

Abstract
Reactive oxygen species (ROS) are elevated in the heart in response to hemodynamic and metabolic stress, and promote hypertrophic signaling. ROS also mediate the formation of lipid peroxidation-derived aldehydes that may promote myocardial hypertrophy. One lipid peroxidation byproduct, 4-Hydroxy-trans-2-nonenal (HNE), is a reactive aldehyde that covalently modifies proteins thereby altering their function. HNE adducts directly inhibit the activity of LKB1, a serine/threonine kinase involved in regulating cellular growth in part through its interaction with the AMP-activated protein kinase (AMPK), but whether this drives myocardial growth is unclear. We tested the hypothesis that HNE promotes myocardial protein synthesis, and if this effect is associated with impaired LKB1-AMPK signaling. In adult rat ventricular cardiomyocytes exposure to HNE (10 μM for 1 hour) caused HNE-LKB1 adduct formation and inhibited LKB1 activity. HNE inhibited the downstream kinase AMPK, increased hypertrophic mTOR-P70S6K-RPS6 signaling, and stimulated protein synthesis by 27.1 ±3.5%. HNE also stimulated Erk1/2 signaling, which contributed to RPS6 activation but was not required for HNE-stimulated protein synthesis. HNE-stimulated RPS6 phosphorylation was completely blocked using the mTOR inhibitor rapamycin. To evaluate if LKB1 inhibition by itself could promote the hypertrophic signaling changes observed with HNE, LKB1 was depleted in ARVMs using siRNA. LKB1 knockdown did not replicate the effect of HNE on hypertrophic signaling or affect HNE-stimulated RPS6 phosphorylation. Thus, in adult cardiac myocytes HNE stimulates protein synthesis by activation of mTORC1-P70S6K-RPS6 signaling most likely mediated by direct inhibition of AMPK. Because HNE in the myocardium is commonly increased by stimuli that cause pathologic hypertrophy, these findings suggest that therapies that prevent activation of mTORC1-P70S6K-RPS6 signaling may be of therapeutic value.
Keywords: 4-HNE, mTORC1, Protein Synthesis, Cardiac Myocytes, Erk1/2
1. Introduction
Left ventricular hypertrophy (LVH) is a fundamental mechanism of pathological cardiac remodeling that has important implications for both cardiac function and patient outcomes [1]. LVH is due primarily to the increased mass of individual cardiomyocytes as a result of increased protein synthesis, a process regulated by multiple signaling pathways [2]. LVH is associated with increased production of reactive oxygen species (ROS) which play a role in regulating myocardial growth via oxidative post-translational modification (OPTM) of key signaling proteins [3]. In addition to the direct effects of OPTMs, ROS may also act on lipid membranes to generate peroxidation-derived reactive aldehydes that can have important biologic effects [4, 5]. One such reactive aldehyde species generated by lipid peroxidation is 4-hydroxy-trans-2-nonenal (HNE) which can covalently modify proteins via the Michael addition reaction on Cys, His, Lys, and Arg amino acids or through Schiff base formation of Lys or Arg amino acids [6, 7]. HNE and other reactive aldehydes are increased in the plasma of patients with heart failure [8] and myocardial HNE-protein adducts are seen in myocardium from patients with heart failure [9]. Likewise, we found increased myocardial HNE-protein adducts and pathologic remodeling in mouse models of systolic [10] and diastolic heart failure [11] and in both cases HNE adducts and cardiac hypertrophy were ameliorated by an antioxidant intervention.
These observations raise the possibility that HNE is involved in pathologic remodeling by means of protein adducts that promote cardiomyocyte growth signaling. HNE is known to inhibit the LKB1-AMPK signaling cascade [12], a pathway that opposes myocyte hypertrophy [13]. LKB1 is a Ser/Thr kinase that activates the AMP-activated protein kinase (AMPK), a key energy sensor that inhibits cellular functions that consume energy including growth [14]. Active AMPK regulates cellular growth in part via phosphorylation of the Tuberous Sclerosis Complex 2 (TSC2) protein [15], which when associated with TSC1 functions as a GTPase-activating protein (GAP) to prevent activation of the Ras homolog enriched in brain (Rheb) GTPase [16]. GTP-bound Rheb stimulates activity of the mammalian target of Rapamycin (mTOR) [17], which in complex with four additional proteins comprises the rapamycin-sensitive mTOR complex 1 (mTORC1) [18]. Activated mTORC1 increases phosphorylation of the 4E-binding protein 1 (4E-BP1) which becomes dissociated from the eukaryotic initiation factor 4E (eIF4E), thereby permitting cap-dependent protein translation to proceed [19]. Furthermore mTORC1 phosphorylates and activates p70S6 kinase (p70S6K) which in turn phosphorylates the ribosomal protein S6 (RPS6), a component of the 40S ribosomal protein which regulates the initiation of protein translation (see review [20]).
The role of HNE in mediating pathologic hypertrophy was proposed recently by Dolinsky et al., who showed HNE-LKB1 adducts were associated with decreased LKB1 and AMPK activities in hearts of the spontaneously hypertensive rat (SHR) [21]. This thesis was supported by their observations that HNE treatment caused pro-hypertrophic signaling changes in cultured neonatal cardiac myocytes. While these observations suggest that HNE-LKB1 adducts could play an important role in the pathogenesis of myocardial hypertrophy, several questions remain. First, it is not known whether HNE-mediated inhibition of LKB1-AMPK signaling leads to increased protein synthesis. Second, it is possible that pathways other than LKB1-AMPK (e.g., MAPK) mediate or contribute to the overall effects of HNE on growth. Third, since these studies were performed in neonatal myocytes, it is not clear whether HNE exerts a similar growth-promoting effect in adult myocytes which utilize different growth signaling pathways than neonatal cells [22]. Accordingly, the goal of this study was to test the hypothesis that HNE-LKB1 adduction increases protein synthesis in adult rat cardiac myocytes, and if so, to delineate the signaling pathway(s) that mediate this effect.
2. Materials and methods
2.1 Chemicals and materials
4-hydroxynonenal (HNE) was obtained from EMD Millipore (Billerica, MA, USA). The MEK1/2 inhibitor U0126 (9903) was purchased from Cell Signaling Technology and the mTORC1 inhibitor Rapamycin (R-5000) was obtained from LC Laboratories (Woburn, MA). The [3H]-leucine and the [γ-32P]-ATP were purchased from Perkin Elmer (Boston, MA). All other materials were purchased from Thermo Fisher Scientific (Waltham, MA, USA) unless otherwise specified.
2.2 Isolation and culturing of adult rat ventricular myocytes (ARVMs)
The animal protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine. ARVMs were isolated from male Sprague-Dawley rats (Harlan Laboratories, 175–225 grams) as previously described [23–26] and cultured in serum-free media. Briefly, the hearts were rapidly excised and perfused with 37 °C Ca2+ free Krebs-Henseleit Buffer through the aorta using retrograde Langendorff perfusion. The heart was digested with recirculating collagenase (Worthington) and hyaluronidase (Sigma) and then filtered through 100 μm nylon mesh gauze, centrifuged (500 rpm, 3 min) and the cardiomyocyte pellet was gently layered on a bovine serum albumin gradient. The pellet containing viable cardiomyocytes was resuspended and plated on laminin coated dishes in DMEM low glucose media (Invitrogen, 12320-032) supplemented with 0.2% BSA, 2 mM Carnitine, 5 mM Creatine, 5 mM Taurine, 2.5 M Hepes, and 0.1% penicillin-streptomycin for 1 hr to allow for cardiomyocytes to adhere to the laminin coated plates. The media was then replaced and the cells were used for experimentation the following day.
2.3 Culturing conditions of HNE-treated cardiomyocytes
Cardiomyocytes were plated at a density of 50 to 75 cells/mm2 in 6-well plates, 35mm, or 60mm dishes. The following volumes of media were used for each plate: 6-well – 2 mL media/well; 35mm – 2 mL media/dish; 60mm – 3 mL media/dish. The HNE stock was diluted in ddH2O to a working concentration of 10 mM, mixed thoroughly, and then added to the media at a 1:1000 dilution to achieve a final concentration of 10 μM in the media. For each experiment the vehicle treatment indicates the same volume of 100% ethanol was diluted in ddH2O and added to the media at the same dilution as the HNE solution. In all of these experiments the media used for cardiomyocyte isolation and culturing was serum-free.
2.4 [3H]-leucine incorporation assay
ARVMs were treated as noted in the figure legends and incubated with [3H]-leucine (PerkinElmer Life Sciences) at a concentration of 1 μCi/mL of media. After 24 hrs the cells were rinsed twice in ice-cold PBS and the proteins were precipitated using ice-cold 5% TCA for 1 hr at 4 °C. Next the cells were rinsed twice using ice-cold Milli-Q filtered water, all residual water was aspirated, and then the cells were solubilized using 0.4 M NaOH for 1 hr at room temperature. The cellular extracts were collected, mixed with liquid scintillation cocktail, and the incorporated radioactivity was measured using a liquid scintillation counter.
2.5 [3H]-leucine protein degradation assay
ARVMs were pre-loaded with [3H]-leucine at a concentration of 1 μCi/mL of media. After 24 hrs of loading the media was collected, the cells rinsed, and the same volume of media without [3H]-leucine was added back to the cells. The collected media at this point represents the initial time point for the experiment (time = 0 hr). ARVMs were then stimulated with HNE (10 μM) or vehicle and after 1 hr of treatment the media was collected and replaced with fresh media without HNE or vehicle. The collected media at this time point represents 1 hour post-experimentation (time = 1 hr). After 23 hrs the media was again collected and the experiment was terminated. The collected media at this point represents the final measurement after 24 hrs (time = 24 hrs). The collected media was mixed with liquid scintillation cocktail and the [3H]-leucine content was measured using a liquid scintillation counter.
2.6 Antibodies
Antibodies recognizing phosphorylated mTOR (Ser2448), P70S6K (Thr421/Ser424), ribosomal protein S6 (RPS6) (Ser235/236), Akt (Ser473), Erk (Thr202/Tyr204) and antibodies recognizing total LKB1, acetyl-CoA carboxylase (ACC), mTOR, p70S6K, Akt, and Erk were all purchased from Cell Signaling Technology. Rabbit polyclonal antibody recognizing phosphorylated ACC (Ser79) was purchased from Millipore. The antibodies recognizing AMPKα2 (C-20) and LKB1 (M-18) were purchased from Santa Cruz Biotechnology. The goat polyclonal antibody recognizing HNE (ab46544) and the mouse monoclonal antibody recognizing glyceraldehyde 3-phosphate dehydrogenase (GAPDH, ab8245) were both from Abcam. Secondary antibodies coupled to IR 680 or 800 dye were from Li-Cor.
2.7 Western blot analysis
Cells were rinsed with cold PBS and lysed with 1X lysis buffer (Cell Signaling Technology) containing 20 mM Tris-HCl (pH=7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 ug/mL leupeptin, and 100 mM PMSF. The lysates were cleared by centrifugation at 13,000 rpm for 10 min. Proteins (25–50 ug) were separated by SDS-PAGE using polyacrylamide gels and transferred to nitrocellulose membranes. Immunoblotted proteins were visualized using the Li-Cor Odyssey IR imager.
2.8 LKB1 kinase assay
Protein G beads (GE Healthcare) were washed three times with PBS and conjugated to the LKB1 antibody (M-18) for 1 hour at 4 °C. 500 ug protein of each sample was added to 40 μL of the antibody-bead complex, brought up to an equal volume using Homogenization Buffer (62.5 mM Tris-HCl, 5 mM EDTA, 5 mM EGTA) and rotated end on end overnight at 4 °C. The immunopreciptated proteins were washed twice with Homogenization Buffer and twice with Kinase Wash Buffer (40 mM Hepes, 80 mM NaCl, 0.8 mM EDTA, 5 mM MgCl2, 800 μM DTT, 8% Glycerol). The purified proteins were incubated with kinase assay buffer containing the LKBtide synthetic substrate (300 μM, EMD Millipore), HEPES (40 mM), DTT (800 μM), NaCl (80 mM), EDTA (800 μM), Glycerol (8%), ATP (200 μM), and P32-radioactively labeled ATP. After incubation at 37 °C for 20 minutes, the reaction mixture was spotted on Whatman P81 papers, washed four times in 19.5 mM phosphoric acid, and radioactivity was measured by liquid scintillation counting.
2.9 AMPK kinase assay
Protein G beads were washed three times with PBS and conjugated to the AMPKα2 antibody for 1 hour at 4 °C. 500 ug protein of each sample was added to 40 μL of the antibody-bead complex, brought up to an equal volume using Homogenization Buffer and rotated end on end overnight at 4 °C. The immunopreciptated proteins were washed three times with Homogenization Buffer. The purified proteins were incubated with kinase assay buffer containing the SAMS synthetic peptide (200 μM, EMD Millipore), DTT (960 μM), ATP (200 μM), and P32-radioactively labeled ATP. After incubation at 37 °C for 10 minutes, the reaction mixture was spotted on Whatman P81 papers, washed four times in 19.5 mM phosphoric acid, and radioactivity was measured by liquid scintillation counting.
2.10 siRNA knockdown of LKB1
The siRNA constructs used in these experiments were purchased from Dharmacon (siGENOME SMART Pool – Control Non-Targeting siRNA and STK11 siRNA). Freshly cultured ARVMs were transfected with 25 pmol siRNA per p35 tissue culture dish using the Lipofectamine RNAiMAX Reagent (Invitrogen) as described by the manufacturer. 72 hours post-transfection the cells were treated as described in the figure legends.
2.11 Statistical analysis
Statistics were performed with a two-tailed unpaired Student’s t test or ANOVA for multiple comparisons, where appropriate. All data shown represent the results obtained from independent experiments conducted in duplicate or triplicate (as noted in the figure legends) with standard error of the mean (mean ±SEM). Values of p < 0.05 were considered statistically significant.
3. Results
3.1 HNE induces protein synthesis in cultured cardiomyocytes
While prior studies have used HNE concentrations of 40 uM [21] and higher [27] in cardiac myocytes, our previous work has shown that an HNE concentration as low as 10 μM inhibits LKB1 activity through the formation of HNE-adducts [28]. Therefore, cultured adult rat ventricular cardiac myocytes (ARVMs) were treated with 10 μM HNE for 1 hour or Norepinephrine (NE) as a positive control, and the cells were incubated with [3H]-leucine for 24 hours. Under these conditions HNE treatment increased [3H]-leucine incorporation by 27.1 ± 3.5% (S.E.) (Figure 1A), thus suggesting that HNE stimulates protein synthesis in adult cardiac myocytes. As HNE has been shown to stimulate autophagy and protein degradation [29, 30], we next sought to examine if HNE treatment in ARVMs alters protein degradation. ARVMs pre-loaded with [3H]-leucine were assayed for the release of [3H]-leucine containing proteins 24 hours after HNE treatment. HNE stimulation had no effect on the release of degraded peptides containing [3H]-leucine (Figure 1B). These data strongly suggest the observed increase in HNE-stimulated [3H]-leucine incorporation is due to enhanced protein synthesis.
Figure 1. HNE increases protein synthesis in ARVMs.

(A) ARVMs were treated with vehicle or 10 μM HNE for 1 hour, after which the media was removed and replaced with fresh media containing [3H]-leucine. As a positive control, another set of cells were pretreated with Propranolol (2 μM, 30 minutes) and then stimulated with 1 μM Norepinephrine (NE) for 24 hours. After 24 hours the cells were collected and assayed for incorporation of the labeled leucine by liquid scintillation counting (n=6). (B) To examine protein degradation ARVMs were loaded with [3H]-leucine. After 24 hrs of loading the media was collected, replaced, and cells stimulated with HNE (10 μM) or vehicle (time=0 hr). After 1 hr of treatment the media was collected and replaced with HNE-free media (time=1 hr) and after 23 hrs the media was again collected (time=24 hrs). The collected media was assayed for [3H]-leucine content as a measure of degraded proteins (n=3). Each replicate is an independent experiment conducted in duplicate or triplicate using cells obtained from separate preparations of ARVMs. (* = p < 0.05 vs Vehicle, # = p < 0.01 vs HNE, error bars are mean ± standard error).
3.2 HNE causes pro-hypertrophic signaling in the LKB1-AMPK signaling axis
We next investigated the role of the LKB1-AMPK axis in mediating HNE-induced pro-hypertrophic signaling changes. HNE treatment (10 μM, 1 hour) caused HNE-LKB1 adducts (Figure 2A) and in cells treated with HNE there was a concomitant 24.9 ± 12.2% (S.E.) decrease in LKB1 activity (Figure 2B). AMPK, a major substrate for LKB1 in the heart, inhibits myocardial protein synthesis when active [31]. HNE treatment decreased the activity of the AMPK alpha2 isoform by 50.3 ± 5.4% (S.E.) (Figure 2C). Likewise, HNE decreased phosphorylation of acetyl-CoA carboxylase (ACC) (Figure 2D), a downstream substrate of AMPK that regulates lipogenesis via the carboxylation of acetyl-CoA to malonyl-CoA, supporting the observed decrease in AMPK activity. AMPK promotes energetic homeostasis by regulating several metabolic processes, including cell growth, through actions on the mTORC1 – p70S6K – Ribosomal Protein S6 (RPS6) signaling axis. HNE increased the phosphorylation of mTOR, P70S6K, and RPS6 proteins (Figure 3A – C). Thus, HNE inhibited LKB1 and AMPK activities, which were associated with downstream pro-hypertrophic signaling changes via mTORC1, P70S6K, and RPS6.
Figure 2. HNE inhibits LKB1 and AMPK activities.

(A) LKB1 was immunoprecipitated from homogenates of cultured ARVMs incubated for 1 hour with vehicle or 10 μM HNE. Immunoprecipitated LKB1 was analyzed by immunoblotting for HNE adduct formation by densitometry and normalized to total LKB1 as a control for loading (n=6). (B) LKB1 activity in ARVMs treated with HNE (n=6–7). (C) AMPK activity in ARVMs treated with HNE (n=6–7). (D) Levels of phosphorylated ACC (P-ACC) were quantified by densitometry and normalized to total ACC (T-ACC) and GAPDH (n=3). For all of these experiments each replicate is an independent experiment conducted in duplicate using cells obtained from separate preparations of ARVMs, * = p < 0.05, and error bars are mean ± standard error.
Figure 3. Hypertrophic mTOR-P70S6K and RPS6 phosphorylation are induced by HNE.

(A) Levels of phosphorylated mTOR (P-mTOR) were quantified by densitometry and normalized to total mTOR (T-mTOR) and GAPDH (n=3). (B) Levels of phosphorylated p70S6K (P-p70S6K) were quantified by densitometry and normalized to total p70S6K (T-p70S6K) and GAPDH (n=3). (C) Levels of phosphorylated RPS6 (P-RPS6) were quantified by densitometry and normalized to GAPDH (n=4). For all of these experiments each replicate is an independent experiment conducted in duplicate using cells obtained from separate preparations of ARVMs, * = p < 0.05, and error bars are mean ± standard error.
3.3 HNE stimulates Erk1/2
While inhibition of LKB1-AMPK signaling, per se, could explain increased protein synthesis, HNE may form adducts with other potential targets that are known to be involved in growth signaling in cardiac myocytes. For example HNE can inhibit Phosphatase and Tensin Homolog (PTEN) [32], which hydrolyzes phosphatidylinositol 3,4,5,-triphosphate to prevent phosphorylation and activation of Akt by upstream kinases [33]. Considering that Akt inhibits AMPK in the heart [34] and regulates mTORC1 via phosphorylation of Proline Rich Akt Substrate of 40 kDa (PRAS40) [35], activation of Akt could regulate the observed signaling changes. Furthermore HNE induces Erk1/2 activation in both pulmonary epithelial and endothelial cells [36, 37], which could promote protein synthesis independent of LKB1-AMPK-mTORC1 signaling. In ARVMs HNE had no effect on Akt phosphorylation at Ser-473 (Figure 4A), suggesting Akt is not active. However, HNE increased the phosphorylation of Erk1/2 by 82.5 ± 11.7% (S.E.) (Figure 4B). The MEK1/2 inhibitor U0126 decreased basal Erk1/2 phosphorylation, and abolished the effect of HNE on Erk1/2 phosphorylation (Figure 4C), thus confirming that HNE stimulates Erk1/2 via an action at or above the level of MEK1/2 in the pathway.
Figure 4. HNE stimulates Erk1/2 in a MEK-dependent manner.

(A) Homogenates of cultured ARVMs incubated for 1 hour with vehicle or 10 μM HNE were evaluated for levels of phosphorylated Akt (P-Akt) by immunoblotting. Protein loading was normalized to total Akt (T-Akt) and GAPDH (n=3). (B) Levels of phosphorylated Erk1/2 (P-Erk) were quantified by densitometry and normalized to total Erk1/2 (T-Erk) and GAPDH (n=4). (C) ARVMs were incubated with vehicle or U0126 (10 μM, 30 minutes) prior to stimulation by vehicle or HNE. Lysates were immunoblotted for levels of P-Erk and normalized to T-Erk and GAPDH (n=4). For all of these experiments each replicate is an independent experiment using cells obtained from separate preparations of ARVMs, * = p < 0.05, ** = p < 0.01 and error bars are mean ± standard error.
3.4 Erk1/2 signaling is not required for HNE-stimulated protein synthesis
In cardiomyocytes hypertrophic stimuli such as phenylephrine activate RPS6, an effect that is dependent on both MEK-Erk1/2 and mTOR signaling [38, 39]. Furthermore, the RPS6 phosphorylation sites examined (Ser235/236) are activated by MAPK signaling in addition to mTORC1 [40]. Therefore, we next sought to determine if HNE-mediated phosphorylation of RPS6 at Ser235/236 was dependent on the MEK-Erk1/2 signaling cascade. The MEK inhibitor U0126 attenuated HNE-stimulated RPS6 phosphorylation (Figure 5A), suggesting that Erk1/2 signaling is not the primary cause of RPS6 activation yet may mediate the degree of RPS6 phosphorylation. To determine whether Erk1/2 contributes to HNE-stimulated protein synthesis, we examined protein synthesis in the presence of U0126 and found that HNE-stimulated protein synthesis was unaffected by MEK inhibition (Figure 5B). Together these data suggest that activation of Erk1/2 by HNE partially regulates RPS6 phosphorylation, but doesn’t mediate protein synthesis.
Figure 5. Erk1/2 contributes to the activation of RPS6 with HNE treatment.

(A) ARVMs were incubated with DMSO vehicle or U0126 (10 μM, 30 minutes) prior to stimulation by HNE. Lysates were immunoblotted for levels of P-RPS6 and normalized to GAPDH (n=5), * = p < 0.01 vs Veh Con, # = p < 0.05 vs U0126 Con, $ = p <0.05 vs Veh HNE. (B) ARVMs were incubated with vehicle or U0126 (10 μM, 30 minutes) prior to stimulation by vehicle or HNE for 1 hour. After HNE stimulation the media was removed and replaced with fresh media containing [3H]-leucine and vehicle or U0126. After 24 hours the cells were collected and assayed for incorporation of the labeled leucine by liquid scintillation counting (n=5, ns = not significant, * = p < 0.05). For all of these experiments each replicate is an independent experiment using cells obtained from separate preparations of ARVMs and error bars are mean ± standard error.
3.5 mTORC1 is required for the activation of RPS6 by HNE
We next examined the contribution of mTORC1 signaling in HNE-mediated RPS6 activation using the specific inhibitor rapamycin. Rapamycin pretreatment decreased basal RPS6 phosphorylation and abolished the HNE-stimulated RPS6 increase (Figure 6), thereby implicating mTORC1 as a necessary regulator of HNE-stimulated RPS6 activation.
Figure 6. mTORC1 is required for the activation of RPS6 by HNE.
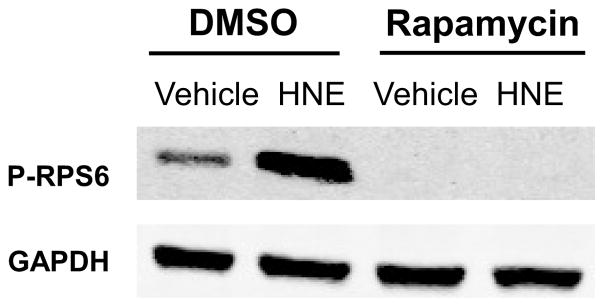
ARVMs were incubated with vehicle or Rapamycin (100 nM, 30 minutes) prior to stimulation by vehicle or HNE. Lysates were immunoblotted for levels of P-RPS6 and GAPDH (n=3). For all of these experiments each replicate is an independent experiment using cells obtained from separate preparations of ARVMs.
3.6 LKB1 knockdown doesn’t recapitulate hypertrophic signaling caused by HNE
Having observed that HNE inhibits LKB1 and promotes hypertrophic signaling through the mTORC1-P70S6K-RPS6 signaling cascade, we next sought to determine if specific inhibition of LKB1 would cause the same signaling changes in ARVMs. Using an siRNA targeted to LKB1, we observed an approximately 68% decrease in LKB1 protein expression 72 hours after siRNA exposure (Figure 7). With LKB1 knockdown we observed no changes in baseline RPS6 phosphorylation, which suggests LKB1 inhibition is not sufficient to promote hypertrophic signaling through this pathway. Furthermore, ARVMs depleted of LKB1 have a similar degree of HNE-mediated RPS6 phosphorylation (Ctrl siRNA: 3.53 ± 0.63 S.E.M., LKB1 siRNA: 2.99 ± 0.58 S.E.M., n=3) suggesting HNE-inhibition of the remaining LKB1 protein fails to promote RPS6 phosphorylation.
Figure 7. Knockdown of LKB1 doesn’t promote hypertrophic RPS6 signaling in ARVMs.


ARVMs were treated with either a non-targeting control siRNA (Ctrl siRNA) or siRNA directed against LKB1 (LKB1 siRNA) for 72 hrs. After 72 hrs of siRNA exposure the ARVMs were treated with vehicle or HNE (10 μM, 1 hr) and the lysates were immunoblotted for protein expression of LKB1, P-RPS6, P-ACC, and GAPDH (n=3). For all of these experiments each replicate is an independent experiment using cells obtained from separate preparations of ARVMs, * = p < 0.05, ** = p < 0.01, *** = p < 0.001 and error bars are mean ± standard error.
4. Discussion
The goal of this study was to determine whether HNE causes increased protein synthesis in adult cardiac myocytes, and if so, to delineate the signaling pathways associated. Prior work in neonatal cardiac myocytes has shown that HNE inhibits LKB1-AMPK signaling which was associated with increased pro-hypertrophic signaling via mTOR and P70S6K [21]. However, it remained to be determined whether these signaling effects lead to increased protein synthesis, which cannot be assumed given the potentially diverse signaling actions of HNE-adduction. Our findings clearly demonstrate that even a limited duration of exposure (1 hour) to a relatively low concentration (10 μM) of HNE causes protein synthesis in ARVMs. The magnitude of the HNE-stimulated protein synthesis is smaller than that of α-AR stimulation (HNE: 27.1 ± 3.5% (S.E.), NE: 70.2 ± 12.8%, Figure 1). However, the conditions of these studies were selected to minimize toxic and pro-apoptotic effects, and likely underestimate the maximal effect of HNE. A dose response curve of HNE treatment suggests that 20 μM may elicit a larger effect on RPS6 phosphorylation, yet higher doses (30 or 40 μM) impair HNE-stimulated RPS6 activation (Supplementary Figure 1A). Increasing amounts of HNE (≥ 10 μM) enhance Erk1/2 phosphorylation, suggesting a transition from a hypertrophic to a stress related signaling response at doses of 30 μM or higher (Supplementary Figure 1B). While we are not aware of others reports demonstrating HNE-stimulated proteins synthesis, HNE has been shown to elicit a mitogenic response in other cell types such as rat aortic smooth muscle cells [41].
HNE can stimulate autophagy and modulate protein degradation [29, 30], which could be a confounding factor in our experimental approach to assess HNE-stimulated protein synthesis utilizing [3H]-leucine incorporation. In order to verify the increased [3H]-leucine incorporation with HNE treatment was due to novel protein synthesis, as opposed to decreased protein degradation, we conducted a [3H]-leucine washout experiment in order to measure the amount of degraded proteins that were secreted. We observed the same HNE treatment conditions that elicited [3H]-leucine incorporation had no effect on the rate of [3H]-leucine washout, which further strengthens our conclusion that HNE promotes protein synthesis in ARVMs. In prior studies examining the effects of HNE on proteasome-mediated protein degradation the dose of HNE was important in determining how HNE altered protein degradation. Lower doses of HNE (10 μM) could stimulate proteasomal degradation [42] but at higher concentrations HNE (> 0.1 mM) inhibits the proteasome itself [43]. The low concentration of HNE used in our study was able to stimulate protein synthesis in ARVMs without altering protein degradation. Perhaps at higher doses of HNE which no longer stimulate RPS6 (40 μM, Supplementary Figure 1A) the presence of excess HNE-modified proteins could lead to increased proteasomal activity.
We next sought to determine if HNE promotes hypertrophic signaling in ARVMs through the inhibition of LKB1-AMPK signaling. Hypertrophic stimuli are known to elicit differential signaling responses in adult and neonatal ventricular cardiomyocytes [22], which is why we decided to confirm the earlier finding in the adult cardiomyocyte. In agreement with the prior study, the formation of HNE-LKB1 adducts was associated with inhibited LKB1 and AMPK kinase activities. The decrease in kinase activity of these proteins was associated with hypertrophic signaling changes through the mTORC1-P70S6K-RPS6 signaling cascade, which demonstrates that the same HNE-mediated pro-hypertrophic signaling changes observed in the neonatal cardiomyocyte persist into the adult cell.
To investigate if other hypertrophic signaling cascades were altered by HNE we examined the phosphorylation status of Akt, which has been shown to modulate mTORC1 independently of LKB1-AMPK signaling [35]. As was previously observed in neonatal cardiomyocytes, we found no change in Akt phosphorylation with HNE treatment, suggesting Akt is not active. We also examined the activation of Erk1/2, as we have previously shown that MAPK signaling is hypertrophic in cardiomyocytes stimulated by α-adrenergic receptor agonists [26]. HNE increased Erk1/2 phosphorylation, and this effect was dependent on the upstream kinase MEK. While our work and other studies have shown that HNE stimulates Erk1/2 activity [36, 37], there are also reports that HNE can form inhibitory adducts of Erk1/2 [44] that decrease Erk1/2 phosphorylation and downstream Elk-1 signaling [45]. The inhibitory effect of HNE on Erk1/2 signaling occurs in the liver and primary hepatocytes using the same HNE dose in this study, demonstrating that disparate tissue-specific signaling changes can occur under otherwise comparable conditions.
Having observed that HNE stimulates Erk1/2 signaling, we next investigated if Erk1/2 signaling contributed to hypertrophic RPS6 phosphorylation and protein synthesis, as both the MAPK and mTORC1 cascades converge on RPS6 via phosphorylation of Ser235/236 [40]. Inhibition of Erk1/2 using U0126 attenuated, but did not prevent HNE-stimulated RPS6 phosphorylation, suggesting HNE-stimulated Erk1/2 signaling partially contributes to the activation of RPS6. In order to address whether this effect is specific to Erk1/2 signaling we utilized another MEK inhibitor, PD98059. Treatment with PD98059 also decreased HNE-stimulated Erk1/2 phosphorylation (data not shown). Since inhibition of Erk1/2 signaling impaired stimulation of RPS6, we sought to determine whether the activation of Erk1/2 by HNE contributes to protein synthesis. We found Erk1/2 activation was not required for HNE-stimulated protein synthesis, which suggests the AMPK-mTORC1 signaling axis is the primary mediator of RPS6 activation and subsequent protein synthesis.
Having evaluated the contribution of MEK-Erk1/2 signaling in RPS6 activation and protein synthesis, we next determined the role of mTORC1 using the specific inhibitor rapamycin. We observed that rapamycin treatment completely blocked HNE-stimulated RPS6 phosphorylation. This observation is in agreement with other work in cardiomyocytes examining protein synthesis using the hypertrophic stimulus phenylephrine, as others have found rapamycin inhibited mTORC1 and downstream RPS6 phosphorylation to partially suppress protein synthesis [39]. Our data suggests that hypertrophic signaling induced by HNE occurs via similar mechanisms, although with HNE treatment RPS6 activation is dependent on mTORC1 which is likely impacted by upstream inhibition of AMPK signaling.
We observed that HNE caused HNE-LKB1 adducts and inhibited LKB1 function. Considering HNE may be modulating a number of other signaling cascades, we sought to test whether inhibition of LKB1 by itself is sufficient to promote hypertrophic signaling through the mTORC1-P70S6K-RPS6 cascade. Using an siRNA targeted to LKB1, we observed a significant decrease in LKB1 expression. In ARVMs depleted of LKB1 we observed no changes in RPS6 phosphorylation, which suggests LKB1 inhibition doesn’t promote hypertrophic signaling through this pathway. Furthermore LKB1-depleted ARVMs treated with HNE had comparable RPS6 phosphorylation. If LKB1 did in fact mediate hypertrophic signaling by HNE through this pathway, we would have expected to see augmented RPS6 phosphorylation with LKB1 knockdown.
Based on these findings we conclude that LKB1 inhibition by itself is not sufficient to promote hypertrophic signaling, and that HNE must be acting either through the inhibition of AMPK or the activation of mTORC1. It has recently been reported that AMPK is a direct target of HNE adduction [46], which if true in ARVMs would agree with the hypertrophic signaling changes we observed in ARVMs depleted of LKB1. While we can’t rule out the possibility that HNE may be stimulating mTORC1 independently of AMPK, the observation that HNE is capable of directly targeting AMPK leads us to propose that HNE promotes hypertrophic signaling via an inhibitory modification of AMPK.
Taken together these findings provide a comprehensive model for HNE-stimulated myocardial protein synthesis (Figure 8). We found HNE inhibits both LKB1 and AMPK activities which is associated with downstream hypertrophic mTORC1-p70S6K-RPS6 activation and increased protein synthesis. HNE also activates MEK-Erk1/2 signaling which doesn’t contribute to the observed protein synthesis. We found that mTORC1 was necessary for the pro-hypertrophic activation of RPS6 by HNE. The inhibition of LKB1 didn’t mediate the expected hypertrophic signaling changes, which suggests that HNE may directly inhibit AMPK to promote hypertrophic signaling in ARVMs. Overall, this study demonstrates oxidant-derived lipid peroxidation byproducts promote myocardial protein synthesis and describes the signaling mechanisms involved, which has important implications in the treatment of pathological cardiac hypertrophy.
Figure 8. Diagram summarizing the effects of HNE on LKB1-AMPK and MEK-Erk1/2 signaling in ARVMs.

HNE inhibits LKB1 and AMPK activities and increases hypertrophic signaling through the mTORC1-P70S6K-RPS6 signaling cascade to promote protein synthesis. Activation of MEK-Erk1/2 signaling didn’t contribute to protein synthesis. The actions of HNE on RPS6 phosphorylation could be prevented with the mTORC1 inhibitor rapamycin but not by knockdown of LKB1 by siRNA, thus suggesting HNE may be acting directly at the level of AMPK.
Supplementary Material
Highlights.
HNE increases protein synthesis in cardiac myocytes at a low concentration of 10 μM.
HNE inhibits LKB1-AMPK signaling which may promote hypertrophic signaling through the downstream mTORC1-P70S6K-RPS6 cascade.
HNE stimulates Erk1/2, 4) Erk1/2 doesn’t mediate HNE-stimulated protein synthesis.
mTORC1 is necessary for HNE-stimulated activation of RPS6, and.
LKB1 inhibition by itself is not sufficient to induce hypertrophic mTORC1-P70S6K-RPS6 signaling. These data demonstrate HNE causes protein synthesis and suggests it may directly inhibit AMPK to mediate downstream hypertrophic signaling via mTORC1-P70S6K-RPS6 signaling in cardiac myocytes.
Acknowledgments
This work was published and presented in abstract form at annual meetings of the Society of Free Radical Biology and Medicine, Free Rad Biol Med 49 Suppl. 1 S15-16, 2010 and Free Rad Biol Med 53 Suppl. 2 S156, 2012. The authors would like to thank Jack Zhu, JinSub Kim, Paul Sali, and Dominique Croteau for their excellent technical assistance with cardiomyocyte preparations. Additionally the authors thank all members of the Colucci lab for helpful discussion and feedback.
Funding
Supported by NIH grant HL-061639 (WSC), the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (WSC, Contract No. N01-HV-28178) and T32 Cardiovascular Training HL-07969 (TDC).
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Velagaleti RS, Gona P, Pencina MJ, Aragam J, Wang TJ, Levy D, et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am J Cardiol. 2014;113:117–22. doi: 10.1016/j.amjcard.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 4.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477:183–95. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pamplona R. Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta. 2008;1777:1249–62. doi: 10.1016/j.bbabio.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 7.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med. 2003;24:149–59. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 8.Mak S, Lehotay DC, Yazdanpanah M, Azevedo ER, Liu PP, Newton GE. Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J Card Fail. 2000;6:108–14. [PubMed] [Google Scholar]
- 9.Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation. 2002;105:2867–71. doi: 10.1161/01.cir.0000018605.14470.dd. [DOI] [PubMed] [Google Scholar]
- 10.Qin F, Lennon-Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, et al. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ Heart Fail. 2010;3:306–13. doi: 10.1161/CIRCHEARTFAILURE.109.864785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin F, Siwik DA, Luptak I, Hou X, Wang L, Higuchi A, et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation. 2012;125:1757–6. doi: 10.1161/CIRCULATIONAHA.111.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolinsky VW, Chan AY, Robillard FI, Light PE, Des RC, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation. 2009;119:1643–52. doi: 10.1161/CIRCULATIONAHA.108.787440. [DOI] [PubMed] [Google Scholar]
- 13.Noga AA, Soltys CL, Barr AJ, Kovacic S, Lopaschuk GD, Dyck JR. Expression of an active LKB1 complex in cardiac myocytes results in decreased protein synthesis associated with phenylephrine-induced hypertrophy. Am J Physiol Heart Circ Physiol. 2007;292:H1460–H1469. doi: 10.1152/ajpheart.01133.2006. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–63. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 15.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 16.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 18.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 19.Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 20.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 21.Dolinsky VW, Chan AY, Robillard FI, Light PE, Des RC, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation. 2009;119:1643–52. doi: 10.1161/CIRCULATIONAHA.108.787440. [DOI] [PubMed] [Google Scholar]
- 22.Schluter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther. 2008;119:311–25. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Claycomb WC, Palazzo MC. Culture of the terminally differentiated adult cardiac muscle cell: a light and scanning electron microscope study. Dev Biol. 1980;80:466–82. doi: 10.1016/0012-1606(80)90419-4. [DOI] [PubMed] [Google Scholar]
- 24.Springhorn JP, Ellingsen O, Berger HJ, Kelly RA, Smith TW. Transcriptional regulation in cardiac muscle. Coordinate expression of Id with a neonatal phenotype during development and following a hypertrophic stimulus in adult rat ventricular myocytes in vitro. J Biol Chem. 1992;267:14360–5. [PubMed] [Google Scholar]
- 25.Takahashi N, Calderone A, Izzo NJ, Jr, Maki TM, Marsh JD, Colucci WS. Hypertrophic stimuli induce transforming growth factor-beta 1 expression in rat ventricular myocytes. J Clin Invest. 1994;94:1470–6. doi: 10.1172/JCI117485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao L, Pimental DR, Amin JK, Singh K, Sawyer DB, Colucci WS. MEK1/2-ERK1/2 mediates alpha1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. 2001;33:779–87. doi: 10.1006/jmcc.2001.1348. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura K, Miura D, Kusano KF, Fujimoto Y, Sumita-Yoshikawa W, Fuke S, et al. 4-Hydroxy-2-nonenal induces calcium overload via the generation of reactive oxygen species in isolated rat cardiac myocytes. J Card Fail. 2009;15:709–16. doi: 10.1016/j.cardfail.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Calamaras TD, Lee C, Lan F, Ido Y, Siwik DA, Colucci WS. Post-translational modification of serine/threonine kinase LKB1 via Adduction of the Reactive Lipid Species 4-Hydroxy-trans-2-nonenal (HNE) at lysine residue 97 directly inhibits kinase activity. J Biol Chem. 2012;287:42400–6. doi: 10.1074/jbc.M112.385831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J. 2008;410:525–34. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Dou X, Gu D, Shen C, Yao T, Nguyen V, et al. 4-Hydroxynonenal differentially regulates adiponectin gene expression and secretion via activating PPARgamma and accelerating ubiquitin-proteasome degradation. Mol Cell Endocrinol. 2012;349:222–31. doi: 10.1016/j.mce.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–9. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 32.Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013;65:680–92. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–7. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 35.Vander HE, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 36.Iles KE, Dickinson DA, Wigley AF, Welty NE, Blank V, Forman HJ. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic Biol Med. 2005;39:355–64. doi: 10.1016/j.freeradbiomed.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem. 2006;281:35554–66. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res. 2002;91:821–9. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 39.Rolfe M, McLeod LE, Pratt PF, Proud CG. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2) Biochem J. 2005;388:973–84. doi: 10.1042/BJ20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruef J, Rao GN, Li F, Bode C, Patterson C, Bhatnagar A, et al. Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation. 1998;97:1071–8. doi: 10.1161/01.cir.97.11.1071. [DOI] [PubMed] [Google Scholar]
- 42.Carbone DL, Doorn JA, Petersen DR. 4-Hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radic Biol Med. 2004;37:1430–9. doi: 10.1016/j.freeradbiomed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 43.Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J Biol Chem. 1999;274:23787–93. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 44.Sampey BP, Carbone DL, Doorn JA, Drechsel DA, Petersen DR. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Mol Pharmacol. 2007;71:871–83. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 45.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J Biol Chem. 2007;282:1925–37. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR. Identification of 5′ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014;289:15449–62. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.