284, 24553–24567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabovic B., Chen Y., Choi J., Vassallo M., Dietz H. C., Ramirez F., von Melchner H., Davis E. C., Rifkin D. B. (2009). Dual functions for LTBP in lung development: LTBP-4 independently modulates elastogenesis and TGF-beta activity. J. Cell. Physiol. 219, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabovic B., Robertson I. B., Zilberberg L., Vassallo M., Davis E. C., Rifkin D. B. (2015). Function of latent TGFβ binding protein 4 and fibulin 5 in elastogenesis and lung development. J. Cell. Physiol. 230, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C., Stewart K. M., Weaver V. M. (2010). The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem A., Röll W., Hashemi T., Dewald O., Djoufack P. C., Fink K. B., Schrickel J., Lewalter T., Lüderitz B., Tiemann K. (2006). Echocardiographic assessment of left ventricular mass in neonatal and adult mice: accuracy of different echocardiographic methods. Echocardiography 23, 900–907. [DOI] [PubMed] [Google Scholar]
- Hanada K., Vermeij M., Garinis G. A., de Waard M. C., Kunen M. G., Myers L., Maas A., Duncker D. J., Meijers C., Dietz H. C., et al. (2007). Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ. Res. 100, 738–746. [DOI] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D., Hilfiker A., Fuchs M., Kaminski K., Schaefer A., Schieffer B., Hillmer A., Schmiedl A., Ding Z., Podewski E., et al. (2004). Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 95, 187–195. [DOI] [PubMed] [Google Scholar]
- Hirai M., Ohbayashi T., Horiguchi M., Okawa K., Hagiwara A., Chien K. R., Kita T., Nakamura T. (2007). Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J. Cell Biol. 176, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi M., Inoue T., Ohbayashi T., Hirai M., Noda K., Marmorstein L. Y., Yabe D., Takagi K., Akama T. O., Kita T., et al. (2009). Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl. Acad. Sci. USA 106, 19029–19034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstra I. K., Birge S., Starcher B., Bailey A. J., Mecham R. P., Shapiro S. D. (2003). Lysyl oxidase is required for vascular and diaphragmatic development in mice. J. Biol. Chem. 278, 14387–14393. [DOI] [PubMed] [Google Scholar]
- Isogai Z., Ono R. N., Ushiro S., Keene D. R., Chen Y., Mazzieri R., Charbonneau N. L., Reinhardt D. P., Rifkin D. B., Sakai L. Y. (2003). Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278, 2750–2757. [DOI] [PubMed] [Google Scholar]
- Kantola A. K., Keski-Oja J., Koli K. (2008). Fibronectin and heparin binding domains of latent TGF-beta binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp. Cell Res. 314, 2488–2500. [DOI] [PubMed] [Google Scholar]
- Kantola A. K., Ryynänen M. J., Lhota F., Keski-Oja J., Koli K. (2010). Independent regulation of short and long forms of latent TGF-beta binding protein (LTBP)-4 in cultured fibroblasts and human tissues. J. Cell. Physiol. 223, 727–736. [DOI] [PubMed] [Google Scholar]
- Kielty C. M., Sherratt M. J., Shuttleworth C. A. (2002). Elastic fibres. J. Cell Sci. 115, 2817–2828. [DOI] [PubMed] [Google Scholar]
- Li D. Y., Brooke B., Davis E. C., Mecham R. P., Sorensen L. K., Boak B. B., Eichwald E., Keating M. T. (1998). Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280. [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lowery J. W., Amich J. M., Andonian A., Rosen V. (2014). N-linked glycosylation of the bone morphogenetic protein receptor type 2 (BMPR2) enhances ligand binding. Cell. Mol. Life Sci. 71, 3165–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäki J. M., Sormunen R., Lippo S., Kaarteenaho-Wiik R., Soininen R., Myllyharju J. (2005). Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 167, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin P. J., Chen Q., Horiguchi M., Starcher B. C., Stanton J. B., Broekelmann T. J., Marmorstein A. D., McKay B., Mecham R., Nakamura T., et al. (2006). Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol. Cell. Biol. 26, 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithieux S. M., Weiss A. S. (2005). Elastin. Adv. Protein Chem. 70, 437–461. [DOI] [PubMed] [Google Scholar]
- Miyazono K., Heldin C. H. (1991). Latent forms of TGF-beta: molecular structure and mechanisms of activation. Ciba Found. Symp. 157, 81–89; discussion 89–92. [DOI] [PubMed] [Google Scholar]
- Nakamura T., Lozano P. R., Ikeda Y., Iwanaga Y., Hinek A., Minamisawa S., Cheng C. F., Kobuke K., Dalton N., Takada Y., et al. (2002). Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415, 171–175. [DOI] [PubMed] [Google Scholar]
- Noda K., Dabovic B., Takagi K., Inoue T., Horiguchi M., Hirai M., Fujikawa Y., Akama T. O., Kusumoto K., Zilberberg L., et al. (2013). Latent TGF-beta binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. USA 110, 2852–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke C. L., Yanagisawa H. (2014). Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol. 37, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F., Sakai L. Y., Dietz H. C., Rifkin D. B. (2004). Fibrillin microfibrils: multipurpose extracellular networks in organismal physiology. Physiol. Genomics 19, 151–154. [DOI] [PubMed] [Google Scholar]
- Ricke-Hoch M., Bultmann I., Stapel B., Condorelli G., Rinas U., Sliwa K., Scherr M., Hilfiker-Kleiner D. (2014). Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc. Res. 101, 587–596. [DOI] [PubMed] [Google Scholar]
- Saharinen J., Taipale J., Monni O., Keski-Oja J. (1998). Identification and characterization of a new latent transforming growth factor-beta-binding protein, LTBP-4. J. Biol. Chem. 273, 18459–18469. [DOI] [PubMed] [Google Scholar]
- Schmelzer C. E., Jung M. C., Wohlrab J., Neubert R. H., Heinz A. (2012). Does human leukocyte elastase degrade intact skin elastin? FEBS J. 279, 4191–4200. [DOI] [PubMed] [Google Scholar]
- Schneider J. E., Wiesmann F., Lygate C. A., Neubauer S. (2006). How to perform an accurate assessment of cardiac function in mice using high-resolution magnetic resonance imaging. J. Cardiovasc. Magn. Reson. 8, 693–701. [DOI] [PubMed] [Google Scholar]
- Seluanov A., Vaidya A., Gorbunova V. (2010). Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 2010, 2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengle G., Charbonneau N. L., Ono R. N., Sasaki T., Alvarez J., Keene D. R., Bächinger H. P., Sakai L. Y. (2008). Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 283, 13874–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sideek M. A., Menz C., Parsi M. K., Gibson M. A. (2014). LTBP-2 competes with tropoelastin for binding to fibulin-5 and heparin, and is a negative modulator of elastinogenesis. Matrix Biol. 34, 114–123. [DOI] [PubMed] [Google Scholar]
- Sterner-Kock A., Thorey I. S., Koli K., Wempe F., Otte J., Bangsow T., Kuhlmeier K., Kirchner T., Jin S., Keski-Oja J., et al. (2002). Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 16, 2264–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic V., Rifkin D. B. (2012). LTBPs, more than just an escort service. J. Cell. Biochem. 113, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torreno-Pina J. A., Castro B. M., Manzo C., Buschow S. I., Cambi A., Garcia-Parajo M. F. (2014). Enhanced receptor-clathrin interactions induced by N-glycan-mediated membrane micropatterning. Proc. Natl. Acad. Sci. USA 111, 11037–11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban Z., Hucthagowder V., Schürmann N., Todorovic V., Zilberberg L., Choi J., Sens C., Brown C. W., Clark R. D., Holland K. E., et al. (2009). Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am. J. Hum. Genet. 85, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachi H., Nonaka R., Sato F., Shibata-Sato K., Ishida M., Iketani S., Maeda I., Okamoto K., Urban Z., Onoue S., et al. (2008). Characterization of the molecular interaction between tropoelastin and DANCE/fibulin-5. J. Biochem. 143, 633–639. [DOI] [PubMed] [Google Scholar]
- Wagenseil J. E., Mecham R. P. (2007). New insights into elastic fiber assembly. Birth Defects Res. C Embryo Today 81, 229–240. [DOI] [PubMed] [Google Scholar]
- Yamauchi Y., Tsuruga E., Nakashima K., Sawa Y., Ishikawa H. (2010). Fibulin-4 and -5, but not Fibulin-2, are associated with Tropoelastin deposition in elastin-producing cell culture. Acta Histochem. Cytochem. 43, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa H., Davis E. C. (2010). Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins. Int. J. Biochem. Cell Biol. 42, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa H., Davis E. C., Starcher B. C., Ouchi T., Yanagisawa M., Richardson J. A., Olson E. N. (2002). Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415, 168–171. [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhöfer Q., Ghanem A., Kim J. S., Kibschull M., Schwarz G., Schwab J. O., Nagy J., Winterhager E., Tiemann K., Willecke K. (2006). Connexin31 cannot functionally replace connexin43 during cardiac morphogenesis in mice. J. Cell Sci. 119, 693–701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.