

Preface
Phosphatidylinositol 3-Kinases (PI3Ks) are critical coordinators of intracellular signaling in response to extracellular stimuli. Hyperactivation of PI3K signaling cascades is one of the most common events in human cancers. In this Review, we discuss recent advances in our knowledge of the roles of distinct PI3K isoforms in normal and oncogenic signaling, the different ways in which PI3K can be upregulated, and the current state and future potential of targeting this pathway in the clinic.
Introduction
Phosphatidylinositol 3-Kinases (PI3Ks) are a family of lipid kinases that integrate signals from growth factors, cytokines, and other environmental cues, translating them into intracellular signals that regulate multiple signaling pathways. These pathways control many physiological functions and cellular processes, including cell proliferation, growth, survival, motility, and metabolism1-3. Activating alterations in PI3K are frequent in a variety of cancers (Table 1; for a fully referenced version see Supplemental Table 1), making this class of enzymes a prime drug target2, 4. Tremendous efforts have been devoted to the development of effective PI3K inhibitors for cancer therapy. Initial PI3K-directed drugs in clinical trials, consisting largely of non-isoform-selective pan-PI3K inhibitors, have not yielded exciting results. However, recent preclinical studies have demonstrated that different PI3K isoforms play divergent roles in cellular signaling and cancer, suggesting that inhibitors targeting individual isoforms may be able to achieve greater therapeutic efficacy. Isoform-selective inhibitors are now emerging in the clinic, and have had promising success. In this Review, we provide an update on what has been learned in recent years about PI3K isoform-specific functions, differences in the modes of PI3K isoform activation, and the progress of isoform-selective inhibitors in preclinical and early clinical studies.
Table 1. Class I PI3K isoform alterations in cancer.
| Alteration Type | Cancer Type | Frequency of Alteration | Sample Size Range |
|---|---|---|---|
| Class IA | |||
|
| |||
| PIK3CA (p110α) | |||
|
| |||
| Mutation | Endometrial | 10.3-53.0% | 29-232 |
| Breast | 7.1-35.5% | 65-507 | |
| Ovarian | 33.0% | 97 | |
| Colorectal | 16.9†-30.6% | 72-195 | |
| Bladder | 5.0-20.0% | 20-130 | |
| Lung | 0.6-20.0% | 5-183 | |
| Cervical | 13.6% | 22 | |
| Glioblastoma | 4.3-11.0% | 91-291 | |
| Head and neck | 8.1-9.4% | 32-74 | |
| Esophageal | 5.5% | 145 | |
| Melanoma | 5.0% | 121 | |
| Prostate | 1.3-3.6% | 55-156 | |
| Sarcoma | 2.9% | 207 | |
| Renal | 1.0-2.9% | 98-417 | |
| Liver | 1.6% | 125 | |
| Megalencephaly‡ | 48.0% | 50 | |
|
| |||
| Copy number gain/amplification | Head and neck | 9.1-100% | 11-117 |
| Cervical | 9.1-76.4% | 22-55 | |
| Lung | 9.5-69.6% | 3-92 | |
| Lymphoma | 16.7-68.2% | 22-60 | |
| Ovarian | 13.3-39.8% | 60-93 | |
| Gastric | 36.4% | 55 | |
| Thyroid | 30.0% | 110 | |
| Prostate | 28.1% | 32 | |
| Breast | 8.7-13.4% | 92-209 | |
| Glioblastoma | 1.9-12.2% | 139-206 | |
| Endometrial | 10.3% | 29 | |
| Thyroid | 9.4% | 128 | |
| Esophageal | 5.7% | 87 | |
| Leukemia | 5.6% | 161 | |
|
| |||
| Increased expression | Prostate | 40.0% | 25 |
|
| |||
| PIK3CB (p110β) | |||
|
| |||
| Mutation | Breast | 0.5% | 183 |
|
| |||
| Copy number gain/amplification | Lung | 56.5% | 46 |
| Thyroid | 42.3% | 97 | |
| Ovarian | 5-26.9% | NA-93 | |
| Lymphoma | 20.0% | 60 | |
| Glioblastoma | 5.8% | 103 | |
| Breast | 4.9-5% | NA-81 | |
|
| |||
| Increased expression | Prostate | 46.7% | 30 |
| Glioblastoma | 3.9% | 103 | |
|
| |||
| PIK3CD (p110δ) | |||
|
| |||
| Copy number gain | Glioblastoma | 40.0% | 10 |
|
| |||
| Increased expression | Neuroblastoma | 52.6% | 19 |
| Glioblastoma | 5.8% | 103 | |
|
| |||
| PIK3R1 (p85α, p55α, p50α) | |||
|
| |||
| Mutation | Endometrial | 19.8-32.8% | 108-243 |
| Pancreatic | 16.7% | 6 | |
| Glioblastoma | 7.6-11.3% | 91-291 | |
| Colorectal | 4.6†-8.3% | 108-195 | |
| Melanoma | 4.4% | 68 | |
| Ovarian | 3.8% | 80 | |
| Esophageal | 3.4% | 145 | |
| Breast | 1.1-2.8% | 62-507 | |
| Colon | 1.7% | 60 | |
|
| |||
| Decreased expression | Breast | 61.8% | 458 |
| Prostate | 17-75%* | NA | |
| Lung | 19-46%* | NA | |
| Ovarian | 22%* | NA | |
| Breast | 18%* | NA | |
| Bladder | 18%* | NA | |
|
| |||
| Copy number loss | Ovarian | 21.5% | 93 |
|
| |||
| PIK3R2 (p85β) | |||
|
| |||
| Mutation | Endometrial | 4.9% | 243 |
| Colorectal | 0.9% | 108 | |
| Megalencephaly‡ | 22.0% | 50 | |
|
| |||
| Amplification | Lymphoma | 23.3% | 60 |
|
| |||
| Increased expression | Colon | 55.0% | 20 |
| Breast | 45.7% | 35 | |
|
| |||
| PIK3R3 (p55γ) | |||
|
| |||
| Copy number gain | Ovarian | 15.0% | 93 |
|
| |||
| Class IB | |||
|
| |||
| PIK3CG (p110γ) | |||
|
| |||
| Copy number gain | Ovarian | 19.3% | 93 |
|
| |||
| Increased expression | Breast | 77.5% | 40 |
| Prostate | 72.4% | 29 | |
| Medulloblastoma | 52.9% | 17 | |
|
| |||
| PIK3R5 (p101) | |||
|
| |||
| Mutation | Melanoma | 38.2% | 68 |
| Gastric | 2.7% | 37 | |
For further detail and references, see the expanded version of this table online (Supplemental Table 1).
Megalencephaly syndromes are a collection of sporadic overgrowth disorders characterized by enlarged brain size and other distinct features.
Combined number of hypermutated and non-hypermutated colon and colorectal patient samples with mutations in the indicated gene.
Represents the percent reduction in gene expression.
NA Sample size not available for this study.
Multiple PI3K classes and isoforms
PI3Ks phosphorylate the 3′;-hydroxyl group of phosphatidylinositides (PtdIns). They are divided into three classes based on their structures and substrate specificities (Figure 1). In mammals, class I PI3Ks are further divided into subclasses IA and IB based on their modes of regulation. Class IA PI3Ks are heterodimers of a p110 catalytic subunit and a p85 regulatory subunit. The genes PIK3CA, PIK3CB, and PIK3CD respectively encode three highly homologous class IA catalytic isoforms, p110α, p110β, and p110δ. These isoforms associate with any of five regulatory isoforms, p85α (and its splicing variants p55α and p50α, encoded by PIK3R1), p85β (PIK3R2), and p55γ (PIK3R3), collectively called p85 type regulatory subunits (reviewed in 1, 5). Class IB PI3Ks are heterodimers of a p110γ catalytic subunit (encoded by PIK3CG) coupled with regulatory isoforms p101 (PIK3R5) or p87 (p84 or p87PIKAP, encoded by PIK3R6). While p110α and p110β are ubiquitously expressed, p110δ and p110γ expression is largely restricted to leukocytes6.
Figure 1. The PI3K family comprises multiple classes and isoforms.
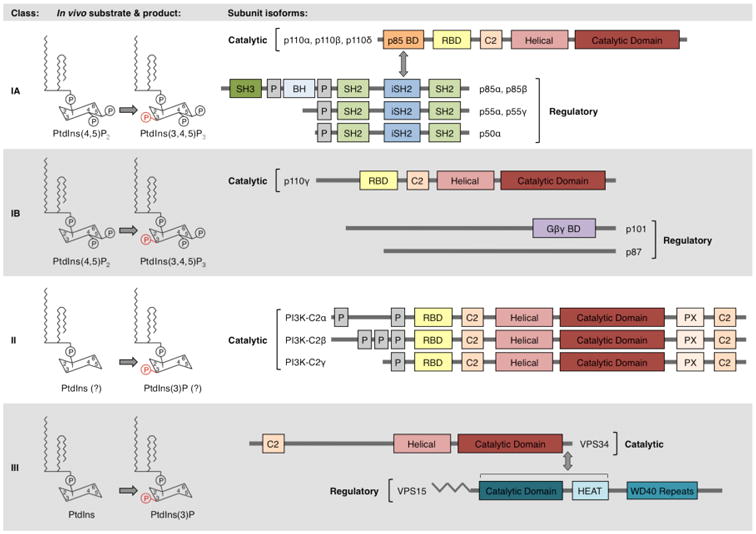
PI3Ks are classified based on their substrate specificities and structures. In vivo, class IA and IB PI3Ks phosphorylate PtdIns(4,5)P2, while class III PI3Ks phosphorylate PtdIns. Some evidence suggests that class II PI3Ks may also preferentially phosphorylate PtdIns in vivo8-10. Class IA PI3Ks are heterodimers of a p110 catalytic subunit and a p85 regulatory subunit. Class IA catalytic isoforms (p110α, p110β, and p110δ) possess a p85-binding domain (p85-BD), RAS-binding domain (RBD), helical domain, and catalytic domain. Class IA p85 regulatory isoforms (p85α, p85β, p55α, p55γ, and p50α) possess an inter-SH2 (iSH2) domain that binds class IA catalytic subunits, flanked by SH2 domains that bind phosphorylated YXXM motifs. The longer isoforms, p85α and p85β, additionally possess N-terminal SH3 and breakpoint cluster homology (BH) domains. Class IB PI3Ks are heterodimers of a p110γ catalytic subunit and a p101 or p87 regulatory subunit. p110γ possesses an RBD, helical domain, and catalytic domain. The domain structures of p101 and p87 are not fully known, but a C-terminal region of p101 has been identified as binding Gβγ subunits120. The monomeric class II isoforms (PI3K-C2α, PI3K-C2β, and PI3K-C2γ) possess an RBD, helical domain, and catalytic domain. VPS34, the only class III PI3K, possesses helical and catalytic domains. VPS34 forms a constitutive heterodimer with the myristoylated, membrane-associated VPS15 protein. Other indicated domains include proline-rich (P) domains and membrane-interacting C2 domains. Modified with permission from Reference 2.
In the absence of activating signals, p85 interacts with p110, inhibiting p110 kinase activity. Upon receptor tyrosine kinase (RTK) or G-protein coupled receptor (GPCR) activation, class I PI3Ks are recruited to the plasma membrane, where p85 inhibition of p110 is relieved and p110 phosphorylates PtdIns 4,5-bisphosphate (PtdIns(4,5)P2) to generate PtdIns(3,4,5)P3 (Figure 2A). This lipid product acts as a second messenger, activating AKT-dependent and –independent downstream signaling pathways (reviewed in 1-3). The phosphatase and tensin homolog (PTEN) lipid phosphatase removes the 3′ phosphate from PtdIns(3,4,5)P3 to inactivate PI3K signaling.
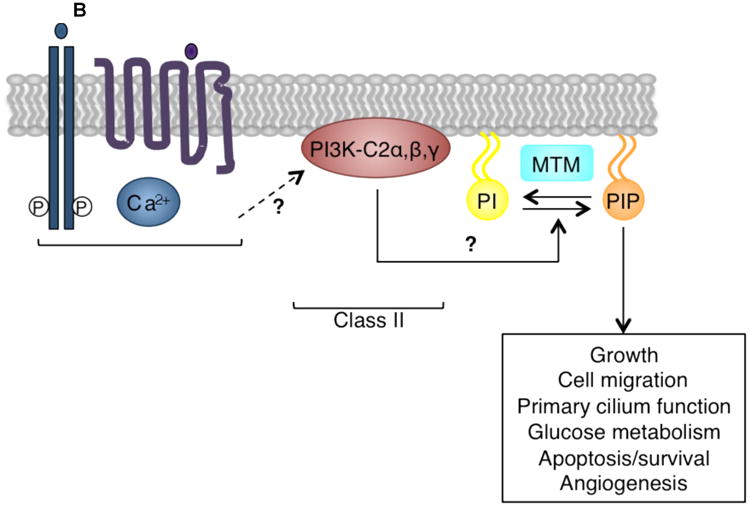
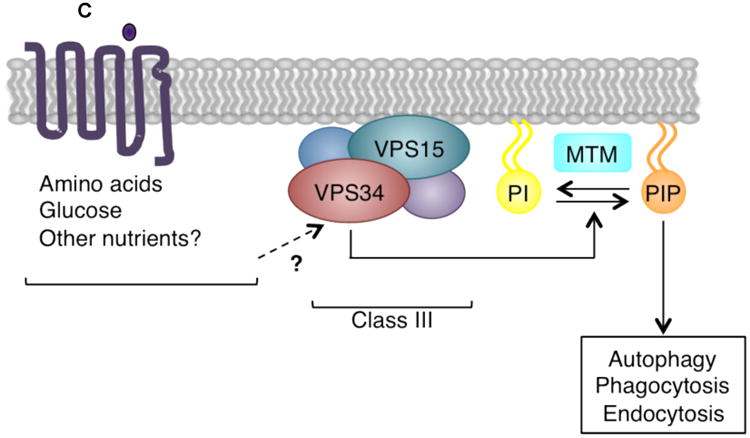
Figure 2. Signaling by class I, II, and III PI3K isoforms.
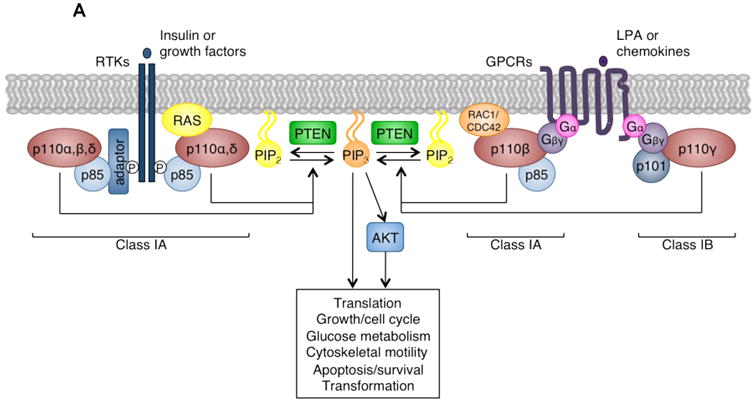
(A) Upon receptor tyrosine kinase (RTK) or G-protein coupled receptor (GPCR) activation, class I PI3Ks are recruited to the plasma membrane by interaction with phosphorylated YXXM motifs on RTKs or their adaptors, or with GPCR-associated Gβγ subunits. There they phosphorylate PtdIns(4,5)P2 (PIP2) to generate PtdIns(3,4,5)P3 (PIP3), a second messenger which activates a number of AKT-dependent and –independent downstream signaling pathways regulating diverse cellular functions including growth, metabolism, motility, survival, and transformation. The phosphatase and tensin homolog (PTEN) lipid phosphatase removes the 3′ phosphate from PtdIns(3,4,5)P3 to inactivate class I PI3K signaling. Modified with permission from Reference 2.
(B) Class II PI3Ks are not well understood, but may be activated by a number of different stimuli, including hormones, growth factors, chemokines, cytokines, phospholipids, and calcium (Ca2+). Although in vitro class II PI3Ks can phosphorylate both PtdIns and PtdIns(4)P, in vivo this class may preferentially phosphorylate PtdIns (PI) to generate PtdIns(3)P (PIP)8-10. Class II PI3Ks regulate cellular functions including glucose transport, endocytosis, cell migration, and survival. Myotubularin (MTM) family phosphatases remove the 3′ phosphate from PtdIns(3)P to inactivate class II PI3K signaling.
(C) The class III VPS34-VPS15 heterodimer is found in distinct multiprotein complexes, which perform specific cellular functions. VPS34 may be activated by stimuli including amino acids, glucose, and other nutrients, and phosphorylates PtdIns (PI) to generate PtdIns(3)P (PIP). It plays critical roles in autophagy, endosomal trafficking, and phagocytosis. MTM family phosphatases remove the 3′ phosphate from PtdIns(3)P to inactivate class III PI3K signaling.
Relatively little is known about class II PI3Ks. There are three class II isoforms, PI3K-C2α, PI3K-C2β, and PI3K-C2γ, respectively encoded by PIK3C2A, PIK3C2B, and PIK3C2G. These monomeric lipid kinases do not possess a regulatory subunit. PI3K-C2α and PI3K-C2β are broadly expressed, while PI3K-C2γ expression is limited to the liver, prostate, and breast7. Although early experiments indicated that PI3K-C2α and PI3K-C2β could phosphorylate both PtdIns and PtdIns(4)P, in vivo PtdIns may be the preferred substrate, generating PtdIns(3)P8-10. The physiological roles of class II PI3Ks are not fully understood, but recent studies suggest that PI3K-C2α is important in angiogenesis10 and primary cilium function11. In addition, PI3K-C2α and PI3K-C2β have been reported to regulate cellular functions including growth and survival (reviewed in 3, 7) (Figure 2B).
The single class III PI3K, VPS34, is encoded by PIK3C3. VPS34 forms a constitutive heterodimer with the myristoylated[G], membrane-associated VPS15 (encoded by PIK3R4), and phosphorylates PtdIns to produce PtdIns(3)P12, 13. In mammals, VPS34 is ubiquitously expressed13. The VPS34-VPS15 dimer is found in distinct multiprotein complexes, which have critical roles in intracellular trafficking and autophagy (reviewed in 3, 14) (Figure 2C). The myotubularin (MTM) family phosphatases MTM1 and MTMR2 remove the 3′ phosphate from PtdIns(3)P, regulating the lipid products of class II and III PI3Ks15-18.
Alterations of PI3K isoforms in cancer
Overactivation of the PI3K pathway is one of the most frequent events in human cancers. The most common mechanism leading to aberrant PI3K signaling is somatic loss of PTEN via genetic or epigenetic alterations (reviewed in 19, 20). The PI3K pathway can also be upregulated by activation of RTKs, or alterations in isoforms of PI3K itself (Table 1).
Class I PI3K catalytic isoform alterations
The transforming potential of class I PI3K catalytic isoforms was first demonstrated by studies in the late 1990s and early 2000s, which showed that fusion of p110α to viral sequences21 or the SRC myristoylation sequence22-24 was activating and highly oncogenic. The 2004 discovery of frequent PIK3CA mutations in human cancers25 brought PI3K to the forefront as a major cancer driver and potential drug target. PIK3CA mutation has since been firmly established as causative in many cancer types (Table 1). Missense mutations occur in all domains of p110α, but the majority cluster in two hotspots, the most common being E542K and E545K in the helical domain and H1047R in the kinase domain. Cell-based analyses confirmed that these hotspot mutations confer transformation via constitutive activation of p110α23, 26, 27. Subsequently, several studies using genetically engineered mouse models (GEMMs) demonstrated roles for mutant PIK3CA in tumor initiation, progression, and maintenance28-32 (Supplemental Table 2). Helical domain mutations reduce inhibition of p110α by p8533-36 or facilitate direct interaction of p110α with insulin receptor substrate 1 (IRS1)37, while kinase domain mutations increase interaction of p110α with lipid membranes33, 36, 38. Other PIK3CA mutations mimic distinct structural conformation changes that occur during activation of PI3K36. Interestingly, some of these mutations in PIK3CA have also been reported in congenital mosaic overgrowth syndromes[G]39-42.
In contrast, mutations in other class I catalytic isoforms are rare. While activating PIK3CD mutations have been described in immune deficiencies43, 44, they have not been linked to cancer. One PIK3CB mutation was detected in a single case of breast cancer45; this helical domain substitution enhances basal PI3K activation, potentially by increasing p110β association with membranes46. Recent structural studies have indicated that p110β may be less inhibited by p8547-49 and thus has higher basal transforming potential. Interestingly, p110δ expression has been detected in some human solid cancer cell lines50, and overexpression of wildtype p110β, p110δ, or p110γ, but not p110α, transforms cells in vitro51. This is consistent with the fact that PIK3CB, PIK3CD, and PIK3CG are generally amplified or overexpressed, but not mutated, in cancers (Table 1).
Class I PI3K regulatory isoform alterations
Recent studies have converged to implicate the p85 regulatory isoforms in tumorigenesis. Since the initial discovery of PIK3R1 mutations in human cancer cell lines and primary tumors52, somatic mutations in PIK3R1 have been identified in a number of different cancers53-57 (Table 1). The majority are substitutions or in-frame insertions or deletions in the inter-SH2 (iSH2) domain[G] of p85α53-56, the region of the protein that makes contact with p11033, indicating this domain as a mutation hotspot53. A number of these iSH2 domain mutants retain the ability to bind and stabilize p110 isoforms, but promote enhanced PI3K activity and transformation due to reduced ability to inhibit p11053-55, 58, 59.
In addition, reduced expression of PIK3R1 has been reported in some cancers57, 60 (Table 1). PIK3R1 mRNA levels inversely correlated with malignancy grade and incidence of metastasis in both breast and liver cancers57, 60. In mice, Pik3r1 ablation increased epithelial neoplasia driven by Pten loss61 and led to spontaneous development of aggressive liver tumors60. This work indicates that p85α can negatively regulate PI3K signaling in cancer, and suggests that p85α has tumor suppressive functions in certain tissues62.
Alterations in genes encoding other regulatory isoforms have also been detected, albeit at a lower frequency. Increased PIK3R2 expression has been reported in breast and colon cancers63 (Table 1). Consistent with this, overexpression of wildtype p85β increased PI3K pathway activation in cells and tumor formation in mice63. Somatic PIK3R2 mutations have been found in endometrial and colorectal cancers53, 55, and causative germline PIK3R2 mutations have been reported in megalencephaly syndromes[G]41. All PIK3R2 mutations described to date are substitutions with no apparent hotspot region, and similar to some p85α mutants, mutations in p85β increase PI3K activation without affecting p110 binding53. Together these studies indicate that PI3K regulatory isoforms may contribute to tumorigenesis by multiple mechanisms.
Class II PI3K isoform alterations
Although class II PI3Ks are not well understood, PIK3C2A or PIK3C2B expression has been implicated in physiological functions important to tumorigenesis9, 64-67. PIK3C2B amplification has been reported in glioblastoma68-70, and somatic PIK3C2B mutations were detected in non-small cell lung cancer71, but the functional consequence of these mutations is unknown. Perhaps the most convincing evidence towards a role for class II PI3Ks in tumorigenesis comes from a recent study demonstrating that mice with Pik3c2a ablation had compromised angiogenesis and vascular barrier integrity, and significant reduction in the size and microvessel density of implanted tumors10. Since mice with embryonic Pik3c2a or Pik3c2b knockout (KO) are viable72, 73, a class II-selective PI3K inhibitor might target tumor angiogenesis with tolerable side effects, although toxicity due to the critical role of PI3K-C2α in maintaining normal renal homeostasis73 would need to be considered.
Type II inositol 3,4-bisphosphate 4-phosphatase (INPP4B), the phosphatase responsible for dephosphorylation of PtdIns(3,4)P2 to PtdIns(3)P74, 75, has also been implicated in cancer. In human mammary cell lines, INPP4B knockdown increased AKT activation and transformation75, 76. INPP4B loss-of-heterozygosity has been detected in cancers75, 77, and reduced INPP4B expression has been correlated with high tumor grade, earlier recurrence, and decreased survival75, 76, 78. Identification of INPP4B as a tumor suppressor suggests that deregulation of the class II PI3K lipid products may contribute to tumorigenesis.
Class III PI3K isoform alterations
There is currently little evidence indicating an oncogenic role for VPS34. One recent study suggested that VPS34 is tyrosine-phosphorylated and activated downstream of SRC, and its lipid kinase activity is required for SRC-mediated transformation79. However, overexpression of wildtype or myristoylated VPS34 was not sufficient to induce cellular transformation80. Another study indicated that VPS34 activity might be decreased in the context of activated epidermal growth factor receptor (EGFR)81. Further investigation is needed to determine whether VPS34 plays a role in transformation.
Divergent roles of class I PI3K catalytic isoforms
Class I PI3K catalytic isoforms share a conserved domain structure. They utilize the same lipid substrates and generate the same lipid products. Despite their similarities, accumulating evidence indicates these isoforms have distinct roles in mediating PI3K signaling in physiological and oncogenic contexts.
GEMMs have been used to elucidate the roles of individual class I PI3K isoforms. Mice with germline KO of Pik3ca or knock-in (KI) of a kinase-dead Pik3ca allele die at day E10.582, 83. Interestingly, Pik3cb KO mice die much earlier at day E3.582, while kinase-dead Pik3cb KI mice develop to maturity with minor defects in size and glucose metabolism, and major defects in male fertility84, 85. These differences suggest an important kinase-independent scaffolding role for p110β84. Germline inactivation of Pik3cd or Pik3cg by KO or KI of kinase-dead alleles yields viable mice that grow to adulthood; however, loss of p110δ results in functional defects in lymphocytes, neutrophils, and mast cells86-89, while loss of p110γ impairs thymocyte development, T cell activation, and neutrophil migration90-92. These studies indicate non-redundant roles in mouse embryonic development for p110α and p110β, the two ubiquitously expressed class I PI3K isoforms, and distinct roles in the immune system and inflammatory response for p110δ and p110γ, the two leukocyte-restricted isoforms.
Technological developments have facilitated further insight into the individual roles of PI3K enzymes. The generation of conditional KO animals using the Cre/loxP recombination system has allowed the functions of each isoform to be studied in different tissues, stages of development, and pathological settings (Supplemental Table 2). Additional progress has come from studies using RNA interference (RNAi) and a new generation of isoform-selective PI3K inhibitors. These have advanced our understanding of the roles of class I catalytic isoforms in mediating signaling downstream of RTKs, GPCRs, and small GTPases (Figure 3), and in the context of PTEN deficiency (Figure 4A).
Figure 3. Divergent roles of class I PI3K catalytic isoforms in different signaling contexts.
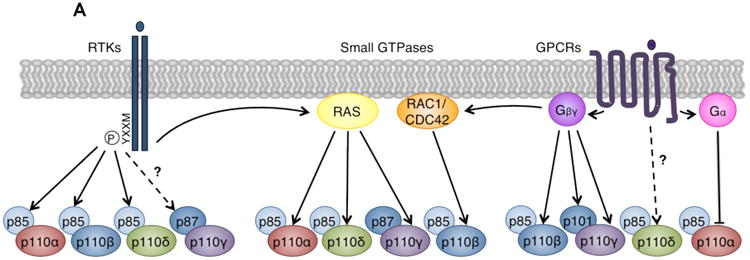
(A) Class I PI3Ks mediate signaling downstream of RTKs, GPCRs, and small GTPases. Left: p85 regulatory subunits bind phosphorylated YXXM motifs on activated RTKs. Because p110α, p110β, and p110δ bind p85, these isoforms mediate signaling downstream of RTKs. Recent evidence also suggests that p87-p110γ may be activated by certain RTKs110. Middle: Small GTPases synergize with RTK and GPCR signals to directly activate PI3Ks by interacting with their RAS-binding domains (RBDs). Isoforms p110α, p110δ, and p110γ bind RAS family GTPases, while p110β binds the RHO family GTPases RAC1 and CDC42143. Right: Gα and Gβγ proteins dissociate from activated GPCRs. Catalytic isoforms p110β and p110γ, and regulatory isoform p101, directly bind and are activated by Gβγ. p110δ may be activated downstream of GPCRs, but the mechanism is unknown126-128. Gα proteins have been reported to directly bind and inhibit p110α129-131. Modified with permission from Reference 3.
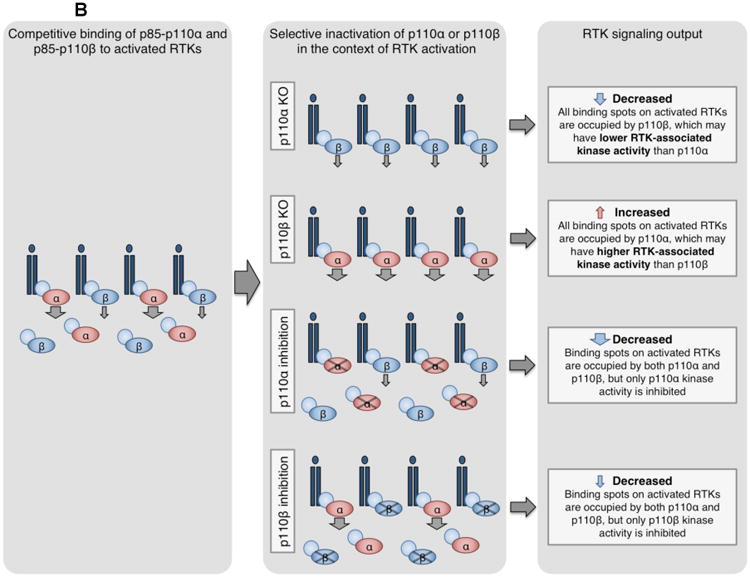
(B) Competition model for p110α and p110β regulation of RTK signaling96. Both p85-p110α and p85-p110β compete for phosphorylated YXXM sites on activated RTKs. However, the maximal specific activity and enzymatic rate of p110α are higher than that of p110β108, 109, and RTK-associated p110α may have higher lipid kinase activity than p110β96. By this model, loss or inactivation of p110α or p110β differentially modulates RTK signaling. Knockout of p110α allows all sites to be occupied by the less active p110β, decreasing RTK output. Conversely, knockout of p110β allows all sites to be bound by the more active p110α, increasing RTK output. Genetically or pharmacologically inactivated p110α or p110β can still bind RTKs but cannot signal, reducing RTK output.
Figure 4. An overview of PI3K inhibitors and their combination with other therapeutics.

(A) Molecular contexts dictating applications for isoform-selective PI3K inhibitors. Light orange boxes: Upregulation or mutation of receptor tyrosine kinases (RTKs), oncogenic RAS mutations, or activating p110α mutations all increase PtdIns(3,4,5)P3 production through p110α, which can be amplified by mutation or loss of PTEN. In these contexts use of p110α-selective inhibitors is effective. Blue boxes: In the absence of other oncogenic alterations, PTEN loss or mutation increases PtdIns(3,4,5)P3 production through p110β, perhaps due to RAC1- or CDC42-mediated p110β activation, or the basal activity of this isoform. In this context use of p110β-selective inhibitors is effective. Dark orange boxes: Upregulation or mutation of B cell receptors (BCRs), cytokine receptors, or other immune cell surface markers increases PtdIns(3,4,5)P3 production through p110δ. In this context use of p110δ-selective inhibitors is effective.
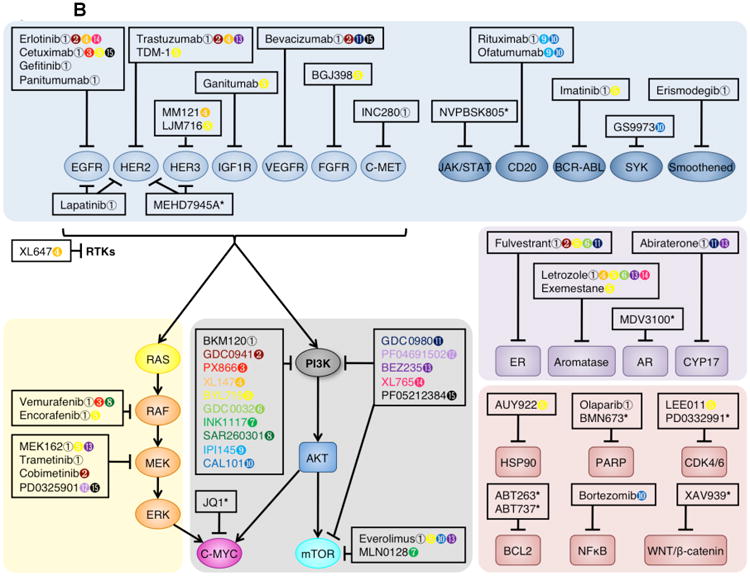
(B) Rational combination of PI3K inhibitors and other targeted therapeutics. Pan-PI3K and dual pan-PI3K and mTOR inhibitors are currently being tested in clinical trials (white box). These agents are being combined with mTOR-selective inhibitors (shown in dark orange), RAS-RAF-MEK-ERK pathway inhibitors (shown in light orange), RTK (shown in grey) or other membrane-associated protein inhibitors (shown in turquoise), hormone signaling inhibitors (shown in dark blue), and other agents inhibiting the cell cycle, apoptosis machinery, or other signaling pathways (shown in purple). Colored symbols indicate targeted therapeutics currently in clinical trials for combination with the designated PI3K inhibitor. For further detail, see Supplementary Table 3.
In mediating RTK signaling
Binding of growth factor ligands induces RTK dimerization, activation, and auto-phosphorylation of tyrosine-containing YXXM motifs on the receptors or their associated adaptor proteins. Class IA p110-p85 heterodimers are then recruited to activated RTKs through direct interaction of p85 SH2 domains[G] with these phosphorylated YXXM motifs93-95 (Figure 2A). Accordingly p110α, p110β, and p110δ can complex with activated RTKs (Figure 3A), and might be expected to mediate growth factor signaling.
Studies using isoform-selective pharmacological inhibitors and genetic inactivation or ablation indicated that loss of p110α activity was sufficient to largely block PI3K signaling in response to a number of growth factors96-101. Notably, genetic ablation or inactivation of p110β had only a modest effect on PI3K signaling following acute RTK activation84, 102, 103. It was suggested that the relative abundance of catalytic isoforms in a particular tissue might dictate which isoforms are dominant in mediating RTK signaling104. This may explain the role of p110δ, which is mainly expressed in leukocytes and is the primary isoform regulating PI3K signaling downstream of certain RTKs in mast cells and macrophages87, 105, 106. However, differential expression does not completely explain isoform dependence, as in many tissues p110β levels are comparable to or even higher than levels of p110α107.
The involvement of p110β in RTK signaling remained puzzling, until a recent study from our group suggested a new model. In mice, while p110α ablation blocked normal mammary development and mammary tumorigenesis driven by polyoma middle T (PyMT) or HER2 (also known as ERBB2), p110β ablation increased mammary gland outgrowth and accelerated tumor formation driven by these oncogenic RTKs96. To explain this negative role of p110β, a competition model was proposed: if p110α has higher RTK-associated lipid kinase activity than p110β, the less-active p110β could compete with p110α for phosphorylated YXXM sites on receptors to modulate PI3K signal strength downstream of RTKs96 (Figure 3B). Although direct comparison of RTK-associated p110α and p110β lipid kinase activity has not been shown, the maximal specific activity and enzymatic rate of p110α are higher than that of p110β108, 109. Biochemical data were consistent with this proposed model, demonstrating that in p110β KO cells, activated RTKs had more bound p110α and higher associated lipid kinase activity96. Furthermore, pharmacologically inactivated p110β could still compete with p110α for binding sites on activated receptors, modestly reducing signaling and tumor growth driven by PyMT or HER296. This model also explains moderately decreased AKT activation, mild hyperglycemia, and delayed HER2-driven tumor formation observed in mice with KI of kinase-dead p110β84, a scenario mimicking p110β-selective kinase inhibition. These studies not only reveal a novel p110β-based regulatory mechanism in RTK-mediated PI3K signaling, but also identify p110α as an important target in cancers driven by oncogenic RTKs.
Initial studies suggested that class IA isoforms mediated signaling downstream of RTKs, while the class IB isoform signaled downstream of GPCRs. Although p110γ activation by GPCRs is well established, a recent report suggested that this class IB isoform might also function downstream of RTKs through regulatory isoform p87 in mouse myeloid cells110 (Figure 3A). Given that p87 and p101 may have distinct tissue distribution111-113 and non-redundant functions110, 111, 113, 114, this suggests that the two class IB regulatory isoforms may mediate p110γ activation in response to specific upstream signals.
In mediating GPCR signaling
GPCRs are a family of seven-transmembrane domain receptors that associate with heterotrimeric G proteins composed of the Gα and Gβγ subunits. Ligand binding to GPCRs results in allosteric activation and disassociation of bound G proteins into their separate subunits, which can then act on intracellular targets.
The single class IB PI3K isoform, p110γ, is activated by G proteins115-117 (Figure 2A). Although association of p110γ with either its p101 or p87 regulatory isoforms increased its activation in response to Gβγ115, 118, 119, recent evidence indicated that p101 is the main regulatory isoform involved in GPCR-mediated p110γ signaling110, 114 (Figure 3A). Both p110γ and p101 interact directly with Gβγ heterodimers, and these contacts are critical for signaling and transformation mediated by p110γ115, 120. Recent studies have shown that in myeloid cells, p110γ can be activated by GPCR and RTK signals in a RAS- or RAP1A-dependent manner to mediate integrin α4β1 activity, leading to tumor inflammation and progression110, 121. Thus p110γ-mediated signaling may contribute to tumorigenesis by controlling both tumor cell characteristics and the tumor microenvironment.
Interestingly, in vitro experiments117, 122-124 and subsequent GEMM studies84, 102, 103 demonstrated a role for p110β in G protein-mediated PI3K signaling (Figure 2A). Recently a region in the C2-helical domain linker of p110β was shown to bind Gβγ subunits (Figure 3A); this region is not present in other class IA isoforms125, and is similar to the region of p110γ that binds Gβγ120. Abrogation of p110β-Gβγ interaction blocked p110β-mediated signaling and transformation downstream of GPCRs, and inhibited the proliferation and invasiveness of cancer cells125. Although p110δ does not directly interact with G proteins, a non-redundant role for this isoform in GPCR-mediated leukocyte migration has been demonstrated in certain contexts126-128; however, the mechanism of p110δ activation downstream of GPCRs is unknown. It has also been reported that some Gα proteins directly bind and inhibit p110α129-131. Clearly, class I PI3K isoforms cooperate with GPCRs in a number of different ways to regulate signaling and transformation.
Downstream of RAS and other small GTPases
RAS superfamily proteins[G] are direct activators of the PI3K pathway. All class I PI3K catalytic isoforms possess an N-terminal RAS-binding domain (RBD) (Figure 1) allowing them to interact with RAS GTPases[G] or other RAS superfamily members (Figure 3A).
Activated or oncogenic mutant RAS proteins directly bind and increase the enzymatic activity of both p110α132, 133 and p110γ134-136. Cellular and structural studies suggest that p110γ association with RAS might both increase its membrane translocation114, 135 and allosterically increase p110γ kinase activity135. Interestingly, RAS is required for activation of p110γ bound to regulatory isoform p87, but not p101114. In vitro, the transforming capability of both helical domain p110α mutants34, 137 and of overexpressed wildtype p110γ51, 138 are dependent on their association with RAS. GEMM studies using KI of Pik3ca with an RBD mutation or KO of endogenous Pik3ca revealed that the p110α-RAS interaction is critical for both the initiation and maintenance of lung tumors139, 140 and the development of myeloid leukemia141 driven by oncogenic KRAS. In mice, p110γ-RAS binding is required for inflammation-induced PtdIns(3,4,5)P3 accumulation142 and inflammation-associated tumor progression110, 121. These studies highlight the importance of p110α or p110γ interaction with RAS in both normal PI3K signaling and transformation.
Although p110δ was shown to bind RAS in vitro143, 144, some studies indicated that p110δ kinase activity was not stimulated by HRAS, NRAS, or KRAS, but instead by RRAS and TC21 (also known as RRAS2)145, 146. Furthermore, B and T cells derived from Tc21 KO mice displayed diminished PI3K activity and recruitment of p110δ to T cell receptors (TCRs) and B cell receptors (BCRs), suggesting that TC21 might function upstream of p110δ147. Thus PI3K signaling through p110δ may be regulated by additional RAS subfamily members.
It was initially anticipated that all p110 isoforms bearing a RBD might interact with RAS GTPases. Surprisingly, in vitro studies determined that p110β kinase activity was not stimulated by any RAS subfamily members146. A recent extensive biochemical study demonstrated that p110β is instead regulated by RAC1 and CDC42 of the RHO GTPase[G] subfamily143 (Figure 3A). Direct interaction between the p110β RBD and RAC1 is important for GPCR-mediated activation of p110β143, indicating cooperative Gβγ and RHO GTPase signaling through p110β. Previous studies reported that an intact RBD was required for signaling and oncogenic transformation by wildtype p110β in cultured cells51, 138, suggesting a potential role for RHO GTPase interaction with p110β in transformation. Notably, RAC1 and CDC42 can also be activated downstream of PI3K by PtdIns(3,4,5)P3-dependent guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs)148-150. The finding of distinct p110β regulation by RAC1 and CDC42 expands PI3K signaling input by GTPases beyond the RAS subfamily, and also supports the notion that PI3K can act both upstream and downstream of GTPases, potentially allowing for positive feedback loops in cancer settings.
In PTEN deficiency
The PTEN lipid phosphatase counteracts class I PI3K activity, making it an important tumor suppressor. Somatic loss of PTEN in human cancers is common. Germline PTEN mutations are also found in several genetic disorders characterized by multiple hamartomas with overgrowth phenotypes, collectively termed PTEN hamartoma tumor syndromes (PHTS)151.
Pten KO mouse models provided a tool to explore the molecular mechanisms underlying diseases caused by PTEN loss. While embryonic Pten KO is lethal152, 153, heterozygous or conditional Pten KO animals recapitulated human disease phenotypes, including development of prostate cancer154-156. Surprisingly, ablation of p110β, but not p110α, blocked prostatic intraepithelial neoplasia (PIN) induced by PTEN loss102. Subsequent studies demonstrated a correlation between PTEN deficiency and sensitivity to p110β knockdown or inhibition in human cancer cell lines both in vitro and in mouse xenografts157-159. However, the mechanism governing the specific importance of p110β in the context of PTEN loss remains elusive. Perhaps the unique role for p110β as a convergence point for GPCR and RAC1 or CDC42 signals (Figure 4A) contributes to transformation induced by PTEN deficiency. Structural studies have also suggested that compared to p110α, p110β is less inhibited by p85, and may supply a basal level of PtdIns(3,4,5)P3 47-49. This may explain why wildtype p110β can be oncogenic when it is overexpressed51, 138 or when PTEN is lost.
Although p110β is the primary PI3K isoform involved in many cases of tumorigenesis driven by PTEN loss, studies have shown that depending on the tissue type and pathology both p110α and p110β may be involved160-162. Mice with Pten ablation in the basal epidermal compartment require both p110α and p110β for the development of hyperproliferative epidermal lesions closely resembling PHTS163, 164. In this model, spatially distinct roles for these isoforms in epidermal compartments were identified: p110α is responsible for RTK signaling in and survival of suprabasal cells, whereas p110β is important for GPCR signaling in and proliferation of basal cells164. In mice with thymocyte-specific Pten KO, not surprisingly both p110δ and p110γ were required for the development of T cell acute lymphoblastic leukemia (T-ALL)165. This suggests that in certain contexts, transformation driven by PTEN loss may be governed by the PI3K isoforms that are dominant in that tissue or compartment.
Since PTEN loss removes one mechanism of PI3K pathway negative regulation, the specific roles of p110 isoforms in this pathogenic context can be influenced by other activating inputs. These can be cues from the tissue microenvironment, or other coexisting genetic events. A recent GEMM study demonstrated that concomitant activation of oncogenic KRAS in ovarian endometrioid adenocarcinoma driven by Pten ablation shifted the PI3K isoform reliance from p110β to p110α162 (Figure 4A). Consistent with this, a subset of PTEN-mutant human endometrioid endometrial cancer cell lines harboring other PI3K-activating mutations were found to be resistant to p110β inhibition166. It is also possible that other genetic events downstream of PI3K or in PI3K-independent pathways may render PTEN-null tumors less reliant on PI3K. Thus determination of isoform dependency in PTEN-deficient tumors remains a challenge.
Therapeutic targeting of PI3K isoforms in cancer
The central role of PI3K in cancer makes it an attractive therapeutic target. Enormous efforts have focused on the development of drugs targeting PI3K, many of which are undergoing clinical evaluation (Tables 2-4). Unlike drugs targeting other oncogenic kinases, such as EGFR, BRAF, and ALK, PI3K inhibitors have shown limited efficacy as mono-therapies in early trials on patients with tumors harboring PI3K pathway activation167. The effectiveness of these early PI3K inhibitors may have been limited by their lack of specificity, and by compensatory signaling feedback loops and co-existing genetic and epigenetic alterations. The development of novel isoform-selective PI3K inhibitors (Figure 4A) and their rational combination with other therapeutics (Figure 4B and Supplemental Table 3) may substantially improve therapeutic outcomes.
Table 2. Pan-PI3K inhibitors and their clinical applications.
| Agent | Company | Target | Trial stage* | Tumor types* |
|---|---|---|---|---|
| BKM120 | Novartis | Class I PI3Ks | I, II, and III |
|
| GDC0941 | Genentech | Class I PI3Ks | I and II |
|
| BAY80-6946 | Bayer | Class I PI3Ks | I and II |
|
| ZSTK474 | Zenyaku Kogyo Co. | Class I PI3Ks | I and II |
|
| PX866 | Oncothyreon | Class I PI3Ks | I and II |
|
| XL147 | Exelixis/Sanofi-Aventis | Class I PI3Ks | I and II |
|
| CH5132799 | Chugai Pharma Europe | Class I PI3Ks | I |
|
Data taken from an April 2014 search of http://www.clinicaltrials.gov.
NSCLC, non-small cell lung carcinoma; CRPC, castration-resistant prostate cancer; GIST, gastrointestinal stromal tumor; SCCHN, squamous cell carcinoma of the head and neck; GBM, glioblastoma multiforme
Table 4. Isoform-selective PI3K inhibitors and their clinical applications.
| Agent | Company | Target | Trial stage* | Tumor types* |
|---|---|---|---|---|
| BYL719 | Novartis | p110α | I and II |
|
| GDC0032 | Genentech | p110α | I |
|
| INK1117 | Intellikine/Millenium | p110α | I |
|
| AZD8186 | Astra-Zeneca | p110β | I |
|
| GSK2636771 | GlaxoSmithKline | p110β | I and II |
|
| SAR260301 | Sanofi | p110β | I |
|
| IPI145 | Infinity | p110δ and p110γ | I, II, and III |
|
| AMG319 | Amgen | p110δ | I | Lymphoid malignancies |
| CAL101 (GS101) | Gilead Sciences | p110δ | I, II, and III |
|
| GS9820 | Gilead Sciences | p110β and p110δ | I |
|
Data taken from an April 2014 search of http://www.clinicaltrials.gov.
SCCHN, squamous cell carcinoma of the head and neck; ESCC, esophageal squamous cell carcinoma; GIST, gastrointestinal stromal tumor; CRPC, castration-resistant prostate cancer; sqNSCLC, squamous non-small cell lung cancer; TNBC, triple-negative breast cancer; CLL, chronic lymphocytic leukemia; SLL, small lymphocytic leukemia; ALL, acute lymphoblastic leukemia; INHL, indolent non-Hodgkin lymphoma; MCL, mantle cell lymphoma; AML, acute myeloid leukemia; MM, multiple myeloma
Emerging isoform-selective PI3K inhibitors
Most PI3K inhibitors in early clinical trials are ATP-competitive agents that target all class I isoforms with similar potencies. These include pan-PI3K inhibitors (Table 2) such as GDC0941168 and dual pan-PI3K and mTOR inhibitors (Table 3) such as BEZ235169. Though these drugs display potent preclinical anti-tumor activity, their success in clinical trials as single agents has been modest167. The therapeutic window and efficacy of pan-PI3K inhibitors are limited in some cases by adverse effects arising from a broader spectrum of off-target effects170. Furthermore, while both pan-PI3K and isoform-selective inhibitors have on-target effects from suppression of essential PI3K functions, for example glucose homeostasis, pan-PI3K inhibitors likely have additional on-target effects from inhibiting isoforms that are not contributing to tumorigenesis. Isoform-selective inhibitors may achieve greater efficacy with fewer toxic effects, and are emerging in the clinic (Table 4).
Table 3. Dual pan-PI3K and mTOR inhibitors and their clinical applications.
| Agent | Company | Target | Trial stage* | Tumor types* |
|---|---|---|---|---|
| GDC0980 | Genentech | PI3K and mTOR | I and II |
|
| PF04691502 | Pfizer | PI3K and mTOR | I |
|
| BGT226 | Novartis | PI3K and mTOR | I and II |
|
| BEZ235 | Novartis | PI3K and mTOR | I and II |
|
| XL765 | Sanofi | PI3K and mTOR | I |
|
| GSK2126458 | GlaxoSmithKline | PI3K and mTOR | I |
|
| DS7423 | Daiichi Sankyo | PI3K and mTOR | I | Solid tumors |
| PWT33597 | Pathway Therapeutics | PI3K and mTOR | I | Adv. solid tumors |
| SF1126 | Semafore Pharmaceuticals | PI3K and mTOR | I | Adv. solid tumors |
| PF05212384 | Pfizer | PI3K and mTOR | I and II | Adv. solid tumors |
Data taken from an April 2014 search of http://www.clinicaltrials.gov.
TCC, transitional cell carcinoma; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CML, chronic myelogenous leukemia; GBM, glioblastoma multiforme
The most effective single agent PI3K-based therapy to date is idelalisib (CAL101 or GS1101), a p110δ-selective inhibitor. Idelalisib has achieved notable success in early trials for patients with chronic lymphocytic leukemia or indolent lymphoma, and is currently in phase III clinical trials171, 172. Interestingly, this dramatic response is not due to genetic activation of the PI3K pathway, as neither PI3K mutation nor PTEN loss is common in these malignancies. Given the important role of p110δ in signaling downstream of BCRs86, 88, 89 and the fact that leukemic B cells have been shown to be dependent on BCR signaling, it is likely that idelalisib functions by blocking essential BCR signals. Two recent articles provide great insight into the success of idelalisib trials (see 173 and 174).
In addition to the role of p110δ in B cell malignancies, a recent preclinical study showed that this isoform also contributes to PTEN-null T-ALL165. However, p110δ-selective inhibition in this study was insufficient to suppress tumorigenesis; combined inhibition of both p110δ and p110γ was required for effective anti-PI3K therapy165. The involvement of p110δ and p110γ in leukocyte signaling and hematological malignancies has drawn great attention, and new inhibitors that target both isoforms simultaneously are in clinical trials for B and T cell lymphomas (Table 4). These isoforms may also mediate immune responses that support the growth of solid tumors. In a mouse model, p110γ inhibition blocked myeloid cell recruitment to tumors, thus suppressing malignancy by targeting the tumor microenvironment110. Another study indicated that p110δ inhibition impaired tumor growth by disrupting regulatory T cell-mediated immune tolerance175. These findings indicate potential new applications for p110δ- or p110γ-selective therapies in cancer.
The frequency of PIK3CA mutations in solid tumors has generated great interest in the potential for p110α-selective inhibitors in targeting these cancers. Data presented at the 2013 San Antonio Breast Cancer Symposium (SABCS) indicated promising early clinical activity of p110α-selective inhibitors BYL719 or GDC0032 as single agents in patients with PIK3CA-mutant advanced breast tumors176. Recent preclinical findings that HER2-or KRAS-driven tumors rely on p110α96, 139-141, 162 underscore the need for clinical evaluation of p110α-selective drugs in these disease settings. In these studies, growth of HER2- or KRAS-driven solid tumors is inhibited similarly by pan- and p110α-selective inhibitors96, 140, but only modestly by p110β-selective inhibition96, 162. However, further study is needed to determine the contexts in which simultaneous inhibition of p110α and p110β can improve outcomes of KRAS- or HER2-driven disease.
One drawback of p110α-selective inhibitors is their inevitable on-target adverse effects on insulin signaling and glucose metabolism, since p110α is the major isoform mediating these functions98, 100. In the clinic, the effect of p110α-selective inhibitors on glucose homeostasis must be carefully managed177, and is in some cases limiting167. To circumvent this, inhibitors are being developed that specifically target p110α harboring hotspot mutations. Such agents might be used at high doses with low toxicity, similar to mutant-selective BRAF inhibitors that have had great clinical success178, 179. A major obstacle to this approach is the heterogeneity of oncogenic PIK3CA mutations. Some progress has been made with the discovery of GDC0032, which was reported at the 2013 SABCS to have enhanced potency in PIK3CA mutant breast cancer models180; one preclinical study also reported success using stapled peptides to specifically disrupt the interaction of p110α-E545K with IRS137. However, devising strategies to selectively interrupt mutant-specific function remains challenging. If developed, this class of inhibitor will likely be most effective in early stage tumors with PIK3CA mutations, as advanced PIK3CA-mutant tumors may have escaped their dependency on oncogenic p110α29. Such drugs would also be ideal for treating congenital overgrowth syndromes caused by PIK3CA mutations occurring during early embryonic development39-42. In these contexts, p110α mutant-selective inhibitors may yield improved therapeutic index.
Several preclinical studies have documented that certain PTEN-deficient tumors depend on p110β102, 157, 159, prompting a new clinical trial with the p110β-selective inhibitor GSK2636771 in patients with PTEN-deficient advanced solid tumors (NCT01458067). However, since PTEN is a negative regulator of PI3K, isoform-dependency of PTEN-deficient tumors can be complicated as it can be affected by tissue type, co-existing genetic events, and microenvironmental cues that fuel cancer cells. In model systems where PTEN-deficient tumors are found to be dependent on p110β, addition of oncogenic RTKs, RAS, or mutant PIK3CA can shift dependency partially or totally to p110α (Figure 4A). Recent studies also show that prolonged treatment of PTEN-deficient tumor cells with p110β-selective inhibitors can shift isoform dependency from p110β to p110α (N. Rosen, unpublished observations). Therefore in most PTEN-deficient solid tumors, both p110α and p110β should be targeted.
Although development of dual p110α- and p110β-selective inhibitors has proven difficult98, combination of individual p110α- and p110β-selective inhibitors might offer flexibility in the dosing of each isoform-selective inhibitor to further reduce toxicity and increase the therapeutic window. One approach could involve continuous inhibition of p110β to suppress elevated basal PI3K activity due to PTEN loss, combined with pulsatile inhibition of p110α to avoid toxicity due to glucose elevation. Such a strategy might also avoid the reported shift in isoform dependency of tumors from p110α to p110β after prolonged treatment with the p110α-selective inhibitor BYL719 (J.A. Engelman, unpublished observations). Ultimately, the success of targeting PI3K in cancer will likely require better understanding of which PI3K isoforms to target in a given disease setting, improved inhibitors, and more careful dosing strategies.
Resistance mechanisms and combination therapeutic strategies
PI3K-based therapeutic approaches have encountered a number of roadblocks in the form of intrinsic and acquired resistance mechanisms. A large body of work has identified multiple signaling feedback loops, compensatory parallel signaling pathways, and modes of downstream pathway activation that may result in clinical resistance to PI3K inhibitors. Consequently, combination therapies are being developed and evaluated in both preclinical and clinical settings (Figure 4B and Supplemental Table 3), and will be necessary to maximize clinical efficacy of PI3K inhibitors.
The first indication of feedback loops in the PI3K pathway came from experiments with mTOR inhibitors. In early studies mTOR inhibition led to p70 ribosomal protein S6 kinase (S6K) suppression, IRS1 upregulation, and PI3K-AKT activation181. This prompted the development of dual pan-PI3K and mTOR inhibitors that are currently in clinical trials (Table 3). Interestingly, feedback loops can also arise from dual PI3K and mTOR inhibition. A recent preclinical report suggested that PI3K and mTOR blockade activated the Janus kinase 2 (JAK2)-signal transducer and activator of transcription 5 (STAT5) signaling axis via IRS1, generating resistance to PI3K and mTOR inhibition, which could be overcome by targeting JAK2182. Similarly, in another preclinical study treatment with BEZ235 increased phosphorylation of multiple signaling molecules, including STAT3, STAT5, JUN, and p90 ribosomal S6 kinase (p90RSK)183. Isoform-selective PI3K inhibitors can also generate feedback loops: in a recent study of PIK3CA mutant breast tumors, mTOR complex 1 (mTORC1) reactivation by insulin-like growth factor 1 (IGF1) and neuregulin 1 (NRG1) was associated with tumor resistance to the p110α-selective agent BYL719, necessitating concurrent mTORC1 inhibition using RAD001184. Inhibiting both PI3K and mTOR, possibly in conjunction with additional signaling pathways, may be required to achieve effective anti-tumor activity.
Another important resistance mechanism to PI3K pathway inhibition is increased expression of RTKs, such as HER3, IGF1R, insulin receptor (IR), and EGFR, via forkhead box O (FOXO)-mediated transcriptional upregulation185. Robust HER3 induction in response to PI3K inhibition has been reported in several tumor types183, 186, 187. While HER3 itself does not possess tyrosine kinase activity, it dimerizes with EGFR, HER2, or HER4, hyperactivating the PI3K pathway and dampening the efficacy of PI3K drugs. A preclinical study demonstrated that combination of the HER3-neutralizing antibody LJM716 and the p110α-selective inhibitor BYL719 potently blocked PI3K signaling and growth of HER2-positive breast tumor xenografts, even without a direct HER2 antagonist188. Similarly, combination of the dual EGFR and HER3 inhibitor MEHD7945A with a PI3K inhibitor (GDC0941) or AKT inhibitor (GDC0068) effectively blocked the growth of triple-negative breast cancer cells in vitro and in xenografts in a preclinical study189. Blockade of PI3K along with upstream RTKs may therefore circumvent certain PI3K therapy resistance mechanisms (Figure 4B).
Activation of convergent signaling pathways, for example the RAS-RAF-MEK-ERK pathway, can also lead to PI3K pathway inhibition resistance. Mutant RAS can activate both the RAF-ERK and PI3K-AKT-mTOR pathways in cancer cells; blocking the PI3K pathway in such cells leads to upregulation of the ERK pathway190. Inhibition of both PI3K and ERK pathways successfully suppressed the growth of cancer cells in mouse models28, 140, 191, and combinations of MEK inhibitors and pan- or isoform-selective PI3K agents are being evaluated in clinical trials. However, there is preclinical evidence that some of these combinations may be limited due to synergistic toxicity140. Preclinical studies indicate that pulsatile inhibition of both PI3K and ERK pathways may provide more effective anti-tumor activity while limiting toxic effects191, suggesting that optimization of such combinations in the clinic will require careful dosing strategies.
Another mode of resistance to PI3K-directed therapies arises from the activation of transcription downstream or outside of the PI3K pathway. Several reports have indicated MYC amplification or overexpression29, 192 or activation of the Notch and WNT/β-catenin pathways193, 194 as mechanisms of resistance to PI3K inhibition. Recently, the bromodomain and extraterminal (BET) inhibitor JQ1 has been shown to downregulate transcription of MYC, among other targets195. XAV939 has also been identified as an inhibitor of WNT/β-catenin-mediated transcription196. Combination of PI3K inhibition with these agents is being actively pursued in preclinical settings.
Other combination therapies have been suggested by assessing pathways that may synergize with PI3K (Figure 4B). As presented at the 2012 and 2013 SABCS, anti-estrogen therapies are being tested in combination with PI3K inhibitors in clinical trials for breast cancer patients176, 197, 198. In a brain tumor study, coordinate activation of sonic hedgehog (SHH) and PI3K signaling was found in PTEN-deficient glioblastoma; combination of BKM120, a pan-PI3K inhibitor, and LED225, a smoothened (SMO) inhibitor that blocks SHH signaling, resulted in synergistic anti-tumor effects199. Poly-(ADP-ribose) polymerase (PARP) and PI3K inhibitors have been found to cooperate in prostate and triple-negative breast cancers200-202. It appears that PI3K inhibition downregulates BRCA1 and BRCA2, impairing homologous recombination and sensitizing BRCA-wildtype cancer cells to PARP inhibition. Another attractive approach is combination of PI3K-targeted agents with drugs that suppress anti-apoptotic factors. B cell lymphoma 2 (BCL2), myeloid cell leukemia sequence 1 (MCL1), and other pro-survival proteins are frequently upregulated in cancer, and may explain why PI3K inhibition is often cytostatic in tumor cells. BCL2 or MCL1 suppression may induce cytotoxicity in response to PI3K inhibition203. Finally, an emerging approach is to combine PI3K inhibitors with agents that disrupt cell cycle machinery204. The p16-Cyclin D-cyclin-dependent kinase 4 (CDK4)-CDK6 pathway is frequently dysregulated in cancer. A number of CDK4 and CDK6 inhibitors, including LEE011 and palbociclib (PD0332991), are entering clinical trials for combination with pan- or p110α-selective inhibitors. Such rational combination therapies will be required to increase the success of PI3K inhibitors.
Conclusions and perspective
Targeting the PI3K pathway remains both an opportunity and a challenge for cancer therapy. Recent advances have provided the framework and rationale for inhibiting select class I PI3K catalytic isoforms. We have learned a great deal about the divergent roles of these isoforms in different signaling contexts, and are beginning to understand the importance of each isoform in various tissues, compartments, and cancer types. These findings have informed preclinical and clinical studies with isoform-selective PI3K agents, which offer improved specificity and reduced toxicity over first-generation pan-PI3K drugs. Isoform-selective PI3K inhibitors have seen promising success in early- and late-stage clinical trials for solid and hematological malignancies, highlighting the potential for isoform-selective PI3K therapeutics.
Although we have made substantial progress, further efforts are needed. We have only recently begun to appreciate the importance of class I regulatory isoforms in tumorigenesis. The different ways in which p85 subunits contribute to cancer, and the effective means to pharmacologically inhibit these mechanisms, are still not fully understood. Similarly, while a recent study indicates that class II isoform PI3K-C2α is important for pathophysiological angiogenesis, the roles of class II and III PI3Ks in cancer remain unclear.
For the class I catalytic isoforms, we must continue to precisely define the disease settings in which different PI3K isoforms will need to be targeted. To better inform isoform-selective therapeutic strategies, a set of biomarkers to predict the active p110 isoforms in a given tumor would be ideal, but development of this will require systematic studies. Continued work to understand the underlying cellular programs that protect tumors with aberrant PI3K activation from PI3K-targeted therapy will also be important. This will allow for better rational design of combination therapies, which will be necessary to overcome compensatory pathway activation and acquired resistance mechanisms and maximize the anti-tumor activity of PI3K inhibitors. Dosing strategies will also need to be carefully considered, as recent studies suggest that in some cases pulsatile inhibition may reduce toxicity without sacrificing efficacy. Progress in these areas should increase the effectiveness of PI3K-directed therapies in the clinic.
Supplementary Material
Key points.
Oncogenic mutation of PI3K catalytic isoform p110α is frequent in human cancers, while catalytic isoforms p110β, p110δ, and p110γ are rarely mutated but can be overexpressed. Mutation or loss of expression of regulatory isoform p85α is also associated with cancer.
Although class IA PI3K catalytic isoforms share structural and substrate similarities, they have distinct roles in mediating PI3K signaling in different physiological and oncogenic contexts.
Cancer cells with upregulation or mutation of receptor tyrosine kinases (RTKs), oncogenic RAS mutations, or activating p110α mutations are highly dependent on p110α, even in the presence of mutation or loss of PTEN.
In many cases, tumorigenesis driven by PTEN loss depends on p110β. However, PI3K isoform dependence in PTEN-deficient transformation may be governed by other PI3K isoforms that are dominant in a tissue or compartment, or shifted by coexisting oncogenic mutations.
Isoforms p110α, p110δ, and p110γ bind to and are activated by RAS subfamily GTPases, while p110β binds and is activated by RHO subfamily GTPases RAC1 and CDC42.
Non-isoform-selective pan-PI3K inhibitors have not yielded exciting clinical results, but second-generation PI3K drugs targeting individual PI3K isoforms may be able to achieve greater therapeutic efficacy by offering improved specificity and reduced toxicity.
The p110δ-selective inhibitor idelalisib has been remarkably effective in clinical trials for patients with B cell malignancies, while p110α-selective inhibitors have shown promise in early phase trials for patients with solid tumors bearing PIK3CA mutations or HER2 amplification.
Intrinsic and acquired resistance mechanisms are an ongoing challenge for PI3K-directed therapeutic approaches. To overcome this, combination therapies or alternative dosing strategies are being developed and evaluated in both preclinical and clinical settings.
Acknowledgments
We would like to thank T.M. Roberts for critical reading of the manuscript, T.M. Roberts and L.C. Cantley for helpful discussions, and N. Rosen and J.A. Engelman for sharing relevant and important unpublished observations. We also thank the reviewers for their helpful suggestions. We apologize to the many colleagues whose excellent work we were unable cite due to space limitations. Research in the laboratory of J.J.Z. is supported by US National Institutes of Health (NIH) Grants CA172461-01, P50 CA168504-01A1, P50 CA165962-01A1 and Stand Up to Cancer Dream Team Translational Research Grant SU2C-AACR-DT0209.
Glossary
- Myristoylated
irreversible co-translational modification of proteins in which a myristoyl group is covalently attached to an N-terminal amino acid of a nascent polypeptide, promoting membrane localization of the modified protein
- Congenital mosaic overgrowth syndromes
a clinically heterogeneous group of genetic disorders characterized by abnormal progressive localized growth. They are caused by diverse somatic mutations and associated with increased cancer risk
- Inter-SH2 (iSH2) domain
the domain of p85 regulatory PI3K isoforms that is located between the C- and N-terminal SH2 domains and directly interacts with class IA p110 catalytic isoforms
- Megalencephaly syndromes
a collection of sporadic overgrowth disorders characterized by enlarged brain size and other distinct features
- SH2 domain
SRC homology 2 domain; a structurally conserved protein–protein interaction domain that facilitates interaction with phosphorylated tyrosine residues on other proteins
- RAS superfamily proteins
small monomeric membrane-associated GTPases, which are divided into the RAS, RHO, RAB, ARF, and RAN subfamilies based on structure and function
- RAS GTPases
subfamily of RAS superfamily GTPases that plays critical roles in signal transduction. In mammals, the three major RAS subfamily members are HRAS, KRAS, and NRAS
- RHO GTPases
subfamily of RAS superfamily proteins that shares similar roles in signal transduction to RAS GTPases and is best characterized for the regulation of cell shape, movement, and polarity
Biographies
Lauren M. Thorpe received her BS in Biological Sciences in 2008 from Carnegie Mellon University, and is now completing her PhD at Harvard University in the lab of Dr. Jean J. Zhao. There, her work has focused on understanding the role of p85 regulatory isoforms in modulating physiological and pathophysiological PI3K signals.
Haluk Yuzugullu is a postdoctoral fellow in the laboratory of Dr. Jean J. Zhao at the Dana-Farber Cancer Institute and Harvard Medical School Department of Biological Chemistry & Molecular Pharmacology, Massachusetts, USA. His research focuses on cancer cell signaling and discovery of potential drug targets using mouse models of cancer.
Jean J. Zhao is an Associate Professor in the Department of Cancer Biology at Dana-Farber Cancer Institute (DFCI) and the Department of Biological Chemistry & Molecular Pharmacology at Harvard Medical School, Boston, USA. She received her PhD from Tufts Medical School, Boston, USA, and did postdoctoral research with Thomas Roberts at DFCI. Her research centers on understanding the roles of key kinases, phosphatidylinositol 3-kinase in particular, in normal tissue physiology and cancer pathogenesis. Her lab's homepage can be found at http://jeanzhao.dfci.harvard.edu/.
Footnotes
Links to web sites: https://www.clinicaltrials.gov/
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–41. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 4.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 5.Mellor P, Furber LA, Nyarko JN, Anderson DH. Multiple roles for the p85alpha isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem J. 2012;441:23–37. doi: 10.1042/BJ20111164. [DOI] [PubMed] [Google Scholar]
- 6.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–30. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 7.Falasca M, Maffucci T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J. 2012;443:587–601. doi: 10.1042/BJ20120008. [DOI] [PubMed] [Google Scholar]
- 8.Falasca M, et al. The role of phosphoinositide 3-kinase C2alpha in insulin signaling. J Biol Chem. 2007;282:28226–36. doi: 10.1074/jbc.M704357200. [DOI] [PubMed] [Google Scholar]
- 9.Maffucci T, et al. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol. 2005;169:789–99. doi: 10.1083/jcb.200408005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshioka K, et al. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med. 2012;18:1560–9. doi: 10.1038/nm.2928. [DOI] [PubMed] [Google Scholar]
- 11.Franco I, et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell. 2014;28:647–58. doi: 10.1016/j.devcel.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schu PV, et al. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- 13.Volinia S, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995;14:3339–48. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 15.Blondeau F, et al. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. 2000;9:2223–9. doi: 10.1093/oxfordjournals.hmg.a018913. [DOI] [PubMed] [Google Scholar]
- 16.Lu N, et al. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012;10:e1001245. doi: 10.1371/journal.pbio.1001245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velichkova M, et al. Drosophila Mtm and class II PI3K coregulate a PI(3)P pool with cortical and endolysosomal functions. J Cell Biol. 2010;190:407–25. doi: 10.1083/jcb.200911020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao C, Backer JM, Laporte J, Bedrick EJ, Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–46. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol. 2004;15:171–6. doi: 10.1016/j.semcdb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 20.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 21.Chang HW, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–50. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 22.Klippel A, et al. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–27. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao JJ, et al. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–8. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao JJ, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–95. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 25.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 26.Isakoff SJ, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 27.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–7. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu P, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011;17:1116–20. doi: 10.1038/nm.2402. In ref. 29, an inducible GEMM of PIK3CAH1047R-driven mammary tumors was used to identify potential mechanisms of resistance to PI3K-targeted therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan W, et al. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene. 2013;32:318–26. doi: 10.1038/onc.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinross KM, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–7. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu R, et al. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. Am J Pathol. 2013;182:1391–9. doi: 10.1016/j.ajpath.2012.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 34.Zhao L, Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010;9:596–600. doi: 10.4161/cc.9.3.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miled N, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 36.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA) Proc Natl Acad Sci U S A. 2012;109:15259–64. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hao Y, et al. Gain of interaction with IRS1 by p110alpha-helical domain mutants is crucial for their oncogenic functions. Cancer Cell. 2013;23:583–93. doi: 10.1016/j.ccr.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–7001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orloff MS, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. 2013;92:76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurek KC, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–15. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riviere JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rios JJ, et al. Somatic gain-of-function mutations in PIK3CA in patients with macrodactyly. Hum Mol Genet. 2013;22:444–51. doi: 10.1093/hmg/dds440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Angulo I, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–71. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lucas CL, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15:88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 46.Dbouk HA, et al. Characterization of a tumor-associated activating mutation of the p110beta PI 3-kinase. PLoS One. 2013;8:e63833. doi: 10.1371/journal.pone.0063833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dbouk HA, Pang H, Fiser A, Backer JM. A biochemical mechanism for the oncogenic potential of the p110beta catalytic subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2010;107:19897–902. doi: 10.1073/pnas.1008739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, et al. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell. 2011;41:567–78. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogt PK. PI3K p110beta: more tightly controlled or constitutively active? Mol Cell. 2011;41:499–501. doi: 10.1016/j.molcel.2011.02.017. In refs. 47 and 48, biochemical and structural studies were used to demonstrate that p85 inhibition of p110β is different from that of p110α. Ref. 49 provides commentary on how these two studies together may indicate that p110β is a more basally active isoform. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer C, et al. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003;63:1667–75. [PubMed] [Google Scholar]
- 51.Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289–94. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Philp AJ, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–9. [PubMed] [Google Scholar]
- 53.Cheung LW, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–85. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urick ME, et al. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011;71:4061–7. doi: 10.1158/0008-5472.CAN-11-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaiswal BS, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–74. doi: 10.1016/j.ccr.2009.10.016. Ref. 55, along with refs. 53 and 58, demonstrates the transforming potential of cancer-associated iSH2 domain p85α mutants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cizkova M, et al. PIK3R1 underexpression is an independent prognostic marker in breast cancer. BMC Cancer. 2013;13:545. doi: 10.1186/1471-2407-13-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu H, et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci U S A. 2009;106:20258–63. doi: 10.1073/pnas.0902369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010;107:15547–52. doi: 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taniguchi CM, et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 2010;70:5305–15. doi: 10.1158/0008-5472.CAN-09-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo J, et al. Modulation of epithelial neoplasia and lymphoid hyperplasia in PTEN+/- mice by the p85 regulatory subunits of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2005;102:10238–43. doi: 10.1073/pnas.0504378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it's implication in cancer. Cell Cycle. 2005;4:1309–12. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 63.Cortes I, et al. p85beta phosphoinositide 3-kinase subunit regulates tumor progression. Proc Natl Acad Sci U S A. 2012;109:11318–23. doi: 10.1073/pnas.1118138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biswas K, et al. Essential role of class II phosphatidylinositol-3-kinase-C2alpha in sphingosine 1-phosphate receptor-1-mediated signaling and migration in endothelial cells. J Biol Chem. 2013;288:2325–39. doi: 10.1074/jbc.M112.409656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Katso RM, et al. Phosphoinositide 3-Kinase C2beta regulates cytoskeletal organization and cell migration via Rac-dependent mechanisms. Mol Biol Cell. 2006;17:3729–44. doi: 10.1091/mbc.E05-11-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elis W, et al. Down-regulation of class II phosphoinositide 3-kinase alpha expression below a critical threshold induces apoptotic cell death. Mol Cancer Res. 2008;6:614–23. doi: 10.1158/1541-7786.MCR-07-0262. [DOI] [PubMed] [Google Scholar]
- 67.Diouf B, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–303. doi: 10.1038/nm.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003;13:507–18. doi: 10.1111/j.1750-3639.2003.tb00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rao SK, Edwards J, Joshi AD, Siu IM, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol. 2010;96:169–79. doi: 10.1007/s11060-009-9959-4. [DOI] [PubMed] [Google Scholar]
- 70.Nobusawa S, et al. Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol. 2010;20:936–44. doi: 10.1111/j.1750-3639.2010.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu P, et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33:1270–6. doi: 10.1093/carcin/bgs148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harada K, Truong AB, Cai T, Khavari PA. The class II phosphoinositide 3-kinase C2beta is not essential for epidermal differentiation. Mol Cell Biol. 2005;25:11122–30. doi: 10.1128/MCB.25.24.11122-11130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris DP, et al. Requirement for class II phosphoinositide 3-kinase C2alpha in maintenance of glomerular structure and function. Mol Cell Biol. 2011;31:63–80. doi: 10.1128/MCB.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Norris FA, Atkins RC, Majerus PW. The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J Biol Chem. 1997;272:23859–64. doi: 10.1074/jbc.272.38.23859. [DOI] [PubMed] [Google Scholar]
- 75.Gewinner C, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–25. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fedele CG, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010;107:22231–6. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stjernstrom A, et al. Alterations of INPP4B, PIK3CA and pAkt of the PI3K pathway are associated with squamous cell carcinoma of the lung. Cancer Med. 2014 doi: 10.1002/cam4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodgson MC, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–82. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirsch DS, Shen Y, Dokmanovic M, Wu WJ. pp60c-Src phosphorylates and activates vacuolar protein sorting 34 to mediate cellular transformation. Cancer Res. 2010;70:5974–83. doi: 10.1158/0008-5472.CAN-09-2682. [DOI] [PubMed] [Google Scholar]
- 80.Denley A, Gymnopoulos M, Kang S, Mitchell C, Vogt PK. Requirement of phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol Cancer Res. 2009;7:1132–8. doi: 10.1158/1541-7786.MCR-09-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wei Y, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–84. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13:169–72. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 83.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274:10963–8. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 84.Ciraolo E, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3. doi: 10.1126/scisignal.1161577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ciraolo E, et al. Essential role of the p110beta subunit of phosphoinositide 3-OH kinase in male fertility. Mol Biol Cell. 2010;21:704–11. doi: 10.1091/mbc.E09-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clayton E, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–63. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ali K, et al. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature. 2004;431:1007–11. doi: 10.1038/nature02991. [DOI] [PubMed] [Google Scholar]
- 88.Okkenhaug K, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297:1031–4. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 89.Jou ST, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–91. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sasaki T, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–6. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 91.Yum HK, et al. Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol. 2001;167:6601–8. doi: 10.4049/jimmunol.167.11.6601. [DOI] [PubMed] [Google Scholar]
- 92.Martin AL, Schwartz MD, Jameson SC, Shimizu Y. Selective regulation of CD8 effector T cell migration by the p110 gamma isoform of phosphatidylinositol 3-kinase. J Immunol. 2008;180:2081–8. doi: 10.4049/jimmunol.180.4.2081. [DOI] [PubMed] [Google Scholar]
- 93.Rameh LE, Chen CS, Cantley LC. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell. 1995;83:821–30. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- 94.Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1998;273:30199–203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 95.Yu J, et al. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–87. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Utermark T, et al. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 2012;26:1573–86. doi: 10.1101/gad.191973.112. Ref. 96 uses GEMMs of oncogenic RTK-driven mammary tumors to identify a novel mechanism in which p110β competes with p110α for RTK binding to modulate lipid kinase activity in response to RTK activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao JJ, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103:16296–300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knight ZA, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Foukas LC, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–70. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 100.Sopasakis VR, et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010;11:220–30. doi: 10.1016/j.cmet.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Graupera M, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–6. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 102.Jia S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guillermet-Guibert J, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A. 2008;105:8292–7. doi: 10.1073/pnas.0707761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaussade C, et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J. 2007;404:449–58. doi: 10.1042/BJ20070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Papakonstanti EA, et al. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121:4124–33. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 106.Vanhaesebroeck B, et al. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat Cell Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 107.Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A. 2007;104:7809–14. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meier TI, et al. Cloning, expression, purification, and characterization of the human Class Ia phosphoinositide 3-kinase isoforms. Protein Expr Purif. 2004;35:218–24. doi: 10.1016/j.pep.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 109.Beeton CA, Chance EM, Foukas LC, Shepherd PR. Comparison of the kinetic properties of the lipid- and protein-kinase activities of the p110alpha and p110beta catalytic subunits of class-Ia phosphoinositide 3-kinases. Biochem J. 2000;350 Pt 2:353–9. [PMC free article] [PubMed] [Google Scholar]
- 110.Schmid MC, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715–27. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bohnacker T, et al. PI3Kgamma adaptor subunits define coupling to degranulation and cell motility by distinct PtdIns(3,4,5)P3 pools in mast cells. Sci Signal. 2009;2:ra27. doi: 10.1126/scisignal.2000259. [DOI] [PubMed] [Google Scholar]
- 112.Voigt P, Dorner MB, Schaefer M. Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J Biol Chem. 2006;281:9977–86. doi: 10.1074/jbc.M512502200. [DOI] [PubMed] [Google Scholar]
- 113.Shymanets A, et al. p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J Biol Chem. 2013;288:31059–68. doi: 10.1074/jbc.M113.508234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kurig B, et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc Natl Acad Sci U S A. 2009;106:20312–7. doi: 10.1073/pnas.0905506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brock C, et al. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J Cell Biol. 2003;160:89–99. doi: 10.1083/jcb.200210115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stoyanov B, et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–3. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- 117.Maier U, Babich A, Nurnberg B. Roles of non-catalytic subunits in gbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem. 1999;274:29311–7. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- 118.Suire S, et al. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr Biol. 2005;15:566–70. doi: 10.1016/j.cub.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 119.Stephens LR, et al. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–14. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 120.Vadas O, et al. Molecular determinants of PI3Kgamma-mediated activation downstream of G-protein-coupled receptors (GPCRs) Proc Natl Acad Sci U S A. 2013;110:18862–7. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schmid MC, et al. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS One. 2013;8:e60226. doi: 10.1371/journal.pone.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kubo H, Hazeki K, Takasuga S, Hazeki O. Specific role for p85/p110beta in GTP-binding-protein-mediated activation of Akt. Biochem J. 2005;392:607–14. doi: 10.1042/BJ20050671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kurosu H, et al. Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem. 1997;272:24252–6. doi: 10.1074/jbc.272.39.24252. [DOI] [PubMed] [Google Scholar]
- 124.Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase beta in signaling from G protein-coupled receptors to Akt. J Biol Chem. 2000;275:12069–73. doi: 10.1074/jbc.275.16.12069. [DOI] [PubMed] [Google Scholar]
- 125.Dbouk HA, et al. G protein-coupled receptor-mediated activation of p110beta by Gbetagamma is required for cellular transformation and invasiveness. Sci Signal. 2012;5:ra89. doi: 10.1126/scisignal.2003264. Ref. 125 uses HDX-MS to identify the site on p110β responsible for Gβγ binding, which is not conserved among other class IA p110 isoforms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saudemont A, et al. p110gamma and p110delta isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc Natl Acad Sci U S A. 2009;106:5795–800. doi: 10.1073/pnas.0808594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reif K, et al. Cutting edge: differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173:2236–40. doi: 10.4049/jimmunol.173.4.2236. [DOI] [PubMed] [Google Scholar]
- 128.Durand CA, et al. Phosphoinositide 3-kinase p110 delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol. 2009;183:5673–84. doi: 10.4049/jimmunol.0900432. [DOI] [PubMed] [Google Scholar]
- 129.Ballou LM, Chattopadhyay M, Li Y, Scarlata S, Lin RZ. Galphaq binds to p110alpha/p85alpha phosphoinositide 3-kinase and displaces Ras. Biochem J. 2006;394:557–62. doi: 10.1042/BJ20051493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ballou LM, Lin HY, Fan G, Jiang YP, Lin RZ. Activated G alpha q inhibits p110 alpha phosphatidylinositol 3-kinase and Akt. J Biol Chem. 2003;278:23472–9. doi: 10.1074/jbc.M212232200. [DOI] [PubMed] [Google Scholar]
- 131.Yeung WW, Wong YH. Galpha16 interacts with Class IA phosphatidylinositol 3-kinases and inhibits Akt signaling. Cell Signal. 2010;22:1379–87. doi: 10.1016/j.cellsig.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 132.Rodriguez-Viciana P, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 133.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–51. [PMC free article] [PubMed] [Google Scholar]
- 134.Rubio I, Rodriguez-Viciana P, Downward J, Wetzker R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem J. 1997;326(Pt 3):891–5. doi: 10.1042/bj3260891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pacold ME, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–43. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 136.Suire S, Hawkins P, Stephens L. Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol. 2002;12:1068–75. doi: 10.1016/s0960-9822(02)00933-8. [DOI] [PubMed] [Google Scholar]
- 137.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–7. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561–74. doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- 139.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 140.Castellano E, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–30. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gritsman K, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J Clin Invest. 2014;124:1794–809. doi: 10.1172/JCI69927. Refs. 140 and 141 use GEMMs to demonstrate the specific importance of p110α in the development and maintenance of RAS-driven cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Suire S, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–9. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- 143.Fritsch R, et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153:1050–63. doi: 10.1016/j.cell.2013.04.031. In ref. 143, extensive biochemical studies identify RHO but not RAS small GTPases as directly binding and activating p110β via its RBD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vanhaesebroeck B, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94:4330–5. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murphy GA, et al. Involvement of phosphatidylinositol 3-kinase, but not RalGDS, in TC21/R-Ras2-mediated transformation. J Biol Chem. 2002;277:9966–75. doi: 10.1074/jbc.M109059200. [DOI] [PubMed] [Google Scholar]
- 146.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–54. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Delgado P, et al. Essential function for the GTPase TC21 in homeostatic antigen receptor signaling. Nat Immunol. 2009;10:880–8. doi: 10.1038/ni.1749. [DOI] [PubMed] [Google Scholar]
- 148.Klarlund JK, et al. Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science. 1997;275:1927–30. doi: 10.1126/science.275.5308.1927. [DOI] [PubMed] [Google Scholar]
- 149.Welch HC, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–21. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 150.Krugmann S, et al. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol Cell. 2002;9:95–108. doi: 10.1016/s1097-2765(02)00434-3. [DOI] [PubMed] [Google Scholar]
- 151.Liaw D, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 152.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 153.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 154.Kwabi-Addo B, et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A. 2001;98:11563–8. doi: 10.1073/pnas.201167798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang S, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 156.Alimonti A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ni J, et al. Functional characterization of an isoform-selective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2012;2:425–33. doi: 10.1158/2159-8290.CD-12-0003. Ref. 157 identifies a novel p110β-selective inhibitor and demonstrates its effectiveness against PTEN-deficient tumors in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Torbett NE, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97–110. doi: 10.1042/BJ20080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wee S, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105:13057–62. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jia S, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Berenjeno IM, et al. Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J. 2012;442:151–9. doi: 10.1042/BJ20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schmit F, et al. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A. 2014;111:6395–400. doi: 10.1073/pnas.1323004111. Ref. 162 uses GEMMs to demonstrate that concomitant expression of oncogenic RAS can shift the isoform reliance of PTEN-deficient tumors from p110β to p110α. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang Q, Weisberg E, Zhao JJ. The gene dosage of class Ia PI3K dictates the development of PTEN hamartoma tumor syndrome. Cell Cycle. 2013;12:3589–93. doi: 10.4161/cc.26812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Q, et al. Spatially distinct roles of class Ia PI3K isoforms in the development and maintenance of PTEN hamartoma tumor syndrome. Genes Dev. 2013;27:1568–80. doi: 10.1101/gad.216069.113. Ref. 164 identifies distinct roles for both p110α and p110β in epidermal compartments of a GEMM of PTEN hamartoma tumor syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Subramaniam PS, et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell. 2012;21:459–72. doi: 10.1016/j.ccr.2012.02.029. In ref. 165, GEMMs are used to show that both p110δ and p110γ contribute to T-ALL driven by PTEN loss. [DOI] [PubMed] [Google Scholar]
- 166.Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013;19:3533–44. doi: 10.1158/1078-0432.CCR-12-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–53. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 168.Raynaud FI, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009;8:1725–38. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 170.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–56. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Furman RR, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gopal AK, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–18. doi: 10.1056/NEJMoa1314583. Refs. 171 and 172 report dramatic success in the treatment of patients with B cell malignancies with the p110δ-selective inhibitor idelalisib. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fruman DA, Cantley LC. Idelalisib--a PI3Kdelta inhibitor for B-cell cancers. N Engl J Med. 2014;370:1061–2. doi: 10.1056/NEJMe1400055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vanhaesebroeck B, Khwaja A. PI3Kdelta inhibition hits a sensitive spot in B cell malignancies. Cancer Cell. 2014;25:269–71. doi: 10.1016/j.ccr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 175.Ali K, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014 doi: 10.1038/nature13444. Ref. 175 demonstrates that p110δ inactivation inhibits the growth of certain solid tumors by blocking regulatory T cells-mediated immune suppression in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Juric D, et al. Abstract PD1-3: Ph1b study of the PI3K inhibitor GDC-0032 in combination with fulvestrant in patients with hormone receptor-positive advanced breast cancer. Cancer Research. 2013;73:PD1–3. [Google Scholar]
- 177.Busaidy NL, et al. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 2012;30:2919–28. doi: 10.1200/JCO.2011.39.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sampath D, et al. Abstract P4-15-02: The PI3K inhibitor GDC-0032 enhances the efficacy of standard of care therapeutics in PI3K alpha mutant breast cancer models. Cancer Research. 2013;73:P4-15-02. [Google Scholar]
- 181.O'Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Britschgi A, et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 183.Muranen T, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Elkabets M, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. Ref. 184 identifies mTOR pathway activation as a mechanism of resistance to p110α-selective therapy that can be overcome by mTORC1 inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chandarlapaty S, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. Ref. 185 shows that PI3K-AKT pathway inhibition induces a feedback loop leading to overexpression and activation of multiple RTKs that can be overcome by combined PI3K and RTK inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sergina NV, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Garrett JT, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108:5021–6. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Garrett JT, et al. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013;73:6013–23. doi: 10.1158/0008-5472.CAN-13-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tao JJ, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7:ra29. doi: 10.1126/scisignal.2005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Serra V, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–57. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Will M, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014;4:334–47. doi: 10.1158/2159-8290.CD-13-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Muellner MK, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–93. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tenbaum SP, et al. beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med. 2012;18:892–901. doi: 10.1038/nm.2772. In ref. 194, WNT/β-catenin pathway hyperactivation is identified as a potential mechanism contributing to resistance to PI3K pathway inhibition and metastasis in colon cancer. [DOI] [PubMed] [Google Scholar]
- 195.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 197.Juric D, et al. Phase I study of BYL719, an alpha-specific PI3K inhibitor, in patients with PIK3CA mutant advanced solid tumors: preliminary efficacy and safety in patients with PIK3CA mutant ER-positive (ER+) metastatic breast cancer (MBC) Cancer Research. 2012;72:P6-10-07. [Google Scholar]
- 198.Juric D, et al. Abstract P2-16-14: Preliminary safety, pharmacokinetics and anti-tumor activity of BYL719, an alpha-specific PI3K inhibitor in combination with fulvestrant: Results from a phase I study. Cancer Research. 2013;73:P2-16-14. [Google Scholar]
- 199.Gruber Filbin M, et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat Med. 2013;19:1518–23. doi: 10.1038/nm.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Juvekar A, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048–63. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ibrahim YH, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–47. doi: 10.1158/2159-8290.CD-11-0348. Refs. 200 and 201 demonstrate that PI3K inhibition leads to BRCA1/2 downregulation and subsequent sensitization to PARP inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gonzalez-Billalabeitia E, et al. Vulnerabilities of PTEN-p53-deficient prostate cancers to compound PARP/PI3K inhibition. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rahmani M, et al. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3-and Bim-dependent mechanism. Cancer Res. 2013;73:1340–51. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vora SR, et al. CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell. 2014;26:136–49. doi: 10.1016/j.ccr.2014.05.020. In ref. 204, a combinatorial drug screen identifies synergistic antitumor activity of CDK4/6 and p110α-selective inhibitors in PIK3CA mutant breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.