

Abstract
Purpose
In multiple cell metazoans, the ability of polarized epithelial cells to convert to motile mesenchymal cells in order to relocate to another location is governed by a unique process termed epithelial-mesenchymal transition (EMT). While being an essential process of cellular plasticity for normal tissue and organ developments, EMT is found to be involved in an array of malignant phenotypes of tumor cells including proliferation and invasion, angiogenesis, stemness of cancer cells and resistance to chemo-radiotherapy. Although EMT is being extensively studied and demonstrated to play a key role in tumor metastasis and in sustaining tumor hallmarks, there is a lack of clear picture of the overall EMT signaling network, wavering the potential clinical trials targeting EMT.
Methods
In this review, we highlight the potential key therapeutic targets of EMT linked with tumor aggressiveness, hypoxia, angiogenesis and cancer stem cells, emphasizing on an emerging EMT-associated NF-κB/HER2/STAT3 pathway in radioresistance of breast cancer stem cells.
Results
Further definition of cancer stem cell repopulation due to EMT-controlled tumor microenvironment will help to understand how tumors exploit the EMT mechanisms for their survival and expansion advantages.
Conclusions
The knowledge of EMT will offer more effective targets in clinical trials to treat therapy-resistant metastatic lesions.
Keywords: epithelial-mesenchymal transition, metastasis, cancer stem cell, tumor aggressiveness, therapeutic resistance
Epithelial-mesenchymal transition (EMT)
EMT is a fundamental biological process by which epithelial cells undergo biochemical shifts to become mesenchymal cells in order to generate or regenerate tissues that have different polarization from the original epithelia (Larue and Bellacosa 2005; Thompson et al. 2005). Upon EMT, epithelial cells, the apicobasal polarized adherent cells with intracellular adherent complexes, undergo multiple biological changes that enable them to become non-polarized elongated mesenchymal cells which lack intercellular junctions and can move throughout the extracellular matrix (Thiery and Sleeman 2006). Three types of EMT have been described: A, EMT type 1, associated with implantation, embryo formation, and organ development in embryonic stage after fertilization; B, EMT type 2, associated with wound healing, tissue regeneration and organ fibrosis; and C, EMT type 3, related to cancer progression and metastasis (Kalluri 2009; Kalluri and Weinberg 2009). Though having a similar group of governing genes and biochemical elements, different EMT is distinct to one another by cell types and tissues and occur at different developmental stages of an organism (Baum et al. 2008; Micalizzi et al. 2010). Cells, proteins, as well as stimuli, vary with certain overlaps in phenotypic similarities and pathway mechanisms. An epithelial cell undergoing EMT may present morphological changes from a cobblestone monolayer stationary cell to a motile spindle-shape cell switching differentiation markers from cell-cell junction proteins and cytokeratin intermediate filaments to Fibronectin and Vimentin filaments. Degradation of underlying basement membrane and abilities to migrate and invade through extracellular matrix of epithelial cells are the hallmark consequences of the EMTs (Kalluri and Weinberg 2009; Yang and Weinberg 2008). Such an epithelial gain-of-function phenomenon is a reversible process, i.e., mesenchymal-epithelial transition (MET), its countermeasure reverting the mesenchymal cells back to epithelial cells (Hugo et al. 2007; Thiery and Sleeman 2006). While relatively little is known regarding the function of MET, a large number of proteins and pathways governing EMT have been identified. For example, the building-up of mesenchymal markers and losing of epithelial markers such as accumulation of N-cadherin with degradation of E-cadherin are major features of EMT. The EMT markers include genes and proteins of cell surface, cytoskeleton, extracellular proteins matrix, and transcription factors. As far, there are more than 70 protein markers identified and used to determine cells expressing or leaning towards epithelial or mesenchymal phenotype. To target EMT, a number of microRNAs (miRNAs) are shown to be accountable for the transition driving and reversing EMT processes (Kalluri and Weinberg 2009; Lamouille et al. 2013; Zeisberg and Neilson 2009). However, the challenge is that certain EMT markers can synchronously exert more than a single role to facilitate EMT forward. For example, the EMT-related transcription factor marker Ets-1 induces glomerular reorganization or vascular inflammation, and Snail is involved in inflammation, wound healing and hyperplasia; both contribute to the regulation of microenvironment and gene expression levels to actuate EMT (Du et al. 2010; Hotz et al. 2010; Mizui et al. 2006; Zhan et al. 2005). This review focuses on the overall signaling network of EMT (specifically, type three EMT) in tumor aggressiveness and metastasis with an emphasis on EMT-associated NF-κB/HER2/STAT3 pathways in radioresistance of breast cancer stem cells. Further testing the potential therapeutic elements in EMT interception will necessary for inventing new therapeutic target to control metastatic tumors.
EMT in development
The earliest EMT events occur during the implantation of the embryo into the uterus, the extravillous trophoectoderm cells undergo EMT in order to invade the endometrium and subsequently anchor itself in the placenta. Synchronously, during the gastrulation, a group of epiblast cells moves to midline and forms a primitive streak as the first sign of gastrulation. These cells then undergo EMT further generate mesoderm and endoderm. Mesoderm and ectoderm of an adult organism underwent several cycles of EMT and MET in order to form various tissues within the body. Otochord, somites, nephritic ducts, splanchnopleure, and somatopleure derived from epithelial mesodermal cells that underwent MET; while liver, pancreas and cardiac valves are examples of internal organs derived from endodermal cells underwent EMT/MET (Acloque et al. 2009; Johansson and Grapin-Botton 2002; Tanimizu and Miyajima 2007). Neural crest formation is also another EMT-related event. The epithelial neuroectoderm cells form a neural tube then undergo EMT to generate migratory neural crest cells, which then disperse throughout the embryo to undergo differentiation for different cell types, such as melanocytes and glial cells (Duband and Thiery 1982). Formation of these derivative cell types often requires MET to aggregate post-migratory neural crest cells and form derivative cells such as sensory ganglia (Acloque et al. 2009).
EMT in tissue regeneration and organ fibrogenesis
Under inflammation stress, injury-damaged epithelial cells undergo EMT to avoid apoptosis as an adaptive response from the injury. These mesenchymal cells then move through the underlying basement membrane and become an additional source of fibroblast cells for reparation of the injured tissue (Liu 2010; Zeisberg et al. 2007b). Under fibrosis, however, overproduction of extracellular matrix by myofibroblasts derived from fibroblast and stromal cell results in accumulation of fibrotic matrix rich in collagen, especially type I and III. Initiation of myofibroblasts is accompanied with α-smooth muscle actin (α-SMA) expression and activation of TGF-β signaling from the injury site (Desmouliere et al. 2003; Guarino et al. 2009). Regeneration and fibrogenesis of certain organs such as kidney, heart, lung, liver and intestine are associated with EMT (Kalluri 2009; Kim et al. 2006; Potenta et al. 2008; Zeisberg et al. 2007a; Zeisberg et al. 2007b). Continuation of organ fibrogenesis, as a self-sustaining mechanism with perpetual generation of cytokines, could result in organ failure and, thus, has received attentions in research. EMT has been underlined as a process responsible for invasive fibrosis and proposed as a target for inhibition for the treatment of organ fibrosis (Guarino et al. 2009; Yang and Liu 2002).
EMT in tumor proliferation and metastasis
Tumor heterogeneity and plasticity have long been recognized; however, EMT has only been identified and described as the underlying process for tumor metastasis in the last two decades (Birchmeier et al. 1996; Fidler 1975; Fidler 1978). So far, Type Three EMT is classified as the course that customarily converts primary tumor epithelial cells into invasive and metastatic mesenchymal tumor cells with enhanced mobility. The tumor mesenchyme can then intravenously migrate to a distant location and reverse back to epithelial cells through MET. During this migratory process, the mesenchymal cancer cells are enhanced with invasiveness and protected from apoptosis and senescence. Upon leaving the primary tumor site to lodging the distant site, MET activation is proceeded potentially due to the absence of EMT-inducing signals from the primary tumor site that stimulated EMT in the first place (Bissell et al. 2002; Kalluri and Weinberg 2009; Thiery 2002; Yang and Weinberg 2008). Importantly, EMT-driven machinery also orchestrates immunosuppression as well as extracellular matrix component and matrix metalloproteinase (MMP) productions to facilitate the completion of tumor metastatic progression (Kalluri and Neilson 2003; Orlichenko and Radisky 2008; Thiery et al. 2009). Changes of tumor microenvironment associated with Ras/ERK signaling could as well contribute to such phenomenon (Turley et al. 2008). Therefore, EMT-mediated tumor metastatic ability is, rather than a single incident of tumor cells gaining motility, attended with a concert of cellular and molecular proceedings since circulating tumor cells do not expand and become a new tumor shortly after dislodgment. Specific proliferation mechanisms are required to be in place for tumor cells in order for expansion to happen; EMT-orchestrated conditions are likely to be a prominent process for this expansion, although it may not be the only route involved in tumor invasion and metastasis (Bragado et al. 2012; Meng and Wu 2012).
Among many factors found to be related to EMT activation, TGF-β was the first found to be able to induce EMT. Upon addition of TGF-β to cell culture, epithelial cells turned into elongated spindle shape with reduced epithelial markers and increased mesenchymal markers (Miettinen et al. 1994; Xu et al. 2009). The transforming growth factor TGF-β has been recognized as a major EMT inducer. Along with its downstream factors, TGF-β has been extensively explored for its functions as a major propeller of TGF-β-induced EMT in cancer progression and metastasis including activation of signaling pathways and transcriptional regulators for both Smad and non-Smad pathways. Transcriptional activities of Snail, ZEB and bHLH families are regulated by TGF-β to activate mesenchymal markers and degrade epithelial markers in a Smad-dependent fashion (Zavadil et al. 2004). In the Smad-independent manner, RhoA, p38 MAPK and PI3K/Akt are involved in the TGF-β-induced EMT. Wnt and Notch signaling also cooperate with TGF-β under certain tissues and conditions for driving EMT (Xu et al. 2009; Zavadil and Bottinger 2005; Zhang 2009).
E-cadherin has been used as a cornerstone marker for epithelial cells (Kalluri and Weinberg 2009). Alteration in E-cadherin expression can result in switching of cell morphology. Induction of EMT by c-Fos oncogene in normal mouse mammary epithelial cells correlates with a decrease in E-cadherin expression (Eger et al. 2000). On the contrary, ectopically expression of E-cadherin in the cells that underwent EMT causes cells to lose their mesenchymal phenotype (Eger et al. 2000; Reichmann et al. 1992). Reduction of E-cadherin expression induced by β-catenin accumulation in the nucleus, where it becomes part of Tcf/LEF complexes, is subject to EMT and acquisition of an invasive phenotype (Kim et al. 2002; Thiery 2002). The levels of E-cadherin expression are inversely associated with patient survival (Hirohashi 1998). Mutations in the E-cadherin gene have been identified in cancer cells that are more susceptible to induction of EMT and cancer metastasis (Muta et al. 1996; Saito et al. 1999). Studies on the molecular mechanisms underlying E-cadherin loss in the EMT program revealed that transcription factors, such as Snail, Slug induced by TGF-β exposure, facilitate acquisition of a mesenchymal phenotype by repression of E-cadherin expression (Medici et al. 2008).
Accumulating numbers of proteins are shown to be involved in EMT. Following the EMT activation, alterations of EMT markers detecting phenotypical changes, such as E-cadherin, N-cadherin, Laminin-1, ZO-1, Cytokeratin, Vimentin and Fibronectin are indicated to be the evidence whether the cells are undergoing or leaning towards the stage of EMT or MET (Kalluri and Weinberg 2009). However, these presently available markers appear not be able to indicate invasive properties or relate to tumor invasion in case of cancer EMT. Further identification of EMT markers, especially those responsible for or closely reflecting the degree of tumor invasiveness and aggressiveness are desirable in order to predict and target cancer EMT and metastasis as EMT appears to be the key feature underlying metastasis of cancer (Chaffer and Weinberg 2011; Kudo-Saito et al. 2009).
Interestingly, in addition to alteration in cellular mechanisms, new evidence indicates that existence of mesenchymal cells contained within tumor may play a role in EMT-induced tumor metastasis. The mixture between mesenchymal stem cells and metastatic tumor cells greatly enhances tumor metastatic potential through the communication between the two types of cells utilizing CCR5, a chemokine receptor involved in binding interaction with the gp120 HIV-1 envelope glycoprotein (Karnoub et al. 2007). Similarly, circulating EMT tumor cells which are capable to invade into adjacent connective tissues by themselves failed to establish metastatic nodules; while circulating non-EMT tumor cells are capable of doing so but lack the ability to invade connective tissues. The mixed between the two, however, can achieve the spontaneous metastasis process by complementing each other's properties for lodging and invading the secondary site, as observed that the majority of circulating cancer cells are positive for EMT markers (Aktas et al. 2009; Tsuji et al. 2009). The metastatic enhancement of EMT and non-EMT cells, with different morphologic properties, augments the complexity in dealing with EMT-mediated cancer metastasis. Simply inhibition of EMT-related molecules, in hope for lessening EMT-mediated tumor invasion, may not be sufficient to prevent metastasis. As non-EMT cells may play a part in promoting tumor metastasis; a potential communication between EMT and non-EMT cells may provide additional important information on cancer metastasis. Thus, EMT alone could not drive the full process of tumor metastasis; similarly, neither all fibrogenesis events are occurred via EMT (Christiansen and Rajasekaran 2006; Fragiadaki and Mason 2011; Garber 2008; Grabias and Konstantopoulos 2012; Kriz et al. 2011; Rock et al. 2011; Tarin et al. 2005; Taura et al. 2010). Therefore, the concept of complementary EMT or incomplete EMT should be advised when assessing EMT in tumor metastasis.
EMT in vascularization
Vascularization is an essential process for tumor sustenance and expansion since tumorigenesis as well as tumor growth require additional blood vessel formation for nutrient and oxygen supplies. Although there is yet evidence to support that EMT directly induces or associates with signals for angiogenesis, neither for tumor nor normal organ, TGF-β could promote angiogenesis by directly inducing capillary formation of endothelial cells in addition to EMT activation (Akhurst and Derynck 2001; Derynck et al. 2001; Kumar-Singh et al. 1999). The dual functions of TGF-β provide additional survival advantages for tumor by both providing blood supply and escaping route for tumor cells in case of starvation. Blood cells and circulatory components can influence EMT and motility of tumor cells. Direct interaction between platelets or CD8 T cells with cancer cells is shown to promote tumor metastasis through the TGF-β/Smad and NF-κB signaling pathways and cells capable of undergoing EMT show cancer stem cell-like characteristics (Labelle et al. 2011; Santisteban et al. 2009). PDGF-D is potentially the factor responsible for the platelet-cancer cells interaction triggering EMT (Kong et al. 2009). Moreover, activation of tumor EMT by the blood components can result in generation of CSCs as a byproduct for those tumors that have direct access to blood vessel. Tumor cells are also likely to relocate towards blood supply and lymph node, rather than following a Brownian motion (Wu et al. 2014). Thus, tumor access to blood supply may increase the likelihood of tumor cells undergoing to the evil transition process and subsequent metastasis as a nature of the transition (Singh and Settleman 2010).
EMT interconnects with chemotherapy resistance
EMT-mediated therapeutic resistance has been observed in several types of cancer and could limit treatment options for patients. Mesenchymal cells are more resistant to EGFR and PI3K/Akt pathway inhibitors in general (Byers et al. 2013). Resistance of chemotherapeutic reagents including gemcitabine, 5-FU (5-fluorouracil), cisplatin and adriamycin corresponds with ZEB-1 and Twist levels and inversely correlates with E-cadherin and epithelial markers EVA1 and MAL2 expressions. Likewise, c-Met activation and MDR (multidrug resistance) induction are also linked with their expressions in the same manner (Arumugam et al. 2009; Li et al. 2009a). Acquired resistance of gefitinib in lung cancer drives cells towards EMT, as a part of chemo-adaptive response, as well as develops resistance to several tyrosine kinase inhibitors (Rho et al. 2009). Ovarian cancer resistant to paclitaxel also have increased expression of Snail and Twist (Kajiyama et al. 2007). Depletion of synergistic effects in cetuximab is observed in head and neck tumors with c-myc upregulation and EMT progression (Skvortsova et al. 2010). Similarly, colorectal cancer cells resistant to oxaliplatin induced EMT through Vimentin induction (Yang et al. 2006b). Overexpression of Twist, Snail, and FOXC2 in breast cancer cells not only provide tumor cells EMT activation but also ABC transporter upregulation and subsequent MDR (Saxena et al. 2011). Expression levels of several proteins have been tested and verified as prognostic factors for cancer patients depending on tissue types and pathological settings. Concerning EMT, expressions of Twist, Vimentin, Snail, Slug, FOXC1, β-catenin, E-cadherin are examples of markers used to indicate patient prognosis in chemotherapy (Al-Saad et al. 2008; Fanelli et al. 2008; Taube et al. 2010; Uchikado et al. 2011; Xie et al. 2009). Sensitivity of concurrent chemoradiation therapy in non-small cell lung cancer depends upon expressions of EMT marker proteins (E-cadherin, cytokeratin, N-cadherin, and Vimentin). Tumors with positive EMT markers can develop a subpopulation with resistant phenotype rendering them difficult to treat (Shintani et al. 2011). Concurrent chemoradiation therapy, thus, may not be able to overcome EMT-mediated tumor resistance but could be the root for tumor resistant stem cells and metastasis through EMT, as observed in recurrent ovarian cancer (Ahmed et al. 2010). These reports collectively indicate that EMT and induction of EMT-inducing genes are an integral part of adaptive response to therapeutic reagents by advancing cells towards mesenchymal features for their gain-of-function and resistance to evade cell death. In order to overcome such resistance, disruption of EMT pathway specific in tumor cells is desirable.
Hypoxia induces EMT
In addition to the relationship between blood components and cancer EMT, induction of EMT by angiogenic factors can synchronically increase tumor aggressiveness as well as facilitate nutrient supply for tumor. Angiogenesis factor VEGF and its receptor, VEGFR-1, are capable to elicit EMT of cancer cells through TGF-β and EMT-associated pathways through Snail, Twist and Slug (Gonzalez-Moreno et al. 2010; Yang et al. 2006a). However, in case of blood supply inadequacy such as hypoxia, HIF-1 becomes a key factor in stimulating EMT by activating Twist, Snail and uPAR expressions to drive EMT forward (Cannito et al. 2008; Higgins et al. 2007; Lester et al. 2007; Luo et al. 2006; Yang and Wu 2008; Yang et al. 2008; Zhang et al. 2013a). Hypoxic condition could also induce TGF-β, GLIPR-2 and Notch signaling while PER2 is down regulated to advance cells to EMT (Huang et al. 2013; Hwang-Verslues et al. 2013; Ishida et al. 2013; Matsuoka et al. 2013). As observed that loss of oxygen supply could transform tumor epithelial cells into spindle shape with lost cell to cell contacts (Theys et al. 2011). Inhibition of GSK-3β and activation of PI3K/Akt pathway are also involved in the hypoxia-mediated EMT possibly through transient intracellular increased generation of reactive oxygen species (ROS) and HIF-1/VEGF-dependent pathway (Cannito et al. 2008; Yan et al. 2009; Zhou et al. 2004). Introduction or production of ROS, particularly mitochondrial ROS, appears to be an important inducer of EMT as an integral part of TGF-β-mediated EMT (Rhyu et al. 2005; Yoon et al. 2005; Zhang et al. 2007; Zhou et al. 2009). Wnt/β-catenin pathway also plays a role in this hypoxia-mediated EMT as β-catenin is a repressor of E-cadherin transcription (Zhang et al. 2013b). As illustrated in Fig. 1, GSK-3β regulates Snail so as to both controlling its subcellular localization and its repression of E-cadherin transcription (Zhou et al. 2004). The accumulating evidence has tight-knitted hypoxia and EMT in tumor aggressiveness, which highlights the potential alternative survival modalities under the circumstance of oxygen and nutrient deficiency which may be targeted to inhibit tumor cells (Brizel et al. 1996; Pugh and Ratcliffe 2003; Shweiki et al. 1992; Sullivan and Graham 2007).
Fig. 1.
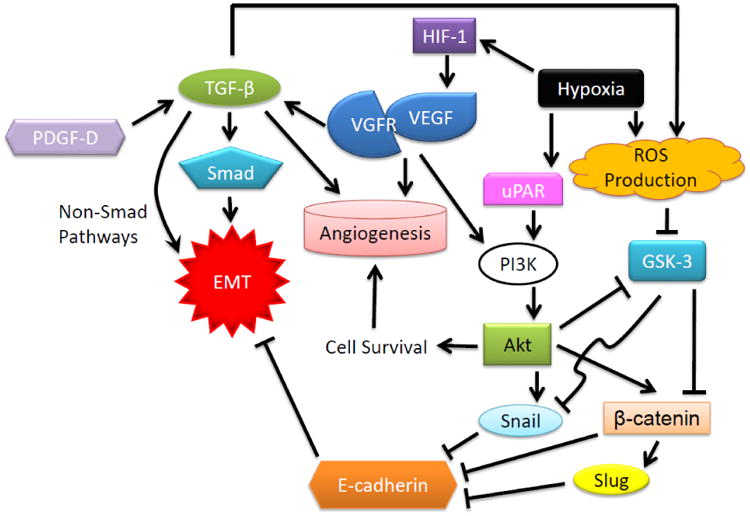
Illustration of the relationship between EMT, angiogenesis and hypoxia. Hypoxia stimulates EMT factors by activating a similar set of genes for hypoxic responses and angiogenesis through HIF-1 and ROS generation resulting in the activation of angiogenic pathway. Binding of VEGF to VEGFR signals TGF-β and PI3K to stimulate EMT in both Smad and non-Smad dependent manners. Both hypoxic condition and EMT activation contribute to tumor aggressiveness and metastasis.
EMT in behavior of cancer stem cells
Although still controversial, the concept of cancer stem cells (CSCs, or called tumor initiating cells, TICs) is accepted to describe the general heterogeneity of tumor cell population responsible for establishment, growth and sustenance of a tumor (Baker 2008; Reya et al. 2001). CSCs ranked at the top of tumor cell hierarchy, having elevated capacity of therapy resistance, angiogenesis and metastasis in addition to high tumor formation potential and self-renewability, as shown that only a few hundred of CSCs or less of CSC-like cells are sufficient to form a new tumor in recipient rodents (Al-Hajj et al. 2003; Bao et al. 2006b; Hermann et al. 2007). Such phenomenon has brought attentions to cancer stem cell investigation as well as its origination. Two major hypotheses regarding the origin of CSCs are that tumor cells are originated from either transformation of a group of normal cells, which a few of them turned into CSCs through their plasticity, or neoplastic transformation of normal stem cells which has been demonstrated to be a potential origin of tumor and CSCs (Bonnet and Dick 1997; Fujimori et al. 2012; Reya et al. 2001; Sell and Pierce 1994). Although the underlying mechanisms of such conversion from normal to tumor stem cells are largely unknown, loss of contact inhibition of human mesenchymal stem cells was shown to be first step in neoplastic transformation (Serakinci et al. 2004). The discovery is further confirmed by evidence that EMT is a mesenchymal cell generating process allowing acquisition of cancer stem cell characteristics (Mani et al. 2008; Morel et al. 2008). Therefore, it is rational to hypothesize that during the shifting course from epithelial to mesenchymal cells, EMT may mechanistically enhance cancer cell tumorigenesis or renewability of tumor cells, as indicated that in both normal and carcinoma human breasts epithelial cells with CD44+CD24- express far greater levels of EMT transcription factors than in CD44-CD24+ cells (Mani et al. 2008; Polyak and Weinberg 2009). In addition to improving tumor cell mobility, invasiveness and death resistance, a variety of genes has been observed to be up-regulated during or as a part of the EMT process including those of stem cell related genes. Up-regulation of TGF-β, TNF-α, FOXQ1, FOXC2, FOXM1, Oct4, Wnt, Notch, Hedgehog and Nanog, demonstrated to be activators of EMT, could give rise to stem cell-like cancer cells or cancer cells with stem cell properties. Suppression of these genes also reduces tumor aggressiveness as well as EMT (Asiedu et al. 2011; Bao et al. 2011; Chiou et al. 2010; Hollier et al. 2013; Huber et al. 2005; Qiao et al. 2011; Reiman et al. 2010). Many of these genes have been identified to be associated with cell stemness as well as stem cell characteristics, which implicate that EMT and stemness machineries are overlapped. Loss of mitochondrial DNA also activates EMT and generates CSCs through calcineurin in a reversible manner while activation of Akt and β-catenin is indispensable for maintaining EMT-modulated cancer stem cell characteristics (Guha et al. 2013; Li and Zhou 2011). Additionally, external stimuli could prove to be able to induce CSC generation through EMT as demonstrated that CD8 T cells could signal generation of breast cancer stem-like cells (Santisteban et al. 2009). This evidence has pointed out the significance of EMT not only in the cell transitioning process but also in CSC generation from tumor as observed that circulating tumor cells underwent EMT often express cancer stem cell markers and characteristics (Hennessy et al. 2009). Elimination of CSCs, the primary goal for CSC and cancer research, may be accomplished through inhibition of EMT program or domination of MET within cancer cells.
EMT and CSCs in therapy resistance
One of the prominent characteristics of CSCs is their resistant phenotype to many anti-cancer modalities (Donnenberg and Donnenberg 2005; Rich 2007). Besides deriving tumor cells towards becoming the stemness feature, many EMT-associated genes are behind other mechanisms inducing cell resistance to therapy. Up-regulation of p21 from Notch slowed down cell growth providing cell survival advantages against therapeutic insults (Nefedova et al. 2004). Nanog also provides protection of prostate and breast cancer cell resistance in addition to enhancing expressions of CXCR4, IGFBP5, CD133 and ALDH1 for cancer stem cell features (Jeter et al. 2011). Expectedly, inhibition of FOXQ1, shown to be a poor prognostic indicator, could lessen both metastatic potential and resistance of tumor cells through the reversal of EMT (Feng et al. 2012; Sehrawat et al. 2012; Zhu et al. 2013). Although several key factors remain to be investigated, it is appropriate to assume that the signaling networks among metastasis, vascularization, CSC generation and tumor resistance to therapy are consolidated by the EMT pathway. Thus, it is reasonable to propose that disturbance to one of the EMT elements may affect more than a single feature of tumor, and thus could be used as a strategy for exploitation in cancer therapy.
EMT in cancer resistance to chemo-radiotherapy
A group of breast cancer MCF-7 clones that can survive long-term therapeutic radiation with increased DNA repair gene expression was isolated from irradiated MCF-7 cell population and identified to contain breast cancer stem cells with enhanced radioresistance (Ahmed et al. 2006; Duru et al. 2012; Li et al. 2001). Although the exact mechanism of the radioresistant cancer stem cells is to be elucidated, as mentioned earlier that EMT can produce CSCs which are generally known to be resistant to therapies, with elevated levels of free radical scavenging proteins, DNA repair as well as drug transporting capacity (Bao et al. 2006a; Dean et al. 2005; Diehn et al. 2009; Rich 2007). The process of EMT also contributes to tumor chemo- and radio-resistance via activation of EMT-mediated genes related to suppression of cell death mechanisms, as inhibition of Snail expression could sensitize tumor cells to genotoxic stress (Kajita et al. 2004). Activation of Notch signaling pathway through EMT and suppression of p53-mediated apoptosis by Snail and Slug provide examples of EMT-mediated tumor resistance to therapies (Kurrey et al. 2009; Wang et al. 2009). Notch signaling pathway also links between EMT and gemcitabine resistance in pancreatic cancer (Wang et al. 2009). Initiation of EMT, therefore, requires activation of an array of genes to alter the cell biochemically and physiologically, simultaneously; such activation influences other genes into defining other cell phenotypes. Both normal and tumor tissues have deployed the EMT mechanism in order to evade cell death. Type two EMT is the chief example of normal tissue avoiding cell death with enhanced repair capacity of fibroblastic cells derived from EMT. In cancerous cells, acquisition of EMT minimizes cell death by both reducing lateral membrane and contacting apoptosis signals due to loose structure of mesenchymal cells and enhancing pro-survival mechanisms. One of the pro-survival factors closely involved in EMT is NF-κB and, perhaps, the most influential transcription factor which orchestrates resistance characteristic. NF-κB is labelled as an essential gene for both induction and maintenance of EMT independent of TGF-β and considered to be directly connected with EMT by activating ZEB1, ZEB2 and Vimentin expressions which suppress E-cadherin protein level. Activation of NF-κB can also be achieved by TGF-β during EMT as a feedback response to carry out EMT (Chua et al. 2007; Huber et al. 2004a; Huber et al. 2004b; Huber et al. 2005; Maier et al. 2010). Treatment of proteasome inhibitor NPI-0052 mediated expression of RKIP (Raf kinase inhibitor protein) through NF-κB inhibition which resulted in Snail inhibition and subsequent EMT repression (Baritaki et al. 2009). Exposure to ionizing radiation which invokes NF-κB signaling pathways improved cell motility and proceeded cells towards EMT (Jung et al. 2007). Identification of NF-κB-mediated Twist activation further linked and broadened our understanding of EMT to NF-κB-arbitrated cancer cell properties including adaptive response, apoptosis resistance and metastatic properties (Yu et al. 2014). As NF-κB effector genes are responsible for a variety of cellular responses ranging from inflammation, immune response, cell adhesion, cell proliferation, anti-apoptotic response, oncogenesis to metastasis (Ahmed and Li 2008; Mayo and Baldwin 2000; Orlowski and Baldwin 2002). Further investigation in the mechanisms underlying coordination of NF-κB and its effectors in EMT process may shed light on novel therapeutic approaches as well as potential cancer prevention.
EMT in radioresistant breast cancer stem cells with activated NF-κB/HER2/STAT3 signaling pathway
Enrichment of CSCs through therapy is evidenced as tumor, rather than a collective group of homogenous cells, behaves in an organ-like manner with heterogeneity subtypes and hierarchical cellular organization (Eyler and Rich 2008; Li et al. 2008). Exposure to ionizing radiation is known to induce tissue remodeling and changes in microenvironment (Rodemann and Blaese 2007). Whether the roles of TGF-β is still debatable whether it is responsible for tumor migration; modulation of matrix degrading enzymes including uPA and MMPs is affected by ionizing radiation (Moncharmont et al. 2014). Enhancement of tumor migration and invasion is another imminent changes made by radiation through EMT and other mechanisms related to ECM proteins, which leads to eventual tumor resistance to radiation (Zhou et al. 2011). On top of the radiation-enhanced tumor survival and invasiveness, we found that exposure to therapeutic fractionated radiation can promote the selection of breast cancer stem cells (BCSCs) through induction of HER2 expression through radiation-induced NF-κB binding to HER2 gene promoter and activating transcription of the gene (Ahmed and Li 2008; Cao et al. 2009). However, as it has been noticed that HER2+ BCSCs can be detected not only in HER2+ tumor cells, but also in radioresistant HER2-/low breast cancer (Duru et al. 2012). The side population harbored within tumors regardless of the status of the majority of cells is often observed having CSC-like properties (Chiba et al. 2006; Ho et al. 2007). In this instance, newly repopulated cells from the surviving BCSCs express HER2 in order to meet with the challenge of the stressor. Expression of HER2, as a receptor molecule, then activates STAT3, a latent cytoplasmic transcription factor that conveys signals from cell surface to nucleus by cytokines or growth factors through both JAK2 and Src-dependent manners as supported by the data showing that HER2 and STAT3 are co-expressed in radioresistant HER2+/CD44+/CD24-/low but not in HER2-/CD44+/CD24-/low BCSCs (Cheng et al. 2008; Duru et al. 2012; Ren and Schaefer 2002). Additionally, we recently demonstrated dramatic increase in STAT3 phosphorylation at Tyrosine-705 (Y-705) residue in fractionated-irradiated MDA-MB-231 cells and MCF7-HER2 cells but not in their original parental cells. Moreover, evidence shows that STAT3 phosphorylation at Y705 was observed in MCF7-HER2 cells but not in MCF7 wild-type (Chung et al. 2014). These results further confirm the serial involvement of NF-κB/HER2/STAT3 to EMT and stemness of tumor cells, as shown in Fig. 2. Additional studies show that the stem cell markers, Oct-4 and SOX-2, were expressed in MCF7-HER2 cells, but not in the MCF7 wild-type cells and knocking-down of STAT3 also down-regulated the two stemness markers (Chung et al. 2014). In agreement with the cancer stem cell markers, the protein levels of mesenchymal markers Vimentin and Slug increased significantly while epithelial marker E-cadherin decreased dramatically in MCF7-HER2 compared with control MCF7 wild-type cells. Inhibition of STAT3 activation with stattic, a non-peptidic small molecule that inhibits STAT3 activation by selectively inhibiting dimerization and nuclear translocation of STAT3, diminished expression of EMT markers Vimentin and Slug and concurrently dwindled the expression of stem cell markers SOX-2 and Oct-4, suggesting an intrinsic relationship between EMT and cancer stem cells (Schust et al. 2006). An emerging evidence also suggests EMT-inducers as bona fide regulators of mammary stem cell state and commitment (Ansieau 2013). The evidence that Slug is present in mammary stem cells and it cooperates with SOX-9 in orchestrating stem cell state of the cells further defines that EMT, cell stemness, and therapy resistance are closely interrelated (Guo et al. 2012).
Fig. 2.
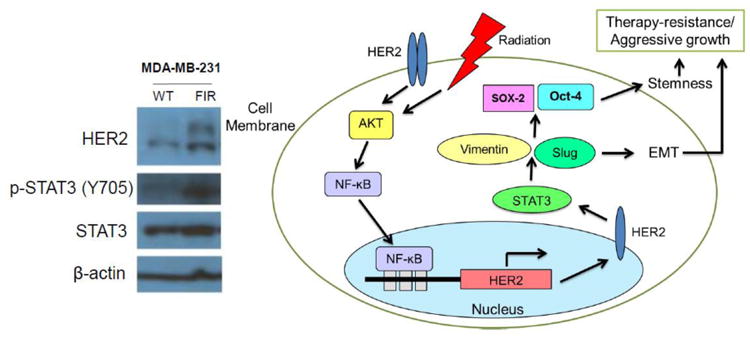
STAT3 phosphorylation at Y705 is increased in radiation-induced HER2-upregulated radioresistant breast cancer cells. Immunoblot analyses of HER2, STAT3 and pY705-STAT3 were performed in cancer cell lines MDA-MB-231 and its radioresistant cells (FIR). The activated phosphorylation of STAT3 is enhanced in the FIR cells (Left panel). Schematic diagram presenting EMT-associated radioresistance and aggressiveness of radioresistance breast cancer stem cells with activated HER2 and STAT3. Therapeutic resistance is a result of adaptive response which activates EMT and stemness governing genes. STAT3 activation is an intermediary in the activation of EMT (Right panel).
ROS-mediated EMT activation and tumor radioresistance
Although ROS has been considered a toxic reagent, ROS signaling is an important intracellular signaling pathway altering redox state of the cell to induce physiological responses ranging from growth factor stimulation to the generation of inflammatory response (Finkel 2011). The sources of ROS are from both intracellular origins and environmental factors. The major source of cellular ROS is derived from electron transport chains within mitochondria, as a part of aerobic respiration. This ROS source is demonstrated to be primarily linked with the undertaking of EMT. Production of mitochondrial ROS can be prolonged by TGF-β-mediated disruption of mitochondrial complex IV activity; thus, cell growth is delayed as a mechanism for cell death resistance (Yoon et al. 2005). The excess mitochondrial ROS then stimulates activation of Snail transcription factor and Smad pathway through MAPK/ERK cascade resulting in EMT (Radisky et al. 2005; Rhyu et al. 2005). Activation of the redox sensitive transcription factor NF-κB by ROS is also another route for ROS-mediated EMT as described earlier of NF-κB contributions to EMT (Chua et al. 2007; Julien et al. 2007).
Exposure to therapeutic dose of radiation is well-defined to impose both cellular and mitochondrial generation of ROS and led to alterations of gene expressions including those responsible for radioresistance and potential EMT as radiation is known to activate TGF-β (Barcellos-Hoff 1993; Leach et al. 2001; Li et al. 2001). Radiation-induced TGF-β is, therefore, an apparent and prominent pathway for EMT activation as non-malignant human tumor cells can undergo EMT under the influence of radiation-activated TGF-β (Andarawewa et al. 2007; Zhou et al. 2011). Under the stress of radiation, transcription factor FoxM1 upregulates JNK1 expression which then further activates TGF-β for the progression of EMT (Balli et al. 2013). TGF-β-mediated EMT, in accordance with ATM, is capable to activate Homeobox B9 (HOXB9) for enhancement of DNA damage and repair responses leading to radioresistance of breast cancer (Chiba et al. 2012). Loss of E-cadherin due to hypoxic condition contributes to radioresistance of tumor cells in addition to the lack of oxygen which is known to attenuate radiation-induced DNA damages (Theys et al. 2011). Down-regulation of serine protease HtrA1 is also linked with ATM-mediated DNA damage response and EMT activation through miR-200 depletion (Wang et al. 2012). Interestingly, EMT could be a part of low dose radiation-induced radioadaptive response as observed that EMT is activated in breast cancer cells receiving low dose radiation (Zhang et al. 2011). These connections of EMT and radioresistance strongly suggest that induction of EMT is a part of adaptive response to ionizing radiation as tumor cells may apply apoptosis and cell death resistance properties of mesenchymal cells in order to survive the challenges of radiation. EMT is, accordingly, a pathway behind radio-adaptive response and tumor resistance to therapy.
microRNAs targeting EMT
Recent research has turned attention to non-coding RNAs (ncRNAs) as it is taking the center stage in studies of many aspects of human diseases and cancer research including EMT. The roles of miRNAs in EMT have been explored, which leads to miR-10b and miR-21 being used as markers for mesenchymal phenotype (Kalluri and Weinberg 2009). Many studies provide further evidence regarding functions of microRNA in regulating EMT. Several miRNAs including miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and miR-429), miR-30, miR-34, miR-138, miR-192, miR-194 and miR-205 are demonstrated to target EMT-inducing transcription factors and proteins to enforce epithelial features and keep invasiveness and aggressiveness of tumor quiescent. Ectopic expressions of these miRNAs can also induce MET or reduce EMT through various mechanisms predominantly including suppression of Snail, TGF-β, ZEB1/ZEB2, EZH2, BMI-1, and EMT expression marker, N-cadherin, as illustrated in Fig. 3 (Burk et al. 2008; Dong et al. 2011; Gregory et al. 2008b; Kim et al. 2011a; Kim et al. 2011b; Liu et al. 2011; Meng et al. 2010). Expression of E-cadherin is also cooperatively regulated under the same set of miRNAs by the subduction of ZEB1 and ZEB2 which are the transcriptional repressors of E-cadherin. Down-regulation of E-cadherin is identified as a spontaneous factor in progression, aggressive phenotype and poor prognosis of cancer (Perl et al. 1998; Wijnhoven et al. 2000). MiR-29a and miR-155, however, could degrade cell polarity by targeting tristetraproline (TTP) and RhoA GTPAse, respectively, as both proteins are regulators of cellular polarity and tight junction moderators (Bullock et al. 2012). Similarly, overexpression of miR-9, an E-cadherin regulator, could reduce E-cadherin expression by 70% while reciprocally induced Vimentin expression (Ma et al. 2010). Providing this information, the discovery of miRNA in cancer EMT regulation is rapidly expanding and could be useful in specific drug development for suppression of cancer EMT.
Fig. 3.
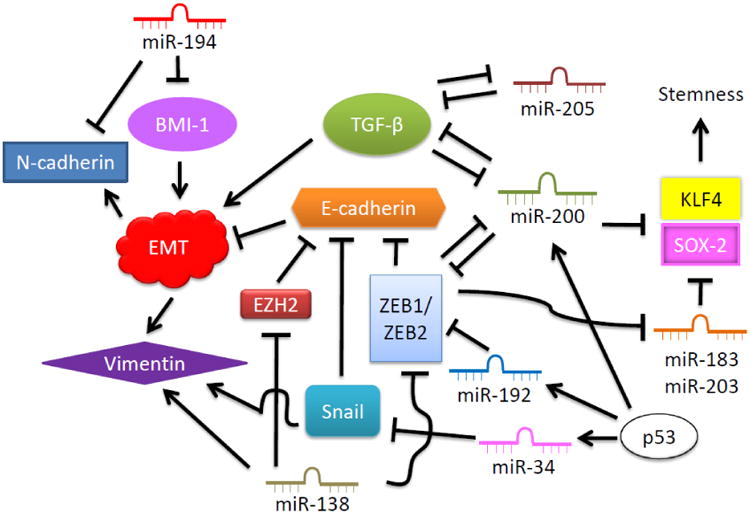
Relationship between microRNAs with EMT and EMT-related factors. Various miRNAs involve in regulations of EMT through inhibition of EMT initiating factors including TGF-β, Snail, ZEB1/2 and BMI-1. These factors also provide negative feedback loop to suppress miRNAs and reconstitute miRNA profile to allow EMT and mesenchymal elements as well as down-regulate epithelial marker proteins. P53 is a master regulator of miR-34, miR-192 and miR-200 in impeding EMT through targeting of ZEB1/2 and Snail transcription factors.
One of the most studied and, perhaps, the most targeted miRNAs involved in EMT regulation is miR-200 family. Inhibition of miR-200 family lifts TGFβ-2 and ZEB-1 expressions and activities; thus, allowing EMT proceeding and E-cadherin suppression in the process (Braun et al. 2010; Gregory et al. 2008a; Korpal et al. 2008). MiR-200 is also linked with suppression of stem cell factors, SOX-2 and KLF4, along with the miRNAs, miR-183 and miR-203 (Wellner et al. 2009). Tumor suppressor p53 has also been highlighted in regulating transcriptions of EMT inhibitory miRNAs especially in regards to miR-200 (Chang et al. 2011; Kim et al. 2011b). These functions of miR-200 in preventing EMT and sequential cancer metastasis have been extensively explored in several types of cancer, which lead to miR-200 being branded as the guardian against pluripotency and cancer progression (Peter 2009). The balance switch between ZEB1-ZEB2 and miR-200/miR-138 in controlling cancer EMT could be a target for exploitation in preventing tumor metastasis and improve clinical prognosis.
As aforementioned, miRNAs play an essential role in regulating EMT affecting metastatic status of a tumor. On the same hand, miRNAs are involved in tumor resistance to several chemotherapeutic agents such as docetaxel by miR-192, miR-424 and miR-98 and 5-FU and methotrexate by miR-140 (Wang et al. 2010). In terms of EMT-mediated resistance, it appears that artificially introduced expression of miRNAs including miR-200 and treatment of natural product isoflavone to activate miR-200 and let-7 could reverse EMT and potentially reduce chemotherapeutic resistance of pancreatic cancer (Li et al. 2009b). Further exploration of miRNAs functions in EMT and EMT-mediated cancer resistance may provide critical approaches to improvement of therapy for resistant as well as metastatic cancer.
Targeting EMT to treat tumor aggressiveness and metastasis
EMT has been proposed to be a potential target for clinical application to control cancer initiation, maintenance and expansion (Findlay et al. 2014). The idea of suppressing EMT to subside cancer metastatic potential and enhance malignant cell killing has already been tested using various approaches by inhibiting EMT prominent pathways. As discussed earlier that activation STAT3 by fractionated irradiation or chemotherapeutic reagents closely engages with both tumor EMT and cell stemness; therefore, inhibition of the transcription factor along with the other EMT-related transcription factors is proposed, although it could be unfavorable for certain conditions (Davis et al. 2014). Epigallocathechin gallate (EGCG) with quercetin inhibiting EMT-related genes such as Vimentin, Snail, Slug and nuclear β-catenin was proved to be effective in impairing EMT as well as the subpopulation with cancer stem cell like characters in prostate cancer cells. Self-renewal, invasion and migration were all inhibited with enhanced apoptosis (Tang et al. 2010). Inhibition of CSC generation and EMT by Resveratrol was also shown to be useful in preventing EMT, tumor growth and development of pancreatic cancer (Shankar et al. 2011). Receptor tyrosine kinase Axl inhibitor, SGI-7097, also provided sensitivity of mesenchymal cells in non-small cell lung cancer (Byers et al. 2013). Several other compounds chiefly targeting TGF-β, Wnt, Notch, Hedgehog, JAK-STAT and PDGFR are currently undergoing Phase I and II trials for several cancer types (Pattabiraman and Weinberg 2014). These reagents, with further studied and developed for precise targeting and delivery could become beneficial in both studies of clinical EMT and practical treatment of malignant cancer.
The idea of utilizing miR-200 family to suppress ZEB1/ZEB2, maintain epithelial polarization and induce MET for tumor mesenchyme could also be beneficial in the near future for preventing aggressive tumor metastasis. Manipulation of miRNAs such as miRNA-200 family and miRNA-205 could also prove to be worthwhile in reducing EMT for better prognosis. Targeting EMT-inducing genes with miRNAs or shRNAs has been observed in reducing EMT as well as metastatic and resistant potential of tumor cells. However, applying these molecules in the clinics remains impractical. The idea of activating certain proteins regulating a set of EMT-regulated miRNAs has been studied. Repression of ZEB1/ZEB2 expressions with miR-192 and miR-200 mediated by tumor suppressor p53 appears to be effective in blocking EMT (Chang et al. 2011; Kim et al. 2011b). Further investigations of EMT-silencing miRNAs and their modulating transcription factors are necessary for overcoming EMT and enhancing cancer therapeutic outcomes especially for high grade tumors or those with high metastatic capability. The approach of EMT repression could provide additional benefits in cancer therapy as adverse effects to normal tissue would be minimal.
Conclusions and perspectives
The functions of EMT in a tumor involve several key cancer hallmarks: origination (generation of CSCs), maintenance (therapy resistance and activation of multiple cancer pathways) and expansion (angiogenesis and metastasis; Fig. 4). New targets of EMT-mediated tumor aggressiveness and resistant phenotype are emerging. For instance, EMT is activated in HER2-positive breast cancer stem cells that can survive long-term therapeutic radiation and are linked to resistant phenotype and poor prognosis of breast cancer patients. In such case, inhibition of EMT could be effective in treating the resistant current/metastatic tumors with HER2-overexpression. However, simplification of the complex network by using a single molecule to suppress EMT could prove to be ineffectual as there are redundancies and bypasses in biological setting within tumors in which adaptive responses can clearly be observed. Therefore, inhibition of EMT in combination with other modalities such as anti-HER2 approaches in breast cancer may be effective in suppression of tumor growth while preventing them from dislocation. New challenges will appear by using different EMT-targeting modalities which requires further elucidating EMT-mediated signaling network, especially in the pathways overlapping with tumor microenvironments such as hypoxia as well as intrinsic resistance of tumor cells and the repopulation of cancer stem cells, which is informative in treatment of resistant metastatic tumors.
Fig. 4.
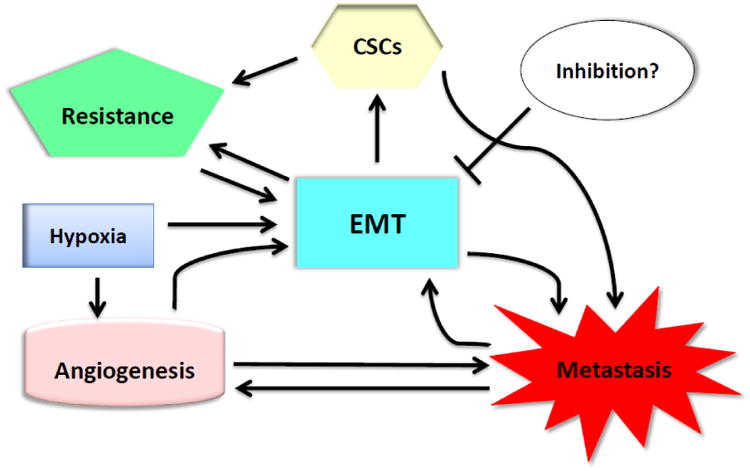
EMT-associated tumor perpetuation. EMT takes the central role in sustaining tumor by integrating hallmarks of cancer involving several processes of tumor maintenance, expansion, motility and migration to secondary locations. In combination with neoplastic formation, EMT leads to generation of stem-like group of cells which cancer stem cell properties which provide further therapy resistance phenotypes for tumor cells. EMT is also associated with hypoxic condition in driving angiogenesis. Suppression of EMT may provide alternatives in approaching solutions for cancer therapy.
Acknowledgments
We apologize for not being able to cite many important articles due to space restrictions. We thank Dr. Colleen Sweeney at University of California Davis and Dr. Max Wicha at University of Michigan for constructive discussion on breast cancer research. DN is supported by Chulabhorn Foundation of Thailand. The research projects in the laboratory of the authors have been supported by NIH NCI Grants CA133402 and CA152313, as well as a grant from the US Department of Energy Office of Science DE-SC0001271 to JL.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
References
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed KM, Dong S, Fan M, Li JJ. Nuclear factor-kappaB p65 inhibits mitogen-activated protein kinase signaling pathway in radioresistant breast cancer cells. Mol Cancer Res. 2006;4:945–955. doi: 10.1158/1541-7786.MCR-06-0291. [DOI] [PubMed] [Google Scholar]
- Ahmed KM, Li JJ. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic Biol Med. 2008;44:1–13. doi: 10.1016/j.freeradbiomed.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Abubaker K, Findlay J, Quinn M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr Cancer Drug Targets. 2010;10:268–278. doi: 10.2174/156800910791190175. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11:R46. doi: 10.1186/bcr2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Saad S, Al-Shibli K, Donnem T, Persson M, Bremnes RM, Busund LT. The prognostic impact of NF-kappaB p105, vimentin, E-cadherin and Par6 expression in epithelial and stromal compartment in non-small-cell lung cancer. Br J Cancer. 2008;99:1476–1483. doi: 10.1038/sj.bjc.6604713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andarawewa KL, Erickson AC, Chou WS, Costes SV, Gascard P, Mott JD, Bissell MJ, Barcellos-Hoff MH. Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 2007;67:8662–8670. doi: 10.1158/0008-5472.CAN-07-1294. [DOI] [PubMed] [Google Scholar]
- Ansieau S. EMT in breast cancer stem cell generation. Cancer Lett. 2013;338:63–68. doi: 10.1016/j.canlet.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71:4707–4719. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Melanoma in mice casts doubt on scarcity of cancer stem cells. Nature. 2008;456:553. doi: 10.1038/456553a. [DOI] [PubMed] [Google Scholar]
- Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32:231–244. doi: 10.1038/emboj.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112:2296–2306. doi: 10.1002/jcb.23150. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006a;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006b;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff MH. Radiation-induced transforming growth factor beta and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res. 1993;53:3880–3886. [PubMed] [Google Scholar]
- Baritaki S, Chapman A, Yeung K, Spandidos DA, Palladino M, Bonavida B. Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: pivotal roles of Snail repression and RKIP induction. Oncogene. 2009;28:3573–3585. doi: 10.1038/onc.2009.214. [DOI] [PubMed] [Google Scholar]
- Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19:294–308. doi: 10.1016/j.semcdb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Brand-Saberi B. Epithelial-mesenchymal transitions in cancer progression. Acta Anat (Basel) 1996;156:217–226. doi: 10.1159/000147848. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012;195:25–39. doi: 10.1007/978-3-642-28160-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J, Hoang-Vu C, Dralle H, Huttelmaier S. Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene. 2010;29:4237–4244. doi: 10.1038/onc.2010.169. [DOI] [PubMed] [Google Scholar]
- Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, Dewhirst MW. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941–943. [PubMed] [Google Scholar]
- Bullock MD, Sayan AE, Packham GK, Mirnezami AH. MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol Cell. 2012;104:3–12. doi: 10.1111/boc.201100115. [DOI] [PubMed] [Google Scholar]
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, Wistuba II, Coombes KR, Minna JD, Heymach JV. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, Cravanzola C, Bravoco V, Colombatto S, Parola M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29:2267–2278. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]
- Cao N, Li S, Wang Z, Ahmed KM, Degnan ME, Fan M, Dynlacht JR, Li JJ. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat Res. 2009;171:9–21. doi: 10.1667/RR1472.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, Liu M, Chen CT, Yu D, Hung MC. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008;283:14665–14673. doi: 10.1074/jbc.M707429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba N, Comaills V, Shiotani B, Takahashi F, Shimada T, Tajima K, Winokur D, Hayashida T, Willers H, Brachtel E, Vivanco MD, Haber DA, Zou L, Maheswaran S. Homeobox B9 induces epithelial-to-mesenchymal transition-associated radioresistance by accelerating DNA damage responses. Proc Natl Acad Sci U S A. 2012;109:2760–2765. doi: 10.1073/pnas.1018867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, Taniguchi H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240–251. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, Wu CW. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433–10444. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–8326. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- Chung SS, Giehl N, Wu Y, Vadgama JV. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int J Oncol. 2014;44:403–411. doi: 10.3892/ijo.2013.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci. 2014 doi: 10.1016/j.tips.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest. 2003;83:1689–1707. doi: 10.1097/01.lab.0000101911.53973.90. [DOI] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong P, Kaneuchi M, Watari H, Hamada J, Sudo S, Ju J, Sakuragi N. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol Cancer. 2011;10:99. doi: 10.1186/1476-4598-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- Du F, Nakamura Y, Tan TL, Lee P, Lee R, Yu B, Jamora C. Expression of snail in epidermal keratinocytes promotes cutaneous inflammation and hyperplasia conducive to tumor formation. Cancer Res. 2010;70:10080–10089. doi: 10.1158/0008-5472.CAN-10-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duband JL, Thiery JP. Appearance and distribution of fibronectin during chick embryo gastrulation and neurulation. Dev Biol. 1982;94:337–350. doi: 10.1016/0012-1606(82)90352-9. [DOI] [PubMed] [Google Scholar]
- Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, Wen Y, Xiao K, Eldridge A, Chromy BA, Li S, Spitz DR, Lam KS, Wicha MS, Li JJ. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res. 2012;18:6634–6647. doi: 10.1158/1078-0432.CCR-12-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Schaffhauser B, Beug H, Foisner R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J Cell Biol. 2000;148:173–188. doi: 10.1083/jcb.148.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanelli MA, Montt-Guevara M, Diblasi AM, Gago FE, Tello O, Cuello-Carrion FD, Callegari E, Bausero MA, Ciocca DR. P-cadherin and beta-catenin are useful prognostic markers in breast cancer patients; beta-catenin interacts with heat shock protein Hsp27. Cell Stress Chaperones. 2008;13:207–220. doi: 10.1007/s12192-007-0007-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhang X, Zhu H, Wang X, Ni S, Huang J. FoxQ1 overexpression influences poor prognosis in non-small cell lung cancer, associates with the phenomenon of EMT. PLoS One. 2012;7:e39937. doi: 10.1371/journal.pone.0039937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975;35:218–224. [PubMed] [Google Scholar]
- Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978;38:2651–2660. [PubMed] [Google Scholar]
- Findlay VJ, Wang C, Watson DK, Camp ER. Epithelial-to-mesenchymal transition and the cancer stem cell phenotype: insights from cancer biology with therapeutic implications for colorectal cancer. Cancer Gene Ther. 2014;21:181–187. doi: 10.1038/cgt.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Pathol. 2011;92:143–150. doi: 10.1111/j.1365-2613.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori H, Shikanai M, Teraoka H, Masutani M, Yoshioka K. Induction of cancerous stem cells during embryonic stem cell differentiation. J Biol Chem. 2012;287:36777–36791. doi: 10.1074/jbc.M112.372557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst. 2008;100:232–233. 239. doi: 10.1093/jnci/djn032. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Moreno O, Lecanda J, Green JE, Segura V, Catena R, Serrano D, Calvo A. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Exp Cell Res. 2010;316:554–567. doi: 10.1016/j.yexcr.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Grabias BM, Konstantopoulos K. Epithelial-mesenchymal transition and fibrosis are mutually exclusive reponses in shear-activated proximal tubular epithelial cells. FASEB J. 2012;26:4131–4141. doi: 10.1096/fj.12-207324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008a;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008b;7:3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40:1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, Mendoza A, Khanna C, Van Winkle T, Avadhani NG. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene. 2013 doi: 10.1038/onc.2013.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, Tam WL, Mani SA, van Oudenaarden A, Weinberg RA. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, He X, Weigman V, Fan C, Palazzo J, Hortobagyi GN, Nolden LK, Wang NJ, Valero V, Gray JW, Perou CM, Mills GB. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–4124. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–4833. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, Miura N, Rosen JM, Mani SA. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73:1981–1992. doi: 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz B, Visekruna A, Buhr HJ, Hotz HG. Beyond epithelial to mesenchymal transition: a novel role for the transcription factor Snail in inflammation and wound healing. J Gastrointest Surg. 2010;14:388–397. doi: 10.1007/s11605-009-1068-3. [DOI] [PubMed] [Google Scholar]
- Huang SG, Zhang LL, Niu Q, Xiang GM, Liu LL, Jiang DN, Liu F, Li Y, Pu X. Hypoxia promotes epithelial--mesenchymal transition of hepatocellular carcinoma cells via inducing GLIPR-2 expression. PLoS One. 2013;8:e77497. doi: 10.1371/journal.pone.0077497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004a;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Beug H, Wirth T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle. 2004b;3:1477–1480. doi: 10.4161/cc.3.12.1280. [DOI] [PubMed] [Google Scholar]
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC, Hsieh TH, Su FY, Lin LC, Abbondante S, Yang CY, Hsu HM, Yu JC, Chang KJ, Shew JY, Lee EY, Lee WH. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A. 2013;110:12331–12336. doi: 10.1073/pnas.1222684110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Hijioka H, Kume K, Miyawaki A, Nakamura N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol Lett. 2013;6:1201–1206. doi: 10.3892/ol.2013.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–3845. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson KA, Grapin-Botton A. Development and diseases of the pancreas. Clin Genet. 2002;62:14–23. doi: 10.1034/j.1399-0004.2002.620102.x. [DOI] [PubMed] [Google Scholar]
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- Jung JW, Hwang SY, Hwang JS, Oh ES, Park S, Han IO. Ionising radiation induces changes associated with epithelial-mesenchymal transdifferentiation and increased cell motility of A549 lung epithelial cells. Eur J Cancer. 2007;43:1214–1224. doi: 10.1016/j.ejca.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol. 2004;24:7559–7566. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–283. [PubMed] [Google Scholar]
- Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463–476. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011a;195:417–433. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, Alder H, Liu CG, Dejean A, Croce CM. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011b;208:875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Sarkar FH. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27:1712–1721. doi: 10.1002/stem.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh S, Weyler J, Martin MJ, Vermeulen PB, Van Marck E. Angiogenic cytokines in mesothelioma: a study of VEGF, FGF-1 and -2, and TGF beta expression. J Pathol. 1999;189:72–78. doi: 10.1002/(SICI)1096-9896(199909)189:1<72::AID-PATH401>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27:2059–2068. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–207. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005;24:7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61:3894–3901. [PubMed] [Google Scholar]
- Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–436. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhou BP. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. doi: 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009a;15:2657–2665. doi: 10.1158/1078-0432.CCR-08-2372. [DOI] [PubMed] [Google Scholar]
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- Li Y, VandenBoom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009b;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Xia L, Lee LM, Khaletskiy A, Wang J, Wong JY, Li JJ. Effector genes altered in MCF-7 human breast cancer cells after exposure to fractionated ionizing radiation. Radiat Res. 2001;155:543–553. doi: 10.1667/0033-7587(2001)155[0543:egaimh]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang C, Chen Z, Jin Y, Wang Y, Kolokythas A, Dai Y, Zhou X. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem J. 2011;440:23–31. doi: 10.1042/BJ20111006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, He DL, Ning L, Shen SL, Li L, Li X. Hypoxia-inducible factor-1alpha induces the epithelial-mesenchymal transition of human prostatecancer cells. Chin Med J (Engl) 2006;119:713–718. [PubMed] [Google Scholar]
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214–228. doi: 10.1016/j.canlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka J, Yashiro M, Doi Y, Fuyuhiro Y, Kato Y, Shinto O, Noda S, Kashiwagi S, Aomatsu N, Hirakawa T, Hasegawa T, Shimizu K, Shimizu T, Miwa A, Yamada N, Sawada T, Hirakawa K. Hypoxia stimulates the EMT of gastric cancer cells through autocrine TGFbeta signaling. PLoS One. 2013;8:e62310. doi: 10.1371/journal.pone.0062310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19:4875–4887. doi: 10.1091/mbc.E08-05-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Wu G. The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metastasis Rev. 2012;31:455–467. doi: 10.1007/s10555-012-9379-3. [DOI] [PubMed] [Google Scholar]
- Meng Z, Fu X, Chen X, Zeng S, Tian Y, Jove R, Xu R, Huang W. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology. 2010;52:2148–2157. doi: 10.1002/hep.23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–134. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizui M, Isaka Y, Takabatake Y, Sato Y, Kawachi H, Shimizu F, Takahara S, Ito T, Imai E. Transcription factor Ets-1 is essential for mesangial matrix remodeling. Kidney Int. 2006;70:298–305. doi: 10.1038/sj.ki.5001541. [DOI] [PubMed] [Google Scholar]
- Moncharmont C, Levy A, Guy JB, Falk AT, Guilbert M, Trone JC, Alphonse G, Gilormini M, Ardail D, Toillon RA, Rodriguez-Lafrasse C, Magne N. Radiation-enhanced cell migration/invasion process: A review. Crit Rev Oncol Hematol. 2014 doi: 10.1016/j.critrevonc.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muta H, Noguchi M, Kanai Y, Ochiai A, Nawata H, Hirohashi S. E-cadherin gene mutations in signet ring cell carcinoma of the stomach. Jpn J Cancer Res. 1996;87:843–848. doi: 10.1111/j.1349-7006.1996.tb02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25:593–600. doi: 10.1007/s10585-008-9143-9. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Baldwin AS., Jr NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–852. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011;71:3076–3086. doi: 10.1158/0008-5472.CAN-10-2787. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann E, Schwarz H, Deiner EM, Leitner I, Eilers M, Berger J, Busslinger M, Beug H. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–1116. doi: 10.1016/s0092-8674(05)80060-1. [DOI] [PubMed] [Google Scholar]
- Reiman JM, Knutson KL, Radisky DC. Immune promotion of epithelial-mesenchymal transition and generation of breast cancer stem cells. Cancer Res. 2010;70:3005–3008. doi: 10.1158/0008-5472.CAN-09-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Schaefer TS. ErbB-2 activates Stat3 alpha in a Src- and JAK2-dependent manner. J Biol Chem. 2002;277:38486–38493. doi: 10.1074/jbc.M112438200. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, Kim CH, Lee JC. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009;63:219–226. doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodemann HP, Blaese MA. Responses of normal cells to ionizing radiation. Semin Radiat Oncol. 2007;17:81–88. doi: 10.1016/j.semradonc.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Saito A, Kanai Y, Maesawa C, Ochiai A, Torii A, Hirohashi S. Disruption of E-cadherin-mediated cell adhesion systems in gastric cancers in young patients. Jpn J Cancer Res. 1999;90:993–999. doi: 10.1111/j.1349-7006.1999.tb00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011;2:e179. doi: 10.1038/cddis.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Sehrawat A, Kim SH, Vogt A, Singh SV. Suppression of FOXQ1 in benzyl isothiocyanate-mediated inhibition of epithelial-mesenchymal transition in human breast cancer cells. Carcinogenesis. 2012;34:864–873. doi: 10.1093/carcin/bgs397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6–22. [PubMed] [Google Scholar]
- Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrodder H, Jensen T, Kassem M. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004;23:5095–5098. doi: 10.1038/sj.onc.1207651. [DOI] [PubMed] [Google Scholar]
- Shankar S, Nall D, Tang SN, Meeker D, Passarini J, Sharma J, Srivastava RK. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PLoS One. 2011;6:e16530. doi: 10.1371/journal.pone.0016530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, Matsumoto T, Matsuura N, Ohta M, Okumura M. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann Thorac Surg. 2011;92:1794–1804. doi: 10.1016/j.athoracsur.2011.07.032. discussion 1804. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsova I, Skvortsov S, Raju U, Stasyk T, Riesterer O, Schottdorf EM, Popper BA, Schiestl B, Eichberger P, Debbage P, Neher A, Bonn GK, Huber LA, Milas L, Lukas P. Epithelial-to-mesenchymal transition and c-myc expression are the determinants of cetuximab-induced enhancement of squamous cell carcinoma radioresponse. Radiother Oncol. 2010;96:108–115. doi: 10.1016/j.radonc.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Graham CH. Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev. 2007;26:319–331. doi: 10.1007/s10555-007-9062-2. [DOI] [PubMed] [Google Scholar]
- Tang SN, Singh C, Nall D, Meeker D, Shankar S, Srivastava RK. The dietary bioflavonoid quercetin synergizes with epigallocathechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J Mol Signal. 2010;5:14. doi: 10.1186/1750-2187-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimizu N, Miyajima A. Molecular mechanism of liver development and regeneration. Int Rev Cytol. 2007;259:1–48. doi: 10.1016/S0074-7696(06)59001-1. [DOI] [PubMed] [Google Scholar]
- Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. discussion 6000-5991. [DOI] [PubMed] [Google Scholar]
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–1036. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theys J, Jutten B, Habets R, Paesmans K, Groot AJ, Lambin P, Wouters BG, Lammering G, Vooijs M. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiother Oncol. 2011;99:392–397. doi: 10.1016/j.radonc.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5995. doi: 10.1158/0008-5472.CAN-05-0616. discussion 5995. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135–7139. doi: 10.1158/0008-5472.CAN-09-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression? Nat Clin Pract Oncol. 2008;5:280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikado Y, Okumura H, Ishigami S, Setoyama T, Matsumoto M, Owaki T, Kita Y, Natsugoe S. Increased Slug and decreased E-cadherin expression is related to poor prognosis in patients with gastric cancer. Gastric Cancer. 2011;14:41–49. doi: 10.1007/s10120-011-0004-x. [DOI] [PubMed] [Google Scholar]
- Wang N, Eckert KA, Zomorrodi AR, Xin P, Pan W, Shearer DA, Weisz J, Maranus CD, Clawson GA. Down-regulation of HtrA1 activates the epithelial-mesenchymal transition and ATM DNA damage response pathways. PLoS One. 2012;7:e39446. doi: 10.1371/journal.pone.0039446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, Sarkar FH. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13:109–118. doi: 10.1016/j.drup.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- Wijnhoven BP, Dinjens WN, Pignatelli M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br J Surg. 2000;87:992–1005. doi: 10.1046/j.1365-2168.2000.01513.x. [DOI] [PubMed] [Google Scholar]
- Wu PH, Giri A, Sun SX, Wirtz D. Three-dimensional cell migration does not follow a random walk. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1318967111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Li K, Ouyang X. Twist, an independent prognostic marker for predicting distant metastasis and survival rates of esophageal squamous cell carcinoma patients. Clin Exp Metastasis. 2009;26:1025–1032. doi: 10.1007/s10585-009-9292-5. [DOI] [PubMed] [Google Scholar]
- Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Fu Y, Tian D, Liao J, Liu M, Wang B, Xia L, Zhu Q, Luo M. PI3 kinase/Akt signaling mediates epithelial-mesenchymal transition in hypoxic hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009;382:631–636. doi: 10.1016/j.bbrc.2009.03.088. [DOI] [PubMed] [Google Scholar]
- Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ, Ellis LM. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006a;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006b;12:4147–4153. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96–107. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7:2090–2096. doi: 10.4161/cc.7.14.6324. [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- Yoon YS, Lee JH, Hwang SC, Choi KS, Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- Yu L, Mu Y, Sa N, Wang H, Xu W. Tumor necrosis factor alpha induces epithelial-mesenchymal transition and promotes metastasis via NF-kappaB signaling pathway-mediated TWIST expression in hypopharyngeal cancer. Oncol Rep. 2014;31:321–327. doi: 10.3892/or.2013.2841. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007a;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007b;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, Oettgen P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest. 2005;115:2508–2516. doi: 10.1172/JCI24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Jia Z, Guo X, Yang T. Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am J Physiol Renal Physiol. 2007;293:F723–731. doi: 10.1152/ajprenal.00480.2006. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1alpha in hepatocellular carcinoma. BMC Cancer. 2013a;13:108. doi: 10.1186/1471-2407-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, Liang T. Wnt/beta-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1alpha signaling. Carcinogenesis. 2013b;34:962–973. doi: 10.1093/carcin/bgt027. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li X, Zhang N, Yang Q, Moran MS. Low doses ionizing radiation enhances the invasiveness of breast cancer cells by inducing epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2011;412:188–192. doi: 10.1016/j.bbrc.2011.07.074. [DOI] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1120–1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, Qiu LB, Ding GR, Su XM, Mei S, Guo GZ. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys. 2011;81:1530–1537. doi: 10.1016/j.ijrobp.2011.06.1956. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Pang Z, Xing Y, Wan F, Lan D, Wang H. Short hairpin RNA targeting FOXQ1 inhibits invasion and metastasis via the reversal of epithelial-mesenchymal transition in bladder cancer. Int J Oncol. 2013;42:1271–1278. doi: 10.3892/ijo.2013.1807. [DOI] [PubMed] [Google Scholar]