

Abstract
Yeast cells have developed complex mechanisms to cope with extracellular insults. An increase in external osmolarity leads to activation of the stress-activated protein kinase Hog1, which is the main regulator of adaptive responses, such as gene expression and cell cycle progression, that are essential for cellular survival. Upon osmostress, the G1-to-S transition is regulated by Hog1 through stabilization of the cyclin-dependent kinase inhibitor Sic1 and the downregulation of G1 cyclin expression by an unclear mechanism. Here, we show that Hog1 interacts with and phosphorylates components of the core cell cycle transcriptional machinery such as Whi5 and the coregulator Msa1. Phosphorylation of these two transcriptional regulators by Hog1 is essential for inhibition of G1 cyclin expression, for control of cell morphogenesis, and for maximal cell survival upon stress. The control of both Whi5 and Msa1 by Hog1 also revealed the necessity for proper coordination of budding and DNA replication. Thus, Hog1 regulates G1 cyclin transcription upon osmostress to ensure coherent passage through Start.
INTRODUCTION
Tight regulation of critical transitions through the cell cycle is a conserved characteristic from yeast to mammals. One such transition is the G1-to-S transition. In yeast cells, the commitment to enter into S phase, known as Start, is the point when the cyclin CLN gene and some CLB genes are strongly induced. Furthermore, transcriptional positive-feedback loops govern this transition (1–3). Two transcription factors are responsible for G1-specific transcription: the Swi4 cell cycle box binding factor (SBF) (4–6) and the MluI cell cycle box binding factor (MBF) (7). These factors are composed of specific DNA binding proteins, i.e., Swi4 and Mbp1, respectively, and the common subunit Swi6 (8). SBF is required for the transcription of a subset of G1-specific genes, which includes the cyclin CLN1 and CLN2 genes (9–11). On the other hand, MBF triggers the transcription of the so-called S-phase cyclins CLB5 and CLB6 (12, 13). The G1 cyclins Cln1 and Cln2 are required for bud formation, whereas the S-phase cyclins Clb5 and Clb6 are essential for the initiation of DNA replication. In early G1, the SBF transcription factor is kept inactive by the transcriptional repressor Whi5, which is the yeast functional ortholog of the human pocket protein Rb. Whi5 is strongly bound to SBF-dependent promoters, which keeps transcription blocked. However, Cdc28 (the main cyclin-dependent kinase [CDK] in yeast) and Pho85, in association with the G1 cyclin Cln3 and Pcl9, respectively, can phosphorylate Whi5, causing its eviction from these promoters (14–16), thereby allowing its active export from the nucleus by the karyopherin Msn5 (17, 18). These effects on Whi5 enable RNA polymerase (Pol) II to trigger SBF-dependent transcription, generating an initial burst of Cln2. Subsequently, Cdc28-Cln2 can reinforce Whi5 phosphorylation and fully alleviate its repression of SBF (14, 15). Cln2 activity is further required for the activation of MBF, which leads to the production of Clb5. The CDK inhibitor Sic1 specifically blocks CDK-Clb5 activity during progression of G1 (19, 20). However, Cdc28-Cln2 also phosphorylates Sic1, targeting it for ubiquitination and degradation (21–23). This degradation is required for the sudden activation of Cdc28-Clb5, which phosphorylates the prereplicative complex components Sld2 and Sld3 (24, 25), thereby licensing DNA replication origins for firing. It has been recently demonstrated that Clb5 is also involved in the phosphorylation of Sic1, creating another positive-feedback loop (26).
Other proteins have been shown to play a role in G1-specific transcription. For instance, Stb1 and Nrm1 are required for correct regulation of MBF activity (8, 27, 28), whereas Msa1 interacts with and can be found at SBF and MBF promoters. Msa1 is a phosphoprotein with cell cycle-dependent nucleocytoplasmic shuttling (17) that was isolated as a high-copy suppressor of Dpb11 and Sld2 mutants (29) and as an interactor with the SBF and MBF transcription factors (30). Msa1 has been proposed to act as a coactivator of G1 transcriptional machinery, since its overexpression leads to advancement of the timing of G1-specific transcription (30). Msa1 has also been recently shown to interact with other transcription factors such as Tec1 and Ste12 to promote transcription in developmental processes (31). The Msa2 protein is a homolog of Msa1 that has been shown to participate in the same processes as Msa1 (30, 30, 31). Although both Msa1 and Msa2 have been proposed to function as transcriptional regulators, the molecular mechanisms governing the functions of these proteins remain unclear.
The stress-activated protein kinase (SAPK) Hog1/p38 is at the bottom of a prototypical mitogen-activated protein kinase (MAPK) cascade signal transduction pathway that is activated upon increase of extracellular osmolarity (32, 33). Upon osmostress, Hog1 exerts a plethora of adaptive responses in the cell (33), such as the control of gene expression (34) and the regulation of cell cycle progression (35–40) for full adaption to stress. Hog1 induces a transient delay in the G1-to-S transition upon osmostress by a dual mechanism that ensures a proper and robust delay in cell cycle progression before DNA replication. First, Hog1 phosphorylates the CDK inhibitor Sic1 at threonine 173, which blocks its interaction with the E3 ligase Cdc4 and therefore inhibits its degradation. Stabilization of Sic1 blocks the S-CDK activity that is required for DNA replication (39). Second, Hog1 can also inhibit G1 cyclin transcription by an unknown mechanism (36, 39). Mathematical modeling has shed light on the importance of both apparently redundant mechanisms, showing that Sic1 stabilization has a predominant role in G1 regulation when cells receive the stress after Start, whereas cyclin transcription regulation is more important earlier in G1 (41, 42). Indeed, the overexpression of Clb5 can overcome Sic1-dependent arrest in G1 upon stress (41).
In this report, we show that G1 cyclin downregulation is mediated by Hog1 through direct regulation of the transcriptional machinery. Population and single-cell analyses revealed that this inhibition is exerted at the promoter of cyclins and depends on Hog1 phosphorylation of Whi5 and Msa1. Both regulators are required for the coordination of budding and DNA replication upon osmostress (see Fig. S1 in the supplemental material). Finally, we show that proper downregulation of cyclins reveals an additional level of control for adequate passage through Start upon osmostress.
MATERIALS AND METHODS
Yeast strains and plasmids.
A complete list of the yeast strains and plasmids used in this study is provided in the supplemental material. Genomic disruptions were made by long flanking homology PCR-based gene disruption. Tagging of genomic open reading frames (ORFs) with hemagglutinin (HA) or fluorescent epitopes was performed by a PCR-based strategy, with plasmids from the Toolbox collection as templates (43). All mutated genes were sequenced and integrated into the genome at their native loci and under the control of their own promoters. Unless otherwise indicated, all cells were grown in rich yeast extract-peptone-dextrose (YPD) medium at 30°C.
G1 synchronization.
Yeast cells were grown in rich medium to an optical density at 660 nm (OD660) of 0.6. Synthetic α-factor was then added to the cultures, and the cultures were grown at the appropriate temperature for 2 h. Cells were then washed twice with fresh medium, and the pellet was resuspended in rich medium with or without 0.4 M NaCl. For bar1Δ mutant cells, α-factor was used at a final concentration of 0.4 μg ml−1, whereas for non-bar1Δ mutant cells α-factor was used at a final concentration of 20 μg ml−1.
HOG pathway activation.
Genetic activation of Hog1 was achieved by growing yeast cells with the sln1ts-4 allele (39) at 37°C for 1 h. Release from α-factor arrest maintaining Hog1 activation was performed as follows. Synthetic α-factor was added to cells growing exponentially in YPD medium at 25°C. After 1 h, the cultures were shifted to 37°C and grown for a further 60 min. α-Factor was washed out twice with warm YPD medium. The cell pellet was finally resuspended in warm YPD medium and grown at 37°C for the times indicated.
Northern blot analysis.
Yeast strains were grown in rich medium to an OD660 of 0.6, synchronized with α-factor, released into rich medium, and immediately subjected to osmotic shock (0.4 M NaCl) for the time indicated or left untreated. Total RNA was extracted, and expression of the following genes was detected by hybridization of the membranes with radiolabeled PCR fragments containing the entire ORF of CLN2 (1.6 kbp), CLB5 (1.3 kbp), ACT1 (1.1 kbp), or ENO1 (1.3 kbp). Signals were quantified with a Fujifilm BAS-5000 phosphorimager.
In vitro kinase assays.
Glutathione S-transferase (GST) fusion proteins made with the wild-type and mutant versions of Whi5, Msa1, Hog1, and Pbs2EE were expressed in Escherichia coli DH5α and purified with glutathione-Sepharose beads (GE Healthcare) in STET buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA [pH 8.0], 5% Triton X-100, 2 mM dithiothreitol [DTT], 1 mM phenylmethanesulfonyl fluoride [PMSF], 1 mM benzamidine, 2 mg ml−1 leupeptin, 2 mg ml−1 pepstatin). One milligram of Hog1 was activated with 0.5 mg of Pbs2EE in the presence of kinase buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 2 mM DTT) and 50 μM ATP. After 20 min at 30°C, 2 mg of WT or mutated Whi5 or Msa1 was added to the Hog1-Pbs2EE mixture together with [γ-32P]ATP (0.1 mCi/ml) and the mixture was incubated for 30 min at 30°C. The reaction was terminated by the addition of Laemmli buffer and subsequent boiling. Labeled proteins were resolved by SDS-PAGE, gels were stained with Coomassie blue, dried, and analyzed by autoradiography.
Fluorescent promoter activity reporters.
The CLN2 and CLB5 promoters (800 bp) were PCR amplified from genomic DNA and cloned into plasmids so as to drive the expression of double- or quadruple-Venus fluorescent protein, respectively. The resulting plasmids were linearized and integrated at the LEU2 locus in yeast cells. Venus-expressing cells were grown on YPD medium, synchronized with α-factor, and released under the desired conditions. At the time indicated, 1 ml of cells was taken and cycloheximide (Sigma) was added to a final concentration of 100 μg ml−1. Cells were then incubated in the dark at room temperature for 1 h to allow for Venus protein folding, and samples were stored at 4°C. For analysis of fluorescence content, 100 μl of cells from each time point were added to fluorescence-activated cell sorter (FACS) tubes containing 1 ml of 50 mM sodium citrate. Samples were sonicated and analyzed in a FACScalibur cytometer. Twenty thousand cells were acquired at each time point. Data were analyzed with FlowJo software.
DNA content measurement.
At the times indicated, 100 μl of synchronized cells was released in YPD medium or 0.4 M NaCl, fixed in 1 ml of 70% ethanol for at least 10 min, and kept at 4°C until further processing. Ethanol was removed by centrifugation. Cells were resuspended in a 50 mM sodium citrate solution containing 0.1 mg ml−1 RNase A and incubated overnight at 30°C. A 750-μl aliquot of cells was mixed with 750 μl of a 4 μg ml−1 propidium iodide solution in 50 mM sodium citrate in a FACS tube. Samples were sonicated and analyzed with a BD FACScalibur cytometer. Ten thousand cells were acquired at each time point. Data were analyzed with WinMDI 2.9.
Competition assay.
Cells expressing Eno1-green fluorescent protein (GFP) or Eno1-mCherry at its own locus were generated by homologous recombination of PCR products. The combinations of cells indicated were synchronized in G1 with α-factor and mixed in a 1:1 ratio, released into YPD medium or YPD medium plus 0.4 M NaCl at an initial OD660 of 0.05, and grown at 30°C. At the times indicated, cells were diluted again to an OD660 of 0.05. At these same time points, samples were fixed with 3.7% formaldehyde (Sigma) for 10 min. The fixing solution was washed out, and the cells were resuspended in 1× phosphate-buffered saline and kept in the dark at 4°C until further analysis. Cells were diluted in 50 mM sodium citrate, sonicated, and analyzed with a BD LSRFortessa analyzer. Ten thousand cells were acquired at each time point. Data were processed with FlowJo software.
In vivo phosphorylation assay.
Msa1 and msa19A were tagged with HA. Exponentially growing cells expressing the tagged factors were synchronized in G1 with α-factor and then subjected to a brief osmotic shock (0.4 M NaCl for 10 min) or left untreated. For each condition, 100-ml volumes of synchronized cultures were harvested by centrifugation, and pellets were resuspended in 1 ml of lysis buffer (45 mM HEPES-KOH [pH 7.5], 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 2 mM DTT) supplemented with protease and phosphatase inhibitors (1 mM PMSF, 1 mM benzamidine, 2 mg ml−1 leupeptin, 2 mg ml−1 pepstatin, 25 mM β-glycerophosphate, 1 mM sodium pyrophosphate, 10 mM sodium fluoride, 100 mM sodium orthovanadate). Glass beads were added, and cells were lysed by vortexing. Proteins (5 mg) were incubated overnight with an anti-HA antibody (12CA5) and then incubated with protein G-Sepharose (GE Healthcare) beads for 4 h. Beads were washed five times in lysis buffer, boiled in SDS loading buffer for 10 min, and analyzed by 8% SDS-PAGE, and proteins were transferred to polyvinylidene difluoride membranes. Proteins phosphorylated at SP/TP sites (Hog1 consensus phosphorylation sites) were detected by immunoblotting, for which membranes were incubated overnight at 4°C with an anti phosphoserine threonine antibody (BD Transduction Laboratories).
Coprecipitation assays.
Cells expressing tagged versions of Whi5 or Msa1 were transformed with a plasmid containing GST-Hog1 or GST alone. Cells were grown to mid-log phase on minimal medium for plasmid maintenance. Cells were then treated with 0.4 M NaCl for 10 min and harvested at 4°C. Pellets were resuspended in lysis buffer (45 mM HEPES-KOH [pH 7.5], 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 2 mM DTT) with protease and phosphatase inhibitors, glass beads were added, and cells were vortexed at 4°C. Protein extracts were clarified by centrifugation. Proteins (2 mg) were pulled down with glutathione-Sepharose beads (GE Healthcare) for Msa1-HA–GST-Hog1 interaction or rabbit IgG-agarose beads (Sigma) for Whi5-tandem affinity purification (TAP)–GST-Hog1 interaction. Beads were incubated with extracts overnight and then washed seven times with lysis buffer. Beads were boiled in Laemmli buffer, and proteins were resolved by 10% SDS-PAGE and Western blotted with anti-HA (12CA5), anti-TAP, or anti-GST antibodies. Protein extract (50 μg) was used as the input control.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (44). Briefly, early-log-phase cultures (OD660, 0.6 to 0.9) or synchronized-and-released cultures were exposed to osmostress (0.4 M NaCl) or left untreated for the length of time specified in each experiment. Cells were cross-linked with 1% formaldehyde for 20 min at room temperature, incubated with 330 mM glycine for a further 15 min and washed four times with cold TBS buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl). Pellets were frozen at −20°C. Lysis buffer (50 mM HEPES-KOH [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 1 mM PMSF) and glass beads were added to the pellets, and the cells were vortexed for 15 min at 4°C. Samples were sonicated and clarified by centrifugation. Magnetic beads (Dynabeads; Invitrogen) previously conjugated with the appropriate antibody were added to samples and incubated with rotation at 4°C for 2 to 4 h. Anti-HA (12CA5) and anti-Rpb1 (8WG16; Covance) antibodies were used in this study. Beads were then washed twice with lysis buffer, twice with lysis buffer plus 500 mM NaCl, twice with wash buffer (10 mM Tris-HCl [pH 8.0], 0.25 M LiCl, 1 mM EDTA, 0.5% N-P40, 0.5% sodium deoxycholate), and once with TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). Chromatin was eluted, and cross-linking was reversed by incubation at 65°C overnight. DNA was then phenol extracted and finally resuspended in TE. Quantitative real-time PCR was used to analyze DNA with an Applied Biosystems ViiA7 detector. Specific primers were used for PCR analysis of G1 cyclin promoters (CLN2, −627/−526 before start codon; CLB5, −386/−255 before start codon) and coding regions (CLN2, +325/+475 after start codon; CLB5, +429/+656 after start codon). A telomeric region on the right arm of yeast chromosome VI was used as an internal normalizing control sequence for each DNA analyzed (TELRTa 269437 to 269464 and TELRTb 269615 to 269637).
RESULTS
Osmostress induces a delay in transcription initiation from G1 cyclin promoters.
Hyperosmotic stress causes a delay in entry into S phase (38). This delay is, in part, due to a lag in the transcription of G1 cyclins CLN2 and CLB5 (Fig. 1A). Since osmostress affects many steps of mRNA biology, including processing, transport, and stability (45–47), we checked if the delay in the appearance of these transcripts was due to a defect in transcription initiation or in other steps of mRNA biogenesis. We followed the dynamics of RNA Pol II association with the CLN2 and CLB5 promoters by ChIP with an anti-RNA Pol II (anti-Rpb1) antibody and cells synchronized with α-factor and released into either YPD medium or YPD medium containing 0.4 M NaCl. As shown in Fig. 1B, osmostress produced a delay of approximately 20 to 30 min in the binding of RNA Pol II to both the CLN2 and CLB5 promoters, although the effect was more dramatic with the CLN2 promoter. Of note, RNA Pol II binding dynamics were consistent with the dynamics of mRNA expression shown in Fig. 1A under both control and stress conditions. The reduced amount of RNA Pol II required for the transcription of similar amounts of RNA could be explained by the effect of Whi5 on the CLN2 promoter, in contrast to the CLB5 promoter (see Fig. S2A and B in the supplemental material). These data indicated that osmostress delays the transcription initiation of both cyclins. To confirm that this effect of osmostress on cyclin transcription was mediated through regulation of cyclin promoter activity, we fused a fluorescent reporter to the CLN2 and CLB5 promoters and used these reporters to follow transcription at the single-cell level by flow cytometry (48). Cells were synchronized in G1 with α-factor and then released into fresh medium, which triggered transcription from both cyclin promoters, as reflected by an increase in fluorescence (Fig. 1C). In contrast, an increase in fluorescence was not observed upon osmostress (0.4 M NaCl). Thus, osmostress causes a robust inhibition of transcription initiation of CLN2 and CLB5 in all cells.
FIG 1.

Osmostress delays transcription initiation of G1 cyclins. (A) G1 cyclin expression is delayed upon osmostress. Wild-type cells were released from α-factor arrest into YPD medium (control) or 0.4 M NaCl. Samples were taken at the time points indicated for Northern blot analysis with radioactively labeled probes against CLN2, CLB5, and ENO1 (as a loading control). (B) Binding of RNA Pol II to G1 cyclin promoters is delayed in response to osmostress. Wild-type cells were synchronized in G1 with α-factor and released into either YPD medium (control) or 0.4 M NaCl. Chromatin-bound RNA Pol II was immunoprecipitated with an anti-Rpb1 monoclonal antibody (8WG16; Covance) at the times indicated. The precipitate was analyzed by real-time PCR with primers specific for the CLN2 (left panel) or CLB5 (right panel) promoter region. Graphs represent the averages ± the standard deviations from five independent experiments. (C) Osmostress inhibits transcriptional firing of G1 cyclin promoters. A fluorescent reporter of CLN2 or CLB5 promoter activation was integrated into wild-type cells. Promoter-associated fluorescence of G1-arrested cells (α-factor) or cells released for 40 min into YPD medium (control) or 0.4 M NaCl was analyzed by flow cytometry. A representative experiment with the CLN2 (left panel) and CLB5 (right panel) promoters is shown. Each overlaid histogram represents fluorescence distribution from 20,000 cells.
Global downregulation of gene expression of non-stress-responsive genes occurs through a reduction in RNA Pol II levels (49), which is most likely due to activator eviction from promoters that is caused by stress (50). To assess whether downregulation of cyclin transcription was caused by eviction of activators, we followed promoter association of the HA-tagged transcription factor Swi4 by ChIP (see Fig. S2C in the supplemental material). At the point of arrest by pheromone, HA-Swi4 was already bound to the CLN2 promoter. Osmostress caused immediate eviction of HA-Swi4 from the promoter. Reassociation of HA-Swi4 with the CLN2 promoter was concomitant with induction of transcription. Of note, HA-Swi4 eviction was HOG1 independent (see Fig. S2D in the supplemental material), although in hog1Δ cells, HA-Swi4 reassociation with the CLN2 promoter was less efficient than in wild-type cells, possibly because of inefficient general adaptation.
Hog1 downregulates G1 cyclin transcription.
Activation of the HOG pathway by genetic manipulation has served to unravel the direct role of Hog1 in the cell cycle (37, 39, 41). Growth of sln1ts mutant cells at the restrictive temperature results in the constitutive activation of Hog1 independently of external stimuli (39). Release of sln1ts mutant cells from α-factor arrest at the nonpermissive temperature showed a clear downregulation of CLN2 and CLB5 expression that was totally dependent on Hog1 (39) (Fig. 2A). Strikingly, in contrast to the transcription inhibition that occurs following activation of Hog1 by osmostress, this promoter inhibition was not due to a general eviction of the transcription machinery from the promoters since both Swi4 and RNA Pol II were retained on the promoters upon Hog1 activation (Fig. 2B and C). In contrast, in cells deficient in Hog1, the dynamics of the association of Swi4 and RNA Pol II reflected mRNA transcription. These findings suggested that even though the RNA Pol II machinery is present at the CLN2 and CLB5 promoters, firing of transcription is somehow inhibited.
FIG 2.
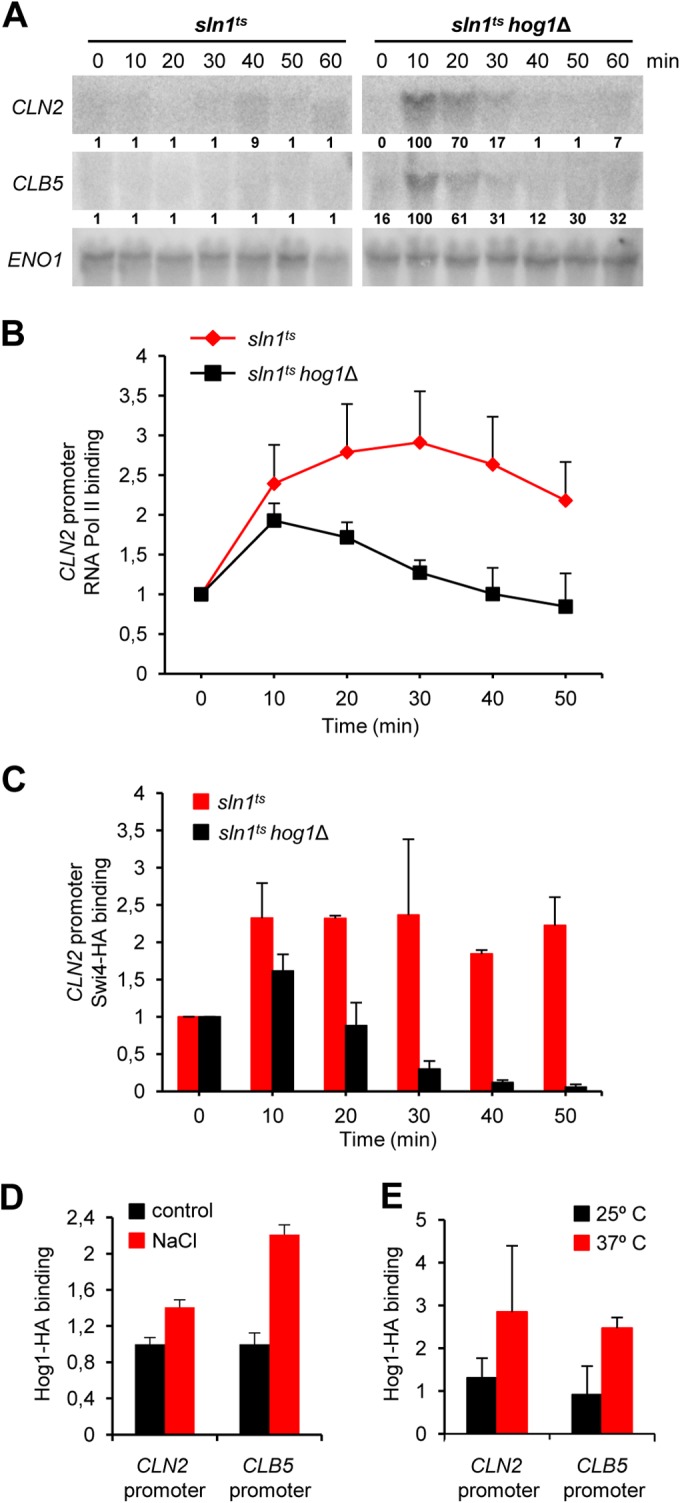
Hog1 is involved in transcriptional downregulation of G1 cyclins. (A) Constitutive activation of the HOG pathway downregulates G1-specific cyclin transcription. sln1ts or sln1ts hog1Δ mutant cells were synchronized in G1 with pheromone and then released into YPD medium at 37°C. RNA was extracted from samples at the times indicated and analyzed by Northern blotting with CLN2, CLB5, and ENO1 probes. Quantification relative to the loading control of each mRNA is depicted below each lane. A value of 100% is assigned to the maximum level of a given transcript. (B) RNA Pol II downregulation depends on Hog1. sln1ts and sln1ts hog1Δ mutant cells were synchronized with α-factor and released into YPD medium at 37°C. RNA Pol II was immunoprecipitated with anti-Rpb1 antibodies at the times indicated after release, and associated chromatin-DNA was analyzed by real-time PCR with primers specific for the CLN2 promoter region. Graphs represent the averages ± the standard deviations from three independent experiments. (C) Swi4 is retained at the CLN2 promoter upon Hog1 activation. Swi4 was tagged with HA in an sln1ts or sln1ts hog1Δ background. The resulting strains were synchronized with α-factor and released into YPD medium at 37°C. Swi4-HA was immunoprecipitated with anti-HA antibodies at the time points indicated after release. Associated chromatin-DNA was analyzed by real-time PCR with primers specific for the CLN2 promoter. Bars represent the averages ± the standard deviations from three independent experiments. (D, E) Hog1 binds to the promoter regions of CLN2 and CLB5. Wild-type cells were incubated for 10 min in the presence or absence (control) of 0.4 M NaCl (D), or sln1ts mutant cells were incubated at a nonpermissive temperature (37°C) for 30 min (E). HA-tagged Hog1 was used for chromatin immunoprecipitation in ChIP assays to monitor its association with the CLN2 and CLB5 promoters as described previously (49). Bars represent the averages ± the standard deviations from three independent experiments.
Hog1 itself associates with stress-responsive loci upon activation to induce gene expression (49, 51, 52). We therefore assessed the association of Hog1 with the CLN2 and CLB5 promoters. ChIP analyses showed that Hog1 associates with both the CLN2 and CLB5 promoters upon stress, albeit to different extents for the different promoters (Fig. 2D). The association of Hog1 with chromatin depends on its interaction with specific transcription factors (45). Swi4 is evicted upon stress and is thus unlikely to be the only factor responsible for Hog1 association. ChIP analysis with Fkh1 (Forkhead protein) showed that this protein is present at the CLN2 and CLB5 promoters upon stress, indicating that alternative transcription factors are present at those promoters in the presence of stress (see Fig. S2E in the supplemental material). Of note, Hog1 also associates, albeit with different kinetics, with the CLN2 and CLB5 promoters upon Hog1 activation in sln1ts mutant cells (Fig. 2E). These combined data indicate that Hog1 might act directly on the CLB5 and CLN2 promoters to regulate their expression.
The transcriptional repressor Whi5 is phosphorylated by Hog1.
To characterize the mechanism by which Hog1 controls CLN2 and CLB5 transcription, we tested the phosphorylation of several components of SBF and MBF (i.e., Swi4, Swi6, Mbp1, and Whi5), which were purified from bacteria as GST-fused proteins, in an in vitro kinase assay with recombinant Hog1. Only one of the proteins tested yielded significant phosphorylation by Hog1, the transcriptional repressor Whi5, which is the yeast homolog of Rb (Fig. 3A). Mutation of the Hog1 consensus sites in Whi5 indicated that Whi5 is phosphorylated in vitro on three residues, Ser88, Thr143, and Thr215. Simultaneous mutation of these three sites to alanine (Whi53A) resulted in total loss of phosphorylation by Hog1 (Fig. 3A).
FIG 3.
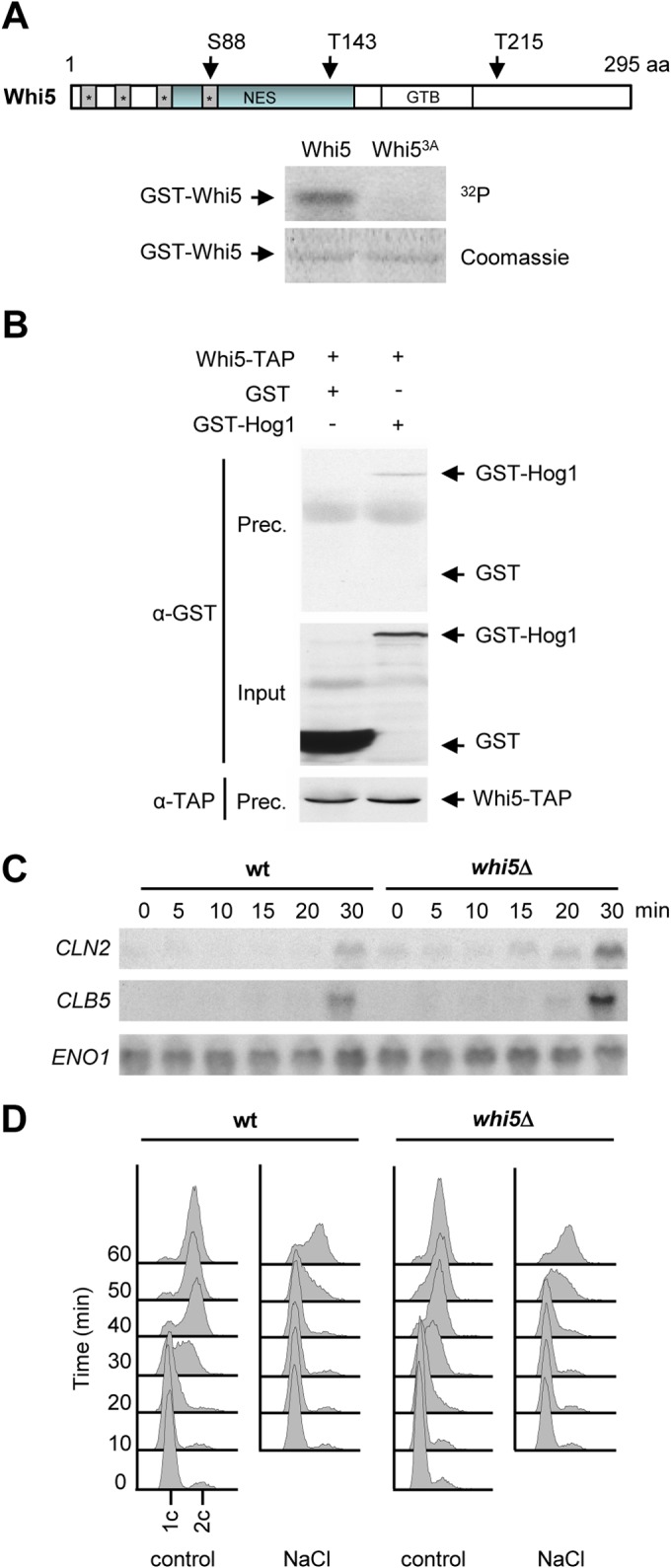
Whi5 is targeted by Hog1 upon osmostress. (A) Mutation of Whi5 Ser88, Thr143, and Thr215 abolishes Hog1 phosphorylation in vitro. Bacterially expressed GST-Whi5 or GST-Whi53A was employed as a substrate in an in vitro kinase assay. Hog1 and the constitutively activated Pbs2EE allele were incubated in kinase buffer containing ATP. Whi5 or Whi53A was then added in the presence of radioactive ATP. Phosphorylated proteins were resolved by SDS-PAGE and detected by autoradiography (top). GST-tagged proteins were detected by staining with Coomassie brilliant blue (bottom). A schematic representation of Whi5 is shown at the top, indicating the location of the three residues phosphorylated by Hog1. The gray boxes denote predicted nuclear localization signals. The blue boxes denote the nuclear export signal (NES), and the GTB box denotes the G1/S transcription factor binding motif. aa, amino acids. (B) Whi5 and Hog1 interact in vivo. Cells expressing Whi5-TAP with either GST-Hog1 or GST alone were exposed to 0.4 M NaCl for 10 min, and anti-TAP antibody was used to immunoprecipitate Whi5. Precipitated proteins (Prec.) were resolved by SDS-PAGE and analyzed by immunoblotting with anti-GST and anti-TAP antibodies as probes. The input represents 2.5% of the immunoprecipitated protein. (C) Absence of Whi5 does not rescue the delay in G1 cyclin transcription induced by osmostress. Wild-type (wt) and whi5Δ mutant cells were synchronized with α-factor and released into 0.4 M NaCl. RNA extracted from samples taken at the time points indicated was analyzed by Northern blotting with probes specific for CLN2 and CLB5 mRNAs. ENO1 mRNA expression was analyzed as a loading control. (D) DNA replication delay induced by osmostress is not dependent on Whi5. Wild-type and whi5Δ mutant cells were synchronized in G1 with α-factor and released into YPD medium (control) or 0.4 M NaCl. DNA content was measured by flow cytometry every 10 min after release.
In vivo interaction between SAPKs and their corresponding substrates can often be detected by their coimmunoprecipitation. We therefore determined if Hog1 and Whi5 physically interact in vivo by testing whether Hog1 coprecipitates with Whi5 from cell extracts. Yeast cells expressing TAP-tagged Whi5 and GST-tagged Hog1 expressed from their genomic loci were subjected to osmostress and Whi5-TAP was immunoprecipitated with specific monoclonal antibodies against the TAP epitope. As shown in Fig. 3B, GST-Hog1 did coprecipitate with Whi5.
To gain further insight into the role of Whi5 in the downregulation of CLN2 and CLB5 upon osmostress, the transcription of both mRNAs was followed in whi5Δ mutant cells after their release from α-factor synchronization in the presence of osmostress. Although there was a clear delay in the expression of cyclins caused by stress in whi5Δ mutant cells, there is a slightly earlier induction of CLN2 expression and, to a lesser extent, of CLB5 than of the wild type (Fig. 3C). Consistent with these findings, whi5Δ mutant cells still displayed the same delay in the initiation of DNA replication upon osmostress as the wild type (Fig. 3D). Thus, the Whi5 transcriptional repressor is a Hog1 substrate, although its role in CLN2 and CLB5 transcription inhibition upon osmostress is modest, suggesting that additional players may be involved in the osmostress-induced downregulation of cyclin expression.
Msa1, a transcriptional coregulator, is another target of Hog1 upon osmostress.
In a quest for alternative players involved in G1 transcriptional regulation that are targeted by Hog1, we applied the above-described in vitro kinase assay to several proteins previously reported to be involved in the control of SBF and MBF promoters (i.e., Whi5, Msa1, Msa2, Mbp1, Swi6, Swi4, Nrm1, and Stb1). We found that, in addition of Whi5, Msa1 was also phosphorylated in vitro by Hog1 (Fig. 4A). Msa1 contains 25 SP/TP potential Hog1 phosphorylation sites. We performed systematic deletion of these Msa1 phosphorylation sites, followed by in vitro kinase assays together with mass spectrometry (MS) analysis. Finally, after systematic mutagenesis, we found that Hog1 phosphorylation of Msa1 in vitro could be abrogated by simultaneously mutating 9 of the 25 SP/TP sites of Msa1 to alanine (Msa19A).
FIG 4.
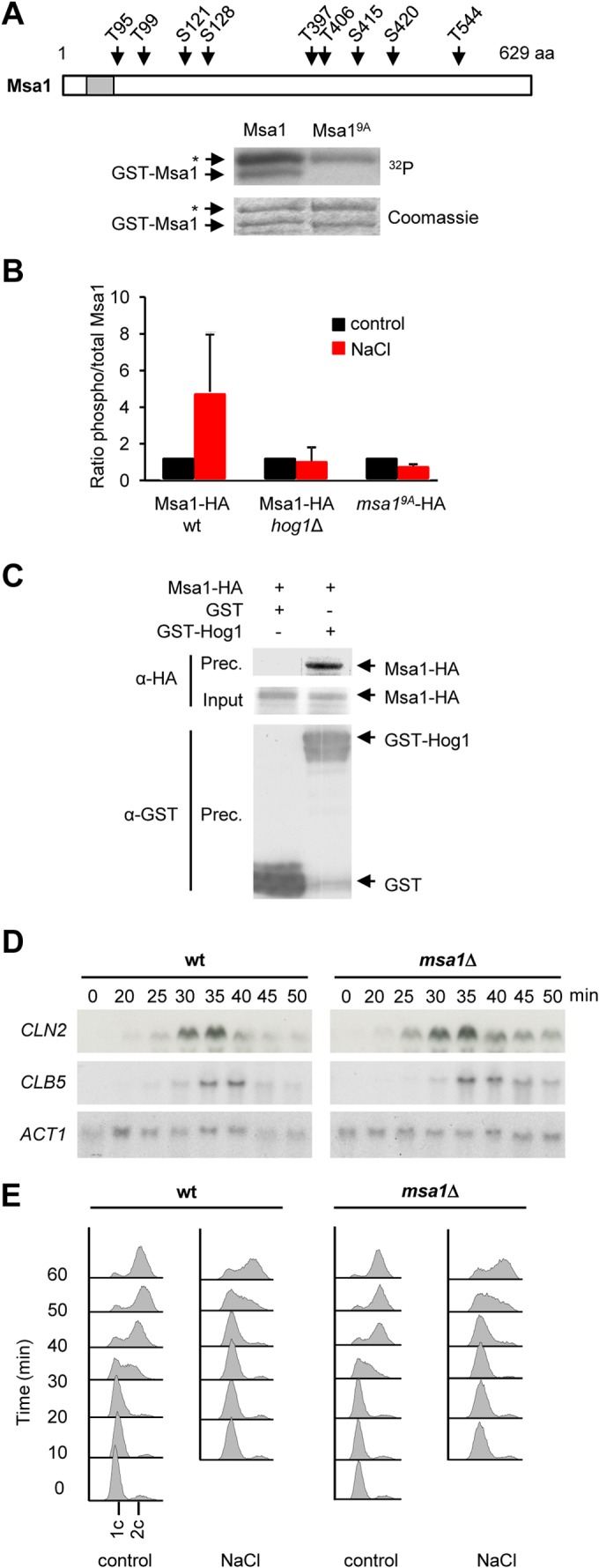
Hog1 targets the Msa1 transcriptional coregulator. (A) Mutation of nine residues in Msa1 abolished Hog1 phosphorylation in vitro. GST-Msa1 or GST-Msa19A was purified from bacteria, subjected to an in vitro kinase assay with activated Hog1, and analyzed as described in the legend to Fig. 3A. Asterisks indicate bands that correspond to purified GST-Pbs2EE. The location of the mapped residues in the Msa1 protein is shown in the cartoon at top. The gray box denotes a predicted nuclear localization signal. aa, amino acids. (B) Msa1 is phosphorylated by Hog1 in vivo. Wild-type (wt) yeast cells expressing Msa1-HA or msa19A-HA, as well as hog1Δ mutant cells expressing Msa1-HA, were synchronized with α-factor and treated with brief osmotic shock (0.4 M NaCl, 10 min) or left untreated (control). Protein extracts were then immunoprecipitated with anti-HA antibody. SP/TP phosphorylated proteins were detected with an anti-phosphoserine-threonine antibody (BD Transduction Laboratories). Bars represent the average ratios of phosphorylated to total Msa1 ± the standard deviations from three independent experiments. (C) Hog1 and Msa1 coimmunoprecipitate in vivo. Cells expressing Msa1-HA and either GST-Hog1 or GST alone were stressed with 0.4 M NaCl for 10 min. Protein extracts were immunoprecipitated with anti-GST antibody. Precipitated proteins (Prec.) were resolved by SDS-PAGE and immunoblotted for Msa1 and Hog1 with anti-HA and anti-GST probes, respectively. The input represents 2.5% of the immunoprecipitated protein. (D) Transcription delay of G1 cyclins is not reduced in msa1Δ mutant cells. Wild-type and msa1Δ mutant cells were synchronized in G1 with α-factor and released into 0.4 M NaCl. Samples were taken at the times indicated after release for RNA extraction for Northern blot analysis. CLN2 and CLB5 mRNAs were detected with specific probes. ACT1 was probed as a loading control. (E) DNA replication is delayed independently of Msa1 upon osmostress. Wild-type or msa1Δ mutant cells were synchronized with α-factor and released into control or 0.4 M NaCl medium. Samples were taken every 10 min after release and analyzed for DNA replication by flow cytometry.
We then determined whether these nine phosphorylation sites in Msa1 were also phosphorylated by Hog1 in vivo. Cells expressing the HA-tagged wild type and the HA-tagged Msa19A mutant were subjected to a brief osmotic shock. Msa1-HA was immunoprecipitated and analyzed by Western blotting with an antibody specific for the detection of phosphorylation at SP/TP sites (BD Transduction Laboratories). Osmostress increased the level of phosphorylation of Msa1-HA >8-fold. This increase in phosphorylation was dependent on Hog1 and was not detected in the Msa19A mutant (Fig. 4B; see Fig. S3 in the supplemental material).
We then assayed Hog1-Msa1 interaction as described for analysis of Hog1-Whi5 interaction. Yeast cells expressing HA-tagged Msa1 and GST-tagged Hog1 expressed from their genomic loci were subjected to osmostress. GST-Hog1 was immunoprecipitated with glutathione beads, and the precipitate was immunoblotted for Msa1-HA. As shown in Fig. 4C, Msa1-HA coprecipitated with GST-Hog1. These data indicate that Msa1 is a bona fide target of Hog1 upon stress.
The effect of Msa1 on the transcription of CLN2 and CLB5 was then assessed by Northern blotting. An approximately similar delay in the transcription of both cyclins upon stress was observed following the release of G1-arrested wild-type and msa1Δ mutant cells at 0.4 M NaCl (Fig. 4D). Of note, although transcription was initiated at the same time, cells deficient in MSA1 showed a prolonged expression of cyclins that did not significantly affect the dynamics of DNA replication (Fig. 4E). Therefore, although Msa1 is also a target of Hog1, Msa1 alone does not mediate the delay in cyclin expression caused by stress.
Whi5 and Msa1/Msa2 act in parallel to regulate CLN2 transcription upon osmostress.
Since both Whi5 and Msa1 are targeted by Hog1 upon stress, we next assessed whether it might be the coordinated action of both transcriptional regulators that mediates cyclin regulation upon stress. Prior to this analysis, we assayed if the Msa1 homolog in S. cerevisiae, named Msa2, might also be involved in the regulation of cyclin transcription upon osmostress. However, we could detect no phosphorylation of Msa2 by Hog1 in vitro and deletion of MSA2 did not alter the pattern of cyclin expression upon osmostress (see Fig. S4A in the supplemental material). However, it could not be formally ruled out that the lack of Msa1 resulted in a compensatory effect exerted by Msa2. We therefore analyzed the potential synergy of Msa1 with Whi5 in the absence of Msa2. We created WHI5 MSA1 MSA2 triple deletion cells and followed cyclin transcription upon osmostress. As shown in Fig. 5 (top and middle), there was only a subtle difference between the wild type and the triple null strain in transcriptional firing after release from synchronization in an unperturbed cell cycle. In clear contrast, the delay in CLN2 transcription caused upon osmostress, and to a lesser extent that in CLB5, was clearly reduced in the whi5 msa1 msa2 mutant strain, which showed an increase in the CLN2 transcript 15 min before an increase was observed in the wild type. This premature induction of CLN2 expression correlates with, albeit displaying similar Swi4 recruitment, an earlier recruitment of RNA Pol II to CLN2 upon stress (see Fig. S2D and S4B in the supplemental material). Therefore, both the Whi5 and Msa1 proteins are important for the downregulation of cyclins observed upon stress.
FIG 5.
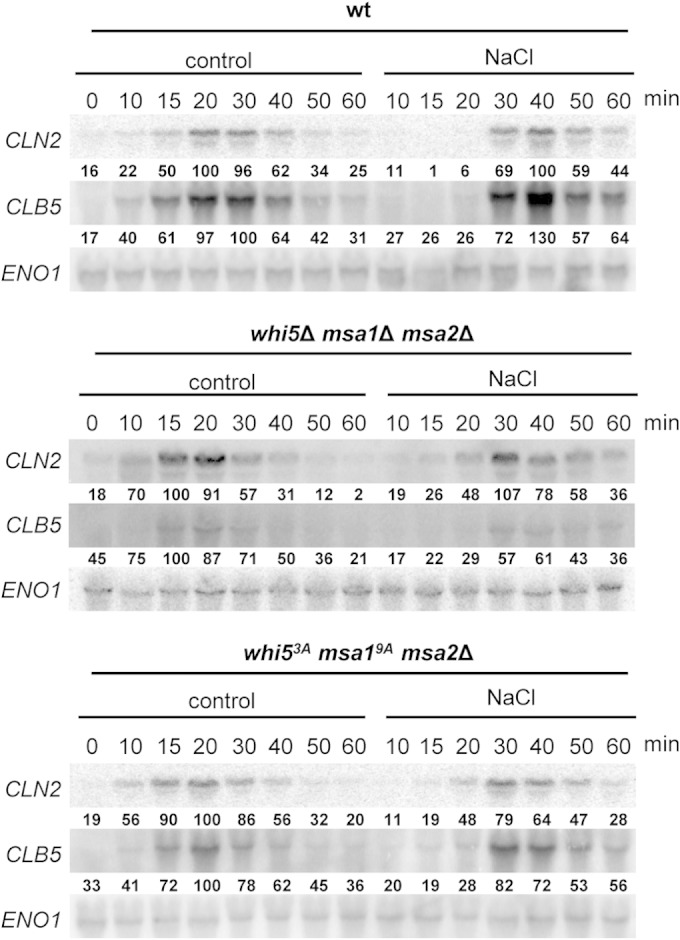
Whi5, Msa1, and Msa2 cooperate to delay CLN2 transcription upon osmostress. Wild-type (wt), triple null (middle), and point mutant (bottom) strains were synchronized in G1 with α-factor and released into YPD medium (control) or 0.4 M NaCl. RNA was extracted from samples taken at the times indicated after release and analyzed by Northern blotting with a radiolabeled probes specific for CLN2, CLB5, or ENO1 mRNA. Quantification relative to the loading control of each mRNA is depicted below each lane. A value of 100% is assigned to the maximum level of a given transcript under control conditions for each strain.
We then assessed whether the phosphorylation of Whi5 and Msa1 by Hog1 was relevant for the prevention of cyclin expression. We constructed a strain bearing Whi5 and Msa1 with all of their previously defined Hog1-dependent phosphorylation sites mutated to Ala (Whi53A and Msa19A) (Fig. 3A and 4A). The mutant genes were integrated into the genome at their own loci and expressed under the control of their own native promoters. The cells were synchronized with α-factor and released under control and osmostress conditions, and cyclin mRNAs were analyzed by Northern blotting (Fig. 5, bottom). The dynamics of cyclin transcription observed upon stress were almost identical to those observed in the triple null strain, suggesting that the stress-induced delay in CLN2 transcription was due to Hog1 phosphorylation of the sites that were mutated in Whi5 and Msa1.
Upon stress, Swi4 is evicted from the CLN2 promoter. Then, we followed promoter association of HA-tagged Whi5 and Msa1 by ChIP. Similarly to Swi4, those factors are evicted upon stress and reassociated with dynamics similar to those of Swi4, both upon stress and in a sln1ts mutant strain, indicating that transcription of cyclins upon reassociation of Swi4 might be prevented by the presence of the two regulators (see Fig. S5 in the supplemental material).
We then assessed the biological impact of earlier cyclin expression upon stress. It is known that Clb5 is required for the initiation of DNA replication whereas Cln2 is required for bud morphogenesis. We therefore reasoned that in the above-described mutant cells, which are unable to delay CLN2 expression, bud emergence should occur earlier than in wild-type cells, but there should be no clear effect on DNA replication. Indeed, DNA replication was delayed to the same extent in wild-type and mutant strains (Fig. 6A). In contrast, the delay in budding caused by stress was almost abolished in both the whi5 msa1 msa2 and the whi53A msa19A msa2 mutant strains, in contrast to the wild type (Fig. 6B). These combined data indicate that Hog1 phosphorylation of Whi5 and Msa1 is important for CLN2 downregulation upon stress and that prevention of this phosphorylation results in the loss of coordination between budding and DNA replication upon osmostress.
FIG 6.

Whi5, Msa1, and Msa2 are involved in the coordination of budding and DNA replication upon osmostress through Hog1 phosphorylation. (A) The delay in DNA replication initiation upon osmostress does not depend on Whi5, Msa1, or Msa2. The wild-type (wt), triple null, or point mutant cells indicated were synchronized in G1 with α-factor and released into YPD medium (control) or 0.4 M NaCl. Samples were taken every 10 min after release, and DNA replication was analyzed by flow cytometry. (B) The delay in budding caused by stress depends on Whi5, Msa1, or Msa2. The same cells as in panel A were synchronized with α-factor and released into either YPD medium (control) or 0.4 M NaCl. At the times indicated after release, the cells were mounted on glass slides and budded cells were counted. Bars represent the averages ± the standard deviations from three independent experiments. In each experiment, at least 100 cells were counted at each time point.
Whi5 and Msa1 Hog1-dependent phosphorylation mediates G1 arrest.
In contrast to the situation under osmostress, downregulation of both CLN2- and CLB5-encoded cyclins occurs when Hog1 is genetically activated in sln1ts mutant cells (39) (Fig. 2). To further characterize the role of Hog1 phosphorylation of Whi5 and Msa1 in the transcriptional downregulation of CLN2 and CLB5, we followed the dynamics of cyclin gene expression in synchronization-and-release experiments with sln1ts mutant cells that contained mutations in WHI5, MSA1, and MSA2. The downregulation of CLN2- and CLB5-encoded cyclins that was observed upon Hog1 activation was partially abolished in the mutant strains (triple null and nonphosphorylatable mutants), in contrast to that in the wild type (Fig. 7A). We then assessed cyclin gene transcription in the sln1ts background at the single-cell level with fluorescent reporters for the CLN2 and CLB5 promoters. Expression from the CLN2 and CLB5 promoters was prevented upon Hog1 activation. In contrast, CLN2 and, to a lesser extent, CLB5 promoter activity was induced in the whi53A msa19A msa2 mutant strain (Fig. 7B and C, see Fig. S6 in the supplemental material). Thus, Hog1 activation represses cyclin promoter activity through Whi5 and Msa1 phosphorylation.
FIG 7.

Phosphorylation of Whi5 and Msa1 by Hog1 prevents G1 arrest. (A) The whi5 msa1 msa2 triple mutant suppress the delay of cyclin expression upon Hog1 activation. The strains indicated were synchronized with α-factor and released at 37°C into YPD medium. RNA was extracted from samples taken at the times indicated after release and analyzed by Northern blotting with radiolabeled probes specific for the CLN2, CLB5, and ENO1 mRNAs. (B, C) Downregulation of both the CLN2 and CLB5 promoters depends on Hog1 phosphorylation of Whi5 and Msa1. sln1ts and sln1ts whi53A msa19A msa2Δ mutant cells were transformed with a fluorescent reporter system for analysis of CLN2 (B) or CLB5 (C) promoter activity. Fluorescence-positive cells were synchronized with α-factor and released at 37°C into YPD medium, and promoter-associated fluorescence was analyzed by flow cytometry in G1-synchronized cells (red lines) or at 90 min (blue lines) or 120 min (orange lines) after release. Each line in the histogram represents the fluorescence distribution from 20,000 cells. (D) G1 arrest depends on the phosphorylation of Whi5 and Msa1 by Hog1. The strains indicated were synchronized with α-factor and released into YPD medium at 37°C. Cell samples were fixed every 10 min after release, and their DNA content was measured by flow cytometry. (E) The Hog1-dependent budding block is mediated by Whi5, Msa1, and Msa2. The same cells as in panel D were microscopically analyzed, and budded cells were counted at the times indicated after release. sln1ts, sln1ts hog1Δ, sln1ts whi5Δ msa1Δ msa2Δ, sln1ts whi53A msa19A msa2Δ, and sln1ts whi53A msa19A msa2Δ hog1Δ mutant cells were analyzed. At least 100 cells of each strain were counted at each time point. Bars represent the averages ± the standard deviations from three independent experiments.
Sustained activation of the HOG pathway causes a prolonged arrest in G1 (39, 53). We tested whether suppression of cyclin downregulation in the above-described mutant cells could suppress this arrest in G1. We synchronized sln1ts or sln1ts mutant cells containing hog1 or whi5 and msa1 mutations with α-factor, released them at the restrictive temperature, and then followed DNA replication and budding. In clear contrast to sln1ts mutant cells, whi5 msa1 msa2 null mutant cells or cells expressing their nonphosphorylatable mutant forms were able to replicate DNA efficiently, showing only a slight delay with respect to sln1ts hog1Δ mutant cells (Fig. 7D). A similar result was observed when the budding index was analyzed; the mutant cells were able to bud with only a 10-min delay compared to the budding of sln1ts hog1Δ mutant cells (Fig. 7E). Of note, the HOG1, whi5 msa1, and msa2 mutations in the sln1ts background yielded results similar to those obtained with sln1ts hog1 mutant cells in both budding and replication, indicating an epistatic relationship between Hog1 and those transcriptional regulators (Fig. 7D and E). Correspondingly, msa1 msa2 whi5 hog1 mutant cells responded similar to hog1 mutant cells upon stress (see Fig. S7 in the supplemental material). Thus, in addition to Sic1 stabilization, regulation of cyclin expression upon Hog1 activation is relevant for cell cycle delay in G1.
The ratio of Sic1 CDKi to Clb5 levels is critical for prevention of replication upon Hog1 activation.
It is known that Hog1 activation results in stabilization of the CDK inhibitor Sic1 and that this stabilization, together with the downregulation of CLB5, is sufficient to inhibit DNA replication inhibition (39, 41). However, overexpression of CLB5 is able to overcome this delay in replication (41). To understand why whi5 msa1 msa2 mutant cells were able to enter replication when Hog1 was activated by genetic means, we quantified the levels of Sic1 and analyzed the ratio of Clb5 to Sic1 proteins over time after release of sln1ts and sln1ts whi5Δ msa1Δ msa2Δ mutant strains from pheromone arrest. This assay showed that whereas the levels of Clb5 cannot overcome those of Sic1 in sln1ts mutant cells, in the triple mutant, the levels of Clb5 were greater than those of Sic1 at 50 min after release (Fig. 8A; see Fig. S8 in the supplemental material). This increase in Clb5 levels compared to Sic1 levels coincides with the entry into replication of the mutant cells. Thus, Hog1 phosphorylation of Whi5 and Msa1 allows the cells to block G1 and S cyclin transcription, budding, and DNA replication initiation, thereby arresting the cells in G1.
FIG 8.
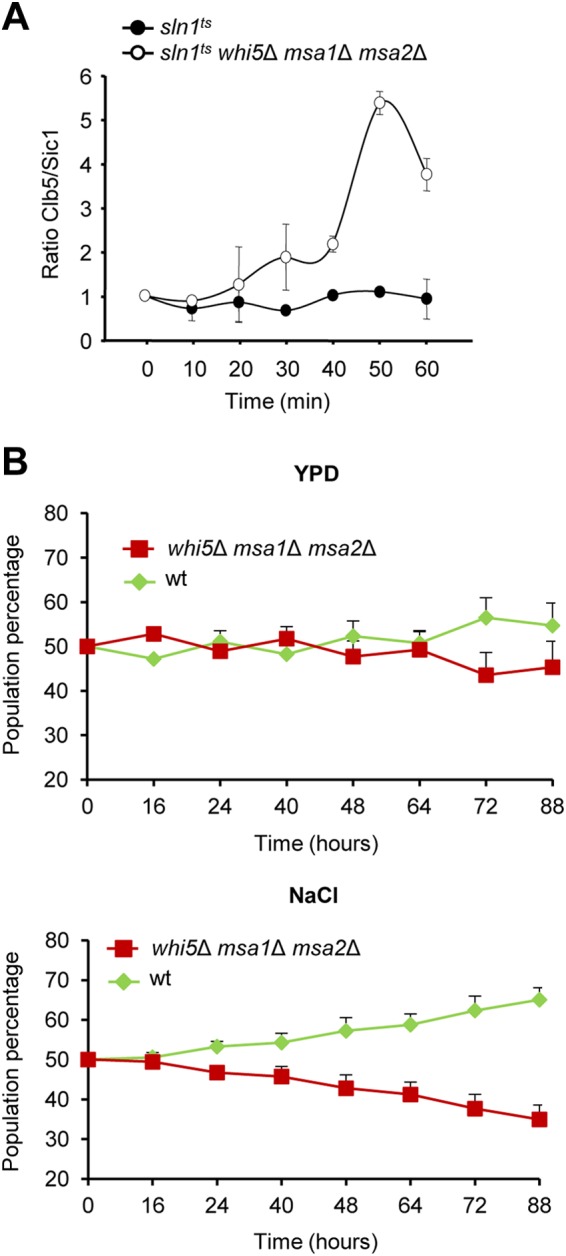
Transcriptional regulation of G1 cyclins is important for cell fitness upon osmostress. (A) Increased Clb5 levels can overcome Sic1-dependent G1 arrest in the absence of Whi5, Msa1, and Msa2. sln1ts and sln1ts whi5Δ msa1Δ msa2Δ mutant cells were synchronized in G1 and released from pheromone arrest into YPD medium at 37°C. Clb5 and Sic1 protein levels were followed over time by Western blotting with specific antibodies (Santa Cruz). Protein levels were normalized to an internal control for each lane. The graphs represent the average ± the standard deviations from three independent experiments. (B) Correct downregulation of G1 cyclins is important for efficient cell survival upon stress. ENO1 was fluorescently tagged in wild-type (wt) cells (with GFP) and in whi5 msa1 msa2 null cells (with mCherry). These cells were mixed in a 1:1 ratio, synchronized with α-factor, and released into YPD medium (top) or 0.4 M NaCl (bottom). At the times indicated, the percentage of each strain present in the culture was assessed by flow cytometry. Data represent the averages ± the standard deviations from three independent experiments.
Downregulation of cyclin expression is important for cell fitness in the presence of stress.
Triple deletion of WHI5, MSA1, and MSA2 has a clear effect on cell cycle reentry and a loss of coordination between budding and DNA replication. We therefore reasoned that these defects could hamper cell fitness upon stress. To test this possibility, we assessed the ability of these mutant cells to compete in the same environment with wild-type cells. To be able to distinguish between the two types of cells, we fluorescently labeled both strains, mixed equal numbers of G1-synchronized cells, and followed the percentage of each strain present over time in control medium or upon osmostress (Fig. 8B). While no difference in cell growth was noted in the absence of stress, a clear enrichment of wild-type over mutant cells was observed upon stress. Similar results were obtained when the fluorescent reporters where expressed in the opposite strain, ruling out any possible effect of the tagging (see Fig. S9 in the supplemental material). These combined data indicate that the downregulation of G1 cyclins through the activity of Whi5 and Msa1 is required for proper cellular fitness upon stress.
DISCUSSION
Yeast cells modify their transcription pattern to cope with changes in external osmolarity (34). Hog1 is a key player in the regulation of gene expression upon osmostress, causing a rapid and transient upregulation of osmoresponsive genes. However, Hog1 also mediates the transcriptional downregulation of specific genes in addition to those globally downregulated by stress (49). For instance, the G1 cyclins are strongly downregulated by Hog1 in response to osmostress and this downregulation is important for cell cycle control (36, 39, 41). Although these previous data showed the relevance of Hog1 cyclin regulation, the mechanism by which this downregulation is achieved is unknown. Here we show that Hog1 downregulates G1 cyclins through, in part, phosphorylation of two cell cycle transcriptional regulators, Whi5 and Msa1. The absence or mutation of both regulators results in less efficient downregulation of cyclins, loss of coordination of CLN2 and CLB5 gene expression, and reduced cellular fitness upon stress.
The activity of the Whi5 transcriptional repressor, which is the ortholog of Rb in mammals, has been shown to be regulated by phosphorylation, and CDK Cdc28 is the major kinase for this regulation of Whi5 (14, 15, 17). Whi5 phosphorylation affects its ability to bind SBF promoters and controls its nuclear localization. Other kinases can also phosphorylate Whi5, on the basis of the fact that non-CDK sites are phosphorylated in vivo (54). We mapped three sites in Whi5 that are phosphorylated by Hog1, and these sites differ from those previously reported to be functionally relevant sites phosphorylated by Cdc28 (54). Briefly, the Hog1 sites mapped in Whi5 are sites 6, 7, and 11 of the 12 SP/TP sites in Whi5. By MS of asynchronously growing cells, Wagner et al. found these sites, in addition to six non-CDK sites, to be phosphorylated (54). However, only CDK sites 8, 9, 10, and 12, which are not coincident with the Hog1 sites, were relevant for Whi5 inactivation and coordination between size and transcription initiation by SBF (54). Interestingly, two of the Hog1 sites in Whi5 mapped in this work (threonines 143 and 215) flank the GTB domain (G1/S transcription factor binding) of Whi5, which is required for binding of this repressor to SBF or Nrm1, which binds to MBF (55). One possible function of Hog1-mediated phosphorylation is that it could alter the binding dynamics or the affinity of Whi5 for the transcription factors, thus altering SBF-dependent transcription upon osmostress. The effect of Hog1 phosphorylation of Whi5 in G1 is rather limited, which suggested additional regulators for cyclin expression. A clear effect on cyclin expression and cell cycle progression was observed when, in addition to Whi5, Msa1 and Msa2 were also deleted. The Msa1 and Msa2 proteins were previously implicated in the cell cycle by acting as a coactivator of G1 transcriptional machinery (30). Here, we have shown a repressive role for these proteins at SBF and MBF promoters when phosphorylated by Hog1. Although the best-studied role of Hog1 in transcription is that of gene induction, here we show how Hog1 can mediate transcriptional repression of cell cycle phase-specific promoters.
Whi5 and Msa1 had different effects on transcription, depending on whether Hog1 was activated by osmostress or by genetic means. Although phosphorylation by Hog1 induced transcriptional downregulation in both cases, this downregulation was observed mainly for CLN2 upon osmostress, whereas upon genetic activation, the HOG pathway blocked transcription from both the CLN2 and CLB5 promoters. These effects were consistent with the suppression of the delay in budding but not in DNA replication upon stress that was observed in the mutant containing nonphosphorylatable (or null) alleles of WHI5, MSA1, and MSA2, whereas both budding and replication were delayed upon genetic activation. The initial response to osmostress is a major eviction of DNA-associated factors from DNA that is independent of Hog1 (50). Indeed, we found that osmostress induces eviction of Swi4 and Pol II from SBF and MBF and that it is only after 15 to 20 min of stress that transcription from these promoters can be induced. The eviction of Swi4 is Hog1 independent, but its reassociation seems to be more efficient in wild-type than in hog1 mutant cells, suggesting either that Hog1 is implicated in its reassociation or that reassociation depends on proper cellular adaptation. In clear contrast, activation of Hog1 by genetic means in the absence of stress does not result in transcription factor release, making this system more suitable to study the direct effects of Hog1 over Whi5 and Msa1 transcription factors. The differential effect seen in both scenarios might suggest a different composition of transcription factor complexes at cyclin promoters under the osmostress, and genetic activation of Hog1 could explain the observed differential effect on SBF and MBF activation. If this is the case, then the complexes associated with the CLN2 promoter appear to be more sensitive to the lack of repression that is due to Hog1-induced transcription factor phosphorylation than CLB5. Alternatively, the differential effect could be caused by a quantitative difference in Cln2 and Clb5 levels and its effect in a transient versus sustained cell cycle delay. Correspondingly, it is only when the Clb5 levels are higher than those of Sic1 upon Hog1 activation that cells can reenter S phase.
Cell cycle delay is important to allow cells time for adaptation before progressing into sensitive phases of the cell cycle (13, 37–39). Cells deficient in WHI5, MSA1, and MSA2 are not osmosensitive, probably because Sic1 can still induce a small delay in replication. However, when mutant cells were grown in competition with wild-type cells, the mutant cells clearly showed reduced fitness upon stress. Thus, this finding supports the notion that efficient transcriptional control over Start by Hog1 confers an important advantage for growth in hypertonic environments.
Control of transcription and cell cycle control are well-known Hog1-dependent adaptive responses to osmostress (45, 56). Although both effects have been studied independently, increasing evidence suggests that Hog1-dependent regulation of transcription and its modulation of cell cycle progression are processes that are intertwined with osmostress. First, osmostress activates a new Hog1-dependent checkpoint in S phase to prevent a collision between replication and transcription machineries, resulting in a delay in DNA replication and therefore in a transient delay in S-phase progression (38). Second, osmostress has been shown to activate a subset of long noncoding RNAs, one of which is an antisense RNA of CDC28 that can regulate Cdc28 levels, resulting in more efficient reactivation of the cell cycle after adaptation to the stress (57). Here, we show that downregulation of cyclin expression is required for the control of cell cycle progression upon stress. In summary, we provide data regarding the role of Hog1 as a transcriptional repressor of CLN2 and CLB5 transcription that is mediated by Hog1 phosphorylation of Whi5 and Msa1/2. This regulation is required for coordinated progression into S phase and to increase cellular fitness upon stress.
Supplementary Material
ACKNOWLEDGMENTS
We thank Laia Subirana and Aida Fernandez for technical support and Alba Duch, Matteo Viganò, and Martí Aldea (IBMB) for cell cycle discussions.
We have no competing financial interests to declare.
This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (BFU2012-33503 and FEDER to F.P. and BFU2011-26722 to E.D.N.) and 2014 SGR 599 (Generalitat de Catalunya). This project is supported by Fundación Botín and by Banco Santander through its Santander Universities Global Division to F.P. F.P. and E.D.N. are recipients of an ICREA Acadèmia award (Generalitat de Catalunya).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01279-14.
REFERENCES
- 1.Dirick L, Bohm T, Nasmyth K. 1995. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. EMBO J 14:4803–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dirick L, Nasmyth K. 1991. Positive feedback in the activation of G1 cyclins in yeast. Nature 351:754–757. doi: 10.1038/351754a0. [DOI] [PubMed] [Google Scholar]
- 3.Skotheim JM, Di TS, Siggia ED, Cross FR. 2008. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454:291–296. doi: 10.1038/nature07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrews BJ, Herskowitz I. 1989. The yeast SWI4 protein contains a motif present in developmental regulators and is part of a complex involved in cell-cycle-dependent transcription. Nature 342:830–833. doi: 10.1038/342830a0. [DOI] [PubMed] [Google Scholar]
- 5.Nasmyth K, Dirick L. 1991. The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66:995–1013. doi: 10.1016/0092-8674(91)90444-4. [DOI] [PubMed] [Google Scholar]
- 6.Ogas J, Andrews BJ, Herskowitz I. 1991. Transcriptional activation of CLN1, CLN2, and a putative new G1 cyclin (HCS26) by SWI4, a positive regulator of G1-specific transcription. Cell 66:1015–1026. doi: 10.1016/0092-8674(91)90445-5. [DOI] [PubMed] [Google Scholar]
- 7.Koch C, Moll T, Neuberg M, Ahorn H, Nasmyth K. 1993. A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science 261:1551–1557. doi: 10.1126/science.8372350. [DOI] [PubMed] [Google Scholar]
- 8.de Bruin RA, Kalashnikova TI, Chahwan C, McDonald WH, Wohlschlegel J, Yates J III, Russell P, Wittenberg C. 2006. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol Cell 23:483–496. doi: 10.1016/j.molcel.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Cross FR, Hoek M, McKinney JD, Tinkelenberg AH. 1994. Role of Swi4 in cell cycle regulation of CLN2 expression. Mol Cell Biol 14:4779–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eser U, Falleur-Fettig M, Johnson A, Skotheim JM. 2011. Commitment to a cellular transition precedes genome-wide transcriptional change. Mol Cell 43:515–527. doi: 10.1016/j.molcel.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuart D, Wittenberg C. 1994. Cell cycle-dependent transcription of CLN2 is conferred by multiple distinct cis-acting regulatory elements. Mol Cell Biol 14:4788–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lew DJ, Weinert T, Pringle JR. 1997. Cell cycle control in Saccharomyces cerevisiae. Cold Spring Harbor Monogr Arch 21:607–695. doi: 10.1101/087969364.21C.607. [DOI] [Google Scholar]
- 13.Schwob E, Nasmyth K. 1993. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev 7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 14.Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. 2004. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117:899–913. doi: 10.1016/j.cell.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 15.de Bruin RA, McDonald WH, Kalashnikova TI, Yates J III, Wittenberg C. 2004. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117:887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 16.Huang D, Kaluarachchi S, van DD, Friesen H, Sopko R, Ye W, Bastajian N, Moffat J, Sassi H, Costanzo M, Andrews BJ. 2009. Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol 7:e1000188. doi: 10.1371/journal.pbio.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosugi S, Hasebe M, Tomita M, Yanagawa H. 2009. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A 106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taberner FJ, Quilis I, Igual JC. 2009. Spatial regulation of the start repressor Whi5. Cell Cycle 8:3013–3022. doi: 10.4161/cc.8.18.9621. [DOI] [PubMed] [Google Scholar]
- 19.Mendenhall MD. 1993. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science 259:216–219. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- 20.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. 1994. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 21.Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. 1997. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91:221–230. doi: 10.1016/S0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 22.Sheaff RJ, Roberts JM. 1996. End of the line: proteolytic degradation of cyclin-dependent kinase inhibitors. Chem Biol 3:869–873. doi: 10.1016/S1074-5521(96)90174-X. [DOI] [PubMed] [Google Scholar]
- 23.Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. 1997. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka S, Umemori T, Hirai K, Muramatsu S, Kamimura Y, Araki H. 2007. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445:328–332. doi: 10.1038/nature05465. [DOI] [PubMed] [Google Scholar]
- 25.Zegerman P, Diffley JF. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445:281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Lau KY, Sevim V, Tang C. 2013. Design principles of the yeast G1/S switch. PLoS Biol 11:e1001673. doi: 10.1371/journal.pbio.1001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costanzo M, Schub O, Andrews B. 2003. G1 transcription factors are differentially regulated in Saccharomyces cerevisiae by the Swi6-binding protein Stb1. Mol Cell Biol 23:5064–5077. doi: 10.1128/MCB.23.14.5064-5077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho Y, Costanzo M, Moore L, Kobayashi R, Andrews BJ. 1999. Regulation of transcription at the Saccharomyces cerevisiae Start transition by Stb1, a Swi6-binding protein. Mol Cell Biol 19:5267–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JM, Tetzlaff MT, Elledge SJ. 2008. Identification of MSA1, a cell cycle-regulated, dosage suppressor of drc1/sld2 and dpb11 mutants. Cell Cycle 7:3388–3398. doi: 10.4161/cc.7.21.6932. [DOI] [PubMed] [Google Scholar]
- 30.Ashe M, de Bruin RA, Kalashnikova T, McDonald WH, Yates JR, Wittenberg C. 2008. The SBF-and MBF-associated protein Msa1 is required for proper timing of G1-specific transcription in Saccharomyces cerevisiae. J Biol Chem 283:6040–6049. doi: 10.1074/jbc.M708248200. [DOI] [PubMed] [Google Scholar]
- 31.van der Felden J, Weisser S, Brückner S, Lenz P, Mösch HU. 2014. The transcription factors Tec1 and Ste12 interact with coregulators Msa1 and Msa2 to activate adhesion and multicellular development. Mol Cell Biol 34:2283–2293. doi: 10.1128/MCB.01599-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hohmann S. 2009. Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett 583:4025–4029. doi: 10.1016/j.febslet.2009.10.069. [DOI] [PubMed] [Google Scholar]
- 33.Saito H, Posas F. 2012. Response to hyperosmotic stress. Genetics 192:289–318. doi: 10.1534/genetics.112.140863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Nadal E, Ammerer G, Posas F. 2011. Controlling gene expression in response to stress. Nat Rev Genet 12:833–845. doi: 10.1038/nrg3055. [DOI] [PubMed] [Google Scholar]
- 35.Alexander MR, Tyers M, Perret M, Craig BM, Fang KS, Gustin MC. 2001. Regulation of cell cycle progression by Swe1p and Hog1p following hypertonic stress. Mol Biol Cell 12:53–62. doi: 10.1091/mbc.12.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellí G, Gari E, Aldea M, Herrero E. 2001. Osmotic stress causes a G1 cell cycle delay and downregulation of Cln3/Cdc28 activity in Saccharomyces cerevisiae. Mol Microbiol 39:1022–1035. doi: 10.1046/j.1365-2958.2001.02297.x. [DOI] [PubMed] [Google Scholar]
- 37.Clotet J, Escoté X, Adrover MA, Yaakov G, Gari E, Aldea M, de Nadal E, Posas F. 2006. Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. EMBO J 25:2338–2346. doi: 10.1038/sj.emboj.7601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duch A, Felipe-Abrio I, Barroso S, Yaakov G, Garcia-Rubio M, Aguilera A, de Nadal E, Posas F. 2013. Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature 493:116–119. doi: 10.1038/nature11675. [DOI] [PubMed] [Google Scholar]
- 39.Escoté X, Zapater M, Clotet J, Posas F. 2004. Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat Cell Biol 6:997–1002. doi: 10.1038/ncb1174. [DOI] [PubMed] [Google Scholar]
- 40.Yaakov G, Duch A, Garcia-Rubio M, Clotet J, Jimenez J, Aguilera A, Posas F. 2009. The stress-activated protein kinase Hog1 mediates S phase delay in response to osmostress. Mol Biol Cell 20:3572–3582. doi: 10.1091/mbc.E09-02-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adrover MA, Zi Z, Duch A, Schaber J, Gonzalez-Novo A, Jimenez J, Nadal-Ribelles M, Clotet J, Klipp E, Posas F. 2011. Time-dependent quantitative multicomponent control of the G1-S network by the stress-activated protein kinase Hog1 upon osmostress. Sci Signal 4:ra63. doi: 10.1126/scisignal.2002204. [DOI] [PubMed] [Google Scholar]
- 42.Radmaneshfar E, Kaloriti D, Gustin MC, Gow NA, Brown AJ, Grebogi C, Romano MC, Thiel M. 2013. From START to FINISH: the influence of osmotic stress on the cell cycle. PLoS One 8:e68067. doi: 10.1371/journal.pone.0068067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 44.Zapater M, Sohrmann M, Peter M, Posas F, de Nadal E. 2007. Selective requirement for SAGA in Hog1-mediated gene expression depending on the severity of the external osmostress conditions. Mol Cell Biol 27:3900–3910. doi: 10.1128/MCB.00089-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Nadal E, Posas F. 2010. Multilayered control of gene expression by stress-activated protein kinases. EMBO J 29:4–13. doi: 10.1038/emboj.2009.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller C, Schwalb B, Maier K, Schulz D, Dumcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, Martin DE, Tresch A, Cramer P. 2011. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 7:458. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Romero-Santacreu L, Moreno J, Perez-Ortin JE, Alepuz P. 2009. Specific and global regulation of mRNA stability during osmotic stress in Saccharomyces cerevisiae. RNA 15:1110–1120. doi: 10.1261/rna.1435709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pelet S, Rudolf F, Nadal-Ribelles M, de Nadal E, Posas F, Peter M. 2011. Transient activation of the HOG MAPK pathway regulates bimodal gene expression. Science 332:732–735. doi: 10.1126/science.1198851. [DOI] [PubMed] [Google Scholar]
- 49.Nadal-Ribelles M, Conde N, Flores O, Gonzalez-Vallinas J, Eyras E, Orozco M, de Nadal E, Posas F. 2012. Hog1 bypasses stress-mediated down-regulation of transcription by RNA polymerase II redistribution and chromatin remodeling. Genome Biol 13:R106. doi: 10.1186/gb-2012-13-11-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Proft M, Struhl K. 2004. MAP kinase-mediated stress relief that precedes and regulates the timing of transcriptional induction. Cell 118:351–361. doi: 10.1016/j.cell.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Alepuz PM, de Nadal E, Zapater M, Ammerer G, Posas F. 2003. Osmostress-induced transcription by Hot1 depends on a Hog1-mediated recruitment of the RNA Pol II. EMBO J 22:2433–2442. doi: 10.1093/emboj/cdg243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA. 2006. Activated signal transduction kinases frequently occupy target genes. Science 313:533–536. doi: 10.1126/science.1127677. [DOI] [PubMed] [Google Scholar]
- 53.Vendrell A, Martinez-Pastor M, Gonzalez-Novo A, Pascual-Ahuir A, Sinclair DA, Proft M, Posas F. 2011. Sir2 histone deacetylase prevents programmed cell death caused by sustained activation of the Hog1 stress-activated protein kinase. EMBO Rep 12:1062–1068. doi: 10.1038/embor.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner MV, Smolka MB, De Bruin RA, Zhou H, Wittenberg C, Dowdy SF. 2009. Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS One 4:e4300. doi: 10.1371/journal.pone.0004300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Travesa A, Kalashnikova TI, de Bruin RA, Cass SR, Chahwan C, Lee DE, Lowndes NF, Wittenberg C. 2013. Repression of G1/S transcription is mediated via interaction of the GTB motifs of Nrm1 and Whi5 with Swi6. Mol Cell Biol 33:1476–1486. doi: 10.1128/MCB.01333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duch A, de Nadal E, Posas F. 2012. The p38 and Hog1 SAPKs control cell cycle progression in response to environmental stresses. FEBS Lett 586:2925–2931. doi: 10.1016/j.febslet.2012.07.034. [DOI] [PubMed] [Google Scholar]
- 57.Nadal-Ribelles M, Sole C, Xu Z, Steinmetz LM, de Nadal E, Posas F. 2014. Control of Cdc28 CDK1 by a stress-induced lncRNA. Mol Cell 53:549–561. doi: 10.1016/j.molcel.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.