

Abstract
Unbiased proteomic analysis of plasma samples holds the promise to reveal clinically invaluable disease biomarkers. However, the tremendous dynamic range of the plasma proteome has so far hampered the identification of such low abundant markers. To overcome this challenge we analyzed the plasma microparticle proteome, and reached an unprecedented depth of over 3000 plasma proteins in single runs. To add a quantitative dimension, we developed PROMIS-Quan—PROteomics of MIcroparticles with Super-Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) Quantification, a novel mass spectrometry-based technology for plasma microparticle proteome quantification. PROMIS-Quan enables a two-step relative and absolute SILAC quantification. First, plasma microparticle proteomes are quantified relative to a super-SILAC mix composed of cell lines from distinct origins. Next, the absolute amounts of selected proteins of interest are quantified relative to the super-SILAC mix. We applied PROMIS-Quan to prostate cancer and compared plasma microparticle samples of healthy individuals and prostate cancer patients. We identified in total 5374 plasma-microparticle proteins, and revealed a predictive signature of three proteins that were elevated in the patient-derived plasma microparticles. Finally, PROMIS-Quan enabled determination of the absolute quantitative changes in prostate specific antigen (PSA) upon treatment. We propose PROMIS-Quan as an innovative platform for biomarker discovery, validation, and quantification in both the biomedical research and in the clinical worlds.
Biomarker discovery in plasma is one of the holy grails of the proteomic field toward the development of noninvasive diagnostic/prognostic tests (1). To achieve this goal, proteomics necessitates a comprehensive view of the plasma proteome, accurate proteome quantification, combined with relatively short analytical times to enable multiple sample comparisons. However, MS-based biomarker discovery is limited by the vast dynamic range of the plasma, over 11 orders of magnitude (2, 3), which leads to the masking of “tissue leakage” proteins that comprise of potential biomarkers by the core plasma proteins. Two main complementary strategies have been employed to reach identification of low abundance proteins: (i) Targeted proteomics, in which the MS identifies and quantifies only predetermined peptides, thereby circumventing the system's inherent tendency to preferentially detect abundant proteins. This approach is utilized for validation of preselected candidate markers (4–6). (ii) Plasma fractionation, which biochemically reduces the complexity of the proteomes, and enables discovery of novel biomarkers (7, 8).
Targeted MS analysis is dominated by the selected reaction monitoring approach, often in combination with antibody-based enrichment of proteins or peptides and stable isotope labeled standards for quantification (9). This approach benefits from the sensitivity and quantitative capabilities of the triple-quadrupole instruments. Its major limitation is that it relies on prior discovery of candidates within the plasma samples using extensive tissue/cell-line-based analysis and prediction of potential biomarkers. The fractionation strategy reduces both the complexity and the dynamic range of the plasma through depletion of the most abundant plasma proteins, and/or through extensive biochemical separation of proteins and peptides. Although these fractionation approaches enabled identification of thousands of plasma proteins (7), they dramatically reduce the throughput of the method, and thus, the applicability to clinical studies.
A distinct fractionation approach involves the isolation of plasma microparticles and exosomes. Microparticles are large vesicles (100 nm–1 μm), which protrude directly from the plasma membrane, whereas exosomes are smaller (40–100 nm) and originate from endocytic compartments known as the multivesicular endosomes. These microvesicles are constitutively shed from all cell types into the blood, carrying a proteomic signature of their cells of origin (10). Microparticles mediate local and systemic communication in various conditions, in particularly in cancer, where they can promote metastasis, immune evasion of cancer cells and angiogenesis (10–13), but also in other conditions including autoimmune diseases (14) and cardiovascular disorders (15). Therefore, circulating plasma microparticle proteomics can reveal biomarkers of various diseases as the basis for further diagnostic test development.
The profiling of plasma microparticle proteomes initiated by Jin et al. in 2005, with the analysis of 16 samples using two-dimensional (2D)-gels followed by matrix assisted laser desorption ionization- time of flight (MALDI-TOF) MS analysis, which resulted in the identification of 83 proteins (16). In the following years, low resolution MS analysis of plasma microparticles reached up to 229 plasma microparticle proteins and high resolution MS analysis reached 458 proteins (all without false discovery rate (FDR)1 correction)(17, 18). The latest and most comprehensive study of plasma microparticles proteome profiling was published in 2012 by Ostergaard et al., who analyzed 12 samples on the LTQ Orbitrap XL mass spectrometer and identified 536 proteins in total, after 1% FDR correction (19). Other studies have profiled the proteomes of microparticles and exosomes derived from various body fluids other than plasma, including urine (20), saliva (21), cerebral spinal fluid (22), breast milk (23), amniotic fluid (24), seminal fluid (25), and more. However, despite the dramatic reduction of the dynamic range of the analytes, so far it has not yet provided sufficient depth for biomarker discovery. Nevertheless, it has a good prospective for discovering biomarkers. For example, biochemical analysis of breast cancer patient leukocytes-derived microparticles correlated between increased tumor size and increased levels of carcinoembryonic antigen (CEA) and cancer antigen 15-3 (CA15-3), two well-known prognostic markers for colon and breast cancer, respectively (26).
Combining all of the plasma proteomics approaches mentioned above, several prominent surveys of the human plasma proteome have been reported. The first large-scale collaborative study was conducted by the Human Proteome Organization (HuPO) group, which collectively identified 3020 proteins (7). These were later condensed to a list of 889 nonredundant proteins, after taking into account multiple hypotheses control with at least 95% confidence in protein identification (27). The Peptide Atlas team initially combined 91 studies, including the one conducted by HuPO, and altogether produced a list of 1929 proteins (28). Recently this team has elaborated their survey by assembling 127 studies (29) and reached the largest high-confidence list published so far of overall 3677 plasma proteins.
In the current work we applied state of the art proteomics to study the microparticle proteome and developed the PROteomics of MIcroparticles with Super-SILAC Quantification (PROMIS-Quan) method, which combines deep plasma microparticle coverage of more than 3200 proteins in a single run, with dual-mode relative and absolute Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) quantification. We demonstrated its utilization on samples of prostate cancer patients, and calculated the absolute amount of PSA, a well-known prostate cancer biomarker.
EXPERIMENTAL PROCEDURES
Plasma Microparticles Extraction
Blood samples were collected from healthy male donors and from prostate cancer patients that started hormonal anti-androgenic (GnRH agonist) therapy two months prior to radiotherapy treatments. The first blood sample for each patient was taken before radiation therapy; the second blood test was performed 24 h after the first radiation treatment; the third test was performed 2 weeks after the first radiation treatment. In parallel to the radiation therapy the patients resumed with the hormonal treatment. All samples were collected upon institutional ethical approval.
Plasma was separated from the blood samples by centrifugation at 1500 × g for 10 min at 4 °C followed by a second centrifugation of the supernatant and storage of the plasma supernatant at −80 °C. For the isolation of microparticles, plasma samples were thawed on ice to avoid lysis of the microparticles before their separation from the plasma sample, and then centrifuged at 4000 rpm for 20 min at 4 °C. Supernatants were diluted twofold in ice-cold PBS and centrifuged at 20,000 × g at 4 °C for 1 h. Pellets were washed with ice-cold PBS and centrifuged again at 20,000 × g at 4 °C for 1 h. Solubilization of the microparticle pellets was done in lysis buffer containing 6 m urea, 2 m thiourea in 50 mm ammonium bicarbonate. Each microparticle sample from the healthy donors was extracted from ∼3 ml of plasma. Microparticles from prostate cancer patients and the healthy controls of that experiment were extracted from 0.5 ml of plasma.
Preparation of Super-SILAC Mix
SILAC labeling was performed by culturing MDA-MB-231, HeLa, HepG2, RKO, and U2OS cells in SILAC-DMEM, namely DMEM devoid of the natural lysine and arginine and supplemented with 13C615N2-lysine, 13C615N4-arginine, and with dialyzed FBS and antibiotics. LNCaP and Jurkat cells were labeled in SILAC-RPMI with the same supplements. Labeled amino acids were purchased from Cambridge Isotope Laboratories. Cells were cultured for more than 10 doublings in the SILAC medium to attain complete labeling, and the incorporation was examined by separate LC MS/MS analyses. Lysate super-SILAC mix was obtained by lysing cell pellets in 6 m urea, 2 m thiourea in 50 mm ammonium bicarbonate. For both secretome and CLMP extraction, cells were cultured in SILAC serum-free medium for 48 h, followed by centrifugation at 4000 rpm for 20 min at room temperature. The supernatants for secretome samples were diluted 1:1 in 8 m urea prior to trypsin digestion. For CLMP extraction, medium was diluted twofold in ice-cold PBS followed by high-speed centrifugation (1h at 20,000 × g at room temperature). Microparticle pellets were then solubilized in 6 m urea, 2 m thiourea in 50 mm ammonium bicarbonate. Bradford protein determination was used in preliminary experiments, and showed that 1 ml of plasma results in ∼10 μg of microparticle proteins. Combination of the super-SILAC and the microparticle proteins (1:1 ratio) was based on this calculation.
Trypsin Digestion and LC-MS/MS Analysis
All samples (microparticle proteins with or without the super-SILAC standards, PSA calibration curve samples) were reduced with 1 mm DTT, followed by alkylation with 5 mm iodoacetamide and subsequent 3h digestion with endoproteinase Lys-C (Wako Chemicals, Osaka, Japan; 1:100 enzyme to protein ratio). Lysates were diluted fourfold in 50 mm ammonium bicarbonate and digested overnight with sequencing grade modified trypsin (Promega, Madison, WI; 1:50 enzyme to protein ratio). The resulting peptides were acidified with TFA and subsequently purified on C-18 stageTips (30).
LC-MS/MS analysis was performed on the EASY-nLC1000 UHPLC system (Thermo Scientific) coupled to the Q-Exactive or Q-Exactive Plus mass spectrometers (Thermo Scientific) (31) via the EASY-Spray ionization source. Peptides were loaded onto 75 μm i.d. × 50 cm long EASY-spray PepMap columns (Thermo Scientific) packed with 2 μm C18 particles 100 Å pore size, using 4 h gradients at a flow rate of 300 nl/min with buffer A (0.1% formic acid) and separated using a 7–28% buffer B (80% acetonitrile, 0.1% formic acid). MS data were acquired in a data-dependent mode, using a top-10 method. MS spectra were acquired at 70,000 resolution, m/z range of 300–1700 Th, a target value of 3E+06 ions, and a maximal injection time of 20 ms. MS/MS spectra were acquired after HCD fragmentation, with normalized collision energy (NCE) of 25 at 17,500 resolution a target value of 1E+05 ions and maximal injection time of 100 ms. Comparison of super-SILAC types was performed with 5E+05 ions and maximal injection time of 60 ms. Dynamic exclusion was set to 20 or 30 s. All MS measurements were done in the positive ion mode. Single-peptide-based protein identification are listed in supplemental Table S1. MSMS spectra of these peptides are uploaded to Pride (link below).
Computational Analysis
Raw MS files were analyzed with MaxQuant (32) (versions 1.5.0.36 and 1.4.3.2) and the Andromeda search engine (33) integrated into the same versions. MS/MS spectra were searched against the UniprotKB database version Nov2014 including 140,992 entries, a decoy database in which all sequences were reversed and each lysine and arginine were swapped with their preceding amino acid, and a list of common contaminants (247 entries). Search included tryptic peptides with the variable modifications N-terminal acetylation (+42.0106 Da) and methionine oxidation (+15.99491 Da) and the fixed modification of carbamidomethyl cysteine (+57.02146 Da). Maximal number of miscleavages was set to two and maximal number of modifications was set to five. MaxQuant analysis included two search engine steps. The first was used for mass recalibration, and was initiated with a peptide mass tolerance of 20 ppm. The main database search peptide initial mass tolerance was set to 4.5 ppm, and mass tolerance for the fragment ions was set to 20 ppm. Database results were filtered to have a maximal FDR of 0.01 on both the peptide and the protein levels. The minimal peptide length was seven amino acids and a minimum number of peptides per protein was set to one. The “second peptide search” option was enabled to allow identification of two cofragmented peptides. For protein assembly, all proteins that cannot be distinguished based on their identified peptides were assembled into a single protein group. Analysis of SILAC experiments included Lys-8 and Arg-10 as the heavy labels, and enabled the requantify option. For SILAC ratios determination a minimum of two ratio counts between SILAC peptide pairs was required. The “match between runs” option was enabled only in the prostate cancer analysis and PSA calibration curve, for transfer of identification between separate LC-MS/MS runs based on their accurate mass and retention time, with a 1 min match window after retention time alignment. Data analysis was performed on the proteinGroups.txt file after filtration of the proteins that were identified in the reverse database, proteins that were identified only based on their variable modifications and potential contaminants (without excluding potential plasma proteins, such as albumin, hepatocyte growth factor activator, keratins, and thrombospondin).
All bioinformatic analyses were performed on either log2 or log10 scales. Quantification of nonlabeled samples was performed using the intensity values. SILAC-labeled samples were analyzed based on the normalized ratio light to heavy (L/H) after normalization by subtraction of the median value in each sample (to ensure overall comparable protein distribution of samples). Statistical tests and calculations were done using the Perseus program and Matlab. Machine learning algorithms were used to obtain a predictive signature that can distinguish between samples from healthy donors and samples derived from prostate cancer patients. Data were filtered to retain only proteins with numerical values in at least 19 of 28 samples. Ratio values toward the super-SILAC mix were then imputed by replacing missing values with random values that create a normal distribution with a downshift of 1.0 standard deviation and a width of 0.3 of the original distribution. Support vector machine algorithm was used for classification with linear Kernel and ANOVA-based feature ranking. For cross validation the random sampling algorithm was used with 15% of the samples utilized as the test case, and repeating the calculation 250 times. The number of features was chosen based on the lowest error percentage. Classification using the same parameters with the top three ranked features was performed to calculate true positive, true negative, false positive, and false negative rates. Welch's t test was performed with permutation-based FDR 0.05 and S0 = 0.5 (34). Hierarchical clustering was done after z-score normalization of the proteins, and was based on Euclidean distances between averages. Coefficient of variation was determined by comparing protein ratios in three replicates.
PSA Concentration Assays
For MS-based microparticle PSA quantification, equal amounts of super-SILAC mix were combined with final concentrations of 0.5, 2, 10, 50, and 200 ng/ml purified PSA (Prostate specific antigen; Merck Millipore). Calculation of PSA concentration was done by extrapolation from the calibration curve in log2 scale. PSA ELISA kit (R&D Systems, Minneapolis, MN) was utilized according to manufacturer's instructions to determine plasma PSA concentrations.
RESULTS
Deep Coverage of Plasma Microparticle Proteome
Unbiased biomarker discovery in plasma samples requires sufficient depth to achieve “tissue leakage” protein identification, high reproducibility, and short analytical duration. We therefore avoided protein and peptide fractionation, and rather examined the plasma microparticle proteome coverage using single LC-MS/MS runs. Moreover, we optimized the analytical procedure to minimize sample loss and minimize contaminations of core plasma proteins, which are expected to reduce the observable dynamic range. Microparticle isolation was performed by high-speed plasma centrifugation (20,000 × g), which precipitates the microparticles (100 nm–1 μm), but not the exosomes (40–100 nm). To eliminate large quantities of core plasma proteins that may attach to the vesicles, we added a subsequent PBS wash of the pellet followed by an additional centrifugation step. For protein digestion, we selected the in-solution procedure, which ensures minimal sample loss. Microparticle solubilization was performed in urea-based buffer and was followed by an over-night trypsin digestion. Single 4 h LC-MS/MS runs on the Q-Exactive mass spectrometer identified on average 3294 proteins per replicate (peptide and protein FDR = 0.01). Triplicate single-shot analysis identified 28,409 peptides and 3689 proteins (Fig. 1A; supplemental Table S2A; Supplemental Table S3A). In comparison, similar analysis of unfractionated plasma identified only 451 proteins (Fig. 1B; supplemental Table S2A; supplemental Table S3A). Remarkably, the Peptide Atlas database, which is comprised of 127 studies (29), includes a similar number of proteins. Furthermore, these experiments add 2074 plasma-derived proteins to the annotated ones (Fig. 1C).
Fig. 1.
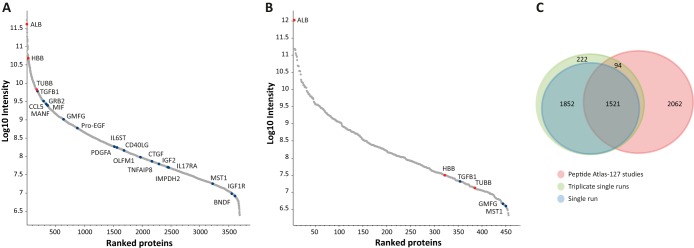
Deep analysis of plasma microparticle proteomes. A, Triplicate single runs of microparticle proteomes identified 3689 proteins ranging from albumin to cytokines. B, Single run of unfractionated plasma identified 451 proteins. Selected tissue-leakage proteins are marked in blue dots; core plasma proteins are marked in red dots. C, Numeric comparison between different methods. Venn diagram showing the overlap between the proteins identified in single LC-MS/MS run, triplicate single runs, and the plasma proteins included in the Peptide Atlas (127 studies) (29).
The dynamic range of the plasma microparticle proteome spanned from albumin and hemoglobin, which are among the most abundant plasma proteins, to cytokines and other secreted factors, such as CCL5 (chemokine ligand 5), MANF (mesencephalic astrocyte-derived neurotrophic factor), GMFG (glia maturation factor, gamma), PDGFA (platelet-derived growth factor A), IGF2 (insulin-like growth factor 2) and MIF (macrophage migration inhibitory factor), among the lowest ones (Fig. 1A). Examination of the plasma concentration of these proteins, as reported in the Plasma Proteome Database (PPD) (35) showed that the microparticle analysis reaches proteins with plasma concentrations of 11 pg/ml (IMPDH2, average PPD concentrations, three peptides), 2.9 ng/ml (Grb2, average PPD concentration, 15 peptides), and additional growth factors/cytokines, which could not be identified in the unfractionated plasma sample (supplemental Fig. S1; selected examples are given in supplemental Table S4). The microparticle proteome identified 276 proteins that were reported to have a concentration lower than 10 ng/ml, whereas the unfractionated plasma, on the other hand, identified only 14 proteins within this range (Fig. 2A). Interestingly, we found low correlation between the intensities of the proteins from the microparticles and the concentrations of the soluble proteins, which shows that the fractionation isolated a distinct sub-proteome of plasma proteins. The ability to reach these low abundance proteins was achieved because of the dramatic reduction in the fraction of abundant proteins. In the unfractionated plasma, 43% of the overall intensity corresponded to albumin, whereas in the microparticle proteome albumin accounted for only 5% of the total intensity. Similarly, 36% of the total intensity originated from the top 10 microparticle proteins, whereas the same number of proteins was responsible for 76% of the intensity of the plasma proteome (Fig. 2B).
Fig. 2.

Proteins abundance in the plasma. A, Intensities of identified proteins as a function of their concentration in the plasma/serum (average values of reported PPD concentrations (35)). B, Cumulative protein intensity of fractionated and unfractionated plasma proteome. Proteins were ranked from the most to the least abundant. Blue, plasma microparticle derived proteins; green, unfractionated plasma proteins.
Among the identified proteins the microparticle proteome provided a rich source of potential biomarkers, as defined by Huttenhain et al. (36). The intensities of these were reproducible and did not concentrate within the lower range of intensities, but rather showed a wide range (Fig. 3A). Furthermore, technical triplicates involving separate microparticle isolation and LC-MS/MS analysis showed an average correlation of 0.93 and 91% overlap between replicates (Fig. 3B), demonstrating the high technical reproducibility of the method. Overall, these results show the potential of microparticle analysis as a platform for unbiased biomarker discovery.
Fig. 3.

Reproducibility of microparticle proteome extraction. A, Potential biomarkers (36) are colored in blue. B, High correlation between technical replicates. The plot is colored according to density.
SILAC-based Relative Quantification Using PROMIS-Quan
Clinical assessment of biomarkers requires their accurate quantification and determination of their absolute amounts. Combination of the microparticle proteins with known amounts of heavy standards can enable quantitative measurement of these specific proteins, but would require remeasurement of the clinical sample for each biomarker of interest. To gain a more comprehensive quantification potential, we established PROMIS-Quan as a two-step approach: First, microparticle proteomes are quantified against a super-SILAC mix that serves as an internal standard (37). Next, the super-SILAC mix is quantified relative to purified proteins of interest, with known absolute amounts (Fig. 4). This dual-mode SILAC quantification approach provides relative quantification of large proportions of the microparticle proteome, and absolute quantification can be determined retrospectively only relative to the super-SILAC standard.
Fig. 4.
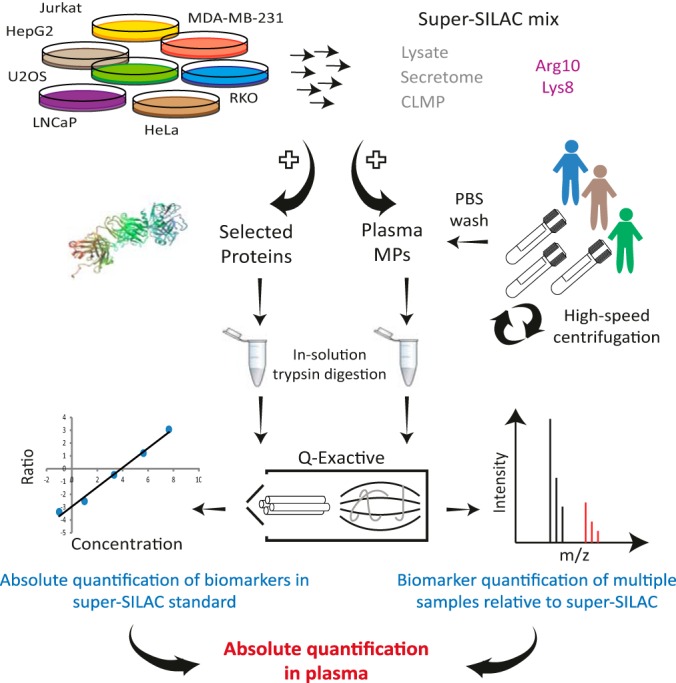
PROMIS-Quan enables relative and absolute microparticle proteome quantification. PROMIS-Quan workflow includes microparticle quantification of multiple samples relative to a super-SILAC mix, followed by absolute quantification of candidate proteins in the super-SILAC standard.
For the development of a SILAC internal standard, we assembled a panel of seven cancer cell lines, which represents various tissues and cancer types to serve as the quantitative reference. As opposed to the previously established super-SILAC mix, which is used for tissue quantification (37, 38), the current super-SILAC is aimed to represent secreted microparticle proteomes. We therefore examined three types of super-SILAC mixes: (1) Cell lysates. (2) Cell culture medium (secretome). (3) Cell line microparticles (CLMPs), namely microparticles isolated from cell culture medium. We combined each of these standards with the plasma microparticles and analyzed them as described above. Single LC-MS/MS runs led to the quantification of 2473, 1992, and 910 proteins for lysate, CLMPs and secretome, respectively (Supplemental Tables S2B and S3B). The fraction of quantified proteins of the total identified proteins was ∼70% for the lysates and CLMPs and ∼60% for the secretome. In the next steps we concentrated on the two finest methodologies, namely CLMPs and lysate standards. Their comparison showed a similar overall distribution, which included a main distribution with the same width for lysate and CLMP standards (FWHM = 2.8). An additional small distribution was highly enriched with core plasma proteins (FDR = 10−30 and 10−6 for lysate and CLMPs, respectively), which are not represented in our standard, yet they are less likely to include candidate biomarkers (Fig. 5; supplemental Fig. S2; supplemental Table S3A; core plasma protein list was taken from Fig. 1B). We further determined the reproducibility of the ratio measurements, and found a median coefficient of variation of 0.25 and 0.26 for the lysates and the CLMP standard, respectively (supplemental Fig. S3; supplemental Tables S2C and S3C). Based on the high similarity of these two standard types, and given the marked difference in protein amounts that are extracted from lysates versus CLMPs, we proceeded using the lysate-based super-SILAC mix.
Fig. 5.
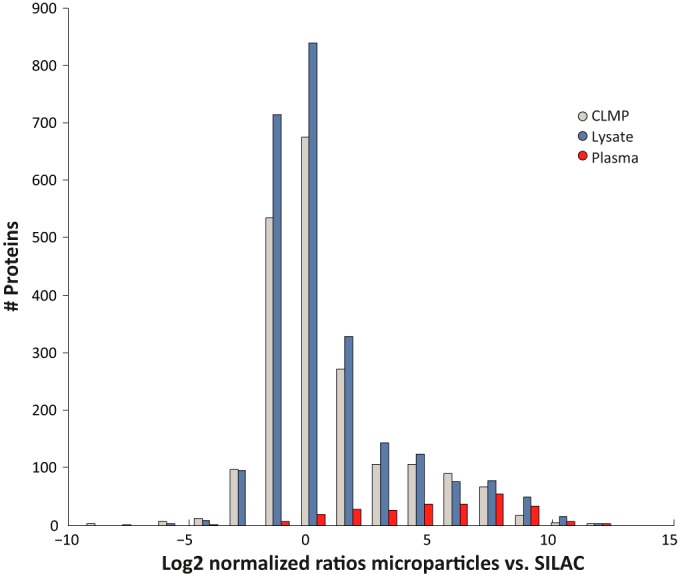
Two super-SILAC standard types. Histogram shows a similar distribution of the normalized ratios in the lysate- and CLMP-based super-SILAC mix. Core plasma proteins, as defined in Fig. 1B, show high ratios toward the super-SILAC mix.
Absolute Quantification of Selected Proteins
The relative quantification enables a direct comparison between any number of samples. Such a comparison can give rise to various potential biomarkers, yet their absolute levels have to be determined for further clinical evaluation. The dual-mode SILAC approach requires determination of the absolute protein amounts only in the super-SILAC standard, and the amounts in each one of the samples can be extrapolated retrospectively. We examined the applicability of PROMIS-Quan for cancer biomarker quantification in plasma samples from prostate cancer patients. As a proof of concept, we quantified the prostate cancer marker Prostate Specific Antigen (PSA/KLK3) (39, 40), and compared microparticles before and after treatment. LC-MS/MS analysis of patient-derived microparticles combined with the super-SILAC lysate enabled quantification of 3076 proteins and specifically identified PSA with two peptides and a 0.24- and 0.17-fold ratio toward the standard before and after treatment, respectively. To determine the absolute amount of PSA in the super-SILAC mix we created a dilution series of unlabeled purified PSA combined with the super-SILAC mix (Fig. 6A). Extrapolation from the calibration curve showed that microparticle PSA levels reduced from 2.3 ng/ml to 1.5 ng/ml upon treatment (Fig. 6B). To validate the MS results, we examined the absolute PSA amounts in the plasma of the same patients using an ELISA assay. Despite differences between the absolute PSA levels in the soluble plasma and the microparticles (as expected from Fig. 2A), we found an identical reduction of 35% upon treatment (Fig. 6B).
Fig. 6.
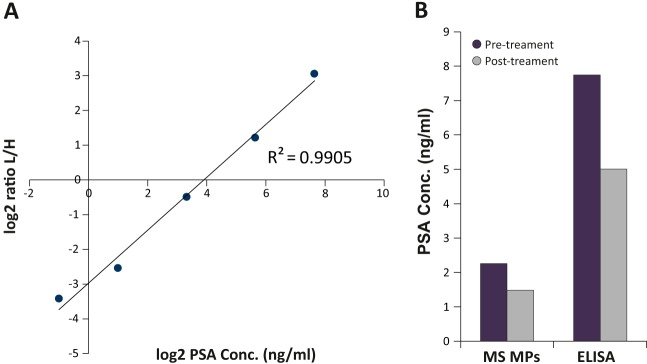
Relative and absolute quantification of PSA. A, dilution series of purified unlabeled protein provides absolute PSA levels in the super-SILAC standard. B, Quantification of treatment-induced reduction in microparticle-PSA using PROMIS-Quan, and the plasma PSA quantification by ELISA.
Biomarker Discovery Using PROMIS-Quan
We examined whether PROMIS-Quan can reveal novel biomarkers in patient-derived microparticles. To that end we compared the plasma microparticle proteome of 12 healthy donors and 16 samples derived from seven prostate cancer patients during the first 2 weeks of radiotherapy treatment (supplemental Tables S2D and S3D). Altogether 5374 different protein groups were identified, 3231 proteins were identified on average in each sample, and 2167 of those were quantified with the super-SILAC mix. We performed a Welch's t test to identify the significantly changing proteins between the healthy and prostate cancer samples. We extracted a signature of 132 proteins that were significantly higher in the prostate cancer patients microparticles and 46 proteins that were higher in healthy donors samples (FDR < 0.05, S0=0.5; supplemental Fig. S4; supplemental Table S5). To further evaluate the predictive value of these proteins, we used support vector machine classification algorithm, ANOVA feature ranking method and random sampling for cross validation, and obtained a signature that includes three proteins that can distinguish between patient- and healthy-derived samples, as shown in the principal component analysis (PCA; Fig. 7A). A receiver operating characteristics analysis resulted in area under the curve of 0.84 (Fig. 7B). The predictive signature included PTPN1, SFXN3, and LPP (p values of 1.36E−05, 3.12E−06, and 7.1E−06, respectively; Fig. 7C). PTPN1 (protein tyrosine phosphatase nonreceptor type 1) was previously found to be correlated with prostate cancer progression (41); LPP (LIM protein) is involved in cell adhesion; SFXN3 (sideroflexin-3) was previously suggested as a serum tumor marker for oral squamous cell carcinoma (42). Altogether, using PROMIS-Quan we were able to capture significant differences between healthy and prostate cancer plasma microparticle proteins and identified candidate markers of the disease.
Fig. 7.
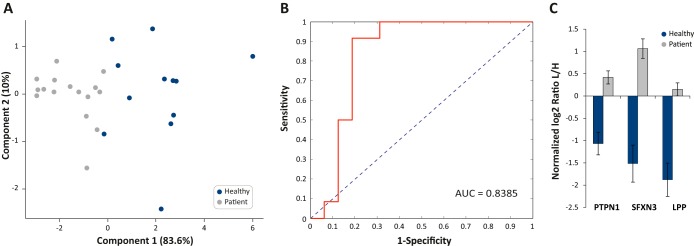
Plasma microparticle-based proteomic signature of prostate cancer patients. A, Principal component analysis (PCA) of healthy donors samples and samples derived from prostate cancer patients segregates between the two groups. B, Receiver operating characteristic curve shows high predictive power for the selected protein signature of biomarkers for prostate cancer diagnosis. C, The fold changes of the three proteins that comprise the predictive biomarkers. Values are in log2 normalized ratio L/H.
DISCUSSION
Biopharmaceuticals analyses of biological specimens mostly rely on ligand binding assays (LBAs) as the standard analytical technique. LBAs utilize antibodies raised against the protein of interest and offer very sensitive and selective results (43). Though LBAs require lower investment in analytical equipment and have straightforward/high-throughput protocols (44), in recent years more and more LC-MS(/MS) techniques, mostly targeted-based methods, began to replace the conventional LBA methods. These MS-based techniques have improved selectivity and linear dynamic range than antibody-based methods, and the use of internal standards can correct for different sources of analytical variability. In the current work we present a novel high-throughput, unbiased, simple, and cost-effective biomarker discovery and quantification platform. In contrast to most alternative techniques, PROMIS-Quan enables biomarker discovery in the plasma samples themselves, and does not rely on mere prediction based on cell line or tissue analyses. For comparison, the well-established SISCAPA technique, which provides absolute measures of candidate peptides that are immunoprecipitated from the plasma, requires multiple method development steps for each protein. These include candidate selection based on extensive tissue analysis and computation predictions, development of peptide-specific antibodies, synthesis of heavy peptide standards and the development of targeted MS-methods (9). Thus, massive investment is required prior to the identification of the protein as a bona fide biomarker. PROMIS-Quan provides an exceptional coverage of the plasma subproteome, and thus enables comprehensive profiling of tissue leakage proteins in the blood. The high coverage reached in single runs provides the throughput necessary for the analysis of large patient cohorts. This high coverage can be further elevated by peptide/protein fractionation in the future.
The current work identified sixfold more proteins than the largest study reported so far, in only single LC-MS/MS runs, and 10-fold more proteins in one entire dataset (5374 proteins; prostate cancer patients). This dramatic improvement lies in the optimization of several analytical steps: (1) Plasma separation involved two 10 min centrifugation steps at 1500 × g at 4 °C. Higher speed (over 2000 × g), which is often used in various studies, was shown to result in a considerable loss of microparticles (45). (2) Efficient microparticle isolation from the plasma was achieved using longer high-speed centrifugation (1h versus 20–30 min). (3) An additional PBS wash of the microparticle pellet reduced the levels of highly abundant plasma proteins that mask the low abundance proteins. In agreement with this, Ostergaard et al. showed that multiple washes of the microparticle pellet reduces the signals of albumin thus affect microparticle protein yield (19). (4) The use of LoBind eppendorf tubes for microparticle extraction provided higher yield per starting volume compared with large-volume, round bottom tubes (supplemental Table S6). (5) In-solution protein digestion dramatically reduced sample loss. Alternative microvesicle extraction protocols include sucrose gradients or filtration steps for cells removal, which may cause large microvesicle loss. Additionally, most previous studies processed the microparticle proteomes using in-gel digestion, which further reduces the yield compared with the in-solution procedure used here. 6) Combination of high resolution MS and high resolution chromatography dramatically increased the number of identified proteins.
Another factor that might influence the number of identified proteins is microvesicles subdivision. Here we chose to use microparticles rather than exosomes because: (1) Microparticle proteins are not limited to the secretory pathway as they originate from the cytoplasm, and thus, better reflect the variety of proteins of the cell of origin. (2) Experimentally, microparticles extraction is simpler than exosomes, as it requires high speed centrifugation rather than ultracentrifugation. Additionally, some of the studies divided the plasma microvesicles population into specific origins (platelets, red blood cells, leukocytes, etc.). For example, a recent study focused on a subset of plasma microparticles, the platelet-derived microparticles. They used in-solution digestion protocol followed by analysis in hybrid LTQ orbitrap XL and identified 603 proteins in nine samples (46). Thus, orienting the analysis toward a subset of the microvesicles leads to less identified proteins.
As a proof of concept, we examined our ability to successfully capture potential biomarkers when analyzing plasma microparticle proteomic profiles. To that end we tested PROMIS-Quan on prostate cancer samples and compared those to healthy donors. We identified a predictive signature that includes three proteins, all of which were higher in patients samples compared with healthy samples. Interestingly, one of the proteins, PTPN1, is a tyrosine phosphatase and a direct target of androgen receptor. PTPN1 is frequently amplified in metastatic tumors and high risk primary tumors (47). Down-regulation of PTPN1 correlates with better prognosis by delaying tumor occurrence, decreasing tumor growth rates and inhibiting cell migration (41). The predictive signature included two other proteins, LPP and SFXN3, which to our knowledge were not previously associated with prostate cancer. Future research can examine the broad applicability of these potential biomarkers for prostate cancer diagnosis.
The unique advantage of PROMIS-Quan is the usage of a dual-mode super-SILAC standard, which provides both relative and absolute quantification. As opposed to quantification using selected candidates with heavy labeled standards, the super-SILAC quantifies thousands of proteins in each sample, and thus enables extraction of a combination of proteins as biomarker signatures. Absolute quantification is achieved in the second step of quantification of super-SILAC proteins using unlabeled standards. The main benefits of this approach are that absolute quantification is performed relative to the standard, therefore it does not require remeasurement of the clinical sample for each biomarker, but can be done retrospectively once the candidate is established as a valuable biomarker. Furthermore, absolute quantification does not involve the synthesis of heavy standards, but rather utilizes nonlabeled purified proteins. The use of super-SILAC mix as an internal standard provides higher accuracy and stability than either label-free quantification or chemical labeling approaches, and thus the same standard can be used in multiple studies, laboratories and clinics, and can serve as the basis for large biomarker meta-analyses. Finally, because microparticles are shed from all cells and tissues, PROMIS-Quan can be applied to a large variety of disease states. We envision that this technology will be utilized in routine blood tests, and will reveal multiple biomarkers in single tests.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD001194.
To access the data please visit: http://tinyurl.com/mv35xpk.
Supplementary Material
Acknowledgments
We thank the members of the Geiger laboratory for fruitful discussions and critical reading of the manuscript. We thank Juergen Cox for statistical advice. We thank Ella Fremder for her assistance in microparticles extraction.
Footnotes
Author contributions: M.H. and T.G. designed research; M.H. and P.O. performed research; O.K. and Y.S. contributed new reagents or analytic tools; M.H. and T.G. analyzed data; M.H. and T.G. wrote the paper; Y.S. and T.G. supervised the research.
* This work was funded by the Israel Science Foundation (grant 1617/12) and by the Israel Center of Research Excellence program (I-CORE, Gene Regulation in Complex Human Disease Center No 41/11).
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S6.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S6.
1 The abbreviations used are:
- FDR
- false discovery rate
- PROMIS-Quan
- PROteomics of MIcroparticles with Super-SILAC Quantification
- SILAC
- stable isotope labeling with amino acids in cell culture
- CLMP
- cell line microparticles
- PSA
- prostate specific antigen
- DMEM
- Dulbecco's Modified Eagle Medium
- RPMI
- Roswell Park Memorial Institute
- FBS
- fetal bovine serum
- UHPLC
- ultra high performance liquid chromatography
- FWHM
- full width at half-maximum.
REFERENCES
- 1. Hanash S. M., Pitteri S. J., Faca V. M. (2008) Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 [DOI] [PubMed] [Google Scholar]
- 2. Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Q., Faca V., Hanash S. (2011) Mining the plasma proteome for disease applications across seven logs of protein abundance. J. Proteome Res. 10, 46–50 [DOI] [PubMed] [Google Scholar]
- 4. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whiteaker J. R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., Yan P., Schoenherr R. M., Zhao L., Voytovich U. J., Kelly-Spratt K. S., Krasnoselsky A., Gafken P. R., Hogan J. M., Jones L. A., Wang P., Amon L., Chodosh L. A., Nelson P. S., McIntosh M. W., Kemp C. J., Paulovich A. G. (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgess M. W., Keshishian H., Mani D. R., Gillette M. A., Carr S. A. (2014) Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol. Cell. Proteomics 13, 1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Omenn G. S., States D. J., Adamski M., Blackwell T. W., Menon R., Hermjakob H., Apweiler R., Haab B. B., Simpson R. J., Eddes J. S., Kapp E. A., Moritz R. L., Chan D. W., Rai A. J., Admon A., Aebersold R., Eng J., Hancock W. S., Hefta S. A., Meyer H., Paik Y. K., Yoo J. S., Ping P., Pounds J., Adkins J., Qian X., Wang R., Wasinger V., Wu C. Y., Zhao X., Zeng R., Archakov A., Tsugita A., Beer I., Pandey A., Pisano M., Andrews P., Tammen H., Speicher D. W., Hanash S. M. (2005) Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins, and a publicly-available database. Proteomics 5, 3226–3245 [DOI] [PubMed] [Google Scholar]
- 8. Whiteaker J. R., Zhang H., Eng J. K., Fang R., Piening B. D., Feng L. C., Lorentzen T. D., Schoenherr R. M., Keane J. F., Holzman T., Fitzgibbon M., Lin C., Cooke K., Liu T., Camp D. G., 2nd, Anderson L., Watts J., Smith R. D., McIntosh M. W., Paulovich A. G. (2007) Head-to-head comparison of serum fractionation techniques. J. Proteome Res. 6, 828–836 [DOI] [PubMed] [Google Scholar]
- 9. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 10. Voloshin T., Fremder E., Shaked Y. (2014) Small but mighty: microparticles as mediators of tumor progression. Cancer Microenviron. 7, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iero M., Valenti R., Huber V., Filipazzi P., Parmiani G., Fais S., Rivoltini L. (2008) Tumor-released exosomes and their implications in cancer immunity. Cell Death Diff. 15, 80–88 [DOI] [PubMed] [Google Scholar]
- 12. Somasundaram R., Herlyn M. (2012) Melanoma exosomes: messengers of metastasis. Nat. Med. 18, 853–854 [DOI] [PubMed] [Google Scholar]
- 13. Thery C., Zitvogel L., Amigorena S. (2002) Exosomes: composition, biogenesis, and function. Nat. Rev. Immunol. 2, 569–579 [DOI] [PubMed] [Google Scholar]
- 14. Buzas E. I., Gyorgy B., Nagy G., Falus A., Gay S. (2014) Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 10, 356–364 [DOI] [PubMed] [Google Scholar]
- 15. Loyer X., Vion A. C., Tedgui A., Boulanger C. M. (2014) Microvesicles as cell–cell messengers in cardiovascular diseases. Circ. Res. 114, 345–353 [DOI] [PubMed] [Google Scholar]
- 16. Jin M., Drwal G., Bourgeois T., Saltz J., Wu H. M. (2005) Distinct proteome features of plasma microparticles. Proteomics 5, 1940–1952 [DOI] [PubMed] [Google Scholar]
- 17. Smalley D. M., Root K. E., Cho H., Ross M. M., Ley K. (2007) Proteomic discovery of 21 proteins expressed in human plasma-derived but not platelet-derived microparticles. Thromb. Haemost. 97, 67–80 [PubMed] [Google Scholar]
- 18. Little K. M., Smalley D. M., Harthun N. L., Ley K. (2010) The plasma microparticle proteome. Semin. Thromb. Hemost. 36, 845–856 [DOI] [PubMed] [Google Scholar]
- 19. Ostergaard O., Nielsen C. T., Iversen L. V., Jacobsen S., Tanassi J. T., Heegaard N. H. (2012) Quantitative proteome profiling of normal human circulating microparticles. J. Proteome Res. 11, 2154–2163 [DOI] [PubMed] [Google Scholar]
- 20. Pisitkun T., Shen R. F., Knepper M. A. (2004) Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. U.S.A. 101, 13368–13373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez-Begne M., Lu B., Han X., Hagen F. K., Hand A. R., Melvin J. E., Yates J. R. (2009) Proteomic analysis of human parotid gland exosomes by multidimensional protein identification technology (MudPIT). J. Proteome Res. 8, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Street J. M., Barran P. E., Mackay C. L., Weidt S., Balmforth C., Walsh T. S., Chalmers R. T., Webb D. J., Dear J. W. (2012) Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J. Transl. Med. 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Admyre C., Johansson S. M., Qazi K. R., Filen J. J., Lahesmaa R., Norman M., Neve E. P., Scheynius A., Gabrielsson S. (2007) Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 179, 1969–1978 [DOI] [PubMed] [Google Scholar]
- 24. Keller S., Rupp C., Stoeck A., Runz S., Fogel M., Lugert S., Hager H. D., Abdel-Bakky M. S., Gutwein P., Altevogt P. (2007) CD24 is a marker of exosomes secreted into urine and amniotic fluid. Kidney Int. 72, 1095–1102 [DOI] [PubMed] [Google Scholar]
- 25. Poliakov A., Spilman M., Dokland T., Amling C. L., Mobley J. A. (2009) Structural heterogeneity and protein composition of exosome-like vesicles (prostasomes) in human semen. Prostate 69, 159–167 [DOI] [PubMed] [Google Scholar]
- 26. Toth B., Nieuwland R., Liebhardt S., Ditsch N., Steinig K., Stieber P., Rank A., Gohring P., Thaler C. J., Friese K., Bauerfeind I. (2008) Circulating microparticles in breast cancer patients: a comparative analysis with established biomarkers. Anticancer Res. 28, 1107–1112 [PubMed] [Google Scholar]
- 27. States D. J., Omenn G. S., Blackwell T. W., Fermin D., Eng J., Speicher D. W., Hanash S. M. (2006) Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat. Biotechnol. 24, 333–338 [DOI] [PubMed] [Google Scholar]
- 28. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmstrom J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110 006353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farrah T., Deutsch E. W., Omenn G. S., Sun Z., Watts J. D., Yamamoto T., Shteynberg D., Harris M. M., Moritz R. L. (2014) State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven Human Proteome Project. J. Proteome Res. 13, 60–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, prefractionation, and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 31. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111 011015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p. p. b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 33. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 34. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nanjappa V., Thomas J. K., Marimuthu A., Muthusamy B., Radhakrishnan A., Sharma R., Ahmad Khan A., Balakrishnan L., Sahasrabuddhe N. A., Kumar S., Jhaveri B. N., Sheth K. V., Kumar Khatana R., Shaw P. G., Srikanth S. M., Mathur P. P., Shankar S., Nagaraja D., Christopher R., Mathivanan S., Raju R., Sirdeshmukh R., Chatterjee A., Simpson R. J., Harsha H. C., Pandey A., Prasad T. S. (2014) Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 42, D959–D965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huttenhain R., Soste M., Selevsek N., Rost H., Sethi A., Carapito C., Farrah T., Deutsch E. W., Kusebauch U., Moritz R. L., Nimeus-Malmstrom E., Rinner O., Aebersold R. (2012) Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4, 142ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 38. Shenoy A., Geiger T. (2014) Super-SILAC: current trends and future perspectives. Expert Rev. Proteomics 12, 13–19 [DOI] [PubMed] [Google Scholar]
- 39. Lilja H., Ulmert D., Vickers A. J. (2008) Prostate-specific antigen and prostate cancer: prediction, detection, and monitoring. Nat. Rev. Cancer 8, 268–278 [DOI] [PubMed] [Google Scholar]
- 40. Makarov D. V., Loeb S., Getzenberg R. H., Partin A. W. (2009) Biomarkers for prostate cancer. Annu. Rev. Med. 60, 139–151 [DOI] [PubMed] [Google Scholar]
- 41. Lessard L., Labbe D. P., Deblois G., Begin L. R., Hardy S., Mes-Masson A. M., Saad F., Trotman L. C., Giguere V., Tremblay M. L. (2012) PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 72, 1529–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murase R., Abe Y., Takeuchi T., Nabeta M., Imai Y., Kamei Y., Kagawa-Miki L., Ueda N., Sumida T., Hamakawa H., Kito K. (2008) Serum autoantibody to sideroflexin 3 as a novel tumor marker for oral squamous cell carcinoma. Proteomics Clin. Appl. 2, 517–527 [DOI] [PubMed] [Google Scholar]
- 43. Bronsema K. J., Bischoff R., van de Merbel N. C. (2012) Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 893–894, 1–14 [DOI] [PubMed] [Google Scholar]
- 44. van den Broek I., Niessen W. M., van Dongen W. D. (2013) Bioanalytical LC-MS/MS of protein-based biopharmaceuticals. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 929, 161–179 [DOI] [PubMed] [Google Scholar]
- 45. Dinkla S., Brock R., Joosten I., Bosman G. J. (2013) Gateway to understanding microparticles: standardized isolation and identification of plasma membrane-derived vesicles. Nanomedicine 8, 1657–1668 [DOI] [PubMed] [Google Scholar]
- 46. Capriotti A. L., Caruso G., Cavaliere C., Piovesana S., Samperi R., Lagana A. (2013) Proteomic characterization of human platelet-derived microparticles. Anal. Chim. Acta 776, 57–63 [DOI] [PubMed] [Google Scholar]
- 47. Wu C., Zhang L., Bourne P. A., Reeder J. E., di Sant'Agnese P. A., Yao J. L., Na Y., Huang J. (2006) Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 66, 1125–1135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.