

Abstract
Clostrubin is a potent antibiotic against methicillin- and vancomycin-resistant bacteria that was isolated from a strictly anaerobic bacterium Clostridium beijerinckii in 2014. This polyphenol possesses a fully substituted arene moiety on its pentacyclic scaffold, which poses a considerable challenge for chemical synthesis. Here we report the first total synthesis of clostrubin in nine steps (the longest linear sequence). A desymmetrization strategy is exploited based on the inherent structural feature of the natural product. Barton–Kellogg olefination forges the two segments together to form a tetrasubstituted alkene. A photo-induced 6π electrocyclization followed by spontaneous aromatization constructs the hexasubstituted B ring at a late stage. In total, 200 mg of clostrubin are delivered through this approach.
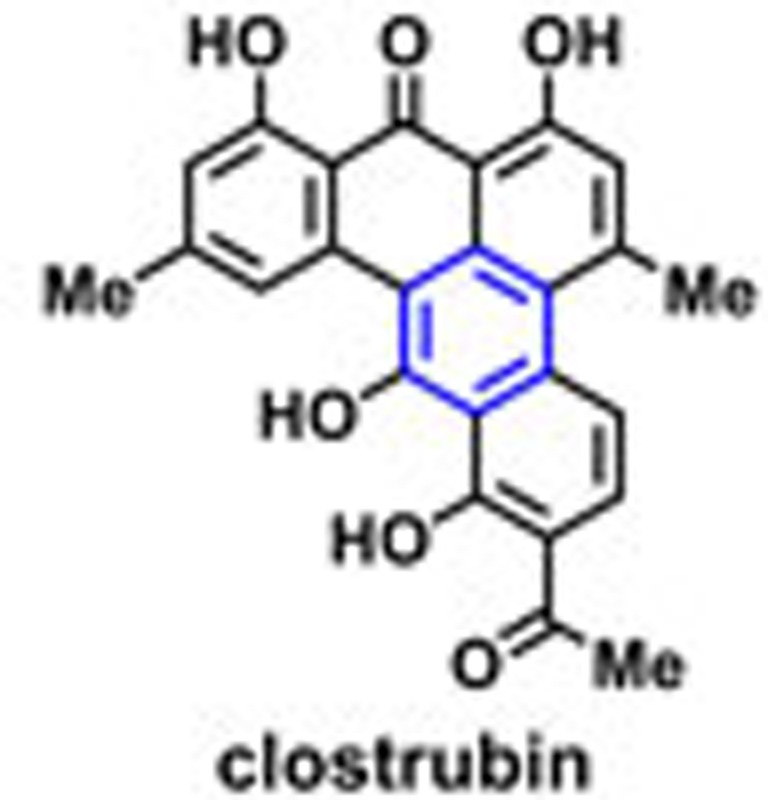 Due to rising resistance, efficient routes to new antibiotics is a vital task for human health. Here, the authors report a short, convergent and elegant synthesis of a very recently reported antibiotic, successfully giving access to this material on scale.
Due to rising resistance, efficient routes to new antibiotics is a vital task for human health. Here, the authors report a short, convergent and elegant synthesis of a very recently reported antibiotic, successfully giving access to this material on scale.
The discovery of effective antibiotic agents is an urgent global demand for combating drug-resistant pathogenic bacterium strains1,2. Secondary metabolites that are produced by microbes as chemical defence have proven to be the most important source of such agents3,4,5,6,7,8,9,10,11,12,13,14. In May 2014, Hertweck and co-workers15 reported the isolation of clostrubin (1, Fig. 1) from the strictly anaerobic bacterium C. beijerinckii. This compound exhibits remarkable potency against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterocci, with minimum inhibitory concentrations of 0.12 and 0.97 μM, respectively. From a structural perspective, clostrubin (1) poses considerable synthetic challenge owing to the fused aromatic ring system and multisubstitution pattern. The potential of 1 as a lead compound for antibiotic development and its limited supply from natural sources stimulated us to launch a chemical synthesis programme immediately.
Figure 1. Retrosynthetic analysis of clostrubin.

The inherent structural symmetry of the molecule inspires a desymmetrization strategy. A Barton–Kellogg olefination would be used for assembling a sterically hindered tetrasubstituted olefin. A 6π electrocyclization/aromatization sequence is envisioned as the key step for the construction of the hexasubstituted aromatic B ring.
In this paper, we report the first total synthesis of clostrubin (1) from commercially available starting materials. This concise synthesis (nine-step for the longest linear sequence) benefits from the inherent structural symmetry of 1. An advanced olefin intermediate was constructed through Barton–Kellogg olefination, and a 6π electrocyclization promoted by ultraviolet light assembled the fully substituted B ring.
Results
Retrosynthetic analysis
We undertook a retrosynthetic analysis of clostrubin (1) taking advantage of the inherent symmetry16 of its molecular architecture, as illustrated in Fig. 1. The initial disassembly of the fully substituted B ring leads to a precursor 2; the recombination of the sterically hindered C8–C9 biaryl bond could rely on a 6π electrocyclization reaction. Electrocyclization has emerged as a powerful tool for the construction of fused ring systems since Nicolaou’s pioneering synthesis of endiandric acids17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32. In particular, the strategy of 6π electrocyclization/aromatization has demonstrated a significant advantage in the synthesis of multisubstituted arenes33,34,35,36,37,38,39,40,41,42,43,44. Recently, we exploited such a strategy in the total syntheses of a series of natural products, such as tubingensin A, daphenylline, xiamycin and oridamycin families, and rubriflordilactone A45,46,47,48. It should be mentioned that the construction of hexasubstituted arenes using such strategy is rare in natural product synthesis. Preparing the symmetrical, tetrasubstituted olefin 2 is indeed of significant challenge with the conventional olefination methods, and Barton–Kellogg reaction is envisioned as a suitable solution49,50,51,52,53,54. Interestingly, this olefination method has found remarkable applications in material science55,56,57 rather than natural product synthesis in recent years. Disconnection at the C15–C16 bond results in stabilized diazo compound 3 and thioester 4. The former may arise from the corresponding anthraquinone precursor, which is further traced back to 2,6-dibromo-1,4-benzoquinone (ref. 58) 5 and Brassard diene (refs 59, 60) 6 in a double retro-Diels–Alder (D-A) manner. The latter would be accessed from commercially available iodide 7 by using lithium chemistry.
Synthesis of the two segments
The synthesis commenced with the preparation of diazoketone 3, as shown in Fig. 2. A sequence of double D-A reactions was carried out: 2,6-dibromo-1,4-benzoquinone 5, prepared in one step from commercially available 1,3,5-tribromophenol58, reacted smoothly with an excess of diene 8 to afford anthraquinone 9 along with a small amount of mono-O-methyl-9, presumably via the intermediacy of 10. The regioselectivity of the D-A reactions may be attributable to the electronic bias induced by the bromine atom. The addition of silica gel may accelerate the hydrolysis of the silyl ether and thus led to a rapid aromatization along with release of HBr. Notably, when a single equivalent of 8 was used for the D-A reaction, a bromonaphthoquinone was readily prepared, presumably with the intermediacy of a mono cycloadduct61. The crude 9 was treated with K2CO3 and MeI to give compound 11 (45% overall yield from 5). 11 underwent Clemmensen-type reduction in the presence of SnCl2 and HCl to give the corresponding monoketone62, which instantaneously tautomerized to anthranol 12. Exposure of crude 12 to 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and TsN3 furnished diazoketone 3 in 89% overall yield.
Figure 2. Synthesis of the anthraquinone segment.

Reagents and conditions: (a) tetrahydrofuran (THF), −78 °C, 6 h; then silica gel, 22 °C, 2 h. (b) K2CO3 (3.0 eq), MeI (2.5 eq), N,N-dimethylformamide (DMF), 22 °C, 18 h, 45% (two steps). (c) SnCl2·H2O (6.0 eq), aqueous HCl (37 wt%), AcOH, 22 °C, 30 min. (d) DBU (3.0 eq), TsN3 (1.1 eq), methylene chloride (CH2Cl2), 22 °C, 20 h, 89% (two steps). TMS, trimethylsilyl.
We observed unexpected reactivity of anthraquinone 11 (Fig. 3) during the above studies, which influenced the overall strategy of the synthesis. In theory, the C9 carbonyl of 11 should be sterically more hindered for nucleophilic attack due to two neighbouring methoxy groups. From an electronic perspective, this upper ketone could be considered as an equivalent of a double vinylogous carbonate than is also rather unreactive as an electrophile. To our surprise, we obtained hydrazone 13 in 61% yield when treating 11 with TsNHNH2; the anticipated regioisomer was not detected. The structure of 13 was determined by X-ray crystallographic analysis. This observation interrupted our initial plan of exploiting the C10 tosylhydrazone of 11 as the potential precursor for the Barton–Kellogg olefination. We further examined other types of nucleophiles such as benzyl Grignard reagent for the addition reaction with 11. In this case, two regioisomeric alcohols 14 and 15 were isolated in 32% and 40% yields, respectively. Both structures were confirmed by nuclear Overhauser effect (NOE) studies. The enhanced reactivity of the C9 carbonyl may be attributable to inductive effects from the o-methoxy substitutents or relief of 1,3-allylic strain that occurs on nucleophilic additions. Thus, the strategy involving direct olefination of C10 carbonyl of anthraquinone 11 (for example, with functionalized benzylic metal species or phosphonate carbanion) had to be abandoned due to the poor regioselectivity.
Figure 3. Unusual reactivity of the anthraquinone intermediate.

Reagents and conditions: (a) TsNHNH2 (1.0 eq), TsOH·H2O (10 mol%), EtOH, 60 °C, 2 h, 61%. (b) BnMgBr (1.0 eq), tetrahydrofuran (THF), 22 °C, 15 min, 32% for 14 and 40% for 15. ORTEP, Oak Ridge Thermal Ellipsoid Plot.
We then focused on the synthesis of the thioester segment as the electron donor in the devised Barton–Kellogg olefination, as shown in Fig. 4. Aldehyde 16 was prepared in one step from commercially available 2-iodophenol63. Treatment with K2CO3 and MeI gave methyl ether 17 (99% yield), which underwent MeMgBr addition followed by silylation to provide compound 18 (94% yield) in one pot. Hexamethylphosphoramide (HMPA) was found to be crucial to enhance the nucleophilicity of the magnesium alkoxide. 18 was subjected to the magnesium–halogen exchange conditions (EtMgBr) to generate a functionalized Grignard reagent45,47,64,65,66, which was quenched by CS2 and MeI to give dithioester 19. It is noteworthy that lithium–halogen exchange did not lead to a satisfactory result. The desilylation took place spontaneously during acid workup to deliver alcohol 20 in 67% yield from 18. Oxidation of 20 with Dess–Martin periodinane (DMP) afforded ketone 21 (83% yield) without destroying the sulfur-containing functionalities, and the subsequent methanolysis furnished thioester 4 in 66% yield.
Figure 4. Synthesis of the thioester segment.

Reagents and conditions: (a) K2CO3 (1.5 eq), MeI (1.5 eq), N,N-dimethylformamide (DMF), 22 °C, 20 h, 99%. (b) MeMgBr (1.05 eq), 0 °C, 10 min; then HMPA (2.0 eq), triethylamine (Et3N; 1.5 eq), chlorotrimethylsilane (TMSCl; 1.5 eq), tetrahydrofuran (THF), 22 °C, 10 min, 94%. (c) EtMgBr (1.5 eq), THF, 50 °C, 1 h; then CS2 (30 eq), 70 °C, 3 h; then MeI (4.0 eq), 22 °C, 8 h, 67%. (d) DMP (1.2 eq), methylene chloride (CH2Cl2), 22 °C, 1 min, 83%. (e) MeONa (10.0 eq), methanol (MeOH), 50 °C, 4 h, 66%.
Completion of the synthesis
With both fragments in hand, we directed our attention to the construction of the aromatic B ring, as depicted in Fig. 5. It is well documented in the literature of Barton–Kellogg olefination that thioketones readily react with diazo compounds without promoters or catalysts55,56,57. After examination of the conventional conditions, we realized that the stabilized diazoketone 3 needed to be activated by Rh2(OAc)4 to form the metal-carbenoid intermediate52,54,67,68,69,70, which was further trapped by relatively unreactive thioester 4. The postulated episulfide intermediate 22 was reduced by Cu powder in situ to afford tetrasubstituted olefin 2 in 85% overall yield. We examined a series of conditions such as heating or FeCl3 to promote the last C–C bond formation but only observed decomposition. Inspired by our synthesis of daphenylline46, we irradiated 2 with ultraviolet light (λ=365 nm). To our delight, this symmetrical olefin underwent a 6π electrocyclization, presumably to provide pentacyclic intermediate 23, which was spontaneously oxidized under an air atmosphere during workup to furnish tetramethyl clostrubin 24 (55% yield from 2). Global deprotection of the methyl groups with aqueous HBr (48 wt%) gave clostrubin (1) with excellent efficiency. The spectra and physical properties of the synthetic 1 are consistent with those reported for the natural product. In total, 200 mg of 1 were obtained through the synthesis.
Figure 5. Completion of the synthesis of clostrubin.

Reagents and conditions: (a) Rh2(OAc)4 (5 mol%), toluene, 50 °C, 55 min; then Cu powder, 110 °C, 1.5 h, 85%. (b) ultraviolet light (λ=365 nm, Hg lamp), CHCl3, 22 °C, 8 h, 55%. (c) aq. HBr (48 wt%), AcOH, 120 °C, 10 h, 95%.
Discussion
In summary, we have accomplished the first total synthesis of clostrubin. The concise and efficient route took advantage of the 6π electrocyclization strategy as well as the inherent structural symmetry of the molecule. The synthesis provides a practical means to obtain this potent antibiotic for further investigations, considering the limited supply and difficult isolation of the naturally occurring sample.
Methods
General
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Tetrahydrofuran was distilled immediately before use from sodium-benzophenone ketyl. Methylene chloride, N,N-dimethylformamide, triethylamine, N,N-diisopropylethylamine and chlorotrimethylsilane were distilled from calcium hydride and stored under an argon atmosphere. Methanol was distilled from magnesium and stored under an argon atmosphere. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Solvents for chromatography were used as supplied by Titan Chemical. Reactions were monitored by thin-layer chromatography carried out on S-2 0.25 mm E. Merck silica gel plates (60F-254) using ultraviolet light as visualizing agent and aqueous ammonium cerium nitrate/ammonium molybdate or basic aqueous potassium permanganate as developing agent. E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. Preparative thin-layer chromatography separations were carried out on 0.25 or 0.50 mm E. Merck silica gel plates (60F-254). Nuclear magnetic resonance (NMR) spectra were recorded on Bruker AV-400 or Agilent 500/54/ASP instrument and calibrated by using residual undeuterated chloroform (δH=7.26 p.p.m.) and CDCl3 (δC=77.16 p.p.m.) or undeuterated dimethylsulfoxide (δH=2.50 p.p.m.) and dimethylsulfoxide-d6 (δC=39.52 p.p.m.), as internal references. Infrared spectra were recorded on a Thermo Scientific Nicolet 380 FT-IR spectrometer. Melting points are uncorrected and were recorded on a Shanghai Jingke SGW X-4 apparatus. High-resolution mass spectra were recorded on a Bruker APEXIII 7.0 Tesla ESI-FT or a Waters Micromass GCT Premier EI mass spectrometer.
For 1H and 13C NMR spectra of compounds, see Supplementary Figs 1–26. For heteronuclear multiple quantum correlation spectroscopy (HMQC) and heteronuclear multiple-bond correlation spectroscopy (HMBC) spectra of compound 13, see Supplementary Figs 27 and 28. For nuclear Overhauser effect spectroscopy (NOESY) spectra of compound 14 and 15, see Supplementary Figs 29 and 30. For the comparisons of 1H and 13C NMR spectra of the natural and synthetic clostrubin, see Supplementary Figs 31 and 32. For the comparisons of 1H and 13C NMR spectroscopic data of the natural and synthetic clostrubin, see Supplementary Tables 1 and 2. For the experimental procedures and spectroscopic and physical data of compounds and the crystallographic data of compound 13, see Supplementary Methods.
Author contributions
A.L., M.Y. and J.L. conceived the synthetic route; A.L. directed the project; M.Y. and J.L. conducted the work; A.L., M.Y. and J.L. analysed the results; and A.L. wrote the manuscript.
Additional information
Accession codes: The X-ray crystallographic coordinates for structure (13) reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1028256. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
How to cite this article: Yang, M. et al. Total synthesis of clostrubin. Nat. Commun. 6:6445 doi: 10.1038/ncomms7445 (2015).
Supplementary Material
Supplementary Figures 1-32, Supplementary Table 1-2, Supplementary Methods, Supplementary References
Acknowledgments
This paper is dedicated to Professor Yong-Qiang Tu for his mentorship to M.Y and J.L. We thank Professor Christian Hertweck for helpful discussions. Financial support was provided by Ministry of Science and Technology (2013CB836900), National Natural Science Foundation of China (21290180, 21172235 and 21222202), Pujiang Program (12PJ1410800) and China Postdoctoral Science Foundation (2014M551480, to M.Y.).
References
- Pearson H. ‘Superbug’ hurdles key drug barrier. Nature 418, 469–469 (2002) . [DOI] [PubMed] [Google Scholar]
- Laxminarayan R. et al. Antibiotic resistance—the need for global solutions. Lancet Infect. Dis. 13, 1057–1058 (2013) . [DOI] [PubMed] [Google Scholar]
- Clardy J., Fischbach M. A. & Walsh C. T. New antibiotics from bacterial natural products. Nat. Biotechnol. 24, 1541–1550 (2006) . [DOI] [PubMed] [Google Scholar]
- Fischbach M. A. & Walsh C. T. Antibiotics for emerging pathogens. Science 325, 1089–1093 (2009) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häbich D. & von Nussbaum F. Platensimycin, a new antibiotic and “superbug challenger” from nature. ChemMedChem 1, 951–954 (2006) . [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C., Li A. & Edmonds D. J. Total synthesis of platensimycin. Angew. Chem. Int. Ed. 45, 7086–7090 (2006) . [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C., Tria G. S. & Edmonds D. J. Total synthesis of platencin. Angew. Chem. Int. Ed. 47, 1780–1783 (2008) . [DOI] [PubMed] [Google Scholar]
- Tiefenbacher K. & Mulzer J. Synthesis of platensimycin. Angew. Chem. Int. Ed. 47, 2548–2555 (2008) . [DOI] [PubMed] [Google Scholar]
- Walker S. et al. Chemistry and biology of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. Chem. Rev. 105, 449–476 (2005) . [DOI] [PubMed] [Google Scholar]
- James R. C., Pierce J. G., Okano A., Xie J. & Boger D. L. Redesign of glycopeptide antibiotics: back to the future. ACS Chem. Biol. 7, 797–804 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Nussbaum F. et al. Structure and total synthesis of lysobactin (katanosin B). Angew. Chem. Int. Ed. 46, 2039–2042 (2007) . [DOI] [PubMed] [Google Scholar]
- Tan Y. X. & Romesberg F. E. Latent antibiotics and the potential of the arylomycins for broad-spectrum antibacterial activity. Med. Chem. Commun. 3, 916–925 (2012) . [Google Scholar]
- Lam H. Y. et al. Total synthesis of daptomycin by cyclization via a chemoselective serine ligation. J. Am. Chem. Soc. 135, 6272–6279 (2013) . [DOI] [PubMed] [Google Scholar]
- King A. M. et al. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 510, 503–506 (2014) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidot S., Ishida K., Cyrulies M. & Hertweck C. Discovery of clostrubin, an exceptional polyphenolic polyketide antibiotic from a strictly anaerobic bacterium. Angew. Chem. Int. Ed. 53, 7856–7859 (2014) . [DOI] [PubMed] [Google Scholar]
- Anderson R. J. & Morris J. C. Total synthesis of variolin B. Tetrahedron Lett. 42, 8697–8699 (2001) . [Google Scholar]
- Nicolaou K. C., Petasis N. A., Zipkin R. E. & Uenishi J. The endiandric acid cascade. Electrocyclizations in organic synthesis. 1. Stepwise, stereocontrolled total synthesis of endiandric acids A and B. J. Am. Chem. Soc. 104, 5555–5557 (1982) . [Google Scholar]
- Nicolaou K. C., Petasis N. A., Uenishi J. & Zipkin R. E. The endiandric acid cascade. Electrocyclizations in organic synthesis. 2. Stepwise, stereocontrolled total synthesis of endiandric acids C–G. J. Am. Chem. Soc. 104, 5557–5558 (1982) . [Google Scholar]
- Nicolaou K. C., Zipkin R. E. & Petasis N. A. The endiandric acid cascade. Electrocyclizations in organic synthesis. 3. "Biomimetic" approach to endiandric acids A–G. Synthesis of precursors. J. Am. Chem. Soc. 104, 5558–5560 (1982) . [Google Scholar]
- Nicolaou K. C., Petasis N. A. & Zipkin R. E. The endiandric acid cascade. Electrocyclizations in organic synthesis. 4. Biomimetic approach to endiandric acids A-G. Total synthesis and thermal studies. J. Am. Chem. Soc. 104, 5560–5562 (1982) . [Google Scholar]
- Beaudry C. M., Malerich J. P. & Trauner D. Biosynthetic and biomimetic electrocyclizations. Chem. Rev. 105, 4757–4778 (2005) . [DOI] [PubMed] [Google Scholar]
- Sklenicka H. M. et al. Stereoselective formal [3+3] cycloaddition approach to cis-1-azadecalins and synthesis of (–)-4a,8a-diepi-pumiliotoxin C. Evidence for the first highly stereoselective 6π-electron electrocyclic ring closures of 1-azatrienes. J. Am. Chem. Soc. 124, 10435–10442 (2002) . [DOI] [PubMed] [Google Scholar]
- Li C., Johnson R. P. & Porco J. A. Total synthesis of the quinone epoxide dimer (+)-torreyanic acid: application of a biomimetic oxidation/electrocyclization/Diels-Alder dimerization cascade. J. Am. Chem. Soc. 125, 5095–5106 (2003) . [DOI] [PubMed] [Google Scholar]
- Parker K. A. & Lim Y.-H. The Total synthesis of (–)-SNF4435 C and (+)-SNF4435 D. J. Am. Chem. Soc. 126, 15968–15969 (2004) . [DOI] [PubMed] [Google Scholar]
- Miller A. K. & Trauner D. Mining the tetraene manifold: total synthesis of complex pyrones from Placobranchus ocellatus. Angew. Chem. Int. Ed. 44, 4602–4606 (2005) . [DOI] [PubMed] [Google Scholar]
- Volgraf M. et al. Biomimetic synthesis of the IDO inhibitors exiguamine A and B. Nat. Chem. Biol. 4, 535–537 (2008) . [DOI] [PubMed] [Google Scholar]
- Chaumontet M., Piccardi R. & Baudoin O. Synthesis of 3,4-dihydroisoquinolines by a C(sp3)-H activation/electrocyclization strategy: total synthesis of coralydine. Angew. Chem. Int. Ed. 48, 179–182 (2009) . [DOI] [PubMed] [Google Scholar]
- Sloman D. L., Bacon J. W. & Porco J. A. Total synthesis and absolute stereochemical assignment of kibdelone C. J. Am. Chem. Soc. 133, 9952–9955 (2011) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Ritson D. J., Burnley J. & Moses J. E. A synthetic approach to kingianin A based on biosynthetic speculation. Chem. Commun. 47, 10605–10607 (2011) . [DOI] [PubMed] [Google Scholar]
- Barcan G. A., Patel A., Houk K. N. & Kwon O. A torquoselective 6π electrocyclization approach to reserpine alkaloids. Org. Lett. 14, 5388–5391 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew S. L., Lawrence A. L. & Sherburn M. S. Total synthesis of kingianins A, D, and F. Angew. Chem. Int. Ed. 52, 4221–4224 (2013) . [DOI] [PubMed] [Google Scholar]
- Lim H. N. & Parker K. A. Total synthesis of kingianin A. Org. Lett. 15, 398–401 (2013) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. A., Alexanian E. J. & Sorensen E. J. Synthesis of the furanosteroidal antibiotic viridin. Angew. Chem. Int. Ed. 43, 1998–2001 (2004) . [DOI] [PubMed] [Google Scholar]
- Ohmori K. et al. Concise total synthesis and structure assignment of TAN-1085. Angew. Chem. Int. Ed. 43, 3167–3171 (2004) . [DOI] [PubMed] [Google Scholar]
- Harrowven D. C., Pascoe D. D., Demurtas D. & Bourne H. O. Total synthesis of (–)-colombiasin A and (–)-elisapterosin B. Angew. Chem. Int. Ed. 44, 1221–1222 (2005) . [DOI] [PubMed] [Google Scholar]
- Fürstner A., Domostoj M. M. & Scheiper B. Total synthesis of dictyodendrin B. J. Am. Chem. Soc. 127, 11620–11621 (2005) . [DOI] [PubMed] [Google Scholar]
- Greshock T. J. & Funk R. L. Synthesis of indoles via 6π-electrocyclic ring closures of trienecarbamates. J. Am. Chem. Soc. 128, 4946–4947 (2006) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greshock T. J. & Funk R. L. An approach to the total synthesis of welwistatin. Org. Lett. 8, 2643–2645 (2006) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley R. J. & Funk R. L. Total syntheses of (±)-cis-trikentrin A and (±)-cis-trikentrin B via electrocyclic ring closures of 2,3-divinylpyrrolines. Org. Lett. 8, 3403–3406 (2006) . [DOI] [PubMed] [Google Scholar]
- Huntley R. J. & Funk R. L. A Strategy for the total synthesis of dragmacidin E. Construction of the core ring system. Org. Lett. 8, 4775–4778 (2006) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staben S. T. et al. Gold(I)-catalyzed cyclizations of silyl enol ethers: application to the synthesis of (+)-lycopladine A. Angew. Chem. Int. Ed. 45, 5991–5994 (2006) . [DOI] [PubMed] [Google Scholar]
- Sloman D. L., Mitasev B., Scully S. S., Beutler J. A. & Porco J. A. Synthesis and biological evaluation of ABCD ring fragments of the kibdelones. Angew. Chem. Int. Ed. 50, 2511–2515 (2011) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Yang M., Chen P., Xiong X. & Li A. Total synthesis of hapalindole-type natural products. Angew. Chem. Int. Ed. 53, 13840–13844 (2014) . [DOI] [PubMed] [Google Scholar]
- Xiong X. et al. Synthesis of indole terpenoid mimics via a functionality-tolerated Eu(fod)3-catalyzed conjugate addition. Chem. Asian J. doi: 10.1002/asia.201403312. [DOI] [PubMed] [Google Scholar]
- Bian M. et al. Total syntheses of anominine and tubingensin A. J. Am. Chem. Soc. 134, 8078–8081 (2012) . [DOI] [PubMed] [Google Scholar]
- Lu Z., Li Y., Deng J. & Li A. Total synthesis of the Daphniphyllum alkaloid daphenylline. Nat. Chem. 5, 679–684 (2013) . [DOI] [PubMed] [Google Scholar]
- Meng Z. et al. Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nat. Commun. 6, 6096 (2015) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yang P., Yao M., Deng J. & Li A. Total synthesis of rubriflordilactone A. J. Am. Chem. Soc. 136, 16477–16480 (2014) . [DOI] [PubMed] [Google Scholar]
- Staudinger H. & Siegwart J. Einwirkungen von aliphatischen diazoverbindungen auf thioketone. Helv. Chim. Acta 3, 833–840 (1920) . [Google Scholar]
- Barton D. H. R. & Willis B. J. Olefin synthesis by twofold extrusion processes. J. Chem. Soc. D 1225–1226 (1970) . [Google Scholar]
- Kellogg R. M. & Wassenaar S. Thiocarbonyl ylides. An approach to “tetravalent sulfur” compounds. Tetrahedron Lett. 11, 1987–1990 (1970) . [Google Scholar]
- Kim G., Chu-Moyer M. Y. & Danishefsky S. J. The total synthesis of dl-indolizomycin. J. Am. Chem. Soc. 112, 2003–2005 (1990) . [Google Scholar]
- Honda T. et al. A synthesis of (+)-nonactic acid by means of the sulfur-ylide rearrangement. Tetrahedron 48, 79–88 (1992) . [Google Scholar]
- Kim G., Chu-Moyer M. Y., Danishefsky S. J. & Schulte G. K. The total synthesis of indolizomycin. J. Am. Chem. Soc. 115, 30–39 (1993) . [Google Scholar]
- Feringa B. L. In control of motion: from molecular switches to molecular motors. Acc. Chem. Res. 34, 504–513 (2001) . [DOI] [PubMed] [Google Scholar]
- Plunkett K. N. et al. Expeditious synthesis of contorted hexabenzocoronenes. Org. Lett. 11, 2225–2228 (2009) . [DOI] [PubMed] [Google Scholar]
- Zhang X. et al. Synthesis, self-assembly, and charge transporting property of contorted tetrabenzocoronenes. J. Org. Chem. 75, 8069–8077 (2010) . [DOI] [PubMed] [Google Scholar]
- Omura K. Rapid conversion of phenols to p-benzoquinones under acidic conditions with lead dioxide. Synthesis (Mass). 1145–1148 (1998) . [Google Scholar]
- Fleming I., Goldhill J. & Paterson I. γ-Sulphenylation of αβ-unsaturated aldehydes, ketones, and esters: the use of O-silylated dienolates. Tetrahedron Lett. 20, 3205–3208 (1979) . [Google Scholar]
- Savard J. & Brassard P. Regiospecific syntheses of quinones using vinylketene acetals derived from unsaturated esters. Tetrahedron Lett. 20, 4911–4914 (1979) . [Google Scholar]
- Bringmann G. et al. A convergent total synthesis of the michellamines. J. Org. Chem. 63, 1090–1097 (1998) . [Google Scholar]
- Prinz H., Burgemeister T., Wiegrebe W. & Müller K. Syntheses of anthracenones. 2. Preparation of 1,8-dimethoxy-(dimethylanthralin) and 4,5-dihydroxy-9(10H)-anthracenone (isoanthralin): a revision. J. Org. Chem. 61, 2857–2860 (1996) . [DOI] [PubMed] [Google Scholar]
- Brady R. M. et al. Synthesis of conformationally constrained benzoylureas as BH3-mimetics. Org. Biomol. Chem. 10, 5230–5237 (2012) . [DOI] [PubMed] [Google Scholar]
- Tamborski C. & Moore G. J. Synthesis of polyfluoroaromatic magnesium compounds through the exchange reaction. J. Organomet. Chem. 26, 153–156 (1971) . [Google Scholar]
- Furukawa N., Shibutani T. & Fujihara H. Preparation of pyridyl Grignard reagents and cross coupling reactions with sulfoxides bearing azaheterocycles. Tetrahedron Lett. 28, 5845–5848 (1987) . [Google Scholar]
- Knochel P. et al. Highly functionalized organomagnesium reagents prepared through halogen–metal exchange. Angew. Chem. Int. Ed. 42, 4302 (2003) . [DOI] [PubMed] [Google Scholar]
- Takano S., Tomita S., Takahashi M. & Ogasawara K. Condensation of a chiral tetrahydro-2-furanthione with diazocarbonyl compounds. Synthesis (Mass). 1116–1117 (1987) . [Google Scholar]
- Padwa A., Kinder F. R. & Zhi L. Generaton of thiocarbonyl ylides from the rhodium(II)-catalyzed cyclization of diazothiocarbonyl compounds. Synlett 287–288 (1991) . [Google Scholar]
- Hürzeler M., Bernet B., Mäder T. & Vasella A. Glyconothio-O-lactones part II. cycloaddition to dienes, diazomethane, and carbenoids. Helv. Chim. Acta 76, 1779–1801 (1993) . [Google Scholar]
- Mlostón G. & Heimgartner H. Carbenoid reactions of dimethyl diazomalonate with aromatic thioketones and 1,3-thiazole-5(4H)-thiones. Helv. Chim. Acta 79, 1785–1792 (1996) . [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-32, Supplementary Table 1-2, Supplementary Methods, Supplementary References