

Abstract
The morbidity and mortality from coronary artery disease (CAD) remain significant worldwide. The treatment for acute myocardial infarction has improved over the past decades, including early reperfusion of culprit coronary arteries. Although it is mandatory to reperfuse the ischaemic territory as soon as possible, paradoxically this leads to additional myocardial injury, namely ischaemia/reperfusion (I/R) injury, in which redox stress plays a pivotal role and for which no effective therapy is currently available. In this review, we report evidence that the redox environment plays a pivotal role not only in I/R injury but also in cardioprotection. In fact, cardioprotective strategies, such as pre- and post-conditioning, result in a robust reduction in infarct size in animals and the role of redox signalling is of paramount importance in these conditioning strategies. Nitrosative signalling and cysteine redox modifications, such as S-nitrosation/S-nitrosylation, are also emerging as very important mechanisms in conditioning cardioprotection. The reasons for the switch from protective oxidative/nitrosative signalling to deleterious oxidative/nitrosative/nitrative stress are not fully understood. The complex regulation of this switch is, at least in part, responsible for the diminished or lack of cardioprotection induced by conditioning protocols observed in ageing animals and with co-morbidities as well as in humans. Therefore, it is important to understand at a mechanistic level the reasons for these differences before proposing a safe and useful transition of ischaemic or pharmacological conditioning. Indeed, more mechanistic novel therapeutic strategies are required to protect the heart from I/R injury and to improve clinical outcomes in patients with CAD.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Enzymesd |
| Angiotensin AT1 receptor | ACE |
| Bradykinin B2 receptor | AMPK |
| Ligand-gated ion channelsb | Cytochrome P450 |
| Ryanodine receptor | Endothelial (e) NOS (NOS3) |
| Ion channelsc | F0/F1 ATPase |
| Connexins | Histone deactylases (HDACs) |
| KATP channels | Inducible (i) NOS (NOS2) |
| Lipoxygenase | |
| Monoamine oxidase (MAO) | |
| MMPs | |
| Neuronal (n) NOS (NOS1) | |
| P38 MAPK | |
| PDE | |
| PI3K | |
| PKA | |
| PKG1 | |
| SERCA | |
| Soluble guanylylcyclase (sGC) |
| LIGANDS | |
|---|---|
| Adenosine | Folic acid |
| Adiponectin | GSH |
| AMP | H2O2 |
| Angiotensin II | Insulin |
| Atorvastatin | NADP |
| ATP | NADPH |
| Bradykinin | Nitric oxide (NO) |
| Calmodulin | ODQ |
| cGMP | PAF |
| Diazoxide | PDGF |
| Endothelin-1 | Prostacyclin (PGI2) |
| Enalaprilat | Tetrahydrobiopterin |
| FAD | Vitamin C |
| FMN |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d,,,).
Introduction
The prevalence of chronic and degenerative diseases, including cardiovascular disease (CVD), is increasing worldwide. Acute coronary syndromes, including myocardial infarction, are the most typical examples of pathologies with a strong redox-sensitive component and remain the leading cause of death in Western countries despite significant progress in primary and secondary prevention and treatment strategies. The only way to treat acute myocardial infarction (AMI) is to promptly restore blood flow to the ischaemic tissue. However, reperfusion is associated with further injury and the duration of the precedent ischaemia is an important determinant of the extent of reperfusion damage (Ferdinandy and Schulz, 2003; Hoffman et al., 2004). Reactive oxygen species/reactive nitrogen species (ROS/RNS) production, which may continue for hours after the beginning of reperfusion, plays an important role in the genesis of reperfusion injury (Bolli et al., 1989,1989) and recruitment of inflammatory cells (Simoons et al., 1986). This is known as ischaemia/reperfusion (I/R) injury (Penna et al., 2009; Pagliaro et al., 2011; Perrelli et al., 2011; Tullio et al., 2013). Oxidative and nitrosative/nitrative stress contribute to the cascade of events leading to cell death in acute coronary syndrome, and increased production of ROS/RNS can modify the expression of several inflammatory mediators during heart injury. Here, we use the word ‘stress’ with a negative connotation, as it will lead to tissue damage. This is somewhat different from the chemical terminology (see Heinrich et al., 2013). When ROS/RNS are protective or involved in physiological mechanisms, we speak of ‘redox signalling’ (see below).
In coronary disease, inflammatory processes are involved in a vicious cycle of deleterious events that lead to cardiac damage, including further oxidative stress, impairing both diastolic and systolic function (Toyokuni, 1999). Thus, not only ischaemic damage, but also reperfusion, can produce cardiac injury and dysfunction. Moreover, I/R injury alters cell excitability and their conduction system, leading to arrhythmias via several mechanisms (Majidi et al., 2009; Lopes et al., 2012). Reperfusion also damages the vascular endothelium, leading to changes in the endothelial structure (e.g. expression of molecules of adhesion) and alignment, with capillary leakage and alterations in blood cells and microembolization, as well as vascular compression due to myocyte swelling.
There is no doubt that acute coronary syndrome and redox biology are entangled and concomitant diseases (co-morbidities), making the condition more complicated. Ageing is a powerful predictor of adverse events following coronary syndrome and reperfusion strategies (Eagle et al., 2004; Schröder et al., 2013). Moreover, it is not clear why older patients continue to have poor outcomes in this syndrome despite improved access to contemporary treatment (Gale et al., 2012). One of the reasons may be the exacerbation of redox-dependent reperfusion injury in these conditions and in the presence of co-morbidities, such as metabolic syndrome, diabetes and cardiac hypertrophy (Giricz et al., 2006; 2009,; Ferdinandy et al., 2007; Görbe et al., 2011; Kocsis et al., 2012; Csonka et al., 2014). Besides increasing ROS/RNS production, ischaemia and reperfusion have also been found to reduce the levels of antioxidant enzymes such as glutathione peroxidase, and superoxide dismutase (SOD) and non-enzymatic antioxidant such as ascorbate and GSH (Vaage et al., 1997). Moreover, I/R alters the function of several enzymes, some of which play an important role in the redox balance of the cell (Penna et al., 2011b; Tullio et al., 2013). Therefore, reduced scavenging and altered enzymatic function further contribute to the development of oxidative stress.
Many hopes were pinned on the use of antioxidants, but results have been disappointing, so far (see the Bench to bedside section). Therefore, there is an urgent need to better understand the biology and the damage caused by I/R and redox stress before considering an appropriate treatment. Clearly, the same level of radical/reactive species may be protective or deleterious, depending on a variety of conditions, including co-morbidities, the protective antioxidant enzyme defence system and reparative process. I/R damage can be reduced by ischaemic preconditioning (I-PreC) (Hoffman et al., 2004; Hausenloy and Yellon, 2007; Hausenloy et al., 2007; 2013,; Murphy and Steenbergen, 2008; Di Lisa et al., 2011) and by ischaemic post-conditioning (I-PostC) (Hausenloy and Yellon, 2007; 2008,; Hausenloy et al., 2007; 2013,; Ivanes et al., 2011; Pagliaro et al., 2011). The mechanisms underlying protection by I-PreC and I-PostC are incompletely understood (Penna et al., 2008a; 2009,; Di Lisa et al., 2011; Pagliaro et al., 2011; Perrelli et al., 2011; Tullio et al., 2013). However, there are no doubts that ROS/RNS signalling plays a role in triggering and mediating these protective phenomena. It is likely that mitochondria have an important role in these processes. For instance, autacoids, formed during conditioning manoeuvres, can trigger protection by inducing activation of the mitochondrial K+ ATP channel (mitoKATP), which then induces the generation of ROS and RNS, both required for conditioning-induced protection (O'Rourke, 2000; Cohen et al., 2001; Forbes et al., 2001; Oldenburg et al., 2002; 2004,; Gucek and Murphy, 2010; Di Lisa et al., 2011; Murphy et al., 2012; 2014,; Penna et al., 2013a) (Figure 1). Notably, I-PreC can be mimicked by pharmacological interventions, including the administration of free radical donors/generators, NO donors, and even nitroxyl anion (HNO/NO−) and ONOO− donors (Wink et al., 1993; Pagliaro, 2003; Pagliaro et al., 2003; du Toit et al., 2008; Tocchetti et al., 2011), whereas pre- and post-conditioning can be blocked by radical scavengers (Penna et al., 2006b; Cohen et al., 2008). Although the cardioprotective effect of conditioning strategies have been proven in several species including humans, it seems that the presence of cardiovascular risk factors, ageing, co-morbidities and other concomitant medications may interfere with cardioprotective signalling pathways (for extensive reviews, see Ferdinandy et al., 2007; Ovize et al., 2013; Hausenloy et al., 2013). Therefore, there is a complex framework in which ROS/RNS can be deleterious (redox stress) or beneficial (redox signalling); a picture that is further complicated by the presence of co-morbidities.
Figure 1.
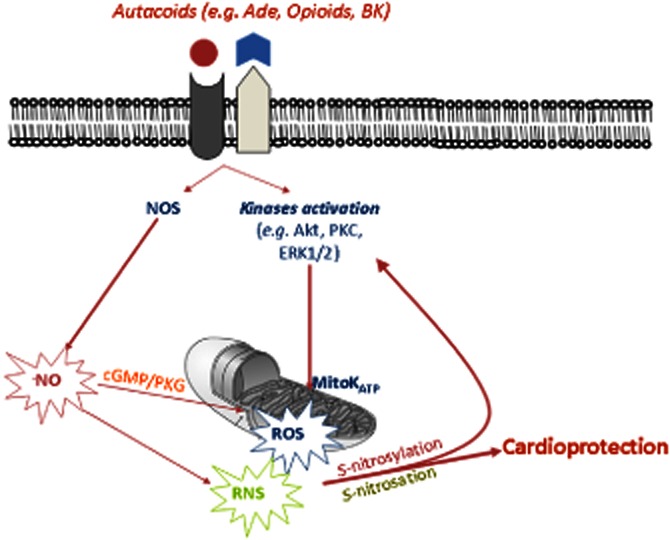
Reactive oxygen and nitrogen species (ROS/RNS) have been identified as a part of cardioprotective signalling molecules, which are essential in pre- and post-conditioning processes. S-nitrosation/S-nitrosylation of enzymes is a specific posttranslational modification that plays an important role in cardioprotection. Mitochondria are of paramount importance in either promoting or limiting ROS/RNS generation and reperfusion injury, and in triggering kinase activation by ROS/RNS signalling in cardioprotection. Ade, adenosine; BK, bradykinin. For other acronyms, see the abbreviation list.
In this review, we will first consider the molecular basis of ROS/RNS generation and the mechanisms of oxidative and nitrosative/nitrative stress. Then, we will consider how ROS/RNS may have a physiological/beneficial role (ROS/RNS signalling) and how ROS/RNS are involved in the process of cardioprotection, namely pre- and post-conditioning. Finally, the possibilities of clinical transition of the conditioning protocols are discussed.
Molecular basis of the biological function of ROS/RNS
The cellular redox status may be considered a continuum that ranges from reductive to oxidative conditions. Reactive species overflow can occur at either extreme (i.e. oxidized or reduced) of the redox potential. Therefore, the biological extremes of the redox spectrum play pivotal roles in disease pathogenesis. Here, we consider oxidative stress and signalling; for reductive stress (i.e. increased ratios of NADPH/NADP and GSH/GSSG), see the review of Brewer et al. (2013).
We propose to revise our approach towards the redox balance and to consider the production of ROS and RNS as a physiological phenomenon that is involved in the regulation of various cellular functions. Only under certain conditions this balanced production can ‘escape’ from the usual control and consequently induce damage via oxidative/nitrosative/nitrative stress. As such, oxidative stress and nitrosative/nitrative stress are disturbances in the oxidation/reduction state of the cell, in which inappropriate ROS/RNS production exceeds antioxidant defences. As a rule, ROS/RNS activity determines toxicity while decreasing signalling ability. However, exceptions exist.
ROS/RNS and oxidative/nitrosative signalling or stress
The reactive species
Before considering the sources of ROS/RNS, here we briefly review the biochemistry and biology of the reactive species. Although reactions to RNS will be considered in more detail, the reader is kindly redirected to more focused reviews on this topic (Espey et al., 2002; Becker, 2004; Martínez and Andriantsitohaina, 2009; Heinrich et al., 2013; Penna et al., 2014).
ROS
A reduction in molecular oxygen (O2) leads to the formation of chemically reactive species (known as ROS). These are the superoxide anion (O2−), hydrogen peroxide (H2O2) and the hydroxyl radical (HO•). Because of their intrinsic chemical properties, each ROS reacts with specific biological targets (amino acid and cysteine residues of proteins, lipids, DNA bases, etc.).
Superoxide anion is a by-product of mitochondrial respiration and is produced mainly by NADPH oxidases. In living cells, the steady-state concentration of O2− is very low (∼10−11 M), which reflects its instability; this is due to not only its reaction with the [Fe–S] cluster but also to spontaneous and SOD-mediated O2− dismutation to H2O2. The instability of O2− and its inability to diffuse through membranes because of its negative charge make this ROS a poor signalling molecule. However, since O2− can cross membranes via anion channels, it has often been suggested that it is responsible for the cardioprotection triggered in the pre- and post-conditioning scenario (see below).
Hydrogen peroxide is formed as a by-product of aerobic metabolism, superoxide formation and dismutation, and as a product of oxidase activity. Both excessive H2O2 and its decomposition product, HO•, formed in a metal-catalysed Fenton-type reaction, are harmful for most cell components. In biological systems, its toxicity is essentially due to its reduction to HO• by Fenton chemistry (Imlay, 2003). In fact, H2O2 is a poor oxidant and reacts mildly with [Fe–S], loosely binds to metals and very slowly to glutathione, Met and free Cys residues (Winterbourn and Metodiewa, 1999). As a consequence, H2O2 is relatively stable (intracellular t1/2 ∼ 1 ms and steady-state levels ∼10−7 M). Since the H2O2 reactivity towards Cys residues can increase significantly depending upon the protein environment, the diffusion of H2O2 might be modulated by changes in membrane permeability or by transport through aquaporins (Bienert et al., 2006). Its selective reactivity and diffusibility makes H2O2 the ideal candidate for redox signalling, as it reacts selectively throughout the cell. It can act as a second messenger in several signal transduction pathways, including immune cell activation, inflammation processes and cell proliferation. It has been suggested that H2O2 is a coronary metabolic dilator and couples myocardial oxygen consumption to coronary blood flow. It produces vasodilatation by the oxidation of intracellular thiols and activation of the p38 MAP kinase. Therefore, physiological coronary metabolic dilatation appears to be mediated by redox-dependent signals (Saitoh et al., 2007).
The reactivity of the hydroxyl radical is mainly non-specific (t1/2 ∼ 10−9 s), which makes it highly toxic and limits its diffusion to sites of production; despite this, HO• may operate in H2O2 sensing (Halliwell and Gutteridge, 1997). Thus, it is possible that HO• has cardioprotective properties, as its effects may be carried downstream by a product of phospholipid oxidation (Garlid et al., 2013).
RNS: nitration, glutathionylation and nitrosylation
RNS are a family of molecules mainly derived from NO, oxygen and superoxide (Figure 2).
Figure 2.
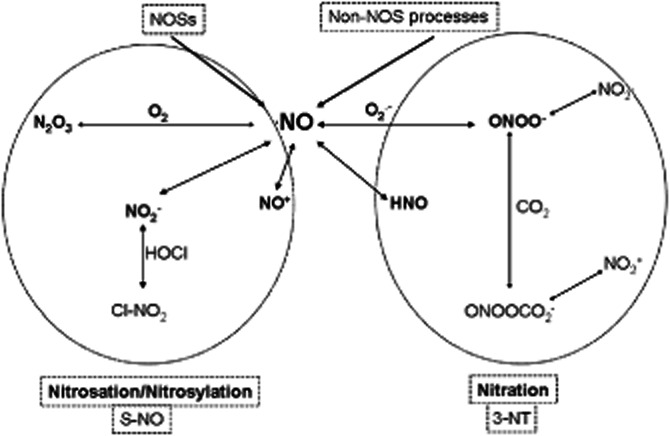
Chemical relationship among reactive oxygen species (ROS) and different reactive nitrogen species (RNS), which can lead to nitrosylation or to nitration. Nitrosylation/Nitrosation refers to the incorporation of the NO moiety (nitroso/nitrosyl group) to a metal and, in protein chemistry, to a sulfur atom to form the S–NO bond (SNO): S-nitrosothiol. Nitration is a term that describes incorporation of a nitro triatomic group (–NO2) and which, in protein chemistry, is used to describe the incorporation of that group at position 3 of the phenolic ring of tyrosine residues (3-NT). ROS chemistry is simplified in the scheme by O2− formation. For other acronyms, see the abbreviation list.
Nitric oxide (nitrogen monoxide, •NO) is synthesized by various NOS enzymes or can be produced by other reactions in the biological systems, which are collectively called ‘non-NOS’ processes (Zweier et al., 2010). Besides endothelial NOS (eNOS or NOS3), many cells constitutively express neuronal NOS (nNOS or NOS1). These two NOSs are Ca2+/calmodulin-controlled isoenzymes, and within the organ and cells, they are localized to different microdomains and are linked to selective signalling (Lim et al., 2008; Zhang and Casadei, 2012). The localization of enzymes and/or antioxidants within the intracellular organelles in the various microdomains has a critical role in the redox biology, determining the final outcome of a nitrosative process. Cells can also express an inducible NOS (iNOS or NOS2), which can produce a large amount of •NO in immune and pathological processes independently of the level of Ca2+ in the cell. Nevertheless, the so-called second window of protection of I-PreC is mediated by increased iNOS activity and the role of iNOS in I/R injury and, specifically, whether this enzyme is protective or detrimental is still a matter of debate. It is likely that iNOS is protective in myocytes but detrimental in inflammatory cells (Guo et al., 2005; 2012,). Although the three isoforms of NOS catalyse the same reactions, they differ in their expression, regulation and physiological/pathophysiological roles (Nathan and Xie, 1994).
The non-NOS process of •NO formation includes non-enzymatic reactions that are favoured by acidic conditions, such as the reduction of nitrite to •NO, and reactions catalysed by non-NOS enzymes, such as cytochrome c, Hb and xanthine oxidoreductase (Zweier et al., 2010). Molecular targets of •NO include metalloenzymes (especially sGC, Hb and cytochromes) and thiols yielding S-nitrosothiols (see also below) (Ferdinandy and Schulz, 2003; Pacher et al., 2007; Penna et al., 2014). The major biological reaction of •NO includes oxidation to nitrite and nitrate. Notably, •NO can react with O2−, yielding peroxynitrite (ONOO−) (Ronson et al., 1999). This reaction leading to peroxynitrite formation depletes the bioactivity of •NO (Guzik et al., 2002). This is important because •NO is a key mediator in many important physiological and cardiovascular functions, including regulation of smooth muscle tone and BP, platelet activation and vascular cell signalling. Therefore, although ONOO− itself is a highly reactive species that can react with various biological targets of the cell including amino acids, DNA, lipids, thiols and low-molecular-weight antioxidants (Pehar et al., 2006), the toxicity of this reaction may be derived from •NO removal. Moreover, this reaction occurs at a relatively slow rate and allows ONOO− to act as a signalling molecule. In fact, ONOO−, similar to O2−, is also able to traverse the cell membranes to some extent through anion channels (Pacher et al., 2007). Yet, peroxynitrite can react with other molecules to form additional types of RNS including nitrogen dioxide (•NO2) and dinitrogen trioxide (N2O3), a nitrosating agent. In fact, important reactions involving RNS, such as nitrosonium (NO+), •NO2 and N2O3, are those leading to S-nitrosothiol production (see below).
Nitration and nitrosation or nitrosylation
Although nitrosylation can be defined as the addition of •NO without a change in the formal charge of the substrate (metal centre or radical species) and nitrosation as the formation of a covalent bond between an NO+ equivalent and a nucleophilic centre (amine or thiol) (Heinrich et al., 2013), nitrosation and nitrosylation are often used interchangeably to refer to the same substrate modification, in biological literature (Murphy et al., 2012; Penna et al., 2014). Actually, nitrosation or nitrosylation of proteins (i.e. the incorporation of NO moieties by covalent bonding to protein groups) is chemically possible in the case of cysteine thiols, leading to the formation of S-nitrosothiol (thionitrite) (Wink et al., 1993; Heinrich et al., 2013). In fact, S-nitrosothiol may be obtained either by the reaction of RNS with a thiol (nitrosation of a thiol) or by •NO with an oxidized thiol (nitrosylation of a thiyl radical) and can be clearly individuated because of the prefix ‘S’, referring to the incorporation of the NO moieties to a sulfur atom to form the S–NO bond. Of note, cysteine residues of proteins may be either reactive (pKa 4–5) or non-reactive (pKa 8.5) (Knock and Ward, 2011; Burgoyne et al., 2012). Therefore, this post-translational modification (S-nitrosothiol formation) can be due to either a S-nitrosation or S-nitrosylation process, which has become popular as protein S-nitrosylation sounds like protein phosphorylation. Indeed, moderate and selective S-nitrosothiol production is emerging as a prototype of redox signalling, which, in biology, is often referred to as S-nitrosylation (Sun et al., 2006a; 2012; 2013,,; Sun and Murphy, 2010; Kohr et al., 2011; Angelone et al., 2012; Murphy et al., 2012; 2014,; Penna et al., 2014) and S-nitrosation (Galkin and Moncada, 2007; Nadtochiy et al., 2007; Chouchani et al., 2010; 2013,; Methner et al., 2013), without any clear evidence that the process leading to S-nitrosothiol production is the former or the latter. Therefore, due the consolidate use of these terms (S-nitrosation/S-nitrosylation), from here on, we will use both terms to refer to the process leading to the formation of S-nitrosothiols within proteins, which are often, although inappropriately, referred to as S-nitrosylated proteins (for the appropriate chemical terminology in this field, see Heinrich et al., 2013).
In contrast, nitration involves an electrophilic addition of a nitro triatomic group, such as an NO2+ equivalent (nitronium, an electron acceptor), to an aromatic ring (the site of electron density) (Espey et al., 2002). In the biological system, the term nitration refers mainly to the incorporation of the NO2+ equivalent at position 3 of the phenolic ring of tyrosine residues (3-nitration of tyrosine or 3-NT). This protein modification is often associated with oxidative/nitrative stress, due to the formation of ONOO−, in the pathology of the CVS (Turko and Murad, 2002; Ischiropoulos, 2003).
However, it must be stressed that, although nitration, nitrosation and nitrosylation, are different types of reactions, they are not mutually exclusive events but are related in a continuum or Yin-Yang process and in many circumstances can result in identical final products (Wink et al., 1993; Penna et al., 2014). For instance, even ONOO− can lead to facilitation of the nitrosylation pathway; indeed, it has been suggested that only when the ratio between •NO and O2− is close to 1:1 the formation of ONOO− can lead to nitration, whereas a small excess of •NO will predominantly lead to nitrosylation via N2O3 formation (Espey et al., 2002). Actually, S-nitrosothiol-mediated protection, through both nitrosation- and nitrosylation-dependent mechanisms, is influenced by the relative rates of •NO and O2− formation. Nevertheless, it has been suggested that 3-NT formation is one of the steps in protective pathways (Kupai et al., 2009; Li et al., 2013).
Glutathionylation is the enzyme-independent addition of GSH, or other low-molecular-weight thiols, to cysteine sulfhydryl residues of proteins. This is a reaction in dynamic equilibrium with S-nitrosation/S-nitrosylation of GSH to form S-nitrosoglutathione and is considered a cellular response to protect essential proteins from permanent loss of function as a consequence of oxidative stress by other ROS/RNS (Klatt and Lamas, 2000; Heinrich et al., 2013).
Therefore, RNS have multiple effects on their potential cellular targets, which include not only proteins but also lipids and DNA and it is likely that RNS have important effects on the shift from a healthy to a disease state (Martínez and Andriantsitohaina, 2009). Indeed, RNS may induce either nitrosative stress via the uncontrolled nitrosation of cysteine residues or nitrative stress through the irreversible nitration of biological molecules, such as tyrosine. Yet, the most important downstream signalling pathway for RNS for conditioning cardioprotection probably involves discrete S-nitrosation/S-nitrosylation of proteins and may be partially independent of NO-mediated cGMP signalling (Penna et al., 2006a; 2011b,; Inserte et al., 2013; Murphy et al., 2014).
Actually, in several conditions, the main NO signalling is cGMP-dependent and stems from the activation of sGC due to the binding of •NO to the haem group of the enzyme, with consequent production of cGMP. The subsequent activation of PKG amplifies the signalling events through protein phosphorylation, including inositol-1,4,5-triphosphate receptor-associated cGMP kinase substrate, the regulator of G protein signalling and the myosin light-chain phosphatase. This ‘classical’ signalling cascade is responsible for the NO-dependent vasodilatation and is regulated at several steps, for example, •NO production, reversible binding to sGC, degradation of cGMP by PDE and dephosphorylation of downstream targets by phosphatases. A plethora of studies can be found on this classical pathway. Here, we focus more on the so-called cGMP-independent NO signalling described earlier. As we will see, this signalling is very important in pre- and post-conditioning.
Sources of ROS and RNS
Besides chemical reactions, several enzymes, pathways and/or mechanisms are associated with the production of reactive species within cells under physiological and pathological conditions. Among these, there are sources of ROS/RNS, including enzymes of mitochondrial respiration, xanthine oxo-reductase (XOR), NADPH oxidases [catalytic subunit of NADPH oxidases (Nox)], and coupled and uncoupled NOSs (Figure 3). Other important oxidase pathways include autoxidation of catecholamines in I/R injury, and the activation of the lipoxygenase and the cytochrome P450 class of enzymes, some of which are present in coronary arteries and myocardium and are known to produce superoxide in response to several stimuli, including bradykinin (Buttery et al., 1996). The majority of these enzymes only produce ROS after they have been damaged by ROS/RNS, as, for example, is the case for uncoupled eNOS and XOR. In contrast, NADPH oxidases produce ROS as their primary function. Here, we describe briefly the role of some of these sources. The reader is redirected to more focused reviews on this topic (Becker, 2004; Zhang and Gutterman, 2007; Costa et al., 2011; Sato et al., 2011; Nickel et al., 2014).
Figure 3.
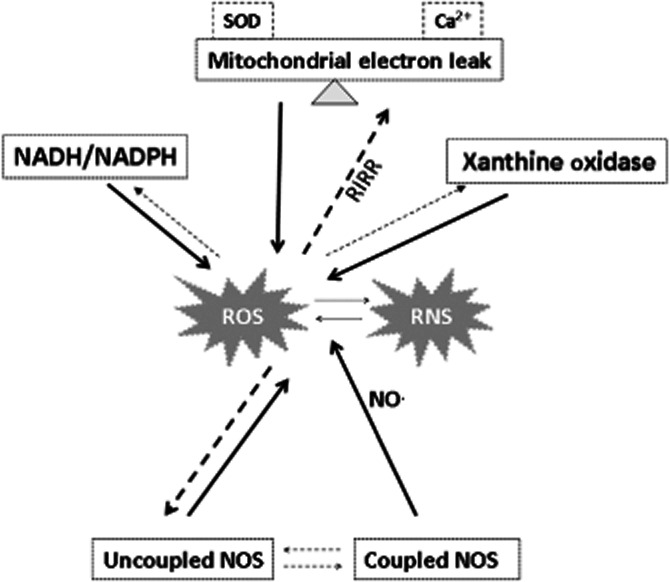
Several interlinked reactions that involve mitochondria respiration, xanthine oxidase, NADPH oxidases, and coupled and uncoupled NOS, which are associated with the production of reactive oxygen and nitrogen species (ROS/RNS) within cells under pathophysiological conditions. The rate of mitochondrial respiration and ROS formation is largely influenced by the internal and external Ca2+ levels and antioxidant activity within mitochondria [i.e. superoxide dismutase (SOD)], and, in turn, by factors such as RIRR. Also, the shift from coupled to uncoupled NOS and the production of ROS by XO are influenced by the levels of ROS/RNS. The dotted arrows indicate this possibility to influence the activity of these enzymes and to sustain the so-called ROS-induced ROS production.
Mitochondrial production of ROS is mainly the net result of O2− production at the electron transport chain and their elimination by antioxidative enzymes, such as manganese SOD in the mitochondrial matrix or copper/zinc SOD in the intermembrane space. The production of O2− by electron transport chain is profoundly influenced by ischaemia and reperfusion and seems exalted by the so-called process of ROS-induced ROS release (RIRR) (Tullio et al., 2013; Nickel et al., 2014). Another mitochondrial source of oxidative signalling and/or stress is monoamine oxidases (MAOs), which are located in the outer mitochondrial membrane. MAO mediates the breakdown of key neurotransmitters, such as noradrenaline, adrenaline and dopamine, and, in the process, generates H2O2. The activity of MAO is altered in I/R and chronic myocardial disease (Kaludercic et al., 2014). Another mitochondrial component that contributes to ROS generation is the p66Shc (for extensive review, see Giorgio et al., 2007; Di Lisa et al., 2009a,b,). Few studies considered p66Shc in the context of I/R injury. In a recent study, the susceptibility to I/R injury was greatly decreased in mice hearts devoid of p66Shc along with a marked reduction of oxidative alterations of proteins and lipids (Carpi et al., 2009). This study suggested that myocardial injury caused by post-ischaemic reperfusion was greatly dependent upon the activity of p66Shc.
The role of XOR in I/R injury has been demonstrated in several heart models of I/R (Berry and Hare, 2004). In the setting of ischaemia, ATP degradation leads to the accumulation of XOR substrates. Moreover, XOR activity is increased after I/R injury. Actually, oxidative stress after reperfusion injury is most likely to be attributable to a combination of local XOR activity and the effects of neutrophils attracted to the region (Harzand et al., 2012).
NOS uncoupling induced by oxidative stress results in further oxidative/nitrosative/nitrative stress. NOS catalytic activity becomes uncoupled when the coupling between the reductase domain and l-arginine oxidation at the active site is lost and electron transfer from NADPH through the flavins to O2 is not inhibited, resulting, in fact, in the formation of O2−• and OH•. NOS uncoupling occurs under certain conditions, such as scarcity or absence of one or more co-factors (calmodulin, FAD, FMN and/or tetrahydrobiopterin), or oxidation of the Zn2+–thiolate centre of the NOS homodimer. It seems that supplementation with tetrahydrobiopterin may reverse NOS uncoupling in some cardiovascular conditions (Carnicer et al., 2013). NOS activity is also profoundly altered in the myocardium after an ischaemic insult (Gonzalez et al., 2009). For instance, after AMI, nNOS expression in the heart is increased (Damy et al., 2003). However, more importantly, it seems that the subcellular localization of nNOS changes from the sarcoplasmic reticulum to the sarcolemma concomitantly with the occurrence of uncoupled NOS activity (Zucchi et al., 2001; Simon et al., 2014). These changes (translocation and uncoupling) explain the different effects of RNS in the post-AMI and failing heart as well as in diabetes-associated myocardial dysfunction (Zhang et al., 2006).
Seven NADPH oxidase family members, which have distinct catalytic subunit (i.e. Nox-1–5 and Duox1 and 2) and different additional protein subunits, have been described (Bedard and Krause, 2007; Santos et al., 2011). The two Nox isoforms that have functional effects in cardiomyocytes are Nox2 and Nox4. It seems that Nox4 has a mitochondrial and endoplasmic reticulum-related perinuclear location and is a slowly inducible isoform. In fact, Nox4 has a low-level of constitutive activity that seems to be regulated largely by changes in its expression level. Nox2 is located predominantly on the plasma membrane and is normally quiescent, but it is acutely activated by various stimuli, including angiotensin II. While Nox2 generates predominantly O2−, Nox4 may generate predominantly H2O2 (Ushio-Fukai, 2009; Ago et al., 2010; Brandes et al., 2010; Nisimoto et al., 2010).
It has been reported that NADPH oxidase subunit expression and activity are increased in both cardiomyocytes and endothelial cells in an animal model of pressure-overload left ventricular hypertrophy (Li et al., 2002). It seems that NADPH oxidase activity is increased in many cardiac disease states. For example, the expression of NADPH subunits, including Nox2, is increased after AMI in both animal models and humans (Krijnen et al., 2003). Using mouse models with selective gain and loss of function for these two Nox isoforms, it has been proposed that a very different role is played by each isoform, roles that themselves vary depending upon the disease condition. Overall, the downstream effects of Nox2 and Nox4 appear to differ, with the former mediating detrimental effects (redox stress), whereas the latter may facilitate beneficial processes (redox signalling) such as angiogenesis and adaptive hypertrophy. For example, deletion of Nox2 was protective against angiotensin II-induced cardiac fibrosis and hypertrophy, whereas deletion of Nox4 was detrimental. However, recent studies suggest that high levels of Nox4 may have detrimental effects via shifting from H2O2 to O2−• production (Bedard and Krause, 2007; Nisimoto et al., 2010; Santos et al., 2011; Zhang et al., 2013). The reasons for this discrepancy remain to be elucidated and the picture may be more complicated than apparent from previous studies.
Before considering the role of ROS/RNS in cardioprotection, let us further define the concepts of redox signalling and stress in the context of cardioprotection.
Redox signals (ROS/RNS signalling)
The redox signalling comprises oxidoreductive chemical reactions that alter proteins post-translationally, thereby creating a coupling between redox balance and cell function. Under normal circumstances, ROS/RNS concentrations are tightly controlled by endogenous antioxidants, keeping them in the picomolar range (Dröge, 2002). However, most cells have been shown to generate a burst of ROS/RNS when stimulated by a plethora of chemicals (e.g. cytokines, angiotensin II, diazoxide, endothelin-1, PDGF, PAF) and physical stimuli (i.e. intermittent shear stress, thermic variations) (Penna et al., 2011a). Then, the ROS/RNS formed play an important role in cellular homeostasis and communication. These transiently increased ROS/RNS act as second messengers in signal transduction for cell signalling and cardiovascular homeostasis.
As said, S-nitrosation/S-nitrosylation leading to S-nitrosothiol production is a selective post-translational protein modification due to targeting of redox-sensitive cysteine residues within proteins. When ROS/RNS are modified, proteins alter their activity, stability, conformation and/or ability to interact with other molecules, resulting in modulations of cellular function. In the heart, redox-modified proteins include proteins involved in calcium handling and contractile function [e.g. calcium calmodulin kinase II, calcium channels, ryanodine receptor, sarco/endoplasmatic reticulum calcium ATPase (SERCA) and phospholamban] (for review, see Burgoyne et al., 2012; Steinberg, 2013; Tullio et al., 2013), as well as proteins involved in various signalling pathways and/or transcriptional activities. Among these are oxidative/nitrosative modifications of enzymes, such as metalloproteinase (MMP) and kinases (i.e. PI3K, AMPK, PKA, PKC and PKG), which may lead to the activation or inhibition of the enzyme depending upon the type of reaction and the site of oxidative/nitrosative modification (see also Knock and Ward, 2011; Burgoyne et al., 2012; Steinberg, 2013; Tullio et al., 2013). The signal may also be indirect; for instance, the H2O2-mediated activation of AMPK is likely mediated via the ROS-induced decrease in the ATP levels. It has also been suggested that hypoxic activation of AMPK is dependent upon mitochondrial ROS but independent of an increase in the AMP/ATP ratio (Emerling et al., 2009). ROS are also important regulators of PKC by reacting with thiol groups associated with the zinc-finger region of the molecule (Korichneva, 2006). Moreover, an RNS-dependent activation of PKC, via a redox-sensitive S-nitrosation/S-nitrosylation process, occurs within the mitochondria (Sun et al., 2006b; Prime et al., 2009). The small monomeric G proteins (ras, rac-1 and RhoA), transcription factors (e.g. NF-κB and HIF-1) and histone deacetylases are also activated by ROS/RNS (Bonello et al., 2007; Morgan and Liu, 2011; Satoh et al., 2011; Surma et al., 2011). Finally, a group of kinases that may be directly or indirectly redox-sensitive, but very important in cardiovascular cell signalling, are MAPKs (see also below) (Johnson and Lapadat, 2002; Martindale and Holbrook, 2002). Also, vasodilatation is, in part, due to a cGMP-independent process of glutathionylation, which protects the cells from further oxidative/nitrosative modifications (Sun et al., 2006b). In fact, the inhibitor of sGC, ODQ (1H-[1,2,4]oxadiazole[4,3,-α]quinoxaline-1-one), does not prevent several NO-dependent effects in many tissues (Busse and Fleming, 2006; Sun et al., 2013).
Moreover, even protein nitration has been suggested to be involved in signalling processes during normal physiological mechanisms (Koeck et al., 2005). In fact, tyrosine nitration by ONOO− may affect tyrosine phosphorylation and MAPK activity in several cell types, thus affecting essential cellular functions (Jope et al., 2000). Therefore, ROS/RNS play a very important role in cell signalling and are thus essential for survival of the organism (Dröge, 2002; Fisher, 2009).
Of course, we must not forget that under pathological conditions and in the presence of co-morbidities, ROS/RNS production is unbalanced by cell defences, inducing deleterious effects in a large number of pathways involved in cell life. The dual role of ROS/RNS in ‘fine-tuning’ the balance between cell death and survival is well illustrated by observations that, during I/R injury, ROS/RNS trigger cell death (apoptosis/necrosis), whereas ROS/RNS generated during preconditioning as well as during post-conditioning manoeuvres prevent cell death (Becker, 2004; Penna et al., 2009; Pagliaro et al., 2011; Tullio et al., 2013). ROS/RNS generated during conditioning can up-regulate pathways able to prevent cell death (see the ROS/RNS and cardioprotection section).
Oxidative and nitrosative/nitrative stress in CVD
As said, redox balance may shift from health (signalling) to disease (stress) in acute events, especially if events occur during ageing and degenerative conditions. Here, we consider in brief the role of redox stress in CVD and in particular in the I/R injury of the myocardium. The reader is kindly redirected to other extensive reviews on this topic (Tocchetti et al., 2011; Zhang et al., 2012; Morales et al., 2014).
Normal cardiovascular performance requires balancing of many complex physiological and biochemical processes, including redox balance. Disturbances of this balance may lead to myocardial dysfunction or, alternatively, a disrupted balance may be a secondary result of structural heart disease such as AMI or cardiomyopathic processes. Altered signalling systems, in turn, contribute to the progression of myocardial dysfunction. Therefore, redox imbalance may be the cause of cardiac disease, which, in turn, may exacerbate redox imbalance. In fact, when excessively produced, or when endogenous antioxidants are depleted, ROS/RNS can inflict damage onto lipids, proteins and DNA. This intracellular reduction–oxidation imbalance, namely oxidative/nitrosative/nitrative stress, can subsequently contribute to the development and/or progression of CVD, such as atherosclerosis, chronic ischaemic heart disease, cardiomyopathy, congestive heart failure, arrhythmias as well as several other degenerative conditions rendering the CVS more susceptible to stress by I/R challenging (Lopes et al., 2012; Tullio et al., 2013). Importantly, ROS/RNS up-regulation leading to cell death is a wanted effect in cancer prevention.
I/R injury is the prototypical pathological example of a situation in which ROS/RNS are produced in amounts far exceeding those that cells and tissues can handle without damage. Redox stress may occur both during ischaemia and reperfusion (Becker, 2004).
At the cellular level, ischaemia is characterized by a virtual lack of O2 and substrates, and ischaemic cells accumulate metabolites such as lactate. Via alterations in pH, Na+ and ATP levels, ischaemia and mainly reperfusion then trigger a cascade of events including massive ROS/RNS generation, loss of nucleotide homeostasis and disruption of Ca2+ homeostasis. Therefore, ROS/RNS are produced during ischaemia, but especially at reperfusion (Becker, 2004).
Oxidative/nitrosative/nitrative stress plays an important role and contributes to the onset and maintenance of post-ischaemic inflammation. Oxidative/nitrosative/nitrative stress after reperfusion injury is most likely attributable to a combination of local enzyme activity and the effects of recruited neutrophils (Hoffman et al., 2004; Zhao, 2004). Increased formation of ROS/RNS is one of the main factors of reperfusion-induced injury. During reperfusion, the O2− production increases, which, along with other ROS/RNS, strongly oxidizes the myocardial fibres, thus favouring cell death (Zhao, 2004). As said, O2− production can be theoretically generated by different enzymatic systems, namely NOX, uncoupled NOS, XOR and complexes of the respiratory chain in the mitochondria. Indeed, mitochondria are key players in I/R injury: basically, disruption of Ca2+ homeostasis results in mitochondrial Ca2+ overload and culminates in the formation of the mitochondrial permeability transition pore (mPTP) and the activation of cell death signalling pathways (Murphy and Steenbergen, 2008; Lemasters et al., 2009; Tullio et al., 2013). Importantly, mPTP opening at reperfusion is a fundamental step of the so-called RIRR that self-sustain ROS and consequently RNS production that inflicts irreversible damage to the cells (Tullio et al., 2013). In fact, it has been reported that mPTP is maintained in a closed state during ischaemia and that in non-protected (naïve) hearts typically opens at reperfusion (Obame et al., 2007; Hausenloy et al., 2009). At the beginning of reperfusion, Ca2+ overload, ROS/RNS stress and pH recovery all occur. These three mechanisms are recognized as potent triggers that increase the open probability of the mPTP. The opening of mPTP will lead to RIRR and cell death by different mechanisms, including mitochondrial disruption from mitochondrial swelling, membrane rupture and/or cytochrome c release, which induces programmed cell death pathways (Hausenloy et al., 2009; Martin et al., 2011; Penna et al., 2013a). This is one of the main reasons why the restoration of blood flow in the infarct-culprit artery may paradoxically result in further damage to the myocardium. This reperfusion injury can significantly reduce the beneficial effects of reperfusion therapy. Therefore, mPTP inhibition at the time of reperfusion appears fundamental to all strategies of cardioprotection thus far envisaged (Tullio et al., 2013). As we will see, S-nitrosation/S-nitrosylation of some putative components of mPTP reduces the probability of pore formation.
ROS/RNS and cardioprotection
Different cardioprotective strategies such as ischaemic and pharmacological preconditioning have been shown to attenuate mitochondrial dysfunction as evidenced by less mitochondrial Ca2+ overload, a better NADH balance and reduced ROS/RNS formation during ischaemia as well as on reperfusion (Riess et al., 2002b; 2003,; Kevin et al., 2003). Although excessive ROS/RNS formation during I/R may contribute to reperfusion injury via nitrative stress by peroxynitrite, ROS/RNS are important elements in the triggering signal of I-PreC during preconditioning manoeuvres. The role of redox balance in I/R injury and in the cardioprotection induced by preconditioning has been extensively reviewed earlier (Cohen et al., 2006; Ferdinandy, 2006; Jones and Bolli, 2006; Tullio et al., 2013; Penna et al., 2014). Therefore, here we discuss redox signalling in preconditioning and focus on more recent studies and especially the involvement of redox signalling in post-conditioning, that has not yet been reviewed in the literature.
Preconditioning
Cardioprotection by I-PreC is obtained by short periods of ischaemia with intervening short periods of reperfusion (a few minutes) prior to an infarcting ischaemia. It requires a complex signalling cascade to be triggered, which includes the opening of mitoKATP (O'Rourke, 2000; Cohen et al., 2001; Oldenburg et al., 2002; 2004,; Yue et al., 2002). Intriguingly, preconditioning can be completely blocked by free radical scavengers, such as N-acetylcysteine (NAC) or mercaptopropionyl glycine (MPG) given during preconditioning manoeuvres (Cohen et al., 2001; Forbes et al., 2001). These results confirmed that oxidative/nitrosative signalling is involved in triggering the cardioprotection induced by preconditioning. In fact, •NO production by activation of different types of NOS and/or by non-NOS processes has been shown to be involved in cardioprotection procedures (Bell and Yellon, 2001; Oldenburg et al., 2004; Cohen et al., 2006; Shiva et al., 2007; Guo et al., 2008; Zweier et al., 2010; Simon et al., 2014). Even donors of HNO (one electron reduction product of •NO) may induce a preconditioning-like effect, which is reversed by NAC (Pagliaro et al., 2003). The ‘classical’ protection induced by •NO in I-PreC is dependent, in part, upon the activation of sGC/cGMP/PKG, which, in turn, leads to the opening of the mitoKATP channel (Jones and Bolli, 2006). Recently, Sun et al. (2013) reported that the I-PreC-induced cardioprotection is not related primarily to the activation of the sGC/cGMP/PKG signalling pathway by •NO, but rather through S-nitrosation/S-nitrosylation signalling. In fact, the infusion of the sGC inhibitor, ODQ, did not completely abolish the cardioprotection induced by I-PreC. Hearts treated with ODQ were protected with a concomitant higher S-nitrosothiol level. These results suggest, at least in some models of cardioprotection, that •NO-mediated cardioprotection is regulated by protein S-nitrosation/S-nitrosylation of cysteine residues rather than through activation of the sGC/cGMP/PKG signalling (Sun et al., 2013). The ease of reversibility and the affirmation of regulated S-nitrosylating and denitrosylating enzymatic and non-enzymatic reactions support the hypothesis that S-nitrosylation regulates the cellular and mitochondrial function through redox mechanisms (Penna et al., 2014). Although the majority of authors demonstrate that •NO is involved in cardioprotection via cGMP-dependent or cGMP-independent mechanisms, others have obtained controversial results using inhibitors of NOS (Jones and Bolli, 2006).
A great part of preconditioning protection is due to the limitation of reperfusion injury with a limitation of ROS/RNS stress, mainly due to the prevention of mPTP opening in the early phase of reperfusion (Hausenloy et al., 2007; 2009,; Hausenloy and Yellon, 2009). The prevention of mPTP opening avoids RIRR and redox stress, whereas oxidative/nitrosative signalling may occur and protect the heart.
The readers can find several reviews describing the protective role of the reperfusion injury salvage kinase (RISK), the survivor activating factor enhancement (SAFE) and the cGMP/PKG pathways (e.g. Penna et al., 2008a; 2013a,; Boengler et al., 2011; Hausenloy et al., 2011; 2013,), which comprise phosphorylation of several target proteins. In brief, enzymes that have been shown to be involved in these pathways include phosphotidylinositol-3-phosphate kinase, extracellular signal-regulated protein kinases, PKB or Akt, PKC, eNOS and JAK-STAT3, a series of kinases that have been termed RISK and SAFE pathways. Once the organ has been preconditioned, these pathways are re-activated at reperfusion, leading, together with other factors, to the prevention of the mPTP formation. The mitochondria in the cell thus continue to be functional and do not release pro-apoptotic factors, preventing cell death in reperfusion. However, it is unclear how the heart ‘remembers’ that it is preconditioned. Similar mechanisms have been observed with regard to I-PostC, mainly implicating the RISK and SAFE pathways and the prevention of mPTP formation (see below). Pharmacological therapy can thus mimic conditioning by targeting the cells at one of these points at the level of the receptors, the signal transduction pathways or the mitochondria.
Here, we focus on recent studies reporting S-nitrosation/S-nitrosylation of critical proteins as a pivotal mechanism of cardioprotection by preconditioning (Murphy and Steenbergen, 2007; Sun et al., 2007; 2012,). Recently, Kohr et al. (2011), using two different methods to measure protein oxidation, showed that preconditioning leads to S-nitrosylation of several proteins and that most of these proteins are protected from further oxidation.
S-nitrosation/S-nitrosylation of proteins involved in calcium handling, such as Ca2+ channels, phospholamban and SERCA2, has been demonstrated (Sun et al., 2006a; Sun and Murphy, 2010; Angelone et al., 2012; Murphy et al., 2014). Moreover, multiple S-nitrosothiol proteins have been shown by proteomic studies in the presence of PreC (Arrell et al., 2006; Shi et al., 2008; Foster et al., 2009). In particular, many of these proteins have been found within the mitochondria, including proteins responsible for mitochondrial metabolism (e.g. αKGDH, glycogen phosphorylase, aconitase). Other important mitochondrial components that are subjected to S-nitrosation/S-nitrosylation during the PreC are the respiratory complexes, including complex I, which is reversibly inhibited when S-nitrosylated (Nadtochiy et al., 2007) or irreversibly inhibited when it is subjected to nitration by ONOO− (Galkin and Moncada, 2007; Sun and Murphy, 2010). Another effect usually observed in conditioning protection is the inhibition of F0-F1-ATPase. This can occur by S-nitrosation/S-nitrosylation, with consequent reduction of ATP consumption by the reverse mode of the F0-F1-ATPase, which typically occurs in I/R of the myocardium (Penna et al., 2004). The inhibition of F0-F1-ATPase preserves ATP levels and reduces the mitochondrial potential, thereby reducing the driving force for Ca2+ uptake into the mitochondria, thus increasing tolerance to I/R (Shiva et al., 2007). Notably, the inhibition of mPTP opening is regulated by ROS, Ca2+ and mitochondrial membrane potential (Heusch et al., 2010; Boengler et al., 2011; Penna et al., 2013a), which are also regulated by S-nitrosation/S-nitrosylation of critical proteins (Piantadosi, 2012). Not only the decrease in Ca2+ loading by increased re-uptake by S-nitrosylated SERCA2, but also the S-nitrosylation of F0-F1-ATPase, reduces indirectly the opening of mPTP, which reduces the breakdown of glycolytic ATP and the acceleration of the fall in the mitochondrial membrane potential. Moreover, S-nitrosylation of cyclophilin D (Nguyen et al., 2011) and/or of voltage-dependent anion channel (Penna et al., 2013a), two putative components of mPTP rich in thiol groups, may occur in cardioprotection. All together, these data support the view that S-nitrosation/S-nitrosylation of mitochondrial and calcium-handling proteins serves as an important mechanism in preconditioning cardioprotection.
Post-conditioning
Cardioprotection by I-PostC is obtained by short periods of reperfusion intervened by short periods of ischaemia (a few seconds) at the beginning of a reperfusion, which follows an infarcting ischaemia. Because I-PostC has the advantage that it can be applied after the ischaemic insult has occurred, this is therapeutically a more favourable approach than preconditioning. It requires a complex signalling cascade to be triggered, which includes the opening of mitoKATP and the activation/inhibition of several enzymes of cardioprotective pathways. With regard to signalling pathways, also for PostC, as for PreC, the greatest attention has focused on the role of the RISK-, the SAFE- and the cGMP/PKG-dependent pathways. Intriguingly, however, I-PostC can be completely blocked by free radical large-spectrum scavengers, such as NAC or MPG given during I-PostC manoeuvres. However, PostC protection is not abolished if the scavenger is given in reperfusion after the PostC manoeuvres have been completed (Downey and Cohen, 2006). More intricate is the relationship with more selective antioxidant enzymes, such as SOD and catalase, whose activity may be influenced by pH (Penna et al., 2011b; 2013c,; Tullio et al., 2013) (see also below). Actually, the gradual normalization of intracellular pH in the initial phase of reperfusion plays a critical role in conditioning strategies. Both in pre- and in post-conditioning, acidosis favours redox signalling and the activation of a complex cascade of signal molecules and prevents the opening of mPTP in the early post-ischaemic phase; a phase in which redox signalling plays a critical role in triggering cardioprotection (Cohen et al., 2007; 2008,; Cohen and Downey, 2011; Inserte et al., 2011b; Penna et al., 2011b; 2013a,; Tong et al., 2014). In particular, acidosis favours the transient formation of S-nitrosylated proteins in post-conditioned hearts (Penna et al., 2011b; Tong et al., 2014).
NO, nitration and nitrosylation may play a finely interconnected role in post-conditioning. It is well known that post-conditioning attenuates endothelial cell dysfunction by increasing eNOS activity and •NO bioavailability in neighbouring cells (Zhao et al., 2003; Ma et al., 2006; Granfeldt et al., 2009). This can be responsible for improved vasodilatation in post-conditioned hearts. Moreover, both pre- and post-conditioning protection can be triggered by pharmacological interventions, including the infusion of exogenous •NO donors, that is, pharmacological PreC or pharmacological PostC (Valen and Vaage, 2005; Gross and Gross, 2006; Jones and Bolli, 2006; Penna et al., 2007; 2008b,; Tissier et al., 2007). Indeed, similar to ischaemic and pharmacological preconditioning, S-nitrosation/S-nitrosylation is also involved in ischaemic and pharmacological post-conditioning. Several studies that used NO donors in reperfusion to induce pharmacological PostC revealed an important role for the S-nitrosation/S-nitrosylation of proteins in the mechanisms of protection (Nadtochiy et al., 2007; Prime et al., 2009; Methner et al., 2013). We and Murphy's group have shown that I-PostC is also mediated by S-nitrosation/S-nitrosylation of several proteins (Penna et al., 2011b; Tong et al., 2014). Due to the abundance of S-nitrosylated proteins, it is also likely that denitrosylation processes are down-regulated. In fact, we have shown that PostC discretely changes the activity of antioxidant enzymes in early reperfusion, slightly decreasing SOD and increasing catalase activity (Penna et al., 2011b; 2013c,). Since SOD may be a denitrosylating enzyme (Sun and Murphy, 2010), these effects may favour the prevalence of S-nitrosothiol proteins, thus reducing injury due to oxidative stress. In fact, it has been proposed that the increase in S-nitrosylation could shield critical cysteine residue(s) from further oxidative damage upon reperfusion (Tullio et al., 2013; Tong et al., 2014).
Importantly, pro-survival enzyme activation may depend upon redox-sensitive reactions. For instance, PKC activation can occur via S-nitrosative processes (Sun et al., 2006b) and the activation of PKC plays a central role in sustaining the cardioprotection induced by post-conditioning (Penna et al., 2006b; Zatta et al., 2006; Cohen and Downey, 2011). The S-nitrosation/S-nitrosylation of the mitochondrial F0-F1-ATPase described for PreC has also been found in PostC (Tong et al., 2014). This is in line with interesting findings reported in a recent study, in which Cys294 of the mitochondrial F0-F1-ATPase was found to form a disulfide bond with another cysteine residue in heart failure, whereas the protective cardiac resynchronization therapy led to S-nitrosation/S-nitrosylation of Cys294 and prevented disulfide formation (Wang et al., 2011).
It has been found that about 50% of those proteins that were S-nitrosylated by PreC were also S-nitrosylated by PostC (Sun et al., 2012; 2013,), suggesting that there might be a common set of proteins targeted by nitrosative signalling with both PreC and PostC. In fact, the S-nitrosation/S-nitrosylation processes are not a random reaction but depend upon a number of conditions. In fact, the instantaneous redox state and ultrastructural accessibility of cysteine residue(s) under low oxygen tension, such as hypoxia, ischaemia and post-conditioning intermittent I/R, may determine whether a particular thiol/thiyl radical in a given protein is subjected to S-nitrosation/S-nitrosylation (Saini et al., 2004; Foster et al., 2012).
During the first minutes of reperfusion, usually a typical large burst of ROS occurs in unprotected (naïve) hearts. The ROS/RNS burst results in the irreversible oxidation/nitration of a number of important proteins. These proteins are damaged and need to be degraded and re-synthesized to regain normal cell function; otherwise, irreversible tissue injury occurs. The shielding effect of S-nitrosothiol could be necessary to trigger protection in early reperfusion and to allow sufficient time for the activation of protective signalling. Since S-nitrosation/S-nitrosylation is a transient readily reversed protein modification, timing seems of essence. This could be of extreme importance during I-PostC manoeuvres. In fact, the ROS/RNS burst is attenuated (not abolished) by I-PostC manoeuvres, and S-nitrosation/S-nitrosylation occurring during post-conditioning may shield modified cysteines from more irreversible states of oxidation until the burst of ROS/RNS vanishes. This point of view is in line with the experimental evidence that a delay in performing PostC manoeuvres results in a loss of protection (Penna et al., 2008a; 2009,; Skyschally et al., 2009; Hausenloy, 2013).
Actually, it has been found that protein nitration may be deleterious in the PostC scenario (Fan et al., 2010; Iliodromitis et al., 2010; Inserte et al., 2013). However, other authors have observed a beneficial effect for this reaction induced by peroxynitrite (Kupai et al., 2009; Li et al., 2013). We have proposed that tyrosine nitration may be a transient initial effect of I-PostC, which is suddenly followed by the prevalence of protein S-nitrosylation, possibly via the so-called secondary reaction described earlier (Penna et al., 2011b). We have shown in rat hearts that after 7 min of reperfusion, I-PostC induces a reduction in the levels of 3-nitrotyrosine formed and a subsequent increase in S-nitrosylated proteins, which persist for at least 120 min of reperfusion (Penna et al., 2011b). In fact, a low level of 3-nitrotyrosine in PostC has often been observed (Kupai et al., 2009; Penna et al., 2011b; Inserte et al., 2013), but a predominant formation of S-nitrosylated proteins has been described (Penna et al., 2011b; Tong et al., 2014). Very recently, we and other authors have shown that protein S-nitrosation/S-nitrosylation occurs mainly in the mitochondria after I-PostC (Penna et al., 2013b; Tong et al., 2014). We have also shown that pharmacological PostC induced by diazoxide [a drug supposed to promote ROS signalling through actions on mitoKATP channels and connexins (Boengler et al., 2007; 2011,; Sánchez et al., 2013)] may induce marked S-nitrosothiol formation in the mitochondrial proteins. In another study, the addition of a mitochondria-targeted NO donor at the start of reperfusion (i.e. pharmacological PostC) has also been found to be cardioprotective (Chouchani et al., 2013). The NO donor used in this study was the so-called MitoSNO, which comprises the NO donor S-nitroso-N-acetylpenicillamine conjugated to a triphenylphosphonium moiety. This lipophilic moiety allows MitoSNO to pass rapidly through membranes driven by the membrane potential and therefore to accumulate several hundred-fold within the mitochondria, where it generates •NO and nitrosothiol in proteins (Chouchani et al., 2013). The S-nitrosation/S-nitrosylation of proteins by MitoSNO and other donors has been confirmed by other authors both in basal conditions and in the context of post-conditioning cardioprotection (James et al., 2007; Tullio et al., 2013 and references therein). Importantly, S-nitrosothiol production is a transitory modification, which is reversed by the so-called denitrosylation processes (Murphy et al., 2012; 2014,; Penna et al., 2014).
It is important to emphasize that phosphorylative pathways may be activated in parallel or in sequence to the nitrosative/nitrosylative processes. For instance, it has been recently reported that the most abundant isoform of PKG (PKGI) within cardiomyocytes is involved in cardioprotection against I/R injury. However, after cardiomyocyte-specific ablation of the PKGI gene in the mouse heart, it was still possible to protect the hearts with several interventions, including I-PostC or pharmacological PostC with the NO donor MitoSNO, via S-nitrosylation of mitochondrial proteins (Methner et al., 2013). Therefore, the authors concluded that PostC may afford protection either by bypassing PKGI or by acting independently or downstream of it. The authors also suggested differences between cGMP/PKGI pathway in myocytes and other cardiac cell types during I-PostC protection in this in vivo study. In fact, they cannot rule out that the exogenous and endogenous •NO may act to protect the heart from I/R injury in a manner that depends upon PKG in other cardiac cell types (Methner et al., 2013). In fact, the PKG pathway has been shown to be involved in PostC protection in different models by several authors (Penna et al., 2006a; Inserte et al., 2011a; 2013,; Methner et al., 2013).
In summary, •NO appears to be an important mediator of cardioprotection. In particular, besides its well-known vasodilator effects, in pre- and post-conditioning, •NO may be involved in both cGMP/PKG-dependent signalling and mitochondrial protein S-nitrosation/S-nitrosylation, thus playing a pivotal role in conditioning cardioprotection. Intriguingly, our recent finding that diazoxide enhances protein S-nitrosylation both in the absence of ischaemia and in the early post-ischaemic phase (Penna et al., 2013b) further supports the idea that an appropriate redox environment is necessary for NO-mediated cardioprotection induced by both pre- and post-conditioning.
Therefore, we can conclude that the oxidative/nitrosative signalling and the increase in S-nitrosation/S-nitrosylation play pivotal roles in cardioprotection against I/R injury, both in pre- and post-conditioning.
Bench to bedside
Of course, oxidative stress is an attractive target for novel therapies, as it represents the common pathway through which different risk factors exert their deleterious effect on the CVS. Included in this spectrum of CVD is AMI. However, we have seen that oxidative steps are necessary for cardioprotection. With no doubt, we need more experimental studies to better understand the mechanisms of stress-related injury and of redox-dependent protection before having a safe and successful transition to clinical scenario of conditioning concepts.
The difficulty in obtaining positive results with conditioning protocols in humans
Although the discovery of I-PreC is of paramount importance from a conceptual viewpoint, the practical significance of preconditioning is limited by the fact that it is a pretreatment.
There is a wealth of evidence supporting the notion that the human heart is amenable to preconditioning-induced protection. Protection can also be obtained with remote conditioning, initiated by ischaemia in a remote organ or tissue and transmitted to the heart. However, as AMI is unpredictable, preconditioning, whether it is applied to the heart or to a remote organ, is limited to scheduled ischaemic events, such as those in patients undergoing cardiac surgery involving cardiac arrest.
In contrast, post-conditioning, applied either to the heart or to a remote organ, may hold greater promise for clinical application. This may be particularly true for ST-segment elevation myocardial infarction (STEMI) patients in whom coronary flow is restored via percutaneous coronary intervention or thrombolysis. In fact, a few years after the first description of I-PostC protection in the canine model by Zhao et al. (2003), Staat et al. (2005) as well as several subsequent phase II studies (Yellon and Hausenloy, 2005; Yang et al., 2007; Thibault et al., 2008; Xue et al., 2010; Heusch, 2013; Ovize et al., 2013) reported a significant reduction in enzyme release in STEMI patients. In these studies, patients were post-conditioned with brief angioplasty balloon inflation/deflation immediately after the reopening of the culprit coronary artery and compared with controls who underwent standard angioplasty or direct stenting of the coronary artery. More recently, however, neutral effects of I-PostC in STEMI patients have been described (Sörensson et al., 2010; Freixa et al., 2012; Tarantini et al., 2012; Limalanathan et al., 2014). Thus, data on the effects of I-PostC in STEMI treated by primary percutaneous coronary intervention are controversial (Laskey, 2005; Laskey et al., 2008; Lønborg et al., 2010; Garcia et al., 2011; Hahn et al., 2013). See Table 1 for a summary of results with PostC in humans.
Table 1.
Outcome of clinical studies that used I-PostC
| Study | Protocola | Patients No. | Results |
|---|---|---|---|
| Staat et al. (2005) | 4 × 60 s | 30 | Positive |
| Laskey (2005) | 2 × 90 s | 17 | Positive |
| Ma et al. (2006) | 3 × 30 s | 94 | Positive |
| Yang et al. (2007) | 3 × 30 s | 41 | Positive |
| Thibault et al. (2008) | 4 × 60 s | 38 | Positive |
| Laskey et al. (2008) | 2 × 90 s | 24 | Positive |
| Lønborg et al. (2010) | 4 × 30 s | 118 | Positive |
| Sörensson et al. (2010) | 4 × 60 s | 76 | Positive |
| Xue et al. (2010) | 4 × 60 s | 43 | Positive |
| Lin et al. (2010) | 3 × 60 s | 75 | Positive |
| Garcia et al. (2011) | 4 × 30 s | 43 | Positive |
| Hahn et al. (2013) | 4 × 60 s | 700 | No differences |
| Limalanathan et al. (2014) | 4 × 60 s | 272 | No differences |
Cycles × Duration of each cycle of ischaemia and reperfusion.
In our opinion, it is not surprising that a variation (ranging from highly protective to neutral and even negative results) in the magnitude of myocardial salvage can be observed among clinical studies. In fact, similar to I/R injury, the I-PostC protection may also be influenced by a number of conditions that are not easy to keep under the control of the physicians in the ‘clinical arena’. Here, particularly relevant are the variables correlated to the duration of ischaemia, the area at risk and the degree/quality of artery reopening in controls and I-PostC-treated patients (see Pagliaro et al., 2011; Heusch, 2013; Przyklenk, 2013).
The possibility of triggering post-conditioning by drugs
Pharmacological PostC would limit the unfavourable consequences linked with the complexity of I-PostC and provides a simple method of myocardial protection, which can be subsequent to all cardiac procedures of artery reopening, including coronary reopening by thrombolysis. Several drugs can act as conditioning agents, which reduce the final myocardial infarct size following ischaemia–reperfusion (Sivaraman and Yellon, 2014). For some of these, there is some evidence supporting the importance of the redox environment, which is the main topic of the present review. For instance, much attention has focused on the cardioprotective effects of volatile anaesthetics, which are largely shown to be protective in many species, including humans (Huhn et al., 2008; Lemoine et al., 2010; Schwiebert et al., 2010). Intriguingly, it has been shown that some of the anaesthetic protective effects can be redox-sensitive (Lemoine et al., 2010). Moreover, NO donors and molecules that activate protective NO-dependent signalling pathways are promising tools for cardioprotection. In particular, organic nitrates and nitrites are effective redox-sensitive cardioprotective agents. However, when nitrate tolerance develops, not only do they lose their protective effects but they may also interfere with endogenous protective mechanisms by increasing nitrative/nitrosative/oxidative stress (Ferdinandy, 2006; Ferdinandy et al., 2007; Zweier et al., 2010).
In this context, a couple of questions arise: (i) is oxidative/nitrosative signalling in cardioprotection a laboratory curiosity, or do these concepts extend to the human heart and clinical scenario? (ii) is it better to trigger signalling or to avoid stress? We should consider the complexity of these questions and recall to attention the fact that it is not trivial to consider the redox status in the context of co-morbidities, mutagenesis, CVD and in particular in I/R injury. Moreover, experimental studies that demonstrated the importance of protein S-nitrosation/S-nitrosylation in cardioprotective signalling have not yet demonstrated a direct link between the protein modifications and cardioprotection. Clearly, however, the approaches aimed simply at avoiding stress are not effective.
The failure of antioxidants
First of all, let us consider the putative role of antioxidants. The term antioxidant refers to any molecule capable of stabilizing or deactivating free radicals and oxidants before they attack cell components. It is now clear that large-spectrum antioxidants can abolish conditioning protection. Moreover, and more importantly, large clinical trials have failed to demonstrate a benefit of large-spectrum antioxidants on cardiovascular outcomes (see Table 2). Studies exploring the possibility that antioxidants such as vitamin A, C, vitamin E, selenium or folic acid may improve the prognosis of patients with CVD have substantially reported neutral and even negative results (Omenn et al., 1996; Sesso et al., 2008; 2012,). In the context of reperfusion, i.v. bolus of either trimetazidine (Downey, 1990) or SOD (Flaherty et al., 1994) showed no beneficial effects on the outcome of patients. Moreover, p.o. administration of vitamin C (Chen et al., 2013) or the effects of combined vitamins C and E, through infusion and capsules (Jaxa-Chamiec et al., 2005; Lee et al., 2005; Cook et al., 2007), did not demonstrate a major effect of these antioxidant treatments on the clinical outcome of patients. Nevertheless, in diabetic patients, a reduction in 30 day cardiac mortality has been reported (Jaxa-Chamiec et al., 2009). Also, in relatively old experimental studies, contradictory results were obtained. For example, some have shown that SOD reduces myocardial infarct size (Werns et al., 1985), whereas others observed no functional or histological protection (Näslund et al., 1992). These contradictory results should suggest a more cautious approach. Intriguingly, there are pieces of evidence that the antioxidants may also limit the benefit due to exercise. For instance, Ristow et al. (2009) found that the subjects who exercised and did not take antioxidant supplements had significant improvements in insulin sensitivity, adiponectin and PPAR-coactivator 1, whereas antioxidants inhibited these metabolic benefits of exercise. Exercise may be a archetypical example of the benefit from transient oxidative signalling (Copp et al., 2009; Ristow and Zarse, 2010; Ristow and Schmeisser, 2011), which also induces an up-regulation of eNOS (Lauer et al., 2005).
Table 2.
Clinical trials investigating the effect of antioxidants on CVD
| Treatment | Patients No. | Result | Ref. |
|---|---|---|---|
| β-Carotene | 39 876 | No benefit | Hennekens et al. (1996) |
| Vitamin A+ β-carotene | 14 641 | No benefit | Omenn et al. (1996) |
| Vitamin E | 22 071 | No benefit | Lee et al. (2005) |
| Vitamin C + E + β-carotene | 73 135 | No benefit | Cook et al. (2007) |
| Vitamin C + E | 8171 | No benefit | Sesso et al. (2008) |
| Vitamin B6 + B12 + folic acid | 6837 | No benefit | Ebbing et al. (2010) |
| Multivitamin | 14 641 | No benefit | Sesso et al. (2012) |
| Vitamin C | Meta-analysis | Adverse effects | Chen et al. (2013) |
Clearly, indiscriminate removal of oxidative stress by an antioxidant is not an effective means to prevent the detrimental processes due to I/R. In fact, oxidative signalling is necessary for several physiological functions, including cell survival against I/R and an array of noxious stimuli. Therefore, aspecific removal of ROS/RNS with large-spectrum antioxidants cannot be considered to achieve clinically meaningful benefit. It seems necessary to consider a site–time-specific and well-timed inhibition of the source of injurious ROS/RNS without affecting redox-sensitive survival signal transduction pathways. This may represent a promising approach to elicit the beneficial effect of drugs affecting (promoting or inhibiting) ROS/RNS formation. Drugs with these characteristics need to be envisaged and studied. However, we can learn something from some medications already in use.
The redox aspects of some effective drugs
Intriguingly, medications such as ACE inhibitors, angiotensin II receptor blockers or statins, which indirectly limit ROS production while favouring •NO production, have been more consistently associated with beneficial effects in both pre-clinical studies and large clinical trials. The redox-dependent reasons for the success of the therapy with these drugs are not specifically studied. It is tempting to speculate that low levels of ROS together with sufficient amount of •NO may favour protective processes. As said, site specificity for antioxidant therapy may play a role of paramount importance. In fact, ACE inhibitors and angiotensin II receptor blockers may limit (not avoid) ROS production by NADPH oxidases. Moreover, ACE inhibitors have the advantage of promoting the bioavailability of bradykinin, which, via B2 receptors, induces the release of vasodilator and antioxidant substances such as •NO and prostacyclin. In fact, bradykinin has been suggested as a conditioning agent in several studies (Penna et al., 2007; 2008b,). The data in humans regarding bradykinin are more controversial (Wang et al., 2009; Pedersen et al., 2011). Although some experimental studies using ACE inhibitors alone have shown cardioprotective effects (Jin and Chen, 2000; Weidenbach et al., 2000; Penna et al., 2010), others have suggested that ACE inhibitors potentiate preconditioning through bradykinin B2 receptor activation, and a further stimulus is needed to enhance ACE inhibitors' protective effects (Morris and Yellon, 1997; Jaberansari et al., 2001). In the context of PostC, the ACE inhibitor, enalaprilat, has been administered directly into the coronary arteries during reperfusion in small clinical trials, with improvement in inflammation, arrhythmias, ST-segment elevation and ventricular repolarization (Bonnemeier et al., 2007; Schaefer et al., 2007; Ungi et al., 2008). Of note, there are several large clinical trials showing good outcomes with the administration of ACE inhibitors after AMI (Kirkpatrick and St John Sutton, 2012).
Also, statins have been consistently shown to improve the prognosis in patients with coronary artery disease, heart failure, hypercholesterolaemia and several other cardiovascular conditions. Similar to ACE inhibitors, statins increase •NO bioavailability by means of several mechanisms (Lu et al., 2004). Further, statins reverse oxidative stress by decreasing the expression and the activity of NADPH oxidase, an effect that overlaps with that described previously for ACE inhibitors (Wassmann et al., 2002; Zhou and Liao, 2009). With regard to I/R injury, it has been shown that statins reduce infarct size in animal models via RISK- and NO-dependent pathways (Bell and Yellon, 2003; Ikeda et al., 2003; Vilahur et al., 2009). Translation to clinical studies has been positive in elective or programmed situations where statins may precondition the heart and may be beneficial in reducing myocardial injury (Vilahur et al., 2009; Nusca et al., 2010). However, the evidence supporting the use of acute high-dose atorvastatin in emergency situations as a post-conditioning agent is somewhat controversial (Kim et al., 2010; Ludman et al., 2011). To the best of our knowledge, there are no large randomized trials using acute high dose of statins in the I/R context.
Although it is impossible to clearly determine which of the properties (antioxidant/pronitrosative, antiproliferative, lipid-lowering effects, haemodynamic, etc.) of these drugs is responsible for their therapeutic impact, it is likely that their tenuous antioxidant and •NO promoter actions may play a pivotal role. In fact, we have seen how protein redox modifications induced by ROS/RNS can regulate and expand protein function under a variety of conditions. While irreversible oxidation usually leads to protein aggregation and degradation, reversible nitrosative signalling that usually occurs on protein cysteine residues can often serve as a sort of switch that regulates protein function and redox signalling pathways upon stress challenges. In the context of tolerance against I/R, including pre- and post-conditioning, a wealth of evidence has revealed that reversible cysteine redox modifications such as S-nitrosation/S-nitrosylation and S-glutathionylation formation can serve as a cellular defence mechanism against tissue I/R injury (see previous discussion). This is in agreement with our observation that the cardioprotective diazoxide induces S-nitrosylation of several proteins either before or after ischaemia (Penna et al., 2013b). In the present review, we have highlighted evidence of selective cysteine redox modifications as a protective measure in I/R injury, demonstrating that protein redox modifications can serve as a therapeutic target for attenuating tissue ischaemic injury. More oxidatively/nitrosatively modified proteins and consequent modulation of their function playing protective roles in tissue I/R injury need to be identified. In particular, we need to ascertain when and how the oxidatively/nitrosatively modifications of such identified proteins can be enhanced/inhibited by pharmacological agents.
Based upon the above considerations, therapy with direct antioxidant drugs might have been a naïve attempt in a complex pathophysiology. Definitely, we need more mechanistic studies to characterize better the sequences of events leading from ROS/RNS to cardiovascular damage or alternatively to cardiovascular protection, and whether there are particular components of these sequences that can be specifically targeted pharmacologically by agents that are available or need to be developed. It is likely that research efforts need to be redirected towards a redox-oriented approach. We know that the redox environment may be profoundly influenced by concomitant diseases and it is likely that each patient may have a different metabolic background in which I/R may trigger a different redox response. Therefore, it is likely that a personalized therapy is necessary to obtain beneficial effects and to redirect the redox response to beneficial rather than deleterious effects.
Summary and conclusions
A plethora of evidence collected throughout the past three decades has shown that ischaemic conditioning, including preconditioning, post-conditioning and remote conditioning, elicits endogenous cardiac protection. There is no doubt that the heart may be redirected to be less vulnerable to I/R injury and that, in this ‘redirection’, a role of paramount importance is played by redox signalling. However, translation of pre- and post-conditioning into clinical protocols is still in its infancy. In our opinion, such translation is, in part, impaired by the complex and intricate role of redox stress and redox signalling in cardiac injury and protection respectively. Here, too, a picture is emerging that induces us to rethink our beliefs: the reactive species are not always good or always bad and also in reperfusion they may play a beneficial role. Clinical application of ischaemic or pharmacological conditioning will depend, at least in part, upon enhancing our understanding of mechanistic and physiological components of redox-dependent myocardial injury and protection. Of course, technical complications in the reperfusion strategies and confounding effects of co-morbidities on the cardiovascular phenotype cannot be ignored and must be taken into consideration to maximize the beneficial effects of reperfusion.
Acknowledgments
We thank Professor Donatella Gattullo for invaluable support. The authors are supported for their research by the MIUR and University of Torino (ex-60%) and are members of the Board of the Italian Society of Cardiovascular Researches and Institute National of Cardiovascular Researches.
Glossary
- AMI
acute myocardial infarction
- CVD
cardiovascular disease
- I/R
ischaemia/reperfusion
- I-PostC
ischaemic post-conditioning
- I-PreC
ischaemic preconditioning
- mitoKATP
mitochondrial ATP-activated K+ channel
- MPG
mercaptopropionyl glycine
- mPTP
mitochondrial permeability transition pore
- NAC
N-acetylcysteine
- ODQ
1H-[1,2,4]oxadiazole[4,3,-α]quinoxaline-1-one
- RIRR
ROS-induced ROS release
- RISK
reperfusion injury salvage kinase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SAFE
survivor activating factor enhancement
- SERCA
sarco/endoplasmatic reticulum calcium ATPase
- SOD
superoxide dismutase
- STEMI
ST-segment elevation myocardial infarction
- XOR
xanthine oxo-reductase
Conflict of interest
The authors have no conflict of interest.
References
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelone T, Quintieri AM, Pasqua T, Gentile S, Tota B, Mahata SK, et al. Phosphodiesterase type-2 and NO-dependent S-nitrosylation mediate the cardioinhibition of the antihypertensive catestatin. Am J Physiol Heart Circ Physiol. 2012;302:H431–H442. doi: 10.1152/ajpheart.00491.2011. [DOI] [PubMed] [Google Scholar]
- Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, et al. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bell RM, Yellon DM. The contribution of endothelial nitric oxide synthase to early ischaemic preconditioning: the lowering of the preconditioning threshold. An investigation in eNOS knockout mice. Cardiovasc Res. 2001;52:274–280. doi: 10.1016/s0008-6363(01)00394-7. [DOI] [PubMed] [Google Scholar]
- Bell RM, Yellon DM. Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up-regulating a pro-survival pathway. J Am Coll Cardiol. 2003;41:508–515. doi: 10.1016/s0735-1097(02)02816-4. [DOI] [PubMed] [Google Scholar]
- Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, et al. Loss of ischemic preconditioning's cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292:H1764–H1769. doi: 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- Boengler K, Heusch G, Schulz R. Mitochondria in postconditioning. Antioxid Redox Signal. 2011;14:863–880. doi: 10.1089/ars.2010.3309. [DOI] [PubMed] [Google Scholar]
- Bolli R, Triana F, Jeroudi MO. Postischemic mechanical and vascular dysfunction (myocardial ‘stunning’ and microvascular ‘stunning’) and the effects of calcium-channel blockers on ischemia/reperfusion injury. Clin Cardiol. 1989;12:III16–III25. [PubMed] [Google Scholar]
- Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EKB, et al. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial ‘stunning’ is a manifestation of reperfusion injury. Circ Res. 1989;65:607–622. doi: 10.1161/01.res.65.3.607. [DOI] [PubMed] [Google Scholar]
- Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1α promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- Bonnemeier H, Schäfer U, Ortak J, Kurz T, Katus HA, Richardt G, et al. Low doses of intracoronary enalaprilat suppress reperfusion-associated ventricular arrhythmias after primary percutaneous coronary interventions for acute myocardial infarction. Pacing Clin Electrophysiol. 2007;30:S160–S165. doi: 10.1111/j.1540-8159.2007.00629.x. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med. 2010;49:687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- Brewer AC, Mustafi SB, Murray TV, Rajasekaran NS, Benjamin IJ. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid Redox Signal. 2013;18:1114–1127. doi: 10.1089/ars.2012.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- Busse R, Fleming I. Vascular endothelium and blood flow. Handb Exp Pharmacol. 2006;176:43–78. doi: 10.1007/3-540-36028-x_2. [DOI] [PubMed] [Google Scholar]
- Buttery LD, Springall DR, Chester AH, Evans TJ, Standfield EN, Parums DV, et al. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab Invest. 1996;75:77–85. [PubMed] [Google Scholar]
- Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal. 2013;18:1078–1099. doi: 10.1089/ars.2012.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi A, Menabò R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. 2009;1787:774–780. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Chen GC, Lu DB, Pang Z, Liu QF. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc. 2013;2:e000329. doi: 10.1161/JAHA.113.000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Hurd TR, Nadtochiy SM, Brookes PS, Fearnley IM, Lilley KS, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Downey JM. Ischemic postconditioning: from receptor to end-effector. Antioxid Redox Signal. 2011;14:821–831. doi: 10.1089/ars.2010.3318. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial K(ATP) channels. Circ Res. 2001;89:273–278. doi: 10.1161/hh1501.094266. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct sparing strategies. Cardiovasc Res. 2006;70:231–239. doi: 10.1016/j.cardiores.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895–1903. doi: 10.1161/CIRCULATIONAHA.106.675710. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol. 2008;103:464–471. doi: 10.1007/s00395-008-0737-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook NR, Albert CM, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, et al. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women's Antioxidant Cardiovascular Study. Arch Intern Med. 2007;167:1610–1618. doi: 10.1001/archinte.167.15.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp SW, Ferreira LF, Herspring KF, Musch TI, Poole DC. The effects of aging on capillary hemodynamics in contracting rat spinotrapezius muscle. Microvasc Res. 2009;77:113–119. doi: 10.1016/j.mvr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Costa VM, Carvalho F, Bastos ML, Carvalho RA, Carvalho M, Remião F. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr Med Chem. 2011;18:2272–2314. doi: 10.2174/092986711795656081. [DOI] [PubMed] [Google Scholar]
- Csonka C, Kupai K, Bencsik P, Görbe A, Pálóczi J, Zvara A, et al. Cholesterol-enriched diet inhibits cardioprotection by ATP-sensitive K+ channel activators cromakalim and diazoxide. Am J Physiol Heart Circ Physiol. 2014;306:H405–H413. doi: 10.1152/ajpheart.00257.2013. [DOI] [PubMed] [Google Scholar]
- Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, et al. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003;17:1934–1936. doi: 10.1096/fj.02-1208fje. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Kaludercic N, Carpi A, Menabò R, Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol. 2009a;104:131–139. doi: 10.1007/s00395-009-0008-4. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Kaludercic N, Carpi A, Menabò R, Giorgio M. Mitochondria and vascular pathology. Pharmacol Rep. 2009b;61:123–130. doi: 10.1016/s1734-1140(09)70014-3. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Canton M, Carpi A, Kaludercic N, Menabò R, Menazza S, et al. Mitochondrial injury and protection in ischemic pre- and postconditioning. Antioxid Redox Signal. 2011;14:881–891. doi: 10.1089/ars.2010.3375. [DOI] [PubMed] [Google Scholar]
- Downey JM. Free radicals and their involvement during long-term myocardial ischemia and reperfusion. Annu Rev Physiol. 1990;52:487–504. doi: 10.1146/annurev.ph.52.030190.002415. [DOI] [PubMed] [Google Scholar]
- Downey JM, Cohen MV. A really radical observation – a comment on Penna et al. in Basic Res Cardiol (2006) 101:180–189. Basic Res Cardiol. 2006;101:190–191. doi: 10.1007/s00395-006-0586-3. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Eagle KA, Guyton RA, Davidoff R, Edwards FH, Ewy GA, Gardner TJ, et al. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1999 Guidelines for Coronary Artery Bypass Graft Surgery) J Am Coll Cardiol. 2004;44:e213–e310. doi: 10.1016/j.jacc.2004.07.021. . Erratum in: J Am Coll Cardiol. 2005: 45:1377. [DOI] [PubMed] [Google Scholar]
- Ebbing M, Bønaa KH, Arnesen E, Ueland PM, Nordrehaug JE, Rasmussen K, et al. Combined analyses and extended follow-up of two randomized controlled homocysteine-lowering B-vitamin trials. J Intern Med. 2010;268:367–382. doi: 10.1111/j.1365-2796.2010.02259.x. [DOI] [PubMed] [Google Scholar]
- Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009;46:1386–1391. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey MG, Miranda KM, Thomas DD, Xavier S, Citrin D, Vitek MP, et al. A chemical perspective on the interplay between NO, reactive oxygen species, and reactive nitrogen oxide species. Ann N Y Acad Sci. 2002;962:195–206. doi: 10.1111/j.1749-6632.2002.tb04068.x. [DOI] [PubMed] [Google Scholar]
- Fan Q, Yang XC, Liu Y, Wang LF, Liu SH, Ge YG, et al. Postconditioning attenuates myocardial injury by reducing nitro oxidative stress in vivo in rats and in humans. Clin Sci (Lond) 2010;120:251–261. doi: 10.1042/CS20100369. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. doi: 10.1038/sj.bjp.0706693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–1356. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89:1982–1991. doi: 10.1161/01.cir.89.5.1982. [DOI] [PubMed] [Google Scholar]
- Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- Foster DB, Van Eyk JE, Marbán E, O'Rourke B. Redox signaling and protein phosphorylation in mitochondria: progress and prospects. J Bioenerg Biomembr. 2009;41:159–168. doi: 10.1007/s10863-009-9217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, et al. Mitochondrial ROMK channel is a molecular component of mitoK(ATP) Circ Res. 2012;111:446–454. doi: 10.1161/CIRCRESAHA.112.266445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freixa X, Bellera N, Ortiz-Pérez JT, Jime'nez M, Paré C, Bosch X, et al. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012;33:103–312. doi: 10.1093/eurheartj/ehr297. [DOI] [PubMed] [Google Scholar]
- Gale CP, Cattle BA, Woolston A, Baxter PD, West TH, Simms AD, et al. Resolving inequalities in care? Reduced mortality in the elderly after acute coronary syndromes. The Myocardial Ischaemia National Audit Project 2003–2010. Eur Heart J. 2012;33:630–639. doi: 10.1093/eurheartj/ehr381. [DOI] [PubMed] [Google Scholar]
- Galkin A, Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem. 2007;282:37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- Garcia S, Henry TD, Wang YL, Chavez IJ, Pedersen WR, Lesser JR, et al. Long-term follow-up of patients undergoing postconditioning during ST-elevation myocardial infarction. J Cardiovasc Transl Res. 2011;4:92–98. doi: 10.1007/s12265-010-9252-0. [DOI] [PubMed] [Google Scholar]
- Garlid AO, Jaburek M, Jacobs JP, Garlid KD. Mitochondrial reactive oxygen species: which ROS signals cardioprotection? Am J Physiol Heart Circ Physiol. 2013;305:H960–H968. doi: 10.1152/ajpheart.00858.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Lalu MM, Csonka C, Bencsik P, Schulz R, Ferdinandy P. Hyperlipidemia attenuates the infarct size-limiting effect of ischemic preconditioning: role of matrix metalloproteinase-2 inhibition. J Pharmacol Exp Ther. 2006;316:154–161. doi: 10.1124/jpet.105.091140. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Görbe A, Pipis J, Burley DS, Ferdinandy P, Baxter GF. Hyperlipidaemia induced by a high-cholesterol diet leads to the deterioration of guanosine-3′,5′-cyclic monophosphate/protein kinase G-dependent cardioprotection in rats. Br J Pharmacol. 2009;158:1495–1502. doi: 10.1111/j.1476-5381.2009.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–195. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görbe A, Varga ZV, Kupai K, Bencsik P, Kocsis GF, Csont T, et al. Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: the role of connexin 43. Am J Physiol Heart Circ Physiol. 2011;300:H1907–H1913. doi: 10.1152/ajpheart.01242.2010. [DOI] [PubMed] [Google Scholar]
- Granfeldt A, Lefer DJ, Vinten-Johansen J. Protective ischemia in patients: preconditioning and postconditioning. Cardiovasc Res. 2009;83:234–246. doi: 10.1093/cvr/cvp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc Res. 2006;70:212–221. doi: 10.1016/j.cardiores.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Gucek M, Murphy E. What can we learn about cardioprotection from the cardiac mitochondrial proteome? Cardiovasc Res. 2010;88:211–218. doi: 10.1093/cvr/cvq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, et al. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and delta1-opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol. 2005;289:H2251–H2227. doi: 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li Q, Wu WJ, Tan W, Zhu X, Mu J, et al. Endothelial nitric oxide synthase is not necessary for the early phase of ischemic preconditioning in the mouse. J Mol Cell Cardiol. 2008;44:496–501. doi: 10.1016/j.yjmcc.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Sanganalmath SK, Wu W, Zhu X, Huang Y, Tan W, et al. Identification of inducible nitric oxide synthase in peripheral blood cells as a mediator of myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2012;107:253. doi: 10.1007/s00395-012-0253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, West NE, Pillai R, Taggart DP, Channon KM. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension. 2002;39:1088–1094. doi: 10.1161/01.hyp.0000018041.48432.b5. [DOI] [PubMed] [Google Scholar]
- Hahn JY, Song YB, Kim EK, Yu CW, Bae JW, Chung WY, et al. Ischemic postconditioning during primary percutaneous coronary intervention: the effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation. 2013;128:1889–1896. doi: 10.1161/CIRCULATIONAHA.113.001690. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Lipid peroxidation in brain homogenates: the role of iron and hydroxyl radicals. J Neurochem. 1997;69:1330–1331. doi: 10.1046/j.1471-4159.1997.69031330.x. [DOI] [PubMed] [Google Scholar]
- Harzand A, Tamariz L, Hare JM. Uric acid, heart failure survival, and the impact of xanthine oxidase inhibition. Congest Heart Fail. 2012;18:179–182. doi: 10.1111/j.1751-7133.2011.00262.x. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ. Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science) Curr Pharm Des. 2013;19:4544–4563. doi: 10.2174/1381612811319250004. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;116:173–191. doi: 10.1016/j.pharmthera.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: new strategies for cardioprotection. Diabetes Obes Metab. 2008;10:451–459. doi: 10.1111/j.1463-1326.2007.00762.x. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: underlying mechanisms and clinical application. Atherosclerosis. 2009;204:334–341. doi: 10.1016/j.atherosclerosis.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol. 2007;102:445–452. doi: 10.1007/s00395-007-0656-1. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol. 2009;104:189–202. doi: 10.1007/s00395-009-0010-x. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Lecour S, Yellon DM. Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal. 2011;14:893–907. doi: 10.1089/ars.2010.3360. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Erik Bøtker H, Condorelli G, Ferdinandy P, Garcia-Dorado D, Heusch G, et al. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2013;98:7–27. doi: 10.1093/cvr/cvt004. [DOI] [PubMed] [Google Scholar]
- Heinrich TA, da Silva RS, Miranda KM, Switzer CH, Wink DA, Fukuto JM. Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol. 2013;169:1417–1429. doi: 10.1111/bph.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekens CH, Buring JE, Manson JE, Stampfer M, Rosner B, Cook NR, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med. 1996;334:1145–1149. doi: 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- Heusch G. Cardioprotection: chances and challenges of its translation to the clinic. Lancet. 2013;381:166–175. doi: 10.1016/S0140-6736(12)60916-7. [DOI] [PubMed] [Google Scholar]
- Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- Hoffman JW, Jr, Gilbert TB, Poston RS, Silldorff EP. Myocardial reperfusion injury: etiology, mechanisms, and therapies. J Extra Corpor Technol. 2004;36:391–411. [PubMed] [Google Scholar]
- Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, Preckel B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br J Anaesth. 2008;100:465–471. doi: 10.1093/bja/aen022. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Young LH, Lefer AM. Rosuvastatin, a new HMG-CoA reductase inhibitor, protects ischemic reperfused myocardium in normocholesterolemic rats. J Cardiovasc Pharmacol. 2003;41:649–656. doi: 10.1097/00005344-200304000-00019. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Andreadou I, Prokovas E, Zoga A, Farmakis D, Fotopoulou T, et al. Simvastatin in contrast to postconditioning reduces infarct size in hyperlipidemic rabbits: possible role of oxidative/nitrosative stress attenuation. Basic Res Cardiol. 2010;105:193–203. doi: 10.1007/s00395-009-0078-3. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Poncelas-Nozal M, Hernando V, Agulló L, Ruiz-Meana M, et al. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J Mol Cell Cardiol. 2011a;50:903–909. doi: 10.1016/j.yjmcc.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Inserte J, Ruiz-Meana M, Rodriguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal. 2011b;14:923–939. doi: 10.1089/ars.2010.3312. [DOI] [PubMed] [Google Scholar]
- Inserte J, Hernando V, Vilardosa Ú, Abad E, Poncelas-Nozal M, Garcia-Dorado D. Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc. 2013;2:e005975. doi: 10.1161/JAHA.112.005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophys Res Commun. 2003;305:776–783. doi: 10.1016/s0006-291x(03)00814-3. [DOI] [PubMed] [Google Scholar]
- Ivanes F, Rioufol G, Piot C, Ovize M. Postconditioning in acute myocardial infarction patients. Antioxid Redox Signal. 2011;14:811–820. doi: 10.1089/ars.2010.3354. [DOI] [PubMed] [Google Scholar]
- Jaberansari MT, Baxter GF, Muller CA, Latouf SE, Röth E, Opie LH, et al. Angiotensin converting enzyme inhibition enhances a subthreshold stimulus to elicit delayed preconditioning in pig myocardium. J Am Coll Cardiol. 2001;37:1996–2001. doi: 10.1016/s0735-1097(01)01232-3. [DOI] [PubMed] [Google Scholar]
- James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- Jaxa-Chamiec T, Bednarz B, Drozdowska D, Gessek J, Gniot J, Janik K, et al. Antioxidant effects of combined vitamins C and E in acute myocardial infarction: the randomized, double blind, placebo controlled, multicenter pilot Myocardial Infarction and VITamins (MIVIT) trial. Kardiol Pol. 2005;62:344–350. [PubMed] [Google Scholar]
- Jaxa-Chamiec T, Bednarz B, Herbaczynska-Cedro K, Maciejewski P, Ceremuzynski L MIVIT Trial Group. Effects of vitamins C and E on the outcome after acute myocardial infarction in diabetics: a retrospective, hypothesis-generating analysis from the MIVIT study. Cardiology. 2009;112:219–223. doi: 10.1159/000151239. [DOI] [PubMed] [Google Scholar]
- Jin ZQ, Chen X. Pretreatment with ramiprilat induces cardioprotection against free radical injury in guinea-pig isolated heart: involvement of bradykinin, protein kinase C and prostaglandins. Clin Exp Pharmacol Physiol. 2000;27:257–262. doi: 10.1046/j.1440-1681.2000.03233.x. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Jope RS, Zhang L, Song L. Peroxynitrite modulates the activation of p38 and extracellular regulated kinases in PC12 cells. Arch Biochem Biophys. 2000;376:365–370. doi: 10.1006/abbi.2000.1728. [DOI] [PubMed] [Google Scholar]
- Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. 2014;73C:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevin LG, Novalija E, Riess ML, Camara AK, Rhodes SS, Stowe DF. Sevoflurane exposure generates superoxide but leads to decreased superoxide during ischemia and reperfusion in isolated hearts. Anesth Analg. 2003;96:949–955. doi: 10.1213/01.ANE.0000052515.25465.35. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kim J, Choi D, Lee CJ, Lee SH, Ko YG, et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: the STATIN STEMI trial. JACC Cardiovasc Interv. 2010;3:332–339. doi: 10.1016/j.jcin.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick JN, St John Sutton M. Assessment of ventricular remodeling in heart failure clinical trials. Curr Heart Fail Rep. 2012;9:328–336. doi: 10.1007/s11897-012-0116-6. [DOI] [PubMed] [Google Scholar]
- Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- Knock GA, Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal. 2011;15:1531–1547. doi: 10.1089/ars.2010.3614. [DOI] [PubMed] [Google Scholar]
- Kocsis GF, Sárközy M, Bencsik P, Pipicz M, Varga ZV, Pálóczi J, et al. Preconditioning protects the heart in a prolonged uremic condition. Am J Physiol Heart Circ Physiol. 2012;303:H1229–H1236. doi: 10.1152/ajpheart.00379.2012. [DOI] [PubMed] [Google Scholar]
- Koeck T, Stuehr DJ, Aulak KS. Mitochondria and regulated tyrosine nitration. Biochem Soc Trans. 2005;33:1399–1403. doi: 10.1042/BST0331399. [DOI] [PubMed] [Google Scholar]
- Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korichneva I. Zinc dynamics in the myocardial redox signaling network. Antioxid Redox Signal. 2006;8:1707–1721. doi: 10.1089/ars.2006.8.1707. [DOI] [PubMed] [Google Scholar]
- Krijnen PA, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, et al. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–199. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupai K, Csonka C, Fekete V, Odendaal L, van Rooyen J, de Marais W, et al. Cholesterol diet induced hyperlipidemia impairs the cardioprotective effect of postconditioning: role of peroxynitrite. Am J Physiol Heart Circ Physiol. 2009;297:H1729–H1735. doi: 10.1152/ajpheart.00484.2009. [DOI] [PubMed] [Google Scholar]
- Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361–367. doi: 10.1002/ccd.20397. [DOI] [PubMed] [Google Scholar]
- Laskey WK, Yoon S, Calzada N, Ricciardi MJ. Concordant improvements in coronary flow reserve and ST-segment resolution during percutaneous coronary intervention for acute myocardial infarction: a benefit of postconditioning. Catheter Cardiovasc Interv. 2008;72:212–220. doi: 10.1002/ccd.21583. [DOI] [PubMed] [Google Scholar]
- Lauer N, Suvorava T, Rüther U, Jacob R, Meyer W, Harrison DG, et al. Critical involvement of hydrogen peroxide in exercise-induced upregulation of endothelial NO synthase. Cardiovasc Res. 2005;65:254–262. doi: 10.1016/j.cardiores.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women's Health Study: a randomized controlled trial. JAMA. 2005;294:56–65. doi: 10.1001/jama.294.1.56. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine S, Bule'on C, Rouet R, Ivascau C, Babatasi G, Massetti M, et al. Bradykinin and adenosine receptors mediate desflurane induced postconditioning in human myocardium: role of reactive oxygen species. BMC Anesthesiol. 2010;10:12. doi: 10.1186/1471-2253-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Loukili N, Rosenblatt-Velin N, Pacher P, Feihl F, Waeber B, et al. Peroxynitrite is a key mediator of the cardioprotection afforded by ischemic postconditioning in vivo. PLoS ONE. 2013;8:e70. doi: 10.1371/journal.pone.0070331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- Lim G, Venetucci L, Eisner DA, Casadei B. Does nitric oxide modulate cardiac ryanodine receptor function? Implications for excitation-contraction coupling. Cardiovasc Res. 2008;77:256–264. doi: 10.1093/cvr/cvm012. [DOI] [PubMed] [Google Scholar]
- Limalanathan S, Andersen GØ, Kløw NE, Abdelnoor M, Hoffmann P, Eritsland J. Effect of ischemic postconditioning on infarct size in patients with ST-elevation myocardial infarction treated by primary PCI results of the POSTEMI (POstconditioning in ST-Elevation Myocardial Infarction) randomized trial. J Am Heart Assoc. 2014;3:e000679. doi: 10.1161/JAHA.113.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XM, Zhang ZY, Wang LF, Zhang L, Liu Y, Liu XL, et al. Attenuation of tumor necrosis factor-alpha elevation and improved heart function by postconditioning for 60 seconds in patients with acute myocardial infarction. Chin Med J (Engl) 2010;123:1833–1839. [PubMed] [Google Scholar]
- Lopes RD, Li L, Granger CB, Wang TY, Foody JM, Funk M, et al. Atrial fibrillation and acute myocardial infarction: antithrombotic therapy and outcomes. Am J Med. 2012;125:897–905. doi: 10.1016/j.amjmed.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Lønborg J, Kelbaek H, Vejlstrup N, Jørgensen E, Helqvist S, Saunamäki K, et al. Cardioprotective effects of ischemic postconditioning in patients treated with primary percutaneous coronary intervention, evaluated by magnetic resonance. Circ Cardiovasc Interv. 2010;3:34–41. doi: 10.1161/CIRCINTERVENTIONS.109.905521. [DOI] [PubMed] [Google Scholar]
- Lu TM, Ding YA, Leu HB, Yin WH, Sheu WH, Chu KM. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am J Cardiol. 2004;94:157–161. doi: 10.1016/j.amjcard.2004.03.052. [DOI] [PubMed] [Google Scholar]
- Ludman AJ, Hausenloy DJ, Babu G, Hasleton J, Venugopal V, Boston-Griffiths E, et al. Failure to recapture cardioprotection with high-dose atorvastatin in coronary artery bypass surgery: a randomised controlled trial. Basic Res Cardiol. 2011;106:1387–1395. doi: 10.1007/s00395-011-0209-5. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhang X, Li C, Luo M. Effect of postconditioning on coronary blood flow velocity and endothelial function and LV recovery after myocardial infarction. J Interv Cardiol. 2006;19:367–375. doi: 10.1111/j.1540-8183.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- Majidi M, Kosinski AS, Al-Khatib SM, Lemmert ME, Smolders L, van Weert A, et al. Reperfusion ventricular arrhythmia ‘bursts’ predict larger infarct size despite TIMI 3 flow restoration with primary angioplasty for anterior ST-elevation myocardial infarction. Eur Heart J. 2009;30:757–764. doi: 10.1093/eurheartj/ehp005. [DOI] [PubMed] [Google Scholar]
- Martínez MC, Andriantsitohaina R. Reactive nitrogen species: molecular mechanisms and potential significance in health and disease. Antioxid Redox Signal. 2009;11:669–702. doi: 10.1089/ars.2007.1993. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Adams NA, Pan Y, Price A, Wong M. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci. 2011;31:359–370. doi: 10.1523/JNEUROSCI.2225-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Methner C, Lukowski R, Grube K, Loga F, Smith RA, Murphy MP, et al. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic Res Cardiol. 2013;108:337. doi: 10.1007/s00395-013-0337-1. [DOI] [PubMed] [Google Scholar]
- Morales CR, Pedrozo Z, Lavandero S, Hill JA. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxid Redox Signal. 2014;20:507–518. doi: 10.1089/ars.2013.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SD, Yellon DM. Angiotensin-converting enzyme inhibitors potentiate preconditioning through bradykinin B2 receptor activation in human heart. J Am Coll Cardiol. 1997;29:1599–1606. doi: 10.1016/s0735-1097(97)00087-9. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2007;75:478–486. doi: 10.1016/j.cardiores.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Kohr M, Sun J, Nguyen T, Steenbergen C. S-nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol. 2012;52:568–577. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, et al. Signaling by S-nitrosylation in the heart. J Mol Cell Cardiol. 2014;73C:18–25. doi: 10.1016/j.yjmcc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Xie QW. Regulation of biosynthesis of nitric oxide. J Biol Chem. 1994;269:13725–13728. [PubMed] [Google Scholar]
- Näslund U, Häggmark S, Johansson G, Pennert K, Reiz S, Marklund SL. Effects of reperfusion and superoxide dismutase on myocardial infarct size in a closed chest pig model. Cardiovasc Res. 1992;26:170–178. doi: 10.1093/cvr/26.2.170. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73C:26–33. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, Lambeth JD. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry. 2010;49:2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusca A, Melfi R, Patti G, Di Sciascio G. Statin loading for acute coronary syndromes. Curr Opin Cardiol. 2010;25:373–378. doi: 10.1097/HCO.0b013e32833987ca. [DOI] [PubMed] [Google Scholar]
- Obame FN, Zini R, Souktani R, Berdeaux A, Morin D. Peripheral benzodiazepine receptor-induced myocardial protection is mediated by inhibition of mitochondrial membrane permeabilization. J Pharmacol Exp Ther. 2007;323:336–345. doi: 10.1124/jpet.107.124255. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Cohen MV, Yellon DM, Downey JM. Mitochondrial K(ATP) channels: role in cardioprotection. Cardiovasc Res. 2002;55:429–437. doi: 10.1016/s0008-6363(02)00439-x. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, et al. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004;286:H468–H476. doi: 10.1152/ajpheart.00360.2003. [DOI] [PubMed] [Google Scholar]
- Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- O'Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- Ovize M, Thibault H, Przyklenk K. Myocardial conditioning: opportunities for clinical translation. Circ Res. 2013;113:439–450. doi: 10.1161/CIRCRESAHA.113.300764. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliaro P. Differential biological effects of products of nitric oxide (NO) synthase: it is not enough to say NO. Life Sci. 2003;73:2137–2149. doi: 10.1016/s0024-3205(03)00593-9. [DOI] [PubMed] [Google Scholar]
- Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, et al. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C. Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal. 2011;14:833–850. doi: 10.1089/ars.2010.3245. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen CM, Schmidt MR, Barnes G, Bøtker HE, Kharbanda RK, Newby DE, et al. Bradykinin does not mediate remote ischaemic preconditioning or ischaemia-reperfusion injury in vivo in man. Heart. 2011;97:1857–1861. doi: 10.1136/heartjnl-2011-300323. [DOI] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Robinson KM, Cassina P, England P, Beckman JS, et al. Peroxynitrite transforms nerve growth factor into an apoptotic factor for motor neurons. Free Radic Biol Med. 2006;41:1632–1644. doi: 10.1016/j.freeradbiomed.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Penna C, Pagliaro P, Rastaldo R, Di Pancrazio F, Lippe G, Gattullo D, et al. F0F1 ATP synthase activity is differently modulated by coronary reactive hyperemia before and after ischemic preconditioning in the goat. Am J Physiol Heart Circ Physiol. 2004;287:H2192–H2200. doi: 10.1152/ajpheart.00327.2004. [DOI] [PubMed] [Google Scholar]
- Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, et al. Post-conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol. 2006a;101:168–179. doi: 10.1007/s00395-005-0543-6. [DOI] [PubMed] [Google Scholar]
- Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, et al. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006b;101:180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P. Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res. 2007;75:168–177. doi: 10.1016/j.cardiores.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Penna C, Mancardi D, Raimondo S, Geuna S, Pagliaro P. The paradigm of postconditioning to protect the heart. J Cell Mol Med. 2008a;12:435–458. doi: 10.1111/j.1582-4934.2007.00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna C, Mancardi D, Tullio F, Pagliaro P. Postconditioning and intermittent bradykinin induced cardioprotection require cyclooxygenase activation and prostacyclin release during reperfusion. Basic Res Cardiol. 2008b;103:368–377. doi: 10.1007/s00395-007-0695-7. [DOI] [PubMed] [Google Scholar]
- Penna C, Mancardi D, Rastaldo R, Pagliaro P. Cardioprotection: a radical view free radicals in pre and postconditioning. Biochim Biophys Acta. 2009;1787:781–793. doi: 10.1016/j.bbabio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Penna C, Tullio F, Moro F, Folino A, Merlino A, Pagliaro P. Effects of a protocol of ischemic postconditioning and/or captopril in hearts of normotensive and hypertensive rats. Basic Res Cardiol. 2010;105:181–192. doi: 10.1007/s00395-009-0075-6. [DOI] [PubMed] [Google Scholar]
- Penna C, Bassino E, Alloatti G. Platelet activating factor: the good and the bad in the ischemic/reperfused heart. Exp Biol Med (Maywood) 2011a;236:390–401. doi: 10.1258/ebm.2011.010316. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Tullio F, Moro F, Parisella ML, Merlino A, et al. Post-ischemic early acidosis in cardiac postconditioning modifies the activity of antioxidant enzymes, reduces nitration, and favors protein S-nitrosylation. Pflugers Arch. 2011b;462:219–233. doi: 10.1007/s00424-011-0970-1. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal. 2013a;18:556–599. doi: 10.1089/ars.2011.4459. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Tullio F, Angotti C, Camporeale A, Poli V, et al. Diazoxide postconditioning induces mitochondrial protein S-nitrosylation and a redox-sensitive mitochondrial phosphorylation/translocation of RISK elements: no role for SAFE. Basic Res Cardiol. 2013b;108:371. doi: 10.1007/s00395-013-0371-z. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Tullio F, Angotti C, Pagliaro P. Acidic infusion in early reperfusion affects the activity of antioxidant enzymes in postischemic isolated rat heart. J Surg Res. 2013c;183:111–118. doi: 10.1016/j.jss.2012.12.029. [DOI] [PubMed] [Google Scholar]
- Penna C, Angotti C, Pagliaro P. Protein S-nitrosylation in preconditioning and postconditioning. Exp Biol Med (Maywood) 2014;239:647–662. doi: 10.1177/1535370214522935. [DOI] [PubMed] [Google Scholar]
- Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:712–721. doi: 10.1016/j.bbagen.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, et al. A mitochondria targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przyklenk K. Reduction of myocardial infarct size with ischemic ‘conditioning’: physiologic and technical considerations. Anesth Analg. 2013;117:891–901. doi: 10.1213/ANE.0b013e318294fc63. [DOI] [PubMed] [Google Scholar]
- Riess ML, Camara AK, Novalija E, Chen Q, Rhodes SS, Stowe DF. Anesthetic preconditioning attenuates mitochondrial Ca2+ overload during ischemia in Guinea pig intact hearts: reversal by 5-hydroxydecanoic acid. Anesth Analg. 2002b;95:1540–1546. doi: 10.1097/00000539-200212000-00013. [DOI] [PubMed] [Google Scholar]
- Riess ML, Novalija E, Camara AK, Eells JT, Chen Q, Stowe DF. Preconditioning with sevoflurane reduces changes in nicotinamide adenine dinucleotide during ischemia-reperfusion in isolated hearts: reversal by 5-hydroxydecanoic acid. Anesthesiology. 2003;98:387–395. doi: 10.1097/00000542-200302000-00019. [DOI] [PubMed] [Google Scholar]
- Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronson RS, Nakamura M, Vinten-Johansen J. The cardiovascular effects and implications of peroxynitrite. Cardiovasc Res. 1999;44:47–59. doi: 10.1016/s0008-6363(99)00184-4. [DOI] [PubMed] [Google Scholar]
- Sánchez JA, Rodríguez-Sinovas A, Barba I, Miró-Casas E, Fernández-Sanz C, Ruiz-Meana M, et al. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia-reperfusion injury and preconditioning protection. Basic Res Cardiol. 2013;108:351. doi: 10.1007/s00395-013-0351-3. [DOI] [PubMed] [Google Scholar]
- Saini HK, Machackova J, Dhalla NS. Role of reactive oxygen species in ischemic preconditioning of subcellular organelles in the heart. Antioxid Redox Signal. 2004;6:393–404. doi: 10.1089/152308604322899468. [DOI] [PubMed] [Google Scholar]
- Saitoh S, Kiyooka T, Rocic P, Rogers PA, Zhang C, Swafford A, et al. Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol. 2007;293:H3720–H3725. doi: 10.1152/ajpheart.00436.2007. [DOI] [PubMed] [Google Scholar]
- Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Yokoyama U, Fujita T, Okumura S, Ishikawa Y. The roles of cytochrome p450 in ischemic heart disease. Curr Drug Metab. 2011;12:526–532. doi: 10.2174/138920011795713715. [DOI] [PubMed] [Google Scholar]
- Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2011;301:H287–H296. doi: 10.1152/ajpheart.00327.2011. [DOI] [PubMed] [Google Scholar]
- Schaefer U, Kurz T, Bonnemeier H, Dendorfer A, Hartmann F, Schunkert H, et al. Intracoronary enalaprilat during angioplasty for acute myocardial infarction: alleviation of postischaemic neurohumoral and inflammatory stress? J Intern Med. 2007;261:188–200. doi: 10.1111/j.1365-2796.2006.01757.x. [DOI] [PubMed] [Google Scholar]
- Schröder J, Müller-Werdan U, Reuter S, Vogt A, Schlitt M, Raaz U, et al. Are the elderly different? Factors influencing mortality after percutaneous coronary intervention with stent implantation. Z Gerontol Geriatr. 2013;46:144–150. doi: 10.1007/s00391-012-0338-y. [DOI] [PubMed] [Google Scholar]
- Schwiebert C, Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, et al. Postconditioning by xenon and hypothermia in the rat heart in vivo. Eur J Anaesthesiol. 2010;27:734–739. doi: 10.1097/EJA.0b013e328335fc4c. [DOI] [PubMed] [Google Scholar]
- Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, MacFadyen J, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300:2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesso HD, Christen WG, Bubes V, Smith JP, MacFadyen J, Schvartz M, et al. Multivitamins in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2012;308:1751–1760. doi: 10.1001/jama.2012.14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Feng J, Qu H, Cheng YY. A proteomic study of S-nitrosylation in the rat cardiac proteins in vitro. Biol Pharm Bull. 2008;31:1536–15340. doi: 10.1248/bpb.31.1536. [DOI] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JN, Duglan D, Casadei B, Carnicer R. Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J Mol Cell Cardiol. 2014;73C:80–91. doi: 10.1016/j.yjmcc.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Simoons ML, Serruys PW, van den Brand M, Res J, Verheugt FW, Krauss XH, et al. Early thrombolysis in acute myocardial infarction: limitation of infarct size and improved survival. J Am Coll Cardiol. 1986;7:717–728. doi: 10.1016/s0735-1097(86)80329-1. [DOI] [PubMed] [Google Scholar]
- Sivaraman V, Yellon DM. Pharmacologic therapy that simulates conditioning for cardiac ischemic/reperfusion injury. J Cardiovasc Pharmacol Ther. 2014;19:83–96. doi: 10.1177/1074248413499973. [DOI] [PubMed] [Google Scholar]
- Skyschally A, Van Caster P, Iliodromitis EK, Schulz R, Kremastinos DT, Heusch G. Ischemic postconditioning: experimental models and protocol algorithms. Basic Res Cardiol. 2009;104:469–483. doi: 10.1007/s00395-009-0040-4. [DOI] [PubMed] [Google Scholar]
- Sörensson P, Saleh N, Bouvier F, Böhm F, Settergren M, Caidahl K, et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart. 2010;96:1710–1715. doi: 10.1136/hrt.2010.199430. [DOI] [PubMed] [Google Scholar]
- Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. 2013;112:393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca+2 channel 1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006a;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006b;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Sun J, Kohr MJ, Nguyen T, Aponte AM, Connelly PS, Esfahani SG, et al. Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid Redox Signal. 2012;16:45–56. doi: 10.1089/ars.2010.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Aponte AM, Kohr MJ, Tong G, Steenbergen C, Murphy E. Essential role of nitric oxide in acute ischemic preconditioning: S-nitros(yl)ation versus sGC/cGMP/PKG signaling? Free Radic Biol Med. 2013;54:105–112. doi: 10.1016/j.freeradbiomed.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma M, Wei L, Shi J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011;7:657–671. doi: 10.2217/fca.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini G, Favaretto E, Marra MP, Frigo AC, Napodano M, Cacciavillani L, et al. Postconditioning during coronary angioplasty in acute myocardial infarction: the POST-AMI trial. Int J Cardiol. 2012;162:33–38. doi: 10.1016/j.ijcard.2012.03.136. [DOI] [PubMed] [Google Scholar]
- Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]
- Tissier R, Waintraub X, Couvreur N, Gervais M, Bruneval P, Mandet C, et al. Pharmacological postconditioning with the phytoestrogen genistein. J Mol Cell Cardiol. 2007;42:79–87. doi: 10.1016/j.yjmcc.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, et al. Playing with cardiac ‘redox switches’: the ‘HNO way’ to modulate cardiac function. Antioxid Redox Signal. 2011;14:1687–1698. doi: 10.1089/ars.2010.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Toit EF, Genis A, Opie LH, Pollesello P, Lochner A. A role for the RISK pathway and K(ATP) channels in pre- and post-conditioning induced by levosimendan in the isolated guinea pig heart. Br J Pharmacol. 2008;154:41–50. doi: 10.1038/bjp.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Aponte AM, Kohr MJ, Steenbergen C, Murphy E, Sun J. Postconditioning leads to an increase in protein S-nitrosylation. Am J Physiol Heart Circ Physiol. 2014;306:H825–H832. doi: 10.1152/ajpheart.00660.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol Int. 1999;49:91–102. doi: 10.1046/j.1440-1827.1999.00829.x. [DOI] [PubMed] [Google Scholar]
- Tullio F, Angotti C, Perrelli MG, Penna C, Pagliaro P. Redox balance and cardioprotection. Basic Res Cardiol. 2013;108:392. doi: 10.1007/s00395-013-0392-7. [DOI] [PubMed] [Google Scholar]
- Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- Ungi I, Pálinkás A, Nemes A, Ungi T, Thury A, Sepp R, et al. Myocardial protection with enalaprilat in patients unresponsive to ischemic preconditioning during percutaneous coronary intervention. Can J Physiol Pharmacol. 2008;86:827–834. doi: 10.1139/Y08-096. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaage J, Antonelli M, Bufi M, Irtun O, DeBlasi RA, Corbucci GG, et al. Exogenous reactive oxygen species deplete the isolated rat heart of antioxidants. Free Radic Biol Med. 1997;22:85–92. doi: 10.1016/s0891-5849(96)00278-x. [DOI] [PubMed] [Google Scholar]
- Valen G, Vaage J. Pre- and postconditioning during cardiac surgery. Basic Res Cardiol. 2005;100:179–186. doi: 10.1007/s00395-005-0517-8. [DOI] [PubMed] [Google Scholar]
- Vilahur G, Casani L, Pena E, Duran X, Juan-Babot O, Badimon L. Induction of RISK by HMG-CoA reductase inhibition affords cardioprotection after myocardial infarction. Atherosclerosis. 2009;206:95–101. doi: 10.1016/j.atherosclerosis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Wang SB, Foster DB, Rucker J, O'Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–757. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei M, Kuukasjärvi P, Laurikka J, Rinne T, Moilanen E, et al. The anti-inflammatory effect of bradykinin preconditioning in coronary artery bypass grafting (bradykinin and preconditioning) Scand Cardiovasc J. 2009;43:72–79. doi: 10.1080/14017430802180449. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–305. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- Weidenbach R, Schulz R, Gres P, Behrends M, Post H, Heusch G. Enhanced reduction of myocardial infarct size by combined ACE inhibition and AT(1)-receptor antagonism. Br J Pharmacol. 2000;131:138–144. doi: 10.1038/sj.bjp.0703544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werns SW, Shea MJ, Driscoll EM, Cohen C, Abrams GD, Pitt B, et al. The independent effects of oxygen radical scavengers on canine infarct size-reduction by superoxide dismutase but not catalase. Circ Res. 1985;56:895–898. doi: 10.1161/01.res.56.6.895. [DOI] [PubMed] [Google Scholar]
- Wink DA, Darbyshire JF, Nims RW, Saavedra JE, Ford PC. Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem Res Toxicol. 1993;6:23–27. doi: 10.1021/tx00031a003. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Xue F, Yang X, Zhang B, Zhao C, Song J, Jiang T, et al. Postconditioning the human heart in percutaneous coronary intervention. Clin Cardiol. 2010;33:439–444. doi: 10.1002/clc.20796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XC, Liu Y, Wang LF, Cui L, Wang T, Ge YG, et al. Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J Invasive Cardiol. 2007;19:424–430. [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nat Clin Pract Cardiovasc Med. 2005;2:568–575. doi: 10.1038/ncpcardio0346. [DOI] [PubMed] [Google Scholar]
- Yue Y, Qin Q, Cohen MV, Downey JM, Critz SD. The relative order of mK(ATP) channels, free radicals and p38 MAPK in preconditioning's protective pathway in rat heart. Cardiovasc Res. 2002;55:681–689. doi: 10.1016/s0008-6363(02)00452-2. [DOI] [PubMed] [Google Scholar]
- Zatta AJ, Kin H, Lee G, Wang N, Jiang R, Lust R, et al. Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signalling. Cardiovasc Res. 2006;70:315–324. doi: 10.1016/j.cardiores.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235–1243. doi: 10.1161/CIRCULATIONAHA.105.581397. [DOI] [PubMed] [Google Scholar]
- Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal. 2013;18:1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tocchetti CG, Krieg T, Moens AL. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med. 2012;53:1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol. 2012;52:341–350. doi: 10.1016/j.yjmcc.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ. Oxidative stress-elicited myocardial apoptosis during reperfusion. Curr Opin Pharmacol. 2004;4:159–165. doi: 10.1016/j.coph.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. . Erratum in: Am J Physiol Heart Circ Physiol. 2004 Jan;286(1):H477. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Liao JK. Statins and cardiovascular diseases: from cholesterol lowering to pleiotropy. Curr Pharm Des. 2009;15:467–478. doi: 10.2174/138161209787315684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi R, Ronca F, Ronca-Testoni S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther. 2001;89:47–65. doi: 10.1016/s0163-7258(00)00103-0. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Li H, Samouilov A, Liu X. Mechanisms of nitrite reduction to nitric oxide in the heart and vessel wall. Nitric Oxide. 2010;22:83–90. doi: 10.1016/j.niox.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]