

Abstract
Wnt signaling stimulates cell proliferation by promoting the G1/S transition of the cell cycle through β-catenin/TCF4-mediated gene transcription. However, Wnt signaling peaks in mitosis and contributes to the stabilization of proteins other than β-catenin, a pathway recently introduced as Wnt-dependent stabilization of proteins (Wnt/STOP). Here, we show that Wnt/STOP regulated by basal Wnt signaling during a normal cell cycle is required for proper spindle microtubule assembly and for faithful chromosome segregation during mitosis. Consequently, inhibition of basal Wnt signaling results in increased microtubule assembly rates, abnormal mitotic spindle formation and the induction of aneuploidy in human somatic cells.
Keywords: aneuploidy, chromosome segregation, mitosis, Wnt signaling
Introduction
Canonical Wnt signaling plays a crucial role in embryonic, somatic and stem cells and is required for normal development, organogenesis and tissue regeneration. Moreover, its deregulation has been implicated in human diseases, most notably in cancer, but also in neurodegenerative diseases 1, 2, 3. The key effector of Wnt signaling is β-catenin, which acts together with its co-factor TCF4 as a Wnt-activated transcription factor 1. In the absence of Wnt signaling, β-catenin is subject to ubiquitin–proteasome-mediated protein degradation, which requires its prior phosphorylation mediated by a cytoplasmic destruction complex comprising glycogen synthase kinase 3 (GSK3), casein kinase 1 (CK1), adenomatous polyposis coli (APC) and Axin-1 1. Upon Wnt ligand binding to frizzled receptors (Fzd) and their co-receptors LRP5 and LRP6, the intracellular domain of LRP6 is phosphorylated by GSK3 and CK1. This is followed by receptor internalization and the formation of so-called signalosomes containing Fzd, phospho-LRP6, disheveled (Dvl), GSK3 and Axin-1 4, 5, 6. Under this condition, GSK3 is inhibited directly by phospho-LRP6 and by sequestration into multivesicular endosomes 7, 8, 9, 10. This prevents β-catenin phosphorylation and, thus, its degradation by the cytoplasmic destruction complex. Accumulated β-catenin can then translocate into the nucleus where it binds TCF4 triggering gene expression 1. Among the various bona fide target genes of β-catenin is AXIN2, a component of the cytoplasmic destruction complex, and C-MYC 11, which mediates the expression of CYCLIN-D, a key driver of the G1/S transition of the cell cycle 12. Thus, Wnt signaling can stimulate cell proliferation at G1/S via triggering gene expression in a β-catenin-dependent manner. Interestingly, when cells enter mitosis, LRP6 is phosphorylated by a mitosis-specific cyclin-dependent kinase (CDK14-cyclin Y), indicating that endogenous Wnt signaling is under cell cycle control peaking at G2/M 13, 14. In line with this, protein levels of β-catenin and Axin-2 also reach their maximum levels at G2/M 15, 16. However, a physiological role for this basal and cell cycle-regulated Wnt signaling has not been revealed so far.
Intriguingly, most recently it was found that Wnt signaling can contribute to the stabilization of proteins other than β-catenin 9, 17. In particular, this occurs at G2/M and is now referred to as Wnt-dependent stabilization of proteins (Wnt/STOP) 18. However, this novel role of Wnt signaling is yet poorly understood and a specific role for the entry into or for the progression of mitosis has not been identified so far.
In addition to that, several Wnt signaling proteins such as APC, Axin-2, Dvl and β-catenin have been implicated as direct regulators of mitosis 13, 19. For instance, APC together with Dvl localizes at the microtubule–kinetochore interface where they might contribute to proper microtubule binding to kinetochores 20, 21, 22. This function seems to be independent of Wnt signaling. However, APC and Dvl2 also associate with the mitotic cell cortex where they might help to anchor astral microtubules to the cortex in order to ensure proper orientation of the mitotic spindle. This function also involves the Wnt receptor Fzd and its co-receptor LRP6 21. Furthermore, β-catenin and Axin-2 are present at mitotic centrosomes where they might be involved in centrosome function, microtubule nucleation and mitotic spindle assembly 23, 24, 25. Thus, Wnt signaling as well as particular Wnt signaling components appear to be involved in the regulation of mitosis, but the nature of their action remains largely elusive.
It is conceivable that the proper progression of mitosis is essential for faithful chromosome segregation and the generation of euploid progenitors in normal somatic cells. On the other hand, aneuploidy as a consequence of mitotic chromosome missegregation is often associated with human diseases including cancer and neurodegenerative diseases 26. In particular, much effort has been undertaken to understand how chromosomes are missegregated in cancer cells, but the underlying mechanisms are still poorly understood 27. Recently, we identified a key mechanism leading to perpetual chromosome missegregation and aneuploidy in human cancer cells 28. In fact, we found that increased microtubule plus end assembly rates in mitosis are directly responsible for the generation of so-called lagging chromosomes during anaphase, which represent a common pre-stage of chromosome missegregation in somatic cells 28, 29. Thus, cells must ensure proper microtubule assembly rates during mitosis in order to maintain a stable karyotype. However, the molecular pathways that ensure proper microtubule plus end assembly during a normal mitosis are ill defined.
In our work presented here, we reveal a requirement for Wnt signaling during mitosis that is independent of canonical Wnt signaling for proper mitotic microtubule plus end assembly and for faithful chromosome segregation in human somatic cells.
Results and Discussion
Inhibition of basal Wnt signaling causes increased mitotic microtubule plus end assembly rates during mitosis
Our previous work established proper microtubule plus end assembly rates during mitosis as an essential determinant for proper mitotic progression and faithful chromosome segregation 28. Therefore, we investigated a potential involvement of non-induced (= basal or baseline) Wnt signaling in this process. We transfected HCT116 and non-transformed human retinal pigment epithelial (hTert-RPE1) cells with siRNAs targeting different Wnt signaling components (Supplementary Fig S1A and B), which did not affect cell proliferation or cell cycle progression (Supplementary Fig S1C). Subsequently, we determined microtubule plus end assembly rates by tracking EB3-GFP fusion proteins 30 in living cells (Supplementary Fig S1D). Interestingly, we found that partial repression of LRP5, LRP6 or DVL2, which led to inhibition of baseline Wnt signaling in the absence of additional Wnt treatment (Supplementary Fig S2A), triggered an increase in microtubule plus end assembly rates in mitotic, but not in interphase cells (Fig1A and B, Supplementary Fig S2B). In contrast, repression of AXIN1 or APC, which activated Wnt signaling (Supplementary Figs S1A and B and S2A), did not alter microtubule assembly rates (Fig1A and B). Interestingly, loss of β-catenin or TCF4 (Supplementary Fig S1A and E), which also inhibits Wnt activity (Supplementary Fig S2A), hardly affected mitotic microtubule assembly rates (Fig1A and B, Supplementary Fig S1F) indicating a β-catenin/TCF4-independent function of Wnt signaling for proper microtubule dynamics in mitosis. It is of note that our former work showed that increased microtubule plus end assembly rates are not associated with alterations in other microtubule dynamics parameters per se 28.
Figure 1.

- Measurements of microtubule plus end assembly rates in living HCT116 cells during mitosis or interphase after siRNA-mediated knockdown of different Wnt pathway components as indicated.
- Microtubule plus end assembly rates in hTERT-RPE-1 cells during mitosis after the indicated siRNA transfections.
- Mitotic microtubule plus end assembly rates in HCT116 cells with low expression of LRP5/6 or DVL2 and after knockdown of CH-TOG/CKAP5.
- Mitotic microtubule plus end assembly rates in HCT116 cells after knockdown of LRP5/6 or DVL2 and in the absence or presence of 0.2 nM Taxol®.
- Mitotic microtubule plus end assembly rates in HCT116 cells in response to treatment with 400 ng/ml sFRP2 or sFRP4.
- Mitotic microtubule plus end assembly rates in HCT116 cells in response to treatment with DKK1.
- Mitotic microtubule plus end assembly rates in HCT116 cells treated with 400 ng/ml of recombinant human Wnt3a for 24 h.
Increased microtubule assembly rates can be efficiently suppressed by partial repression of CH-TOG, a microtubule polymerase involved in catalyzing the incorporation of α/β-tubulin subunits into the growing microtubule plus tip 31. The same effect can be achieved by treatment of cells with sub-nanomolar concentrations of Taxol®, which stabilizes microtubule plus tips 28. Both experimental approaches fully restored proper microtubule plus end assembly rates in cells with reduced expression of LRP5/6 or DVL2 (Fig1C and D, Supplementary Fig S1G).
As an alternative approach to inhibit basal Wnt signaling, we treated cells with purified sFRP and DKK1 proteins 32 (Supplementary Fig S2C and D) and measured microtubule plus end assembly rates. In line with our first results, we found a significant increase in microtubule assembly rates upon sFRP- and DKK-mediated Wnt inhibition (Fig1E and F). However, similar to repression of APC or AXIN1, hyper-activation of Wnt signaling by treatment with additional Wnt3a (Supplementary Fig S2E) did not affect microtubule assembly rates (Fig1G). We conclude that basal Wnt signaling is required to ensure proper microtubule plus end assembly rates in mitotic cells. Thus, the regulation of microtubule dynamics during mitosis might represent an important function of basal and cell cycle-regulated Wnt signaling. Whether this novel mitotic function also involves the cell cycle-regulated and CDK14-cyclin Y-mediated mitotic phosphorylation of LRP6 14 remains to be shown. Similarly, the role of multivesicular endosomes during mitosis that contribute to the inactivation of GSK3 in response to Wnt signaling merits future investigations 9, 10.
Inhibition of Wnt signaling causes mitotic spindle defects and the generation of lagging chromosomes
Next, we investigated the consequences of increased microtubule assembly rates in response to the loss of Wnt signaling during an unperturbed cell cycle. To this end, we generated cell lines stably expressing shRNAs targeting LRP6 or DVL2. Single cell clones were isolated (Supplementary Fig S3A and B), and live cell microscopy experiments verified an increase in mitotic microtubule plus end assembly rates in different cell clones (Fig2A). Analyses of cells synchronized in metaphase (Fig2B, outline) demonstrated an induction of abnormal metaphase spindles associated with incomplete chromosome alignment upon reduced expression of LRP6 or DVL2 (Fig2B). Moreover, repression of LRP5, LRP6 or DVL2, but not of CTNNB1 or AXIN1, caused the generation of lagging chromosomes during anaphase (Fig2C), which typically arise due to erroneous microtubule–kinetochore attachments 33 and represent a common pre-stage of chromosome missegregation in somatic cells 34. Importantly, lagging chromosomes were greatly suppressed upon treatment with low doses of Taxol® (Fig2C), which restores proper microtubule assembly rates (Fig1D). Thus, these findings establish a causal relationship between the inhibition of basal Wnt signaling, an increase in mitotic microtubule assembly and the induction of chromosome missegregation. In contrast, we found that loss of APC caused lagging chromosomes, which were not suppressed by Taxol® treatment (Fig2C). This clearly supports the notion that APC fulfills a role in chromosome segregation independent of microtubule plus end dynamics, most likely in mediating microtubule–kinetochore attachments as suggested earlier 22, 35. It is also of note that both APC and Dvl2 interact and have been implicated not only in mediating microtubule–kinetochore attachments, but also in regulating spindle orientation. Interestingly, in contrast to the microtubule–kinetochore attachments, their functions in spindle orientation appear to be dependent on Wnt signaling 21. Since increased microtubule assembly rates can also cause spindle mis-orientation 28, it is possible that the role of Dvl2 in spindle orientation involves the regulation of microtubule plus end dynamics. On the other hand, our results showing that chromosome missegregation upon loss of DVL2 can be suppressed by low dose Taxol® treatment indicate that an additional role of Dvl2 in microtubule–kinetochore attachments is not responsible for the induction of aneuploidy. From these points, it is clear that further detailed investigations are needed in order to fully understand the various different functions of Wnt signaling components during mitosis.
Figure 2.
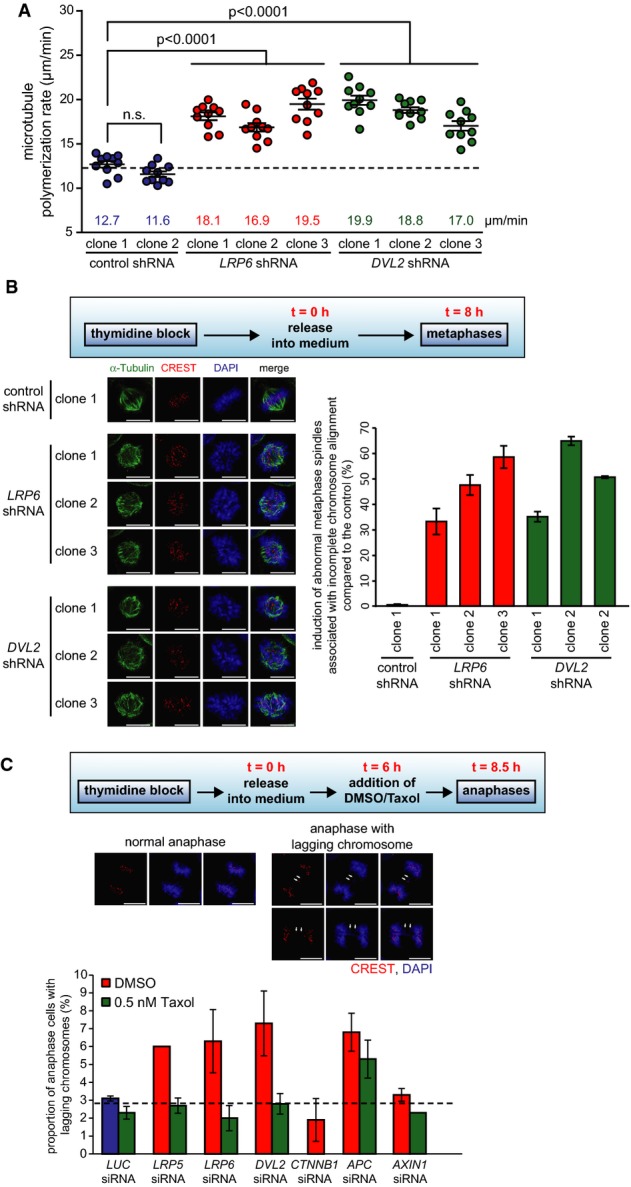
- Mitotic microtubule plus end assembly rates in different single cell clones derived from HCT116 cells stably expressing shRNAs targeting LRP6 or DVL2. Scatter dot plots show average assembly rates (20 microtubules/cell, mean ± SEM, t-test, n = 20 cells).
- Quantification of cells showing abnormal bipolar metaphase spindles. Cells were synchronized in metaphase as indicated. The graph shows the induction of abnormal spindles associated with incomplete chromosome alignment normalized to control cells (mean ± SD, n = 800–1,000 mitotic cells). Representative examples are shown (scale bar, 10 μm).
- Detection of lagging chromosomes in HCT116 cells transfected with siRNAs and synchronized in anaphase. Cells were treated with or without 0.5 nM Taxol®, and the proportion of cells exhibiting lagging chromosomes were quantified (mean ± SD, n = 371–711 anaphase cells). Representative examples are shown (scale bar, 10 μm).
Inhibition of Wnt signaling promotes aneuploidy in human somatic cells that is caused by increased microtubule plus end assembly rates
Next, we asked whether the mitotic abnormalities triggered by inhibition of basal Wnt signaling affect karyotype stability. Chromosome counting from metaphase spreads and interphase FISH analyses were used to determine karyotype variability and the induction of aneuploidy in cell clones derived from HCT116 cells that maintain a near diploid karyotype 36, 37. Clearly, single cell clones stably expressing shRNAs targeting LRP6 or DVL2 and grown for 30 generations exhibited a more than twofold higher karyotype variability and an induction of aneuploidy than clones expressing control shRNAs while maintaining the same modal number of chromosomes (Fig3A, Supplementary Fig S4A and B). To investigate whether the development of aneuploidy is indeed mediated by increased microtubule plus end assembly rates, we generated single cell clones in the absence or presence of low doses of Taxol®. In line with our experiments described above (Fig1D), those single cell clones showed a full restoration of proper microtubule assembly rates upon treatment with Taxol® (Fig3B). Importantly, the high karyotype variability and the generation of aneuploidy induced by repression of LRP6 or DVL2 were significantly suppressed by Taxol® treatment (Fig3C and Supplementary Fig S4C). These results demonstrate that aneuploidy induction is indeed due to increased microtubule assembly triggered by inhibition of Wnt signaling.
Figure 3.
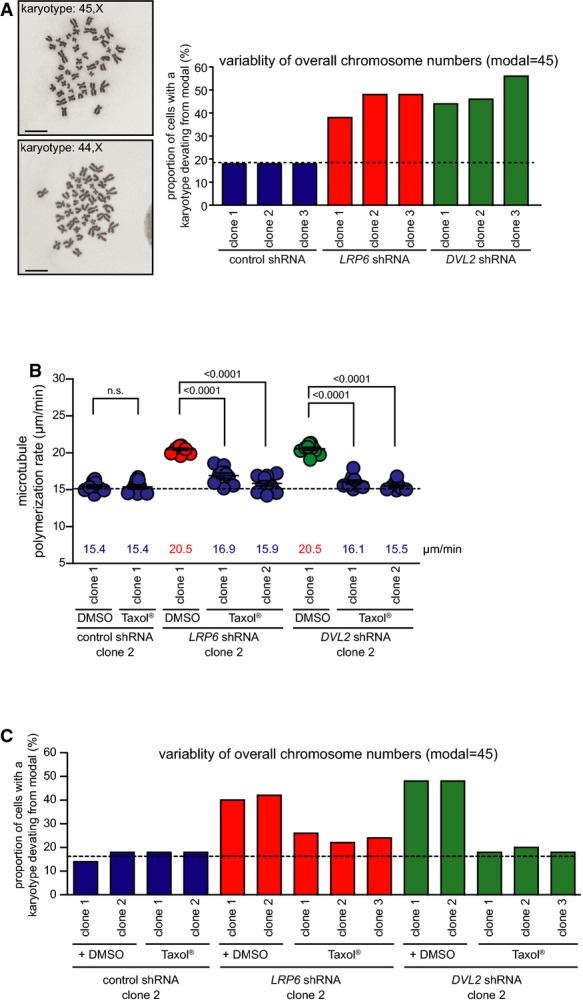
- Chromosome number variability/aneuploidy of different single cell clones derived from HCT116 cells stably expressing control or shRNAs targeting LRP6 and DVL2 and grown for 30 generations. The graph shows the proportion of cells harboring a karyotype with chromosome numbers deviating from the modal (n = 50 cells; modal number = 45). Representative examples of metaphase chromosome spreads showing a normal (45,X) and an aneuploid (44,X) karyotype are shown.
- Mitotic microtubule plus end assembly rates in different single cell clones derived from HCT116 cells stably expressing shRNAs targeting LRP6 or DVL2 and grown for 30 generations in the absence (DMSO) or presence of low doses of Taxol®. Scatter dot plots show average assembly rates (20 microtubules/cell, mean ± SEM, t-test, n = 10 cells).
- Determination of the chromosome number variability/aneuploidy in different single cell clones grown in the absence or presence of Taxol® for 30 generations. The graph shows the proportion of cells harboring a karyotype with chromosome numbers deviating from the modal (n = 50 cells; modal number = 45).
Wnt-mediated protein stabilization is required for proper microtubule assembly during mitosis
Since our data indicate a role for β-catenin/TCF4-independent Wnt signaling at G2/M in the regulation of mitotic microtubule dynamics, we reasoned whether the recently identified Wnt/STOP pathway 18 might be involved in this particular regulation in mitosis. If so, one would expect that increased microtubule assembly rates induced by repression of LRP6 or DVL2 are restored to normal levels by concomitant inhibition of proteasome-mediated protein degradation or upon inhibition of the cytoplasmic Wnt destruction complex. Indeed, proteasome inhibition mediated by an 1-h MG132 treatment on mitotically synchronized cells fully restored normal microtubule assembly rates in HCT116 or RPE-1 cells with reduced levels of LRP6 or DVL2 (Fig4A and B). Moreover, the concomitant repression of LRP6 or DVL2 together with APC or AXIN1 was sufficient to restore proper microtubule assembly rates in mitosis, both, in HCT116 and in RPE-1 cells (Fig4C and D, Supplementary Fig S5A and B). To exclude the possibility that activation of β-catenin/TCF4 activity is involved in the suppression effect, we simultaneously repressed CTNNB1 in APC siRNA-treated cells, but found no reversal effect on the phenotype (Fig4C, Supplementary Fig S5A). Finally, to provide a link between Wnt-mediated protein stabilization and chromosome missegregation, we also analyzed the generation of lagging chromosomes during anaphase. In fact, lagging chromosomes in LRP6- or DVL2-depleted cells were greatly suppressed when AXIN1 was also repressed (Fig4E, Supplementary Fig S5C). Together, these results might point into the direction that Wnt-mediated stabilization of proteins other than β-catenin is required for the maintenance of proper microtubule assembly and for faithful chromosome segregation during a normal mitosis (see model in Supplementary Fig S6). Thus, our work might provide the first physiological role of Wnt/STOP during an unperturbed cell cycle. However, given the fact that apparently many different proteins are subject to Wnt/STOP 9, 17, 18, one might hypothesize that additional key regulatory steps during mitosis, other than microtubule plus end dynamics, might be controlled by the Wnt/STOP pathway. Therefore, it will be of paramount interest to systematically identify the Wnt/STOP target proteins. This will provide important new insights for a possible role of Wnt/STOP as a regulatory pathway for a normal cell cycle and, in particular, for the regulation of a normal mitosis and chromosome segregation.
Figure 4.

- Mitotic microtubule plus end assembly rates in HCT116 cells with low expression levels of LRP6 or DVL2 in the presence or absence of the proteasome inhibitor MG132 (n = 18–20).
- Mitotic microtubule plus end assembly rates in hTERT-RPE-1 cells with repressed LRP6 or DVL2 in the presence or absence of the proteasome inhibitor MG132 (n = 20 cells).
- Mitotic microtubule plus end assembly rates in HCT116 cells with repressed LRP6 or DVL2 and concomitant repression of AXIN1, APC and/or CTNNB1 (n = 18–29 cells).
- Mitotic microtubule plus end assembly rates in hTERT-RPE-1 cells with low expression of LRP6 or DVL2 and concomitant repression of AXIN1 (n = 20 cells).
- Detection of lagging chromosomes in HCT116 cells with low expression of LRP6 or DVL2 and concomitant repression of AXIN1. The proportion of cells exhibiting lagging chromosomes during anaphase were quantified (mean ± SD, n = 600 anaphase cells).
Importantly, abnormal mitosis and the generation of aneuploidy are typical features of various human diseases, most notably cancer and neurodegenerative diseases 26. Interestingly, those diseases have also been associated with de-regulated Wnt signaling. In human cancer, Wnt signaling is frequently over-activated and loss of APC is among the most frequent alterations in colorectal cancer 1. In fact, hyper-active Wnt signaling and loss of APC might contribute to aneuploidy by interfering with spindle checkpoint function and by impairing proper microtubule attachments to kinetochores 14, 20, 21, 22. In our work, we also observed the generation of lagging chromosomes indicating chromosome missegregation upon loss of APC, but this was not dependent on an impairment of mitotic microtubule dynamics.
On the other hand, downregulation of LRP6 or induction of DKK1, both of which are associated with inhibition of Wnt signaling, has been implicated in pathogenesis of Alzheimer′s disease (AD) where aneuploidy is highly prevalent 2, 26, 38. It is intriguing that under these conditions, GSK3, which is under negative control of Wnt/STOP signaling, plays a central role for the hyper-phosphorylation of the microtubule-associated protein tau and for amyloid plaque formation 39. Thus, it is tempting to speculate that hyper-phosphorylation of tau and production of Aβ peptide might be a consequence of loss of Wnt/STOP and could contribute to the induction of aneuploidy in AD brains.
Materials and Methods
Tissue culture
HCT116 cells were cultured at 37°C with 5% CO2 in RPMI1640 containing 10% fetal calf serum, 1% glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin (Invitrogen, the Netherlands). hTert-RPE-1 cells were cultured in DMEM/Nutrient F-10 Ham (1:1 mixture, PAA, Germany).
Transfections
DNA and siRNA transfections were carried out essentially as described 28. Selection of single cell clones was carried out in medium containing 1 μg/ml puromycin. Details are provided in the Supplementary Methods.
Cell treatments
Where indicated, cells were treated with 400 ng/ml recombinant human Wnt3a (R&D Systems, USA) or with up to 600 ng/ml of recombinant human DKK1 or 400 ng/ml secreted Frizzled-related proteins 2 and 4 (sFRP2/sFRP4) (R&D Systems) for 24 h. To stabilize proteins during mitosis, cells were first synchronized in mitosis and then treated with 15 μM of the proteasome inhibitor MG132 (Enzo Life Sciences, Germany) for 1 h.
Western blotting
Western blotting was performed as described 28. Details are provided in the Supplementary Methods.
Microscopy
Microscopy on fixed samples and determination of microtubule plus end assembly rates using tracking EB3-GFP protein in live cell microscopy experiments were carried out as described 28. Details are provided in the Supplementary Methods.
Karyotype analyses
Karyotype analyses on single cell clones using chromosome counting and FISH analyses were performed as described 28. Details are provided in the Supplementary Methods.
Statistical analyses
All data are shown as mean ± standard error of the mean (SEM or SD). Where indicated, unpaired Student's t-tests using the Prism software package, version 4, were applied. Assessment of data was performed blinded by different investigators in independent experiments.
Acknowledgments
We thank Michael Boutros, Iris Augustin, Linda Wordeman, Bert Vogelstein, Matthias Dobbelstein, Tobias Pukrop, Uwe Wolfrum, Marian Grade and Gerald Wulf for cell lines, reagents and advice. We are grateful to Heike Krebber for microscopy support. We also thank Kristina Gamper for help with the generation of stable cell lines and Dennis Vollweiter and Eric Schoger for general lab support. This work was supported by the Deutsche Forschungsgemeinschaft (FOR942) and by a Heisenberg professorship awarded by the DFG to H.B.
Author contributions
AS, KN and NE planned and performed the experiments and analyzed the data. HB coordinated the project, assisted in planning the experiments and analyzed the data. AS, NE and HB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Information
Review Process File
References
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron. 2014;84:63–77. doi: 10.1016/j.neuron.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa CN, Varela-Nallar L, Grabowski PC, Colombres M. Synaptotoxicity in Alzheimer's disease: the Wnt signaling pathway as a molecular target. IUBMB Life. 2007;59:316–321. doi: 10.1080/15216540701242490. [DOI] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi K, Dolan JP, Johnson VG. The low density lipoprotein receptor-related protein 6 interacts with glycogen synthase kinase 3 and attenuates activity. J Biol Chem. 2006;281:4787–4794. doi: 10.1074/jbc.M508657200. [DOI] [PubMed] [Google Scholar]
- Piao S, Lee SH, Kim H, Yum S, Stamos JL, Xu Y, Lee SJ, Lee J, Oh S, Han JK, et al. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS ONE. 2008;3:e4046. doi: 10.1371/journal.pone.0004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinyoles M, Del Valle-Perez B, Curto J, Vinas-Castells R, Alba-Castellon L, de Garcia Herreros A, Dunach M. Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120-catenin/cadherin interaction with LRP5/6. Mol Cell. 2014;53:444–457. doi: 10.1016/j.molcel.2013.12.010. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Niehrs C, Acebron PS. Mitotic and mitogenic Wnt signalling. EMBO J. 2012;31:2705–2713. doi: 10.1038/emboj.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C. Cell cycle control of wnt receptor activation. Dev Cell. 2009;17:788–799. doi: 10.1016/j.devcel.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Hadjihannas MV, Bruckner M, Jerchow B, Birchmeier W, Dietmaier W, Behrens J. Aberrant Wnt/beta-catenin signaling can induce chromosomal instability in colon cancer. Proc Natl Acad Sci U S A. 2006;103:10747–10752. doi: 10.1073/pnas.0604206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmeda D, Castel S, Vilaro S, Cano A. Beta-catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell. 2003;14:2844–2860. doi: 10.1091/mbc.E03-01-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GN, Xu C, Gumbiner MB. Identification of targets of the Wnt pathway destruction complex in addition to beta-catenin. Proc Natl Acad Sci U S A. 2009;106:5165–5170. doi: 10.1073/pnas.0810185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebron SP, Karaulanov E, Berger BS, Huang YL, Niehrs C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell. 2014;54:663–674. doi: 10.1016/j.molcel.2014.04.014. [DOI] [PubMed] [Google Scholar]
- Hadjihannas VM, Behrens J. CIN By WNT: growth pathways, mitotic control and chromosomal instability in cancer. Cell Cycle. 2006;5:2077–2081. doi: 10.4161/cc.5.18.3282. [DOI] [PubMed] [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Niikura Y, Kitagawa K, Kikuchi A. Dishevelled, a Wnt signalling component, is involved in mitotic progression in cooperation with Plk1. EMBO J. 2010;29:3470–3483. doi: 10.1038/emboj.2010.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- Mbom CB, Nelson JW, Barth A. beta-catenin at the centrosome: discrete pools of beta-catenin communicate during mitosis and may co-ordinate centrosome functions and cell cycle progression. BioEssays. 2013;35:804–809. doi: 10.1002/bies.201300045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DD, Meigs ET, Kelly P, Casey JP. Identification of a role for beta-catenin in the establishment of a bipolar mitotic spindle. J Biol Chem. 2004;279:10829–10832. doi: 10.1074/jbc.C400035200. [DOI] [PubMed] [Google Scholar]
- Hadjihannas VM, Bruckner M, Behrens J. Conductin/axin2 and Wnt signalling regulates centrosome cohesion. EMBO Rep. 2010;11:317–324. doi: 10.1038/embor.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia BA, Amon A. Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech. 2014;7:15–20. doi: 10.1242/dmm.013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LS, Bakhoum FS, Compton AD. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuß S, Burfeind P, Aigner A, Wordeman L, Bastians H. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer. Nat Cell Biol. 2014;16:779–791. doi: 10.1038/ncb2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta. 2008;1786:32–40. doi: 10.1016/j.bbcan.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, Stear JH, Noetzel TL, Al-Bassam J, Kinoshita K, Harrison SC, Howard J, Hyman AA. XMAP215 is a processive microtubule polymerase. Cell. 2008;132:79–88. doi: 10.1016/j.cell.2007.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- Silkworth TW, Cimini D. Transient defects of mitotic spindle geometry and chromosome segregation errors. Cell Div. 2012;7:19. doi: 10.1186/1747-1028-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan J, Polakova S, Zhang L, Tolic-Norrelykke IM, Cimini D. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011;21:374–381. doi: 10.1016/j.tcb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum FS, Genovese G, Compton AD. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol. 2009;19:1937–1942. doi: 10.1016/j.cub.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler WK, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Knutsen T, Padilla-Nash HM, Wangsa D, Barenboim-Stapleton L, Camps J, McNeil N, Difilippantonio MJ, Ried T. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosom Cancer. 2010;49:204–223. doi: 10.1002/gcc.20730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martin M, Jurado J, Hernandez F, Avila J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Information
Review Process File