

Abstract
The crystalline cell-surface (S) layer sgsE of Geobacillus stearothermophilus NRS 2004/3a represents a natural protein self-assembly system with nanometer-scale periodicity that is evaluated as a combined carrier/patterning element for the conception of novel types of biocatalyst aiming at the controllable display of biocatalytic epitopes, storage stability, and reuse. The glucose-1-phosphate thymidylyltransferase RmlA is used as a model enzyme and chimeric proteins are constructed by translational fusion of rmlA to the C-terminus of truncated forms of sgsE (rSgsE 131–903, rSgsE331–903) and used for the construction of three principal types of biocatalysts: soluble (monomeric), self-assembled in aqueous solution, and recrystallized on negatively charged liposomes. Enzyme activity of the biocatalysts reaches up to 100% compared to sole RmlA cloned from the same bacterium. The S-layer portion of the biocatalysts confers significantly improved shelf life to the fused enzyme without loss of activity over more than three months, and also enables biocatalyst recycling. These nanopatterned composites may open up new functional concepts for biocatalytic applications in nanobiotechnology.
Keywords: composites, enzyme catalysis, protein engineering, self-assembly, surface layers
1. Introduction
Since the early days of enzyme immobilization the primary goal of using insoluble carriers of ready-made geometric parameters and defined physical and chemical nature has been to insolubilize the enzyme and, thus, to facilitate its separation and reuse with the aim of reducing the production cost by efficient recycling and control over the process[1,2]. There are several other benefits of using immobilized enzymes rather than their soluble counterparts; they may be used as i) stable and reusable devices for analytical and medical applications, ii) selective adsorbents for purification of proteins and enzymes, iii) fundamental tools for solid-phase protein chemistry, and iv) effective microdevices for controlled release of protein drugs.[3] Enzyme reuse in particular has several cost advantages, which are often an essential prerequisite for establishing an economically viable enzyme-catalyzed process.
Immobilized enzymes were first prepared by inclusion in polymeric matrices or binding onto carrier materials.[4] Considerable effort was also put into crosslinking of enzymes by either protein crosslinking or the addition of inert materials.[5] Since then, numerous methods for immobilization on different materials have been developed, including binding to prefabricated carriers, crosslinking of enzyme crystals,[6,7] utilization of cyclodextrin derivatives for enzyme binding to metallic surfaces,[8] exploitation of two-dimensional (2D), crystalline bacterial cell-surface layer (S-layer) proteins as immobilization matrix,[9] or immobilization of enzymes in an inorganic, transparent matrix by the sol–gel process, which is a recently developed and promising approach.[10] Alternatively to conventional (bio)chemical immobilization methods, translational fusion of enzymes to different carrier molecules such as a second enzyme[11] or a particulate form of an S-layer protein[12] has been reported as an interesting novel approach for exploitation in nanobiotechnology. Regardless of its nature or preparation, an immobilized enzyme, by definition, has to perform two essential functions, namely, i) the catalytic function designed to convert the substrate into the desired product with reasonable yield and within reasonable time and space, and ii) the noncatalytic functions designed to aid separation and process control. While the catalytic parameters such as activity, selectivity, and stability are linked to the catalytic function, the non-catalytic parameters of a composite enzyme are related to the physical and chemical nature of the carrier, especially the geometric parameters such as shape and size.[13]
Due to the diversity of nanobiotechnological processes there is no general universally applicable method for enzyme immobilization. Instead, each process requires the design of specific immobilized enzymes that can match the corresponding requirements for the desired process. While it is accepted that immobilization of an enzyme on a carrier can, in some cases, lead to dilution of enzyme activity, the development of novel carriers still constitutes a focus of recent attention.
In this study, the crystalline S-layer protein SgsE from Geobacillus stearothermophilus NRS 2004/3a (GenBank accession number AF328862) is exploited as a carrier for enzyme immobilization in nanobiotechnology. One of the general requirements of nanobiotechnology is the availability of molecular systems designed to have desired properties. SgsE represents a natural protein self-assembly system, which can be used in bottom-up processes as a patterning element for a nanobiotechnological construction kit.[14,15,16] SgsE monomers are naturally aligned by an entropy-driven process into a 2D crystalline array with oblique symmetry exhibiting nanometer-scale periodicity (lattice parameters, a=11.6 nm, b=9.4 nm, γ ≈78°[17,18]). In the SgsE nanolattice one morphological unit corresponds to an SgsE dimer and between the constituent monomers pores of identical shape are present. In general, the unique property of isolated S-layer protein subunits (monomers) to reassemble into 2D crystals identical to those found on intact bacterial cells, in suspension, on diverse solid supports or on liposomes and various interfaces, opens a wide spectrum of S-layer-based applications.[19,20,21,22] In previous studies, it has already been demonstrated that several functional domains introduced into S-layer proteins at distinct positions do not interfere with the intrinsic self-assembly property of the S-layer system.[23,24] As the detailed investigation between structure and function of S-layers is becoming better understood[25,26,27] it is feasible to use a genetic-engineering approach to build novel nanostructured architectures, which combine the benefits of the S-layer protein with a catalytic function. We report on a bottom-up approach to build a multidomain protein from a selected, self-assembling portion of the S-layer protein SgsE from G. stearothermophilus NRS 2004/3a and the glucose-1-phosphate thymidylyltransferase RmlA, originating from the same bacterium.
The RmlA enzyme (EC 2.7.7.24) catalyzes the transfer of a thymidylmonophosphate nucleotide to glucose-1-phosphate, constituting the first step in the biosynthetic cascade of activated sugar metabolites, such as dTDP-β-l-rhamnose[28] or dTDP-α-d-fucose.[29] Considerable interest in this enzyme originates primarily from its involvement in the bio-synthesis of l-rhamnose, which has a confirmed role in pathogenicity of many bacteria.[28] Beyond that, enzymatic oligosaccharide synthesis using recombinant glycosylation enzymes, as utilized in glycobiotechnology, is frequently able to overcome the difficulties associated with chemical methods.[30] Enzyme recycling is again essential. The naturally cytosolic, 33.2 kDa enzyme RmlA was also chosen as a first model because it is under investigation in the context of unraveling the biosynthesis of S-layer glycoprotein glycans in our laboratory.[31] Thus, basic protocols for recombinant enzyme production, enzyme purification, and an enzyme activity assay are readily available. Above that, the 3D structure and the basis of the catalytic mechanism of this enzyme are known.[32,33]
For proof of concept of novel nanostructured biocatalysts based on the self-assembling S-layer protein SgsE and the enzyme RmlA, we specifically deal with i) the characterization of the noncatalytic S-layer matrix, including preparation of truncated protein forms and selection of an appropriate translational fusion site for the biocatalytic function, and ii) the design and activity determination of different types of novel biocatalysts, including soluble (monomeric) and self-assembled biocatalysts as well as liposomes coated with a self-assembled, biocatalytic protein layer.
2. Results and Discussion
2.1. Determination of the Optimal SgsE S-Layer Carrier
We have investigated the feasibility of using SgsE as a carrier for biocatalytic activity. As the presented concept of novel types of S-layer-based biocatalysts relies on the self-assembly property of the S-layer carrier, we have chosen a design approach for a chimeric protein in which mainly the S-layer portion responsible for self-assembly was used. It was of initial interest to investigate i) how much of the SgsE carrier could be truncated without loss of its self-assembly capability (this aspect is also important regarding the length of recombinant protein production) and ii) which terminus of (truncated) SgsE would be spatially accessible for subsequent fusion of the biocatalytic function. SgsE is a 903 amino acid protein, with a leader sequence of 30 amino acids. The mature S-layer protein has a calculated molecular mass of 93.7 kDa and an isoelectric point of 6.1.[18] N-terminal truncation of SgsE was performed based on amino acid sequence alignment and on studies of S-layer proteins from different G. stearothermophilus strains, which revealed that these S-layer proteins are multidomain proteins comprising an N-terminal region involved in anchoring the protein to the cell wall[16,20,34] and a C-terminal region, making up the larger part of the protein, that encodes the self-assembly information.
A 130 amino acid N-terminal truncation (rSgsE131–903, named C_SgsE) and a 330 amino acid N-terminal truncation (rSgsE331–903, named G_SgsE) of SgsE was recombinantly produced in E. coli BL21Star (DE3) and compared to the recombinant full-length rSgsE (SgsA31–903, named A_SgsE). Overexpression of the recombinant proteins, named A_SgsE, C_SgsE, and G_SgsE, as well as either N- or C-terminally hexahistidine-tagged forms of all three proteins, named His-A_SgsE, His-C_SgsE, and His-G_SgsE (for N-terminal hexahistidine tagging) and A_SgsE-His, C_SgsE-His, and G_SgsE-His (for C-terminal His tagging), with expected masses of 98.56, 87.65, and 65.87 kDa, respectively (N-terminally His-tagged forms), and 94.77, 83,86, and 62.07 kDa, respectively (C-terminally His-tagged forms) was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; not shown). Purified proteins were analyzed for their self-assembly property in aqueous solution as well as for their recrystallization property on various supports. Self-assembly products typically appeared in the form of flat sheets (size 1–4 μm2) and double-layered cylinders with diameters decreasing with progressing truncation (65–300 nm). All forms of rSgsE were also able to recrystallize as a closed, monomolecular protein layer on negatively charged, unilamellar liposomes with diameters in the range of 150 to 250 nm. On silicon wafers, with and without poly-L-lysine/hyaluronic acid coating, the crystalline layer fully covered the surface forming monocrystalline domains with different orientation and size (50–200 nm in diameter). Data for the self-assembled and recrystallized protein G_SgsE are shown in Figure 1. According to electron microscopy and atomic force microscopy (AFM) analyses, no differences either in the principal self-assembly behavior nor in the parameters of the oblique rSgsE nanolattice were observed between full-length rSgsE and the two truncated forms; lattice parameters were identical to those reported for the wild-type protein.[18] This implies that deleting 130 or 330 amino acids from the N-terminus of SgsE apparently does not influence S-layer lattice formation, i.e., self-assembly. In a recent study, further truncated forms of SgsE are produced and analyzed with regard to their self-assembly behavior and their physicochemical properties to identify a minimal self-assembly domain of SgsE for further exploitation in nanobiotechnology. At this stage of the study C_SgsE and G_SgsE offered the best compromise when considering the amount of self-assembly and the yield of recombinant protein production and, thus, were used for biocatalyst construction.
Figure 1.

Self-assembly behavior of rSgsE331–903 (G_SgsE) in solution and on different supports. Electron micrographs of A) negatively stained preparations of G_SgsE self-assembled in solution and B) recrystallized on negatively charged liposomes. C) AFM deflection image of G_SgsE after recrystallization on a silicon wafer precoated with poly-l-lysine/hyaluronic acid, measured in contact mode in aqueous solution. The oblique symmetry of the G_SgsE nanolattice can be seen in all preparations; basal vectors are indicated by arrows.
Comparison of the spatial accessibility of the S-layer protein termini by electron microscopy of immuno gold-labeled preparations of hexahistidine-tagged SgsE self-assembled proteins and of liposomes coated with the hexahistidine-tagged SgsE proteins, using an anti-His-tag antibody, indicated that the C-terminus was more surface exposed, while the N-terminus seemed to be buried within the protein mass (not shown). Based on these data, the C-terminus of rSgsE (C_SgsE and G_SgsE) was selected as fusion site for the biocatalytic function.
2.2. Cloning and Expression of the Chimeric Genes Encoding SgsE/RmlA Fusion Proteins
For translational fusion of RmlA to the C-terminus of the N-terminally truncated forms of the S-layer protein rSgsE, i.e., C_SgsE and G_SgsE, the polymerase chain reaction (PCR) product encoding RmlA was ligated with the vector pET28a(+) carrying the corresponding truncated sgsE genes. The chimeric SgsE/RmlA proteins were extracted from biomass of E. coli BL21Star (DE3) expression cultures harboring pET28a-C_SgsE.RmlA and pET28a-G_SgsE.RmlA, respectively, and purified by gel permeation chromatography with 6m urea in enzyme buffer as eluent, which was necessary to keep the chimeric proteins disintegrated (i.e., in their monomeric state). The molecular masses of the chimeric proteins shown on SDS-PA gels and on Western blots using antiSgsE and antiRmlA antibodies, respectively, corresponded well with the calculated molecular mass of 116.0 kDa (C_SgsE.RmlA) and 94.2 kDa (G_SgsE.RmlA) (see Figure 2). Using the established preparation protocol, comprising isolation of chimeric proteins form enzymatically broken E. coli cells by treatment with urea and subsequent purification by gel permeation chromatography, the chimeric proteins were isolated to high purity and an approximately 45-fold enrichment of catalytic activity of the biocatalysts could be achieved when relating the raw extracts and the purified proteins (see below). The overall yield of purified, monomeric protein chimera was 75 mg (C_SgsE.RmlA) and 65 mg (G_SgsE.RmlA) per liter of E. coli BL21Star (DE3) expression culture. The solution of monomeric C_SgsE.RmlA and G_SgsE.RmlA proteins was dialyzed against enzyme buffer and the dialysate was centrifuged to separate monomeric and self-assembled chimeric proteins. The solution of monomers was diluted to a concentration of 0.5 mgmL−1 and stored at 4°C for assessment of storage stability (see below).
Figure 2.

A) SDS-PAGE analysis (8% gel) after Coomassie staining and B) Western-blot analysis using an antiRmlA antibody of biocatalytic proteins. Lane 1: Bench-mark ladder (Invitrogen); lane 2: purified C_SgsE (control); lane 3: purified G_SgsE (control); lane 4: raw extract of a C_SgsE.RmlA expression culture; lane 5: purified C_SgsE.RmlA; lane 6: raw extract of a G_SgsE.RmlA expression culture; lane 7: purified G_SgsE.RmlA; lane 8: purified C_SgsE.RmlA after Western blotting; lane 9: purified G_SgsE.RmlA after Western blotting. 20 μg (raw extracts) and 10 μg (purified proteins) of total protein were loaded on the gel for Coomassie staining; ≈2 μg were loaded for Western blotting.
2.3. Cloning, Expression, and Catalytic Activity of RmlA
Because the RmlA enzyme chosen as fusion partner for introduction of a biocatalytic function into the rSgsE matrix is not commercially available, the enzyme has been cloned into pET28a(+), overexpressed as N-terminally hexahisti-dine-tagged protein and purified by Ni2+ affinity chromatography. Purity of the protein was demonstrated by SDS-PAGE and Western blotting (not shown). The yield of RmlA was 660 mgL−1 of an E. coli BL21Star (DE3) expression culture. Catalytic activity was measured in an in vitro assay by the formation of dTDP-D-glucose from glucose-1-P and dTTP under reaction conditions that have been optimized previously.[31] Over a reaction time of 2 h, 1 mg of enzyme catalyzed the formation of 9.34 μmol of dTDP-D-glucose. This value was set to 100% and enzymatic activities measured for the different types of chimeric biocatalysts are given in relation to this value (see below).
2.4. Comparison of Chimeric Protein Constructs in Terms of Catalytic Activity
Two different forms of chimeric proteins bearing the last 773 or 573 amino acids of rSgsE and the RmlA enzyme at the C-terminus were used for the construction of different types of nanostructured biocatalysts, triggered by the intrinsic self-assembly property of the chimeric rSgsE-based monomers.
2.4.1. Monomeric SgsE-Based Biocatalysts
The results of the in vitro enzyme assay clearly demonstrated that the monomeric biocatalysts C_SgsE.RmlA and G_SgsE.RmlA possessed glucose-1-phosphate thymidylyltransferase activity, with activities reaching 40.8% and 99.7%, respectively, compared to the sole enzyme. These results indicated that the catalytic domains are spatially better accessible when using the more truncated S-layer carrier (G_SgsE), resulting in higher enzymatic activity. As the enzymatic activity of G_SgsE.RmlA perfectly matches the activity of the sole enzyme it is evident that the chosen purification procedure was fully compatible with the nature of the enzyme protein and that no conformational changes had occurred to the catalytic region of RmlA. All purification steps were done in enzyme buffer at 4°C and the concentration of chaotropic agent (urea) was kept at the minimum necessary for the solubilization of the chimeric protein during isolation and purification to prevent harmful effects to the enzyme.
These data furthermore imply that the C-terminal portion of SgsE may be adopted for further developments in the field of nanobiotechnology. As SgsE originates from a thermophilic bacterium and, thus, shows excellent self-assembly behavior even under elevated temperatures (60°C), translational fusion of thermophilic enzymes could constitute an attractive area of future developments.
2.4.2. SgsE-Based Biocatalysts Self-Assembled in Solution
The second type of biocatalyst was obtained by intrinsic self-assembly of C_SgsE.RmlA and G_SgsE.RmlA, triggered by their SgsE portion upon removal of the chaotropic agent through dialysis against pure enzyme buffer. It was crucial to omit CaCl2 in the dialysis solution, which is usually used to promote self-assembly,[18] because of its known negative effect on RmlA activity.[35] However, RmlA activity of self-assembled biocatalysts was surprisingly low, with values reaching only 1% for C_SgsE.RmlA self-assembly protein and 3% for G_SgsE.RmlA self-assembly protein, in relation to sole RmlA. This effect could be explained by electron microscopy evidence of immuno gold-labeled preparations using an antiRmlA antibody. The electron micrographs clearly revealed formation of double-layered self-assembly products with face-to-face inward orientation of the catalytic epitopes (compare with Ref. [17]). Figure 3 shows the results for a cylindrical self-assembly product of G_SgsE.RmlA, on which the gold particles are bound only at the fringes, where the individual layers do not perfectly overlap and, in certain regions, where the outer layer of the cylinder is ruptured.
Figure 3.

Electron micrograph of an immuno gold-labeled (antiRmlA antibody) and negatively stained self-assembly product of G_SgsE.RmlA, revealing only poor spatial accessibility of biocatalytic epitopes due to formation of double-layered cylinders. Accessible regions due to rupture of the outward layer are marked. DL: double-layer; ML: monolayer.
After the biocatalytic property of the chimeric SgsE/RmlA nanolattice was demonstrated, further engineering of the S-layer carrier protein by rational design and directed evolution strategies[36] will create S-layer-based biocatalysts with the potential of forming novel nanostructured systems with control over target epitope display. Such nanopatterned biocatalysts may enable the setting up of novel biocatalytic concepts allowing, for instance, control of substrate and/or product transport. It is further expected that the highly ordered and controllable periodic display of enzyme epitopes within the 2D nanolattice can trigger the selectivity behavior of enzymes. In addition, construction of multifunctional nanopatterned biocatalysts is conceivable by coassembly of different chimeric biocatalysts mediated through their SgsE portion. The principal suitability of the S-layer self-assembly system for such an endeavor has already been demonstrated for the set-up of a three-enzyme biosensor. There, however, the individual enzymes were chemically coupled to the S-layer carrier, followed by deposition on a microfiltration membrane.[37] In particular, the isoporosity of such self-assembled S-layer-based biocatalysts may be advantageous for their integration in flow-through systems.
2.4.3. SgsE-Based Biocatalysts Self-Assembled on the Surface of Liposomes
The third type of biocatalyst was obtained by recrystallization of C_SgsE.RmlA and G_SgsE.RmlA on negatively charged liposomes.[38,39] For recrystallization, the chimeric SgsE/RmlA proteins were applied in great excess in comparison to dipalmitoyl phosphatidylglycerol (DPPC), the major lipid component of the liposomes. As derived from electron microscopy both chimeric proteins were able to recrystallize as a closed protein layer on the liposomes, completely covering the liposome surface. The protein content of the liposome biocatalyst of 6 nmolμmol−1 DPPC, corresponding to a molar ratio of the S-layer portion to DPPC of 1:166, implied complete coverage of the liposomes with a biocatalytic protein monolayer (Figure 4). According to electron microscopy the preparations of liposome-type biocatalysts were essentially free of RmlA/SgsE proteins that self-assemble in solution. Enzyme activities of these biocatalysts were 24.5% (using C_SgsE.RmlA for liposome coating) and 65.4% (using G_SgsE.RmlA for coating) in relation to sole RmlA. This data supports the evidence of better surface display of catalytic epitopes via the shorter S-layer carrier. The overall higher activity of liposome biocatalysts in relation to self-assembled biocatalysts in solution clearly indicated that the S-layer carrier retains its potential for surface display of catalytic epitopes and supports the electron microscopy evidence of self-assembled biocatalysts, which revealed that a lack of enzymatic activity is a consequence of double-layer formation and not of incorrect presentation of biocatalytic epitopes.
Figure 4.

A) Electron micrograph of an immuno gold-labeled (anti-RmlA antibody) and negatively stained liposome covered with a biocatalytic G_SgsE.RmlA monolayer. Inset: a) schematic cross section of a biocatalytic liposome, showing the surface display of the RmlA epitopes within the nanolattice; b) schematic representation of the p2 symmetry of the G_SgsE.RmlA nanolattice. Blue circles: biocatalytic RmlA epitopes (arbitrary positions); yellow symbols: G_SgsE carrier protein. One morphological unit of the nanolattice, corresponding to a G_SgsE.RmlA dimer, is boxed. B) Enzymatic activity of liposome-type biocatalyst was demonstrated by the formation of dTDP-d-glucose as analyzed by HPLC: a) sole RmlA; b) C_SgsE.RmlA, c) G_SgsE.RmlA 1: dTTP; 2: dTDP; 3: dTDP-d-glucose.
To explain the overall lower activity of the biocatalytic chimera when “immobilized” on liposomes in comparison to sole RmlA, parameters that have been suboptimal for the enzyme during liposome coating or during sample manipulation have to be considered. In addition, the enzyme structure is a tetramer[33] and it is likely that through “im mobilization” of RmlA within the S-layer nanolattice (i.e., fusion with rSgsE and self-assembly) tetramerization is prevented. Although enzymatic activity is known to reside in each monomer, the impossibility of gaining the enzymeGs natural state may influence the catalytic efficiency.
Although with the current experimental set-up the liposome type of biocatalyst activities do not match the sole enzyme, the concept of combining a biocatalytic, self-assembling protein with liposome technology offers a wide range of options for simultaneous biocatalysis and targeted drug delivery. In this context it is important to note that it has been shown in our laboratory that S-layer coating has a stabilizing effect on the liposomes with S-layer-coated liposomes being stable for several months at 4°C.[40] Furthermore, by cocrystallization of chimeric rSgsE-based proteins with sole rSgsE, the density of functionality introduced into the nanolattice can be varied over a wide range (not shown). Thus, liposomes that have been functionalized with a biocatalytic S-layer coating could make valuable contributions to the fields of nanomedicine, pharmacy, and nutrition, where engineering of multifunctional nanocarriers combining properties such as targetability, longevity, and loading is of high demand.[41] There is high flexibility for variation of the diameter of the liposome support (in the nano- to micrometer range), so that the size of the biocatalysts can be adjusted to specific needs. For potential applications in the outlined fields, production of endotoxin-free chimeric rSgsE proteins may constitute an important issue. To address this point, the production of A_SgsE in the endotoxin-free background of the Gram-positive bacterium L. lactis has already been demonstrated.[42]
2.5. Storage Stability and Recycling of SgsE-Based Biocatalysts
2.5.1. Storage Stability of Monomeric SgsE-Based Biocatalysts
The results of the RmlA activity assay clearly demonstrated that the SgsE/RmlA proteins possessed thymidylmonophosphate nucleotide transferase activity. We then assessed the stability of the monomeric chimera, which had been stored in enzyme buffer at 4°C without addition of preservatives, by assaying the enzymatic activity under standard reaction conditions over a time span of 17 weeks (Figure 5). Data indicated that storage of rSgsE-based biocatalysts over 14 weeks had no influence on enzyme activity, whereas sole RmlA has already lost 75% of activity over the same time span. Even after 17 weeks, C_SgsE.RmlA still retained 35% of its original activity. In contrast, sole RmlA possessed only 6% of its original activity after 17 weeks.
Figure 5.

Stability of the C_SgsE.RmlA biocatalyst in comparison to the sole enzyme. Enzymatic activity was measured under standardized conditions. The results are expressed as a percentage of the original activity.
We assume that the loss of RmlA activity was rather due to disintegration of the liposomes under the chosen reaction conditions than to shedding of the biocatalytic chimera from the liposomes. In the future, this effect could be minimized by using lipid mixtures for preparation of liposomes of transition temperatures that are adjusted to specific needs and by additional chemical crosslinking of the S-layer-based functional layer.[39]
These preliminary data clearly indicate that the S-layer carrier contributes to the long-term stability of the fused enzyme. Thus, monomeric S-layer-based biocatalysts can be regarded an ideal base for the conception of more complex types of S-layer-based biocatalysts for diverse applications.
2.5.2. Recycling of SgsE-Based Liposome Biocatalysts
To determine whether the liposome type of biocatalyst could be purified away from the product and reused to catalyze the same reaction, C_SgsE.RmlA and G_SgsE.RmlA activities were assayed; coated liposomes were collected by centrifugation and then washed three times with enzyme buffer, and the assay was repeated. After one cycle, for C_SgsE.RmlA 55.2% and for G_SgsE.RmlA 61.0% of the original activity was still detectable.
3. Conclusions
Following the concept of nanobiotechnology, that is, the combination of physical and biological principles with chemical processes for creation of nanoscale structures with novel properties and specific functions, we aimed at engineering the first biocatalytic, self-assembling S-layer protein. The results obtained from chimeric proteins consisting of the 573 or 773 C-terminal amino acids of the S-layer protein rSgsE and the glucose-1-phosphate thymidylyltransferase RmlA fused to the C-terminus showed that this approach is feasible for the conception of novel types of biocatalysts. Due to the intrinsic self-assembly property conferred to the modular protein by the S-layer portion, either in suspension or on various supports, these biocatalysts allow periodic and controllable presentation of biocatalytic epitopes. Furthermore, the S-layer carrier considerably improves the long-term storage stability of the translationally fused enzyme. Proof of concept was carried out by investigation of liposomes covered with a biocatalytic SgsE/RmlA monolayer that were also recyclable.
Despite the fact that none of the constructed nanopatterned biocatalysts represents a fully developed product, it is expected that these novel architectures can be optimized by directed evolution and will open up new functional concepts for biocatalytic applications.
4. Experimental Section
Bacterial strains and growth conditions
G. stearothermophilus NRS 2004/3a was obtained from the N. R. Smith Collection, US Department of Agriculture (Peoria, IL) and was grown on modified S-VIII medium (1% peptone, 0.5% yeast extract, 0.5% meat extract, 0.13% K2HPO4, 0.01% MgSO4, 0.06% sucrose) at 55°C.[43] Escherichia coli DH5α (Invitrogen, Lofer, Austria) was used for cloning; overexpression of proteins was accomplished in E. coli BL21 Star (DE3). Both E. coli strains were grown at 37°C in Luria-Bertani broth (LB broth) supplemented with kanamycin (50 μgmL−1).
General methods
SDS-PAGE and visualization of proteins bands with Coomassie Blue R-250 staining reagent was carried out according to Reference [44]. Western immunoblotting using anti-His-tag monoclonal antibody (Novagen, Madison, WI), anti-SgsE antibody and anti-RmlA antibody, was done as described elsewhere.[45] Protein concentration was determined by the Bio-Rad protein assay (Bio-Rad, Vienna, Austria) using bovine serum albumin (BSA) as standard. Genomic DNA of G. stearothermophilus NRS 2004/3a was isolated with the Qiagen Genomic-tip 100 kit (Qiagen, Valencia, CA) according to the manufacturer’s instruction. Restriction enzymes, calf intestinal alkaline phosphatase, and T4 DNA ligase were purchased from Invitrogen. Qiagen MinElute gel extraction kit was used to purify DNA fragments from agarose gels and Qiagen MinElute reaction cleanup kit was used to purify digested oligonucleotides and plasmids. Plasmid DNA from transformed cells was isolated with the Qiagen Plasmid miniprep kit. Agarose gel electrophoresis was performed as described by Sambrook and Russell.[46] Polymerase chain reaction (PCR Sprint thermocycler, Hybaid, Ashford, UK) was performed using Pwo polymerase (Roche, Vienna, Austria). All primers were synthesized by Invitrogen. PCR conditions were optimized for each primer pair and amplification products were purified using the MinElute PCR purification kit. E. coli transformation was done according to the manufacturer’s instructions. Transformants were screened by in situ PCR reactions using RedTaq ReadyMix PCR Reaction Mix (Sigma–Aldrich, St. Louis, MO); positive clones were analyzed by restriction mapping and confirmed by sequencing (Agowa, Berlin, Germany).
Microscopic methods
Electron microscopy of the different forms of S-layer protein was performed after negative staining with 1% phosphotungstate in Tris-buffered saline (TBS, pH 7.5), using a Philips model CM 12 electron microscope (Philips, Eindhoven, The Netherlands) operated at 80 kV using a 50 μm objective aperture.[43] Immunogold-labeling experiments were carried out using anti-His antibody, or anti-RmlA antibody as primary antibody. After adsorption of samples to 300-mesh copper grids, coated with pioloform and carbon, the grids were incubated on droplets of primary antibody (1:1000 in 1% BSA, 0.5% Tween 20 in TBS) for 30 min. After washing with BSA/Tween/TBS and TBS, grids were incubated for 30 min with 5-nm gold-labeled goat antirabbit IgG as secondary antibody (1:10 in 1% BSA, 0.5% Tween 20 in TBS). After repeated washing with BSA/Tween/TBS and TBS, grids were fixed for 15 min with 2.5% glutaraldehyde in TBS, washed with TBS and negatively stained for 90 s. For labeling of liposomes, Tween 20 was omitted. AFM measurements were performed with a Nanoscope IIIa multimode (Veeco Instruments, Santa Barbara, CA) with a J-scanner (nominal scan size 130 μm). Samples were measured in contact mode in 100 mm NaCl to avoid electrostatic repulsion between tip and sample. For optimal resolution standard 200-μm-long oxide-sharpened silicon nitrite cantilevers with a nominal spring constant of 0.06 Nm−1 (NP-S, NanoProbes, Digital Instruments, Santa Barbara, CA) were used for imaging. The samples were scanned at approximately 4 Hz, the measured scan size varied from 20 μm to 500 nm. The applied force during scanning was minimized to prevent the tip from modifying the sample.
Plasmid construction
Oligonucleotide primers used in the course of this study are summarized in Table 1. Plasmids are listed in Table 2; all plasmids were confirmed by sequencing.
Table 1.
Oligonucleotide primers used for PCR amplification of different forms of sgsE and of rmlA.
| Primer | Nucleotide sequence (5′→3′) |
|---|---|
| A_sgsE for(NcoI) | AATCACC
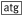 [a]gCGGACGTGGCGACGGTCG
[a]gCGGACGTGGCGACGGTCG |
| C_sgsE for(NcoI) | AATCACC
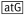 gCAAAATTAGACAAAATGCGCC
gCAAAATTAGACAAAATGCGCC |
| G_sgsE for(NcoI) | AATCACC
 gTAAAATTAGTGGTTGATGGCGC
gTAAAATTAGTGGTTGATGGCGC |
| sgsE_rev(XhoI;+Stop) | AATCA  CTCGAGTTTTGCTACGTTTACAACAGTAGC
CTCGAGTTTTGCTACGTTTACAACAGTAGC |
| sgsE_rev(XhoI;−Stop) | AATCACTCGAGTTTTGCTACGTTTACAACAGTAGC |
| sgsE_rev(KpnI,XhoI;+Stop) | AATCA  CTCGAGGGTACCTTTGCTACGTTTACAACAGTAGC
CTCGAGGGTACCTTTGCTACGTTTACAACAGTAGC |
| C-term. rmlA_for(KpnI) | AATCAGGTAccgcAAGGTATCGTTCTAGCCGG |
| C-term. rmlA_rev(XhoI) | AATCACTCGAGGAAAGGCATTACTAAACTTAGTCTCAAT |
| rmlA_for (NdeI) | AATCACAT
 gAAGGTATCGTTCTAGCCGGCG
gAAGGTATCGTTCTAGCCGGCG |
Artificial restriction sites are underlined; lowercase letters indicate changes in the chromosomal DNA sequence to introduce appropriate restriction sites. The triplets corresponding to the initiation and termination codons in the primer sequence are boxed.
Table 2.
Strains and expression constructs.
| Strain or plasmid | Description | Source |
|---|---|---|
| Geobacillus stearothermophilus NRS 2004/3a | S-layer glycoprotein-covered neotype strain | N. R. Smith collection[43] |
| E. coli DH5α | F-ϕ80 LacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rk−, mk+) phoA supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| E. coli BL21 Star(DE3) | F- ompT hsdSB (rB-mB-) gal dcm rne131 (DE3) | Novagen |
| pET28a(+) | E. coli expression vector, used for introducing a C-terminal hexahistidine-tag to the cloned gene; Kmr | Novagen |
| pET30a(+) | E. coli expression vector, used for introducing an N-terminal hexahistidine-tag to the cloned gene; Kmr | Novagen |
| pET28a-A_SgsE(C) | pET-28a(+)/SgsE31−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a devoid of its signal peptide (aa 1-30), C-terminal hexahistidine-tag; Kmr | This study |
| pET28a-C_SgsE(C) | pET-28a(+)/C_SgsE131−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of aa 1-100 of the mature S-layer protein; C-terminal hexahistidine-tag; Kmr | This study |
| pET28a-G_SgsE(C) | pET-28a(+)/G_SgsE331−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of aa 1-300 of the mature S-layer protein; C-terminal hexahistidine-tag; Kmr | This study |
| pET30a-A_SgsE(N) | pET-30a(+)/SgsE31−903; pET-30a(+) containing sgsE from G. stearothermophilus NRS 2004/3a devoid of its signal peptide (aa 1-30); N-terminal hexahistidine-tag; Kmr | This study |
| pET30a-C_SgsE(N) | pET-30a(+)/SgsE131−903; pET-30a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of aa 1-100 of the mature S-layer protein; N-terminal hexahistidine-tag; Kmr | This study |
| pET30a-G_SgsE(N) | pET-30a(+)/SgsE331−903; pET-30a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of aa 1-300 of the mature S-layer protein; N-terminal hexahistidine-tag; Kmr | This study |
| pET28a-A_SgsE | pET-28a(+)/SgsE31−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of its signal peptide (amino acids 1-30); Kmr | This study |
| pET28a-C_SgsE | pET-28a(+)/SgsE131−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of the first 100 amino acids of the mature S-layer protein; Kmr | This study |
| pET28a-G_SgsE | pET-28a(+)/SgsE331−903; pET-28a(+) containing sgsE from G. stearothermophilus NRS 2004/3a, devoid of the first 300 amino acids of the mature S-layer protein; Kmr | This study |
| pET28a-C_SgsE·RmlA | pET28a-C_SgsE containing RmlA fused to the C-terminus of the S-layer protein; Kmr | This study |
| pET28a-G_SgsE·RmlA | pET28a-G_SgsE containing RmlA fused to the C-terminus of the S-layer protein; Kmr | This study |
| pET28a-RmlA | pET-28a(+) containing rmlA from G. stearothermophilus NRS 2004/3a; C-terminal hexahistidine tag; Kmr | This study |
To determine the spatial accessibility of SgsE termini for translational fusion of the rmlA gene to sgsE, N- and C-terminally hexahistidine-tagged (truncated) forms of the SgsE protein were constructed. The sequences encoding rSgsE31–903 (A_SgsE), rSgsE131–903 (C_SgsE), and rSgsE331–903 (G_SgsE) of G. stearothermophilus NRS 2004/3a were amplified by PCR using the oligonucleotide forward primers A_sgsE for (NcoI), C_sgsE for (NcoI), and G_sgsE for(NcoI), respectively, in combination with the reverse primer sgsE_rev(Xho, +Stop), containing a stop codon, or sgsE_rev (Xho, −Stop), lacking a stop codon for C-terminal fusion. The sgsE fragments were digested with NcoI/XhoI and inserted into the linearized and dephosphorylated expression vectors pET28a(+) and pET30a(+) (Novagen), respectively. The resulting plasmids for overexpression of the rSgsE protein with an N-terminal (N) hexahistidine-tag were named pET30a-A_SgsE(N) and pET30a-C_SgsE(N), pET30a-G_SgsE(N), those with a C-terminal (C) hexahistidine-tag and pET28a-A_SgsE(C), pET28a-C_SgsE(N) and pET28a-G_SgsE(C).
For the construction of chimeric SgsE/RmlA proteins, carrying the catalytic function at the C-terminus, cloning of sgsE393–2709 (C_SgsE), and sgsE993–2709 (G_SgsE) into pET28a(+) was performed as described above, except that oligonucleotide primer sgsE_rev(KpnI,XhoI; +Stop), containing a stop codon, was used. The rmlA fusion partner was PCR-amplified from chromosomal DNA of G. stearothermophilus NRS 2004/3a using the primer pair C-term-rmlA_for(forward primersKpnI)/C-term rmlA_rev(XhoI), digested with KpnI/XhoI, and inserted into the KpnI/XhoI linearized plasmids pET28a-C_SgsE and pET28a-G_SgsE to give a C-terminal in-frame fusion to C_SgsE, and G_SgsE, respectively. Resulting plasmids were named pET28a-C_SgsE.RmlA and pET28a-G_SgsE.RmlA. As a positive control for determination of enzyme activity, the rmlA gene fragment was PCR-amplified from chromosomal DNA of G. stearothermophilus NRS 2004/3a using the primer pair rmlA_for (NdeI)/C-term-rmlA_rev(XhoI) and ligated into pET28a(+) via the NdeI/XhoI sites for expression of RmlA with the His-tag at the N-terminus of rSgsE.
Overexpression and purification of recombinant proteins
Synthesis of recombinant proteins in E. coli BL21Star (DE3) was initiated by the addition of 1 mm IPTG (Fermentas, St. Leon-Rot, Germany) to OD600 ≈0.8 cultures, and cultivation was continued for an additional 4 h at 37°C. Expression of recombinant proteins was monitored by SDS-PAGE. The biomass was collected by centrifugation at 5,000 rpm at 4°C for 20 min. For purification of chimeric SgsE/RmlA proteins (C_SgsE.RmlA and G_SgsE.RmlA), 1 g of biomass (wet weight) was suspended in 10 mL of enzyme buffer (10 mm MgCl2 in 50 mm Tris/HCl, pH 7.5) containing 0.1% Triton X-100, 2 μL of benzonase (250 U/μl; Merck, Darmstadt, Germany) and 40 μL of lysozyme (100 mgmL−1; Merck), and incubated for 20 min at 37°C. The reaction mixture was centrifuged for 15 min at 15000 rpm (4°C), the supernatant was discarded, and the pellet was resuspended at a concentration of 0.5 g (wet weight)mL−1 in enzyme buffer containing 6m urea (Fluka, Buchs, Switzerland) to disintegrate the water-insoluble S-layer protein forms, and stirred on ice for 20 min. After centrifugation, extraction of the remaining pellet was repeated. Both extracts were combined and loaded onto a Superdex 200 prep grade XK16 FPLC-column (1.6×60 cm; GE-Healthcare, Uppsala, Sweden). Elution was performed at 4°C at a flow rate of 1 mLmin−1 using 6m urea in enzyme buffer. Fractions containing the desired protein according to SDS-PAGE analysis were pooled and the protein concentration of the dialysate (representing soluble monomers) was determined. Chimeric SgsE/RmlA proteins were stored as monomers at a concentration of 0.50 mgmL−1 at 4°C without addition of preservatives. As a control, the S-layer matrices rSgsE31–903 (A_SgsE), rSgsE131–903 (C_SgsE), and rSgsE331–903 (G_SgsE) as well as the N-terminally and C-terminally, His-tagged forms thereof were overexpressed, purified, and stored as described for the chimeric proteins. Purification of hexahistidine-tagged RmlA from a 400 mL culture was carried out at 4°C by Ni2+ affinity chromatography using a HisTrap HP column (GE-Healthcare) as described elsewhere.[31] The fractions containing the RmlA protein were pooled, dialyzed against enzyme buffer, and concentrated using centrifugal filter devices (molecular-weight cutoff 10 kDa; Millipore, Vienna, Austria). RmlA was stored at a concentration of 0.66 mgmL−1 at 4°C without addition of preservatives.
Self-assembly of the S-layer matrices and of chimeric SgsE/RmlA proteins in aqueous solution
To promote self-assembly of the different recombinant S-layer matrices and of the chimeric SgsE/RmlA proteins, 2 mL of the monomer solution were dialyzed (dialysis tubing cut-off 20 kDa; Millipore) at 4°C, twice against 10 mm CaCl2 and four times against distilled water (0.5 L each). For dialyzing chimeric SgsE/RmlA proteins, CaCl2 was omitted. Self-assembly formation was examined by electron microscopy.
Recrystallization of the S-layer matrices and of chimeric SgsE/RmlA proteins on silicon wafers
Silicon wafers (p-type, 100 orientation; 7×7 mm; IMEC, Leuven, Belgium) with a native silicon oxide layer were rinsed with 70% ethanol and distilled water and finally dried under nitrogen prior to use. The advancing contact angle of water at the pretreated silicon surface was 56±2° (Kruess contact-angle measurement system G1, Hamburg, Germany). For the preparation of poly-l-lysine/hyaluronic acid coated wafers, wafers were incubated in 1 mgmL−1 of poly-l-Lysine (Sigma–Aldrich) in 0.5m NaCl for 20 min, washed with distilled water, and subsequently incubated in 1 mgmL−1 of hyaluronic acid (Fluka) in 0.5m NaCl for another 20 min. For protein recrystallization, the wafers were immersed in 1 mL of a solution of 10 mm CaCl2, 0.5 mm Tris/HCl, pH 9, containing 0.1 mgmL−1 of protein for 5 h at 25°C.[47] Coated wafers were rinsed with distilled water and stored in distilled water at 4°C until use.
Coating of liposomes with S-layer matrices and chimeric SgsE/RmlA proteins
Negatively charged liposomes were prepared from a mixture of dipalmitoyl phosphatidylcholine, dipalmitoyl phosphatidylglycerol and cholesterol (all lipids were obtained from Sigma–Aldrich) in a molar ratio of 2:2:1, using the dehydration–rehydratation method as described by Mader and co-workers[39] followed by extrusion of liposomes through uniform-pore-size membranes with decreasing pore sizes (400 nm and 200 nm) using a LiposoFast-Pneumatic extruder (Avestin, Ottawa, ON, Canada). The phase-transition temperature of the liposomes is ≈41°C. Integrity of the liposomes was investigated by electron microscopy after negative staining. The resulting liposome preparation was stored in distilled water at 4°C. For coating of liposomes with (chimeric) proteins, 1.5 mL of a solution containing 11.5 nmolmL−1 of monomers in enzyme buffer were mixed with 250 μL of liposome suspension containing 2.8 μmolmL−1 dipalmitoyl phosphatidylcholine. Recrystallization was carried out for 12 h at 22°C in a Test Type Rotator. Excess of non-recrystallized (soluble, monomeric) protein was removed by centrifugation at 13,000 g and 4°C for 10 min in an Eppendorf bench centrifuge and the protein content of the supernatant was determined for calculation of protein loading of the liposomes. The pellet containing the liposome biocatalyst was resuspended in enzyme buffer and assayed for enzyme activity. Integrity of the liposome biocatalyst was checked by electron microscopy.
Glucose-1-phosphate thymidylyltransferase (RmlA) assay
The synthesis of dTDP-d-glucose from the substrates glucose-1-P and dTTP catalyzed by RmlA (provided in its native form and as C_SgsE.RmlA and G_SgsE.RmlA) was assayed according to the procedure described by Lindquist and co-workers,[48] with slight modifications: briefly, 150 μL of reaction mixture containing 50 mm Tris/HCl (pH 7.5), 10 mm MgCl2, 100 nmol glucose-1-P (Sigma–Aldrich) and 100 nmol dTTP (Sigma–Aldrich) as substrates and an appropriate aliquot of biocatalyst (free RmlA, 3.32 μg; soluble (monomeric) SgsE/RmlA forms, 39.5 μg; chimeric SgsE/RmlA proteins self-assembled in solution, 100 μg; chimeric SgsE/RmlA proteins self-assembled on liposomes, 25.0 μg) were incubated at 37°C for 2 h. dTDP-d-glucose was identified by the reverse-phase high-performance liquid chromatography (RP-HPLC) method as described elsewhere.[28] Removal of the different biocatalysts from the reaction product prior to HPLC analysis was performed as follows: i) Monomers and RmlA were removed with Microcon centrifugal filter devices (membrane cutoff 10 kDa, Millipore); ii) self-assembled chimeric SgsE/RmlA protein was separated by centrifugation at 5000 rpm for 5 min in an Eppendorf centrifuge; iii) liposomes coated with SgsE/RmlA fusion protein were removed by centrifugation at 13000 rpm for 5 min in an Eppendorf centrifuge. 50 μL of either the filtrate (experiment i) or of the supernatant (experiments ii, iii) were used for HPLC analysis.
Recycling and shelf-life of SgsE/RmlA fusion proteins
RmlA assays were performed as described above. Subsequently, self-assembled forms of chimeric SgsE/RmlA proteins and of liposomes coated with chimeric SgsE/RmlA protein were washed three times with 150 μL of enzyme buffer. After the last cycle, enzyme activity was determined to calculate the average amount of enzyme lost per washing step. RmlA activity originating from all forms of chimeric SgsE/RmlA proteins was assayed in intervals of three to five weeks over a total time span of 17 weeks. Activity values were compared with those obtained for free RmlA that has been stored under the same conditions.
During the reviewing process of this manuscript a paper dealing with in vivo surface display of AHL-lactonase via the S-layer protein anchor of Bacillus thuringiensis was published by Zhang et al.[49] Another paper exploiting the S-layer protein as a means for binding an extremophilic laminarinase from Pyrococcus furisous to various nanobiotechnologically relevant supports was submitted to J. Biotechnol. by H. Tschiggerl et al.
Acknowledgements
Financial support came from the Austrian Science Fund, projects P19047-B12 (to C.S.) and P18013-B10 (to P.M.), the Hochschuljubiläumsstiftung der Stadt Wien, project H-1809/2006 (to C.S.), and the US Air Force Office of Scientific Research project F49620-03-1-0222 (to U.B. S.).
References
- [1].Bornscheuer U. Adv. Biochem. Eng./Biotechnol. 2005;100:181–203. doi: 10.1007/b136413. [DOI] [PubMed] [Google Scholar]
- [2].Girelli AM, Mattei E. J. Chromatogr. B. 2005;819:3–16. doi: 10.1016/j.jchromb.2005.01.031. [DOI] [PubMed] [Google Scholar]
- [3].Cao L. Curr. Opin. Chem. Biol. 2005;9:217–226. doi: 10.1016/j.cbpa.2005.02.014. [DOI] [PubMed] [Google Scholar]
- [4].Buchholz K, Kasche V. Biokatalysatoren und Enzymtechnologie. Wiley-VCH; Weinheim, Germany: 1997. p. 7. [Google Scholar]
- [5].Silman IH, Katchalski E. Annu. Rev. Biochem. 1966;35:873–908. doi: 10.1146/annurev.bi.35.070166.004301. [DOI] [PubMed] [Google Scholar]
- [6].Tischer W, Kasche V. TIBTECH. 1999;17:326–335. doi: 10.1016/s0167-7799(99)01322-0. [DOI] [PubMed] [Google Scholar]
- [7].Zelinski T, Waldmann H. Angew. Chem. 1997;109:746–748. [Google Scholar]; Angew. Chem. Int. Ed. 1997;36:722–724. [Google Scholar]
- [8].Cao R, Villalonga R, Fargoso A. IEE Proc. Nanobiotechnol. 2005;152:159–164. doi: 10.1049/ip-nbt:20050003. [DOI] [PubMed] [Google Scholar]
- [9].Sára M, Sleytr UB. Appl. Microbiol. Biotechnol. 1989;30:184–189. doi: 10.1007/BF00174459. [DOI] [PubMed] [Google Scholar]
- [10].Lan EH, Dunn B, Zink JI. Methods Mol. Biol. 2005;300:53–79. doi: 10.1385/1-59259-858-7:053. [DOI] [PubMed] [Google Scholar]
- [11].Gilardi G, Meharenna YT, Tsotsou GE, Sadeghi SJ, Fairhead M, Giannini S. Biosens. Bioelectron. 2002;17:133–145. doi: 10.1016/s0956-5663(01)00286-x. [DOI] [PubMed] [Google Scholar]
- [12].Duncan G, Tarling CA, Bingle WH, Nomellini JF, Yamage M, Dorocicz IR, Withers S, Smit J. Appl. Biochem. Biotechnol. 2005;127:95–110. doi: 10.1385/abab:127:2:095. [DOI] [PubMed] [Google Scholar]
- [13].Cao L, van Langen L, Sheldon RA. Curr. Opin. Biotechnol. 2003;14:387–394. doi: 10.1016/s0958-1669(03)00096-x. [DOI] [PubMed] [Google Scholar]
- [14].Debabov VG. Mol. Biol. 2004;4:482–493. [PubMed] [Google Scholar]
- [15].Sleytr UB, Egelseer EM, Ilk N, Pum D, Schuster B. FEBS Lett. 2007;274:323–334. doi: 10.1111/j.1742-4658.2006.05606.x. [DOI] [PubMed] [Google Scholar]
- [16].Sleytr UB, Sára M, Pum D, Schuster B. In: Supramolecular Polymers. 2nd Ed Cifferi A, editor. Taylor & Francis; Boca Raton, FL: 2005. pp. 583–616. [Google Scholar]
- [17].Messner P, Pum D, Sleytr UB. J. Ultrastruct. Mol. Struct. Res. 1986;97:73–88. doi: 10.1016/s0889-1605(86)80008-8. [DOI] [PubMed] [Google Scholar]
- [18].Schäffer C, Wugeditsch T, Kählig H, Scheberl A, Zayni S, Messner P. J. Biol. Chem. 2002;277:6230–6239. doi: 10.1074/jbc.M108873200. [DOI] [PubMed] [Google Scholar]
- [19].Avall-Jaaskelainen S, Palva A. FEMS Microbiol. Rev. 2005;29:511–529. doi: 10.1016/j.femsre.2005.04.003. [DOI] [PubMed] [Google Scholar]
- [20].Schäffer C, Messner P. Glycobiology. 2004;14:31R–42R. doi: 10.1093/glycob/cwh064. [DOI] [PubMed] [Google Scholar]
- [21].Sleytr UB, Messner P, Pum D, Sára M. Angew. Chem. 1999;111:1098–1120. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 1999;38:1034–1054. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [22].Sleytr UB, Sára M, Pum D, Schuster B, Messner P, Schäffer C. In: Biopolymers. Steinbüchel A, Fahnestock SR, editors. Vol. 7. Wiley-VCH; Weinheim, Germany: 2002. pp. 285–338. [Google Scholar]
- [23].Sára M, Egelseer EM, Huber C, Ilk N, Pleschberger M, Pum D, Sleytr UB. In: Biological and Pharmaceutical Nanomaterials. Kumar C, editor. Wiley-VCH; Weinheim, Germany: 2006. pp. 219–252. [Google Scholar]
- [24].Sleytr UB, Huber C, Ilk N, Pum D, Schuster B, Egelseer EM. FEMS Microbiol. Lett. 2007;267:131–144. doi: 10.1111/j.1574-6968.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- [25].Claus H, Akça E, Debaerdemaeker T, Evrard C, Declercq J-P, Harris JR, Schlott B, König H. Can. J. Microbiol. 2005;51:731–743. doi: 10.1139/w05-093. [DOI] [PubMed] [Google Scholar]
- [26].Jarosch M, Egelseer EM, Huber C, Moll D, Mattanovich D, Sleytr UB UB, Sára M. Microbiology. 2001;147:1353–1363. doi: 10.1099/00221287-147-5-1353. [DOI] [PubMed] [Google Scholar]
- [27].Rünzler D, Huber C, Moll D, Köhler G, Sára M. J. Biol. Chem. 2004;279:5207–5215. doi: 10.1074/jbc.M308819200. [DOI] [PubMed] [Google Scholar]
- [28].Giraud MF, Naismith JH. Curr. Opin. Struct. Biol. 2000;10:687–696. doi: 10.1016/s0959-440x(00)00145-7. [DOI] [PubMed] [Google Scholar]
- [29].Zayni S, Steiner K, Pföstl A, Hofinger A, Kosma P, Schäffer C, Messner P. Glycobiology. 2007;17:433–443. doi: 10.1093/glycob/cwl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nahalaka J, Liu Z, Chen X, Wang PG. Chem. Eur. J. 2003;9:372–377. doi: 10.1002/chem.200390038. [DOI] [PubMed] [Google Scholar]
- [31].Graninger M, Kneidinger B, Bruno K, Scheberl A, Messner P. Appl. Environ. Microbiol. 2002;68:3708–3715. doi: 10.1128/AEM.68.8.3708-3715.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Blankenfeldt W, Asuncion M, Lam JS, Naismith JH. EMBO J. 2000;19:6652–6663. doi: 10.1093/emboj/19.24.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dong C, Beis K, Giraud MF, Blankenfeldt W, Allard S, Major LL, Kerr ID, Whitfield C, Naismith JH. Biochem. Soc. Trans. 2003;31:532–536. doi: 10.1042/bst0310532. [DOI] [PubMed] [Google Scholar]
- [34].Sára M. Trends Microbiol. 2001;9:47–49. doi: 10.1016/s0966-842x(00)01905-3. [DOI] [PubMed] [Google Scholar]
- [35].Cowan JA. BioMetals. 2002;15:225–235. doi: 10.1023/a:1016022730880. [DOI] [PubMed] [Google Scholar]
- [36].Kaur J, Sharma R. Crit. Rev. Biotechnol. 2006;6:165–199. doi: 10.1080/07388550600851423. [DOI] [PubMed] [Google Scholar]
- [37].Neubauer A, Hödl C, Pum D, Sleytr UB. Anal. Lett. 1994;27:849–865. [Google Scholar]
- [38].Küpcü S, Sára M, Sleytr UB. Biochim. Biophys. Acta. 1995;1235:263–269. doi: 10.1016/0005-2736(95)80013-6. [DOI] [PubMed] [Google Scholar]
- [39].Mader C, Küpcü S, Sleytr UB, Sára M. Biochim. Biophys. Acta. 2000;1463:142–150. doi: 10.1016/s0005-2736(99)00190-x. [DOI] [PubMed] [Google Scholar]
- [40].Mader C, Küpcü S, Sára M, Sleytr UB. Biochim. Biophys. Acta. 1999;1418:106–116. doi: 10.1016/s0005-2736(99)00030-9. [DOI] [PubMed] [Google Scholar]
- [41].Koo OM, Rubinstein I, Obyuksel H. Nanosist. Nanomater. Nanotekhnol. Nanomedicine. 2005;1:193–212. doi: 10.1016/j.nano.2005.06.004. [DOI] [PubMed] [Google Scholar]
- [42].Novotny R, Scheberl A, Giry-Laterriere M, Messner P, Schäffer C. FEMS Microbiol. Lett. 2005;242:27–35. doi: 10.1016/j.femsle.2004.10.036. [DOI] [PubMed] [Google Scholar]
- [43].Messner P, Hollaus F, Sleytr UB. Int. J. Syst. Bacteriol. 1984;34:202–210. [Google Scholar]
- [44].Altman E, Schäffer C, Brisson J-R, J.-R., Messner P. Eur. J. Biochem. 1995;229:308–315. doi: 10.1111/j.1432-1033.1995.tb20470.x. [DOI] [PubMed] [Google Scholar]
- [45].Steiner K, Novotny R, Patel K, Vinogradov E, Whitfield C, Valvano MA, Messner P, Schäffer C. J. Bacteriol. 2007;189:2590–2598. doi: 10.1128/JB.01592-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd Ed Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- [47].Pum D, Stangl G, Sponer C, Fallmann W, Sleytr UB. Colloids Surf. B. 1997;8:157–162. [Google Scholar]
- [48].Lindquist L, Kaiser R, Reeves PR, Lindberg AA. Eur. J. Biochem. 1993;211:763–770. doi: 10.1111/j.1432-1033.1993.tb17607.x. [DOI] [PubMed] [Google Scholar]
- [49].Zhang L, Ruan L, Wu H, Chen S, Yu Z, Sun M. Appl. Microbiol. Biotechnol. 2007;74:667–675. doi: 10.1007/s00253-006-0696-8. [DOI] [PubMed] [Google Scholar]