

Abstract
The development of asymmetric C–H activation reactions through metal insertions remains in its infancy. The commonly used approach is the desymmetrization of prochiral C–H bonds on the same or different carbons of one achiral molecule using a chiral catalyst. Herein, we report a Pd-catalyzed enantioselective C–H activation reaction via kinetic resolution in which one of the enantiomers of the racemic substrates undergoes faster C–H insertion with the chiral catalysts thereby producing enantioenriched C–H functionalization products that are not accessible via desymmtrization of prochiral C–H bonds. The exceedingly high relative rate (kfast/kslow up to 244) and the subsequent iodination of the remaining enantiomerically enriched starting material using a chiral ligand with the opposite configuration allows for the conversion of both enantiomers of amines into enantiomerically pure iodinated amines.
A wide range of C–H activation reactions have emerged as promising tools for organic synthesis over the past three decades. However, the development of enantioselective C–H activation reactions has met with limited success in terms of efficiency and scope (1). Enantioselective carbene insertions of prochiral methylene C–H bonds adjacent to heteroatoms have been achieved in synthetically useful enantioselectivity (2). Asymmetric nitrene insertion has also been demonstrated in both diastereoselective and enantioselective fashion (3–6). Development of asymmetric C–H activation reactions using organometallic approach has also witnessed limited but encouraging progress. Combining C–H activation with a subsequent asymmetric carbometalation onto double bonds elegantly connects C–H functionalization reactions to asymmetric catalysis (7, 8). An early example of atropselective alkylation in moderate enantioselectivity (49% ee) was reported (9). Recently, Pd-catalyzed desymmetrization of prochiral C–H bonds has been achieved with excellent levels of enantioselectivity (10–14). However, the requirement for the presence of two chemically identical groups necessarily limits the structural diversity of the chiral products, preventing the broad application of this method in asymmetric synthesis.
Chiral amines are one of the most prevalent motifs in bioactive natural products, drug molecules, and chiral catalysts. Despite remarkable progress in development of catalytic enantioselective methods for synthesis of chiral amines (15, 16), the use of methods based on chiral auxiliaries (17), classic resolution, or enzymatic kinetic resolution (18) is often the method of choice in applications. Notably, the nonenzymatic kinetic resolution of amines via asymmetric acyl transfer catalysts remains a significant challenge when compared to the analogous kinetic resolution of alcohols (19–21). In our efforts to develop alternative methods for the asymmetric synthesis of chiral amines, we recently achieved the enantioselective C–H iodination of triflyl-protected benzylamines via desymmetrization (12). However, this approach can only access diarylmethylamines containing two identical aryl groups. We envisioned that a kinetic resolution process via an enantioselective C–H iodination of arylalkylamines could overcome this limitation, providing access to a wide range of chiral α-branched benzylamines. Practically, this type of process would not only lead to the resolution of racemic amines but also concomitantly introduces a new functional handle for the further elaboration of the product. Conceptually, the chiral recognition required in kinetic resolution is fundamentally different to that in the desymmetrization process. The catalyst needs to preferentially recognize one of the enantiomeric substrates in kinetic resolution instead of one of the prochiral groups within the same substrate as in desymmetrization. Despite the landmark success in kinetic resolution via the Jacobsen’s epoxide opening process (22) and the pioneering work on Pd(II)-catalyzed asymmetric oxidation of racemic alcohols (23, 24), kinetic resolution via a Pd-catalyzed C–H activation reaction remains to be established.
Herein we report the discovery of a highly efficient kinetic resolution of chiral amines via Pd-catalyzed C–H iodination with selectivities reaching up to 244 (Fig. 1C). In addition to simple arylalkylamines, a wide range of β-amino acids and β-amino alcohols are compatible with this reaction. The use of ambient temperature provides a significant operational advantage over nonenzymatic acylative kinetic resolution reactions, which often require low temperature operations. We further demonstrate that the remaining starting material can subsequently be iodinated using a MPAA ligand with opposite configuration to give ortho-iodinated amines in high ees, thus rendering this technology a versatile and novel method for converting both enantiomers of the racemic amines into ortho-iodinated chiral benzylamines. The newly introduced ortho-iodides are a useful functional handle, allowing the products to be converted into a broad range of chiral amines. Notably, a de novo synthesis of these ortho-iodinated chiral benzylamines generally relies on the use of ortho-iodinated benzaldehyde derivatives, which require multiple step preparations (25).
Fig. 1.

(A) Carbene and nitrene C–H insertions. (B) Three different organometallic approaches towards asymmetric C–H activation. (C) Enantioselective C–H activation via kinetic resolution.
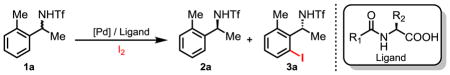
Our experimental design was based on a previous finding that a mono-protected amino acid ligand (MPAA) can effectively control the stereochemistry in Pd-catalyzed asymmetric insertion into prochiral C–H bonds on different carbon centers, leading to desymmetrization (11). This led us to hypothesize that the chiral catalyst assembled from a MPAA and Pd(II) species could preferentially recognize one enantiomer of a racemic substrate during the C–H activation step. If successful, a wide range of C–H activation reactions could potentially be developed into practical tools for asymmetric catalysis via kinetic resolution. Due to its compatibility with low reaction temperatures, we selected our recently developed C–H iodination as a model reaction to investigate the feasibility of achieving kinetic resolution of α-branched benzylamines at room temperature. Thus, 1-(o-tolyl)ethylamine, protected by a triflyl group (1a), was subjected to our iodination conditions in the presence of various mono-protected amino acid ligands (Table 1). We found the use of benzoyl-protected L-2-aminopentanoic acid (norvaline) as the chiral ligand, Pd(II)-catalyzed iodination of 1a proceeds with promising selectivity (entry 1, s = 17.6). A minor increase in steric hindrance on the side chain when leucine is used improves the selectivity to 50 (entries 2–3). However, an even bulkier neopentyl side chain affects the selectivity adversely (entry 4). Interestingly, the introduction of a secondary i-propyl side chain gives good selectivity (entry 5, s = 30.8). Further increase in steric hindrance on the secondary side chain also reduces the selectivity (entry 6). Extensive efforts to improve the selectivity by using a substituted N-benzoyl protecting group were unsuccessful (entries 7–12). We also found that acetyl, trifluoroacetyl and Boc protecting groups are inferior to benzoyl-type protecting group (entries 13–15). We further found that an increase in the reaction concentration improves the selectivity to 62.0 (entry 16). Finally an optimum selectivity of 78.8 is obtained by running the reaction in a mixture of tamyl alcohol and DMSO with the ratio of 5 : 2.2 (entry 17). In this case, both the iodinated product and recovered starting material are obtained with high enantioselectivity (92% ee) at 50% conversion. The reaction also proceeds with 2 mol% Pd catalyst to reach a selectivity of 51.2, albeit at longer reaction times (entry 17).
Table 1.
Optimization of enantioselective C–H iodination.
| ||||||
|---|---|---|---|---|---|---|
| entry | ligand
|
conv. (%)* | ee†
|
s‡ | ||
| R1 | R2 | 2a | 3a | |||
| 1 | C6H5 | nPr | 38 | 50 | 83 | 17.6 |
| 2 | C6H5 | nBu | 48 | 77 | 83 | 24.8 |
| 3 | C6H5 | iBu | 49 | 86 | 88 | 50 |
| 4 | C6H5 | neopentyl | 33 | 31 | 63 | 5.9 |
| 5 | C6H5 | iPr | 48 | 79 | 87 | 30.8 |
| 6 | C6H5 | tBu | 25 | 23 | 70 | 6.8 |
| 7 | 2-CF3C6H4 | iBu | 48 | 80 | 88 | 34.2 |
| 8 | 3,5-(CF3)2C6H3 | iBu | 45 | 39 | 47 | 4.1 |
| 9 | 4-OMeC6H4 | iBu | 57 | 90 | 73 | 15.5 |
| 10 | 4-FC6H4 | iBu | 46 | 70 | 83 | 21.2 |
| 11 | 1-naphthyl | iBu | 39 | 50 | 81 | 13.3 |
| 12 | 2-naphthyl | iBu | 47 | 77 | 88 | 32.9 |
| 13 | Me | iBu | 24 | 15 | 48 | 3.2 |
| 14 | CF3 | iBu | 5 | 1 | 21 | 1.5 |
| 15 | tBuO | iBu | 38 | 24 | 39 | 2.9 |
| 16§ | C6H5 | iBu | 50 | 89 | 90 | 62.0 |
| 17|| | C6H5 | iBu | 50 (50¶) | 92 (89¶) | 92 (89¶) | 78.8 (51.2¶) |
Reaction conditions: 10 mol% Pd(OAc)2, 40 mol% Ligand, 3 eq. Na2CO3, 3 eq. CsOAc, 3 eq. I2, 15 eq. DMSO, 1mL tamyl-OH, air, 20 °C, 24 h
Calculated conversion, c = ee2a/(ee2a + ee3a).
Determined by chiral HPLC analysis.
Selectivity(s) = (rate of fast-reacting enantiomer)/(rate of slow-reacting enantiomer).
0.5 mL tamyl-OH.
0.5 mL tamyl-OH/DMSO (5 : 2.2).
Reaction conditions: 2 mol% Pd(OAc)2, 10 mol% Bz-Leu-OH, 3 equiv. CsOAc, 3 equiv. Na2CO3, 3 equiv. I2, 0.5 mL tamyl-OH/DMSO (5 : 2.2), air, 20 °C, 24 h, then 3 equiv. I2, 20 °C, 24 h.
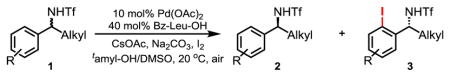
To examine whether this method can be applied to prepare a broad range of chiral ortho-iodinated benzylamines, we subjected amines 1b-l to the optimized conditions. Iodination of benzylamines 1a-c gives good to excellent selectivity whereas the bulkier α-isobutyl and benzyl groups in 1d and 1e lead to a decrease in the selectivity factor to 25.0 and 13.8 respectively (entries 1–5). Iodination of benzylamine 1f containing a cyclopropyl group proceeds with an excellent selectivity factor (entry 6, s 83.5). Arenes containing ortho-methoxy and fluoro groups are also iodinated with outstanding selectivity (entries 7–8, s 148 and 240 respectively). The presence of a para-chloro group on the aryl fragment in 1i was well tolerated (entry 9, s 113). Meta- and para-methyl substituted arenes 1j and 1k are more suitable substrates than the ortho-methylarene 1a affording excellent selectivity (entries 10–11, s 99.5 and 91.7 respectively). Iodination of 1l containing 2-naphthyl group also proceeded with synthetically useful selectivity (entry 12, s 76). In general, the iodinated products were obtained with high levels of enantioselectivity (91–97% ee) with the exception of entries 3–5. To investigate whether the decrease of enantioselectivity with these substrates containing bulkier α-alkyls (entries 3–5) is a general phenomenon, we subjected 1m containing α-butyl to the standard iodination conditions. The reaction proceeds with high enantioselectivity (entry 13, s 124), thus suggesting that the observed adverse effect of the bulky α-alkyl group is only associated with the ortho-methylbenzylamine substrates 1a-e.
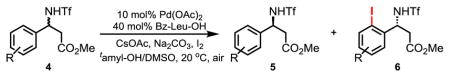
The significance of β-amino acids in pharmaceutical compounds prompted us to investigate whether our newly developed enantioselective iodination can be applied for the production of iodinated chiral β-amino acids. In spite of a number of highly creative asymmetric methods for making enantioenriched β-amino acids (26, 27), resolution is still frequently utilized due to the ease of preparing racemic β-amino acids by the Rodionov reaction. The development of highly efficient catalysts to resolve racemic β-amino acids continues to attract significant attention (28–30). Using our methodology, β-phenyl-β-amino acid 4a is iodinated under the standard conditions to give 6a with excellent selectivity (s, 128). Substrates containing electron-donating groups at the ortho, meta and para positions of the β-phenyl groups are all iodinated with high selectivity factors ranging from 112 to 168 (entries 2–4). Electron-withdrawing groups on the β-phenyl rings are also compatible with this transformation affording selectivity factors as high as 244 (entries 5–7). In all cases, the iodinated amino acid derivatives are obtained with high levels of enantioselectivity (94–99% ee).
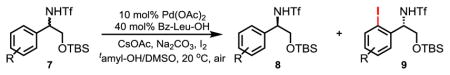
We were pleased to find that this enantioselective C–H activation method is also suitable for preparing ortho-iodinated chiral β-amino alcohols. 2-Phenyl amino alcohol 7a is iodinated with a practically useful selectivity (s, 88). The ortho-methyl group in 7b leads to a slight decrease the selectivity factor (s, 77.2) whereas 2-(ortho-fluoro)-phenyl and 2-naphthyl-amino alcohols were iodinated with excellent selectivity (s, 188 and 112 respectively).
To further demonstrate the versatility of this kinetic resolution process based on enantioselective C–H iodination, we developed a protocol to convert both enantiomers of the racemic amine substrates to the chiral iodinated amines in high enantiomeric purity. Thus, 1.0 gram of 1l is subjected to the standard reaction conditions using the L-amino acid ligand to give 37% iodinated product 3l (maximum 50% yield) with 95% ee (Fig. 2A). The recovered starting material 2l with 69% ee is then iodinated using the D-amino acid ligand to give chiral amine 3l′ in 98% ee (Fig. 2A). The use of ligands possessing the opposite configuration to enantioselectively iodinate the enantiomerically enriched starting material could prove extremely useful when the selectivity factor is lower than 50 and the ee of the starting material is lower than 90% ee. To render this reaction synthetically useful, triflyl protected amine 2l is readily deprotected and converted to benzoyl protected amine 10 under mild conditions without racemization (Fig. 2B). Finally, the chiral iodinated amine 3l is converted to diverse range of amines, illustrating the broad utility of this method to access a diverse range of chiral amines (Fig. 2C).
Fig. 2.

(A) Gram-scale synthesis and reaction with D-amino acid ligand. (B) Deprotection of the triflyl protecting group. (C) Functionalization of iodinated chiral amine 3l.
In summary, we have developed a highly enantioselective C–H iodination reaction for kinetic resolution of arylalkylamines. A wide range of chiral ortho-iodinated α-branched benzylamines, β-amino acids, and amino alcohols can be prepared via this enantioselective C–H iodination reaction using an L-amino acid ligand. The enantiomerically enriched remaining starting material can also be enantioselectively iodinated using a D-amino acid ligand to give the opposite enantiomer of the iodinated amines in excellent enantioselectivity. Conceptually, development of enantioselective C–H activation reactions via kinetic resolution overcomes the limitation imposed by the desymmetrization approach which requires the presence of two chemically identical groups in the substrates.
Supplementary Material
Table 2.
Substrate scope of the enantioselective C–H iodination.
| A | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| entry | 1 | Ar | Alkyl | time (h) | conv.* (yield, %)† | ee (%)‡
|
s§ | |
| 2 | 3 | |||||||
| 1 | 1a | 2-Me-Ph | Me | 24 | 51 (48) | 93 | 91 | 73.6 |
| 2 | 1b | 2-Me-Ph | Et | 24 | 50 (49) | 91 | 91 | 67.3 |
| 3 | 1c | 2-Me-Ph | nBu | 48 | 47 (45) | 77 | 87 | 32.9 |
| 4 | 1d | 2-Me-Ph | iBu | 48 | 51 (49) | 83 | 81 | 25.0 |
| 5 | 1e | 2-Me-Ph | Bn | 48 | 47 (44) | 67 | 75 | 13.8 |
| 6 | 1f | 2-Me-Ph | cyclopropyl | 24 | 42 (42) | 70 | 95 | 83.5 |
| 7 | 1g | 2-OMe-Ph | Me | 24 | 49 (46) | 93 | 97 | 240 |
| 8 | 1h | 2-F-Ph | Me | 24 | 48 (47) | 89 | 96 | 148 |
| 9 | 1i | 2-Me,4-Cl-Ph | Me | 24 | 51 (49) | 97 | 93 | 113 |
| 10 | 1j | 3-Me-Ph | Me | 24 | 45|| (42, mono:di=3:1) | 78 | 92¶ | 99.5 |
| 11 | 1k | 4-Me-Ph | Me | 48 | 45 (35) | 78 | 95 | 91.7 |
| 12 | 1l | 2-naphthyl | Me | 48 | 41 (37) | 67 | 95 | 76.0 |
| 13# | 1m | Ph | nBu | 48 | 42|| (40, mono:di=1:1.4) | 70 | 87¶ (99**) | 124 |
| B | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
| entry | 4 | Ar | time (h) | conv. (yield, %) | ee (%)
|
s | |
| 5 | 6 | ||||||
| 1 | 4a | Ph | 48 | 47* (44, mono:di=2:1) | 85 | 96‡ | 128 |
| 2 | 4b | 2-Me-Ph | 24 | 49 (49) | 93 | 96 | 168 |
| 3 | 4c | 3-Me-Ph | 48 | 50 (44) | 93 | 94 | 112 |
| 4 | 4d | 4-OMe-Ph | 48 | 46* (43, mono:di=3:1) | 82 | 98‡ | 134 |
| 5† | 4e | 4-F-Ph | 48 | 48 (41) | 90 | 98 | 244 |
| 6† | 4f | 4-Cl-Ph | 48 | 49 (40) | 92 | 97 | 152 |
| 7† | 4g | 4-CF3-Ph | 48 | 40 (38) | 65 | 99 | 155 |
| C | |||||||
|---|---|---|---|---|---|---|---|
| |||||||
| entry | 7 | Ar | time (h) | conv. (yield, %) | ee (%)
|
s | |
| 8 | 9 | ||||||
| 1 | 7a | Ph | 48 | 43* (40, mono:di=1:1) | 71 | 87† (99‡) | 88.0 |
| 2 | 7b | 2-Me-Ph | 48 | 50 (49) | 91 | 92 | 77.2 |
| 3 | 7c | 2-F-Ph | 48 | 41 (41) | 68 | 99 | 188 |
| 4 | 7d | 2-naphthyl | 48 | 43 (40) | 72 | 96 | 112 |
Calculated conversion, c = ee2/(ee2 + ee3).
Isolated yield of the iodinated product.
Determined by chiral HPLC analysis.
Selectivity(s) = (rate of fast-reacting enantiomer)/(rate of slow-reacting enantiomer).
Determined by crude 1H-NMR.
ee for the mono product.
3 equiv. I2 was added after 24 h.
ee for the di product.
Determined by crude 1H-NMR.
3 equiv. I2 was added after 24 h.
ee for the mono product.
Determined by crude 1H-NMR.
ee for the mono product.
ee for the di product.
One Sentence Summary.
Enantioselective C–H iodination via kinetic resolution establishes a new avenue for developing asymmetric C–H activation reactions.
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS, 2R01GM084019) for their financial support. J.-Q. Y. and L. C. conceived the concept. L. C. developed the enantioselective C–H iodination. K.-J. X. synthesized the amine substrates. J.-Q. Y. directed the project. We thank Dr J. Spangler for constructive suggestions.
Footnotes
References and Notes
- 1.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Transition metal-catalyzed C–H activation reactions: diastereoselectivity and enantioselectivity. Chem Soc Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- 2.Doyle MP, Duffy R, Ratnikov M, Zhou L. Catalytic carbene insertion into C–H bonds. Chem Rev. 2010;110:704–724. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- 3.Reddy RP, Davies HML. Dirhodium tetracarboxylates derived from adamantylglycine as chiral catalyst for enantioselective C–H aminations. Org Lett. 2006;8:5013–5016. doi: 10.1021/ol061742l. [DOI] [PubMed] [Google Scholar]
- 4.Liang C, Robert-Peillard F, Fruit C, Muller P, Dodd RH, Dauban P. Efficient diastereoselective intermolecular rhodium-catalyzed C–H amination. Angew Chem Int Ed. 2006;45:4641–4644. doi: 10.1002/anie.200601248. [DOI] [PubMed] [Google Scholar]
- 5.Zalatan DN, Du Bois J. A chiral rhodium carboxamidate catalyst for enantioselective C–H amination. J Am Chem Soc. 2008;130:9220–9221. doi: 10.1021/ja8031955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milczek E, Boudet N, Blakey S. Enantioselective C–H amination using cationic ruthenium (II)–pybox catalysts. Angew Chem Int Ed. 2008;47:6825–6828. doi: 10.1002/anie.200801445. [DOI] [PubMed] [Google Scholar]
- 7.Hyster TK, Knorr L, Ward TR, Rovis T. Biotinylated Rh(III) complex in engineered streptavidin for accelerated asymmetric C–H activation. Science. 2012;338:500–503. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye B, Cramer N. Chiral cyclopentadienyl ligand as stereocontrolling element in asymmetric C–H functionalization. Science. 2012;338:504–506. doi: 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]
- 9.Kakiuchi F, Gendre PL, Yamada A, Ohtaki H, Murai S. Atropselective alkylation of biaryl compounds by means of transition metal-catalyzed C–H/olefin coupling. Tetrahedron: Asymmetry. 2000;11:2647–2651. [Google Scholar]
- 10.Shi BF, Maugel N, Zhang YH, Yu JQ. PdII-catalyzed enantioselective activation of C(sp2)–H and C(sp3)–H bonds using monoprotected amino acids as chiral ligands. Angew Chem Int Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 11.Chu L, Wang XC, Moore CE, Rheingold AL, Yu JQ. Pd-catalyzed enantioselective C–H iodination: asymmetric synthesis of chiral diarylmethylamines. J Am Chem Soc. 2013;135:16344–16347. doi: 10.1021/ja408864c. [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi M, Katayev D, Besnard C, Kündig EP. Fused indolines by palladium-catalyzed asymmetric C–C coupling involving an unactivated methylene group. Angew Chem Int Ed. 2011;50:7438–7441. doi: 10.1002/anie.201102639. [DOI] [PubMed] [Google Scholar]
- 13.Anas S, Cordi A, Kagan HB. Enantioselective synthesis of 2-methyl indolines by palladium catalyzed asymmetric C(sp3)–H activation/cyclisation. Chem Comm. 2011;47:11483–11485. doi: 10.1039/c1cc14292e. [DOI] [PubMed] [Google Scholar]
- 14.Saget T, Lemouzy S, Cramer N. Chiral monodentate phosphines and bulky carboxylic acids: cooperative effects in palladium-catalyzed enantioselective C(sp3)–H functionalization. Angew Chem Int Ed. 2012;51:2238–2242. doi: 10.1002/anie.201108511. [DOI] [PubMed] [Google Scholar]
- 15.Nugent TC. Chiral amine synthesis: methods, developments and applications. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- 16.Schmidt F, Stemmler RT, Rudolph J, Bolm C. Catalytic asymmetric approaches towards enantiomerically enriched diarylmethanols and diarylmethylamines. Chem Soc Rev. 2006;35:454–470. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- 17.Robak MT, Herbage MA, Ellman JA. Synthesis and application of tert-butanesulfinamide. Chem Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 18.Paetzold J, Bäckvall JE. Chemoenzymatic dynamic resolution of primary amines. J Am Chem Soc. 2005;127:17620–17621. doi: 10.1021/ja056306t. [DOI] [PubMed] [Google Scholar]
- 19.Arai S, Bellemin-Laponnaz S, Fu GC. Kinetic resolution of amines by a nonenzymatic acylation catalyst. Angew Chem Int Ed. 2001;40:234–236. doi: 10.1002/1521-3773(20010105)40:1<234::AID-ANIE234>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.De CK, Klauber EG, Seidel D. Merging nucleophilic and hydrogen bonding catalysis: an anion binding approach to the kinetic resolution of amines. J Am Chem Soc. 2009;131:17060–17061. doi: 10.1021/ja9079435. [DOI] [PubMed] [Google Scholar]
- 21.Fowler BS, Mikochik PJ, Miller SJ. Peptide-catalyzed kinetic resolution of formamides and thioformamides as an entry to nonracemic amines. J Am Chem Soc. 2011;132:2870–2871. doi: 10.1021/ja9107897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Asymmetric catalysis with water: efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]
- 23.Jensen DR, Pugsley JS, Sigman MS. Palladium-catalyzed enantioselective oxidations of alcohols using molecular oxygen. J Am Chem Soc. 2001;123:7475–7476. doi: 10.1021/ja015827n. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira EM, Stoltz BM. The palladium-catalyzed oxidative kinetic resolution of secondary alcohols with molecular oxygen. J Am Chem Soc. 2001;123:7725–7726. doi: 10.1021/ja015791z. [DOI] [PubMed] [Google Scholar]
- 25.Fustero S, Lazaro R, Aiguabella N, Riera A, Simon-Fuentes A, Barrio P. Asymmetric allylation/Pauson Khand reaction: a simple entry to polycyclic amines. Application to the synthesis of aminosteroid analogues. Org Lett. 2014;16:1224–1227. doi: 10.1021/ol500142c. [DOI] [PubMed] [Google Scholar]
- 26.Wenzel AG, Jacobsen EN. Asymmetric catalytic Mannich reactions catalyzed by urea derivatives: enantioselective synthesis of β-aryl-β-amino acids. J Am Chem Soc. 2002;124:12964–12965. doi: 10.1021/ja028353g. [DOI] [PubMed] [Google Scholar]
- 27.Song J, Wang Y, Deng L. The mannich reaction of malonates with simple imines catalyzed by bifunctional cinchona alkaloids: enantioselective synthesis of β-amino acids. J Am Chem Soc. 2006;128:6048–6049. doi: 10.1021/ja060716f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berkessel A, Cleemann F, Mukherjee S. Kinetic resolution of oxazinones: an organocatalytic approach to enantiomerically pure β-amino acids. Angew Chem Int Ed. 2005;44:7466–7469. doi: 10.1002/anie.200502003. [DOI] [PubMed] [Google Scholar]
- 29.Bumbu VD, Birman VB. Kinetic resolution of N-acyl- β-lactams via benzotetramisole-catalyzed enantioselective alcoholysis. J Am Chem Soc. 2011;133:13902–13905. doi: 10.1021/ja2058633. [DOI] [PubMed] [Google Scholar]
- 30.Zhou S, Wang J, Chen X, Acena JL, Soloshonok VA, Liu H. Chemical kinetic resolution of unprotected β-substituted-β-amino acids using recyclable chiral ligands. Angew Chem Int Ed. 2014 doi: 10.1002/anie.201403556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.