

Abstract
We performed protein pKa calculations and molecular dynamics (MD) simulations of the calcium pump (sarcoplasmic reticulum Ca2+-ATPase (SERCA)) in complex with phospholamban (PLB). X-ray crystallography studies have suggested that PLB locks SERCA in a low-Ca2+-affinity E2 state that is incompatible with metal-ion binding, thereby blocking the conversion toward a high-Ca2+-affinity E1 state. Estimation of pKa values of the acidic residues in the transport sites indicates that at normal intracellular pH (7.1–7.2), PLB-bound SERCA populates an E1 state that is deprotonated at residues E309 and D800 yet protonated at residue E771. We performed three independent microsecond-long MD simulations to evaluate the structural dynamics of SERCA-PLB in a solution containing 100 mM K+ and 3 mM Mg2+. Principal component analysis showed that PLB-bound SERCA lies exclusively along the structural ensemble of the E1 state. We found that the transport sites of PLB-bound SERCA are completely exposed to the cytosol and that K+ ions bind transiently (≤5 ns) and nonspecifically (nine different positions) to the two transport sites, with a total occupancy time of K+ in the transport sites of 80%. We propose that PLB binding to SERCA populates a novel (to our knowledge) E1 intermediate, E1⋅H+771. This intermediate serves as a kinetic trap that controls headpiece dynamics and depresses the structural transitions necessary for Ca2+-dependent activation of SERCA. We conclude that PLB-mediated regulation of SERCA activity in the heart results from biochemical and structural transitions that occur primarily in the E1 state of the pump.
Introduction
The calcium pump (sarcoplasmic reticulum Ca2+-ATPase (SERCA)), a member of the P-type ATPase family, is an integral membrane protein that is responsible for the active transport of Ca2+ from the cytosol into the sarcoplasmic reticulum (SR) lumen of heart and muscle cells (1). Relaxation occurs when SERCA pumps two Ca2+ ions into the SR lumen using energy derived from hydrolysis of one ATP molecule and the countertransport of two protons (2,3). Closely related SERCA isoforms are also responsible for pumping Ca2+ into the endoplasmic reticulum of virtually all nonmuscle cells, potentiating a myriad of Ca2+-dependent cellular activation processes.
In cardiac muscle, SERCA activity is regulated by phospholamban (PLB), a small membrane protein that inhibits SERCA by decreasing its apparent Ca2+ affinity (4,5). Inhibition of SERCA is relieved physiologically either at elevated Ca2+ levels (6) or by phosphorylation of PLB at S16 by protein kinase A (PKA) (7), reversing calcium transport inhibition and greatly augmenting cardiac output (8). Initial attempts to modulate the SERCA-PLB interaction resulted in improved cardiac function in heart-failure animal models (9–13). A deeper understanding of the structural basis of SERCA regulation by PLB is necessary to improve the design of small-molecule and gene therapies for the treatment of human heart failure.
High-resolution spectroscopy, engineered chemical cross-linking, and extensive site-directed mutagenesis have shown that the transmembrane (TM) domain of PLB binds to a large groove formed around TM helices TM2, TM4, TM6, and TM9 of SERCA (14–20). Fluorescence resonance energy transfer (FRET) and NMR spectroscopy studies have shown that in the bound complex, the cytosolic domain of PLB natively undergoes a dynamic equilibrium between inhibitory (T) and noninhibitory (R) structural states, and that a shift between these states is essential for regulation of SERCA activity (21–24). Although the structural equilibrium of PLB is fundamental for SERCA regulation, the atomistic mechanisms by which the T state inhibits SERCA and the phosphorylation-induced R state relieves inhibition remain unknown. Akin et al. (25,26) recently reported a 0.28-nm-resolution crystal structure of SERCA1a bound to the gain-of-function, cross-linkable PLB mutant PLB4 (N27A, N30C, L37A, and V49G-PLB) (25) and in the absence of Ca2+ (26). Although the crystal structure is missing the regulatory cytosolic segment of PLB (residues M1–Q23), it yielded important data on the structural basis of SERCA inhibition by the TM domain of PLB (27). For instance, the crystal structure also revealed that PLB4 binding alters the structure of SERCA around residues V795–A804, resulting in a collapsed Ca2+ binding site I. Based on these observations, it was proposed that PLB inhibits SERCA by populating an E2 state that is incompatible with metal-ion binding (26).
Investigators have traditionally assumed that PLB stabilizes SERCA in E2, as suggested by kinetics, cross-linking, and co-immunoprecipitation. However, the assumption that PLB4 binding stabilizes SERCA in an E2 state is problematic for three reasons. 1) The structure of SERCA in the PLB complex is remarkably similar to the structure of sarcolipin-bound SERCA, which was assigned as an E1 intermediate state due to headpiece structure and Mg2+ binding to the transport sites (28,29). 2) The gating residue E309 points away from residues V304 and A305, allowing the transport sites to be exposed to the cytosol—a feature that is characteristic of E1, but not of E2 (30,31). 3) Experimental studies have shown that PLB binding decreases SERCA’s apparent affinity for Ca2+ by 2- to 3-fold in the submicromolar range (32). This moderate decrease in apparent Ca2+ affinity is incompatible with the millimolar-Ca2+-affinity E2 state of the pump (33).
Two models have been proposed to describe the physical mechanism of PLB regulation: the dissociation model and the subunit model. According to the dissociation model, PLB dissociates from SERCA to relieve inhibition, whereas in the subunit model, inhibition of SERCA is relieved by structural rearrangements within the SERCA-PLB complex, not by dissociation. Spectroscopic measurements obtained with fluorescent and paramagnetic probes have allowed direct detection of the SERCA-PLB interaction in live cells, endoplasmic reticulum membranes, and reconstituted vesicles, supporting the subunit model, with a subtle complex rearrangement in high Ca2+ or PLB phosphorylation (34–36). Cross-linking studies have been interpreted as supporting the dissociation model, but are also consistent with a structural rearrangement of the inhibitory complex (15,17). Major questions remain regarding the nature of the inhibitory interactions between SERCA and PLB: Does PLB-bound SERCA resolved in the crystal structure represents an E1 or an E2 state of the pump? How does PLB binding inhibit Ca2+ binding in the transport sites? What step(s) along the catalytic cycle of the pump is inhibited by PLB? To address these questions, we used the crystal structure of SERCA-PLB4 to obtain a full-length structure of SERCA bound to the inhibitory T state conformation of wild-type (WT) PLB, i.e., with the cytosolic helix of PLB perpendicular to the lipid bilayer normal. We used this model to perform three independent 1 μs MD simulations of the complex in the absence of Ca2+ and in a solution containing physiologically appropriate concentrations of other ions (100 mM K+, 3 mM Mg2+, and 110 mM Cl−). Finally, we determined the structural characteristics of PLB-bound SERCA by comparing the trajectories of the complex with those of the E1 and E2 states of the pump.
Materials and Methods
Protein pKa calculations
We used PROPKA 3.1 (37–40) to calculate the pKa values of acidic residues in the transport sites of SERCA. PROPKA estimates the empirical pKa values of ionizable groups in proteins and protein-ligand complexes based on the 3D structure, and benefits from explicitly incorporating the Coulombic interactions that arise from mutually titrating residues via the Tanford-Roxby procedure (41). PROPKA has previously been used to determine the protonation states of acidic residues in other P-type ATPases, e.g., the Na+-K+ ATPase (42,43). We calculated the pKa values in the E1 state using eight crystal structures retrieved from the Protein Data Bank (PDB): 1SU4, 1VFp, 3W5A, 3W5B, 3AR2, 1T5S, 4H1W, and 3N8G. We also performed pKa calculations on seven crystal structures of the E2 state: 1IWO, 2AGV, 2EAR, 2EAT, 2C8K, 3W5C, and 2EAU. In addition, we used the crystal structure of SERCA-PLB (PDB: 4KYT) to estimate the pKa values of the acidic residues in the complex. A list of the ligands and biochemical state represented by each crystal structure can be found in Table S1 in the Supporting Material.
Modeling of the full-length SERCA-PLB complex
The initial atomic model of WT PLB bound to SERCA was constructed using the crystal structure of SERCA-PLB4 as a template (26). Here, we modeled the complex in two stages: first, we reconstructed four loops of SERCA that were not resolved in the crystal structure of the complex: E45–S48, E79–T86, A241–D245, and N280–I289. Loop reconstruction was performed using MODELER (44). We used the crystal structure of recombinant E1⋅Mg2+ (29) as a template to reconstruct the missing loops of PLB4-bound SERCA, because the structure of the TM domain is virtually identical between the two structures (root mean-square deviation (RMSD) = 0.1 nm), and because TM alignment with E2 was especially poor in the regions where the loops are missing in the crystal structure of SERCA-PLB (Fig. S1). Second, we modeled the structure of the TM domain of rabbit PLB using the coordinates of dog PLB4 bound to SERCA. Only residues A27, C30, A37, and G49 of PLB4 were changed to N27, N30, L37, and V49 to match the sequence of rabbit PLB (Fig. S2). We constructed the structure of the cytosolic domain of PLB using model 14 of the NMR ensemble of the rabbit AFA-PLB mutant (PDB: 2KB7) (45). Details regarding the selection of the most suitable PLB structure and modeling of full-length PLB are provided in Supporting Materials and Methods.
Setting up SERCA-PLB for simulation
We adjusted the ionization states of the acidic residues of Ca2+-binding sites to match the results from our pKa calculations. Therefore, residues E309 and D800 were kept ionized, whereas residues E771 and E908 were modeled as protonated. In addition, we adjusted the pKa of other ionizable residues to a pH value of ∼7.2. The complex was inserted in a pre-equilibrated 12 × 12 nm bilayer of palmitoyl-2-oleoyl-sn-glycerol-phosphatidylcholine (POPC). This initial system was solvated using TIP3P water molecules with a minimum margin of 1.5 nm between the protein and the edges of the periodic box in the z axis. K+, Mg2+, and Cl− ions were added to produce concentrations of 100 mM, 3 mM, and 110 mM, respectively. The size of each protein/lipid/water/ions system was ∼220,000 atoms. CHARMM36 force-field topologies and parameters were used for the protein (80), lipid (81), water, K+, and Cl−. In addition, we used a set of new CHARMM parameters for Mg2+ developed by Allnér et al. (82). This new set of parameters for Mg2+ was chosen to correct the Mg2+-water exchange rate, as previous parameters do not correctly capture the water exchange kinetics between the first coordination shell and bulk water (82).
Setting up E2 SERCA for simulation
We used three crystal structures (PDB 1IWO (48), 2AGV (49), and 3W5C (29)) to simulate the structural dynamics of E2 SERCA. Residues E309, E771, and E908 were modeled as protonated, whereas D800 was left deprotonated. We adjusted the pH of the system to a value of 7.2 by adjusting the side-chain ionization states using PROPKA (39). Each protein system was inserted into a pre-equilibrated 12 × 12 nm POPC bilayer. This initial system was further solvated using TIP3P water molecules with a minimum margin of 1.5 nm between the protein and the edges of the periodic box in the z axis. Finally, K+, Mg2+, and Cl− ions were added in the same concentrations used for the SERCA-PLB systems. The size of each protein/lipid/water/ions system was ∼220,000 atoms. CHARMM36 force-field topologies and parameters with CMAP correction (80,81) were used for the protein, lipid, water, and ions.
Molecular dynamics simulations
We performed molecular dynamics (MD) simulations of SERCA-PLB and E2 SERCA using the program NAMD 2.9 (52) with periodic boundary conditions (53), particle mesh Ewald (54,55), a nonbonded cutoff of 0.9 nm, and a 2 fs time step. The NPT ensemble was maintained with a Langevin thermostat (310 K) and an anisotropic Langevin piston barostat (1 atm). Fully solvated systems were first subjected to energy minimization, and the systems were then gradually warmed up for 200 ps. This procedure was followed by 10 ns of equilibration, with backbone atoms harmonically restrained using a force constant of 1000 kcal mol−1 nm−2. We performed six independent 1 μs MD simulations (three of SERCA-PLB and three of E2 SERCA).
Cartesian principal component analysis
We used Cartesian principal component analysis (cPCA) to compare the structural ensembles of the SERCA-PLB, E1, and E2 states of the pump. cPCA uses the actual dynamics of the protein to generate the appropriate collective coordinates that capture the important structural and dynamic features of the native states of the protein (56). For cPCA, we aligned SERCA structures using the 10-helix TM domain as a reference. We had three purposes in selecting the TM domain as a reference: 1) to determine whether E1 and E2 occupy different structural spaces; 2) to show that E1 and E2 do not spontaneously interchange in the timescales used in this study; and 3) to illustrate the structural dynamics of the headpiece intrinsic to E1 and E2 states. We used Carma (57) to perform cPCA.
Results
Estimation of pKa values of acidic residues in the transport of PLB-bound SERCA
The acidic residues E309, E771, and D800 are essential for metal-ion binding and occlusion in transport sites (58). In particular, changes in the protonation states of E309 and E771 are crucial for the functional transition of SERCA at physiological conditions. For instance, deprotonation of E309 is required for the transition from E2 to E1 (31,59,60), and ionization of E771 is necessary to produce a competent transport site I in E1 (61). Thus, we performed empirical pKa calculations of these residues using the crystal structure of SERCA-PLB (PDB code: 4KYT) and compared the values against high-resolution crystal structures of the E1 and E2 states (Table S1). Although the crystal structures of E1 and E2 were crystallized with one or more bound ligands and under different conditions, experiments indicate that the forward and reverse E2-E1 transition is accompanied by the release and binding of two protons, respectively. Therefore, analysis of multiple crystal structures should be able to capture the protonation states inherent to E1 and E2, and thus help to determine whether PROPKA is sufficiently robust to capture protonation states consistent with experimental data. The pKa values for transport-site residues calculated from individual crystal structures are shown in Table S2.
Predicted pKa values of crystal structures of E2 SERCA indicate that at normal intracellular pH (7.1–7.2 (62)), residues E309, E771, and E908 are protonated. In the E2 state, D800 has a mean pKa value of 7.5, implying that this residue is in equilibrium between protonated and deprotonated states at physiological pH (Table 1). Musgaard et al. (63) found that among charged residues in the transport sites, only D800 is likely to be deprotonated in E2. Hence, we performed additional pKa calculations on MD-relaxed structures of the E2. We found that D800 is deprotonated in solution, in agreement with previous calculations (Table 1). pKa calculations also indicated that only residues E309, E771, and D800 are deprotonated in the E1 state of SERCA (Table 1). Therefore, E2 is protonated at E309, E771, and E908, whereas E1 is protonated only at E908. These findings are in excellent agreement with experimental data showing that the transition from E2 to E1 is coupled with the release of two H+ from the transport sites (2). These findings indicate that despite the inherent limitations of the model (approximate titration state/pKa assignments, implicit solvent limitations, and the use of static structures), PROPKA is sufficiently robust to predict the protonation states that are characteristic of E1 and E2 SERCA.
Table 1.
Predicted pKa values of acidic residues in the transport sites of SERCA
| State | E309 | E771 | D800 | E908 |
|---|---|---|---|---|
| E1 (x ray)a | 5.9 ± 1.0 | 3.9 ± 0.7 | 1.4 ± 0.6 | 8.8 ± 0.2 |
| E2 (x ray)a | 8.6 ± 0.4 | 10.7 ± 0.1 | 7.5 ± 0.2 | 10.0 ± 0.3 |
| E2 (MD)a,c | 9.1 ± 0.3 | 10.1 ± 0.2 | 5.5 ± 0.1 | 10.7 ± 0.3 |
| SERCA-PLB (x ray)b | 4.9 | 9.7 | 6.2 | 8.4 |
| SERCA-PLB (MD)a,c | 5.4 ± 0.3 | 9.2 ± 0.2 | 6.1 ± 0.1 | 7.9 ± 0.2 |
Reported values are mean ± SE.
pKa values calculated using a single crystal structure.
Values calculated at the end of 20 ns MD simulations; n = 5.
Next, we calculated the pKa values of the acidic residues in the transport sites of PLB-bound SERCA. We found that the acidic residues E908 and E771 are protonated at physiological pH (Table 1). This protonation pattern was reproduced in pKa calculations performed using relaxed structures obtained from independent 20 ns MD simulations of the complex in a fully solvated environment (Table 1). During the 20 ns trajectories, we found that the position of residues E771 and E908 remained virtually unchanged compared with crystal structures (RMSD ≤ 0.07 nm). This observation complements the pKa calculations and indicates that in the absence of Ca2+, E771 and E908 are protonated in PLB-bound SERCA.
Structural dynamics of SERCA-PLB in solution
We performed three independent 1 μs MD simulations of the complex to determine the structural stability and dynamics of PLB-bound SERCA protonated at E771 and E908. The structures of the initial model as SERCA-PLB, as well as the structure of the complex at the end of each MD simulation, are shown in Fig. 1 A. We calculated the backbone RMSD for each functional domain of SERCA and PLB to determine the time-dependent structural dynamics of the complex in the 1 μs trajectories (Fig. 1 B). At the beginning of each simulation, the structure of the 10-helix TM domain of PLB-bound SERCA underwent a 0.13 nm drift in the picosecond timescale (Fig. 1 B). This modest change in RMSD is attributed to the relaxation of the TM domain in a lipid/water environment. After this rapid relaxation period, the RMSD values remained virtually unchanged. These observations indicate that the structural arrangement of the TM domain observed in our trajectories is very similar to that determined by x-ray crystallography. Conversely, we observed a large variability in the RMSD values for each domain in the cytosolic headpiece of SERCA-PLB (Fig. 1 B). These findings are in line with previous studies showing that the cytosolic headpiece of SERCA is inherently flexible at physiological conditions (64–68).
Figure 1.
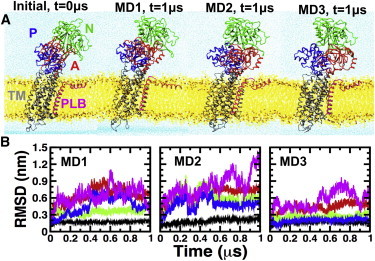
Structural dynamics of SERCA-PLB. (A) Initial and MD-relaxed structures of the SERCA-PLB complex embedded in a lipid bilayer (yellow) and water (blue). SERCA is colored according to its four functional domains: N (green), P (blue), A (red), and TM (gray). PLB is shown in magenta. (B) Time-dependent RMSD evolution of SERCA domains and full-length PLB in SERCA-PLB. The RMSD of the TM domain of SERCA and PLB was calculated using backbone alignment for TM helices of the pump. The RMSD of N, P, and A domains was calculated by aligning the backbone of the cytosolic headpiece with the structure at the beginning of each trajectory. To see this figure in color, go online.
The root mean-square fluctuation (RMSF) values of the main-chain Cα atoms show that the TM helix of PLB in the complex (residues A24–L51) has very low mobility in the complex (Fig. 2 A) and that it exclusively populates an α-helix structure in the microsecond timescale (Fig. S3). In addition, RMSD plots show that the TM helix rapidly settles a plateau around 0.2 nm in all three trajectories (Fig. S4), indicating that the TM helix converged to a position that is virtually identical to that in the crystal structure. Indeed, 77% ± 2% of the native contacts found in the crystal structure of SERCA-PLB4 are present in all three trajectories of the WT PLB-bound complex (Fig. 2 B). Therefore, our simulations validate previous biochemical and crystallographic studies suggesting that PLB4 binds to SERCA in a manner similar to that observed for WT PLB (25,26).
Figure 2.
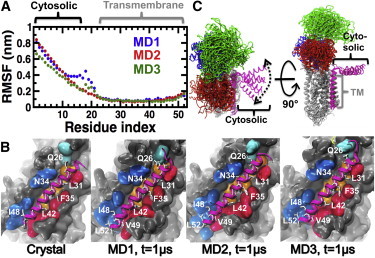
Structural characteristics of PLB in the complex. (A) Cα RMSFs of PLB calculated from each independent MD trajectory. Brackets show the location of the cytosolic and TM helices of PLB. (B) Interactions between the TM domains of SERCA (surface representation) and PLB (cartoon representation) in the crystal and MD structures. PLB is colored in magenta. Residues of PLB that are important for inhibition are shown as white sticks. These residues are located at the SERCA-PLB interface. TM helices of SERCA that are directly involved PLB binding (TM2 (blue), TM4 (cyan), TM6 (orange), and TM9 (red)) are shown. (C) Cartoon representation of the structural ensembles of SERCA-PLB averaged over all three MD trajectories. SERCA and PLB are color-coded as indicated in Fig. 1. To see this figure in color, go online.
We found that although the cytosolic helix of PLB (residues E2–S16) was very mobile (Figs. 2 and S4), this segment populated an α-helix structure in the microsecond-long trajectories (Fig. S1). Although the cytosolic helix diffused through the viscous bilayer surface in the microsecond timescale, we found that this domain of PLB did not interact with SERCA in the trajectories (Fig. 2 C). This finding agrees with solid-state NMR spectroscopy experiments (22). The polar angle between the TM and cytosolic helices of PLB calculated over the combined three trajectories is 90° ± 20°, demonstrating that the cytosolic helix is in contact with the surface of the lipid bilayer in all trajectories.
Comparison between structural ensembles of PLB-bound, E1, and E2 SERCA
We used cPCA to determine whether SERCA-PLB protonated at E771 belongs to the continuum of E1 or E2 structures. We found that the structural changes observed in x-ray structures are well described within the first two principal components, PC1 and PC2. A 2D projection onto PC1 and PC2 revealed the presence of a single cluster containing all seven crystal structures and three clusters of E1 structures (Fig. 3).
Figure 3.
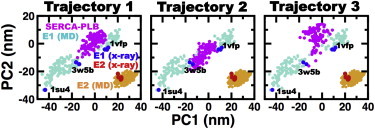
Comparison of the SERCA-PLB ensembles with the crystal and MD structures of E1 and E2. Projection of the first two principal components obtained from the cPCA performed on x-ray and MD ensembles of E1 and E2. The ensemble of SERCA-PLB is shown in magenta. PDB codes are used to indicate the location of open (PDB: 1SU4), partially open (PDB: 3W5B), and closed (PDB: 1VFP) structures of E1 SERCA. Each point represents either a crystal structure or a conformation extracted from the trajectories at time intervals of 0.01 μs. To see this figure in color, go online.
Although a direct comparison between SERCA-PLB and crystal structures could be sufficient to determine whether the complex falls in or near E1 or E2, such an approach could provide incomplete, if not inaccurate, information. For example, recent studies have shown that E1, and to a lesser extent E2, cannot be represented as single structures, because SERCA is structurally very flexible in solution (64,66,67,69). Therefore, we performed a cPCA of ensembles of both x-ray and MD structures of E1 and E2. We generated the MD ensemble of E1 structures by combining three MD trajectories of metal-ion-bound E1 (E1⋅Ca2+, E1⋅K+, and E1⋅Mg2+) into a single trajectory. These trajectories were generated in previous studies performed by our group (64,66). In addition, we performed three 1 μs MD simulations of E2 starting from three different crystal structures of SERCA in this state. We found that despite the relative flexibility of the cytosolic headpiece, the structure of E2 remained unchanged in the MD trajectories (Fig. S5). This finding agrees with previous studies showing that protonation of residues E309, E771, and E908 is sufficient to stabilize the E2 state (60,63). We found that the structural data from MD simulations provide a link between different crystal structures of E1, as well as a more accurate visualization of the structural dynamics of this state at physiological conditions (Fig. 3). Likewise, we found that E2 populates an ensemble of relatively flexible structures, and that crystal structures represent only one possible conformation of E2 in solution (Fig. 3).
The cPCA showed that, despite being protonated at E771, PLB-bound SERCA does not fall within the structural space covered by E2. Instead, we found that the structural ensemble of SERCA-PLB in each independent trajectory falls within the E1 structural space defined by both MD and crystal structures (Fig. 3, magenta). Together with the pKa calculations, this structural analysis indicates that PLB-bound SERCA represents an E1 state of the pump protonated at residues E771 and E908. We designate this state as E1⋅H+771-PLB.
Water access to the cytosolic pathways of E1⋅H+771-PLB
Bublitz et al. (30) recently analyzed water accessibility in high-resolution crystal structures of SERCA and suggested that the ion-exchange mechanism in the pump includes two pathways that connect the transport sites with the cytosol: 1) an N-path, formed by TM helices TM1, TM2, TM4, and TM6, which is primarily used for metal ion binding to the transport sites; and 2) a C-path formed by TM5, TM6, TM7, TM8, and TM10, which is used for H+ release from the transport sites to the cytosol after the E2-to-E1 transition of the pump (30). Analysis of the average water density calculated using MD trajectories showed that, except for E1⋅2Ca2+, where transport sites are almost entirely shielded from bulk water (70), E1 is characterized by an open N-path and a closed C-path (Fig. 4 A); conversely, E2 is characterized by a completely open C-path and a partially open N-path (Fig. 4 B). However, we found that metal ions cannot access the transport sites of E2 despite the presence of a partially open N-path. In addition, we found that the C-path in E2 does not bind metal ions, and the path is occupied by water molecules only. These findings indicate that the C-path of SERCA is used exclusively for H+ countertransport through a chain of hydrogen-bonded water molecules (water wires (71)).
Figure 4.
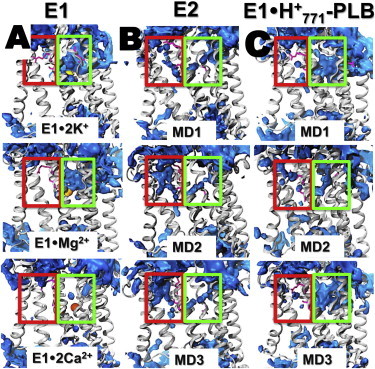
Water density maps of E1, E2, and E1⋅H+771-PLB. (A) Water density map of E1 SERCA in various metal-bound states. (B) Water density of E2 SERCA. (C) Water density calculated for each individual MD trajectory of E1⋅H+771-PLB. The blue surface represents the average weighted atomic density of water molecules using a grid resolution of 0.1 nm. The green and red boxes show the location of the cytosolic N-path and C-path, respectively. SERCA is shown as gray ribbons. To see this figure in color, go online.
To determine whether protonation of E771 affects water accessibility in the N- and C-paths, we calculated the average water density in the trajectories of E1⋅H+771-PLB (Fig. 4 C) and compared it with that of E2 and E1. We found that the transport sites of E1⋅H+771-PLB are connected to the cytosol through a path filled by water molecules, indicating the presence of an open N-path characteristic of other E1 intermediates (e.g., E1⋅2K+ and E1⋅Mg2+). The density maps also revealed the presence of water molecules in the C-path of E1⋅H+771-PLB (Fig. 4, B and C). However, it is unlikely that the partially accessible C-path of E1⋅H+771-PLB is used for H+ countertransport as in E2, for two reasons: 1) unlike E2, the cytosolic side of the C-path is completely sealed in E1⋅H+771-PLB (Fig. 4, B and C), and 2) the water wire necessary for H+ translocation does not exist in E1⋅H+771-PLB (Fig. S6). These findings indicate that the complex does not show E2-like structural characteristics as was suggested in a previous study (26). Instead, it is likely that PLB-induced, temporary structural distortions in the transport sites result in water molecules entering the C-path of E1⋅H+771-PLB.
Metal-ion binding in the transport sites of E1⋅H+771-PLB
Analysis of water accessibility showed that unlike E2, the transport sites of E1⋅H+771-PLB are completely exposed to the cytosol (Fig. 4 C). This finding suggests that water and metal ions can access the transport sites in the complex. Therefore, we analyzed the metal-ion occupancy in the transport sites of E1⋅H+771-PLB. Quantitative analysis revealed that the transport sites of E1⋅H+771-PLB are occupied by one or two K+ ions for 77% ± 1% and 5% ± 2% of the time, respectively. In contrast, the transport sites are metal-ion free only 16% ± 5% of the time. We found that the transport sites of E1⋅H+771-PLB do not bind Mg2+ at physiological ion concentrations, in agreement with x-ray crystallography (26).
Next, we investigated the K+ interactions with the transport sites of E1⋅H+771-PLB. We found that K+ ions bind to nine different positions in the transport sites of E1⋅H+771-PLB (Fig. 5 A), and that the preference for a single site is variable in the trajectories (Fig. 5 B). For instance, K+ interacts with residue D800 (sites 1, 2, and 8) for at least 40% of the time. These findings indicate that metal ions bind in the transport sites of E1⋅H+771. However, the electron density map showed that the complex lacks metal ions bound to the transport sites (26). We found that although the transport sites are occupied by K+ at least 80% of the time, the residence time of K+ in the positions shown in Fig. 5 A is ≤5 ns. This value is much smaller than the residence times of K+ in apo E1 (>0.9 μs) (64), indicating that K+ exchanges frequently in the transport sites. Such a low residence time explains the lack of a positive peak in the Fo − Fc electron density map of the transport sites of PLB-bound SERCA (26).
Figure 5.
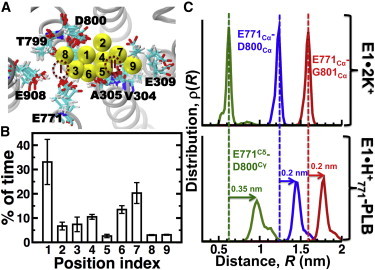
Metal-ion interactions in the transport sites of E1⋅H+771-PLB. (A) Location of the nine different positions occupied by K+ (yellow spheres) in the transport sites of E1⋅H+771-PLB; each position is labeled from 1 to 9. The dashed circles show the approximate location of the transport sites I and II. TM helices are represented by gray ribbons and cation-binding residues are shown as sticks. (B) Percentage of time K+ spends in each position shown in (A). Data are presented as the mean ± SE (n = 3). (C) Distance distributions between residue pairs E771Cα-D800Cα, E771Cα-G801Cα, and E771Cδ-D800Cγ. Distance distributions of E1⋅2K+ (upper panel) were calculated from a 1 μs trajectory reported recently by our group (64). Distance distributions of E1⋅ H+771-PLB (lower panel) were calculated by combining all three trajectories of the complex. To see this figure in color, go online.
E1⋅H+771 binds K+, but is not able to produce competent transport-site geometries (Fig. S7). We analyzed the structural changes in the transport sites induced by PLB to determine why K+ binding is insufficient to produce a structural arrangement in the transport sites associated with SERCA activation (64). Crystallographic data showed that residue N34 of PLB, which plays an essential role in SERCA inhibition (72), induces a deformation in the transport sites though a network of intermolecular contacts involving residues Q108 (TM2), G801 (TM8), and T805 (TM8) of SERCA (26). We analyzed hydrogen bonding by N34 because it has been shown that this residue is directly responsible for the SERCA inhibition (73), whereas the C-terminus and the cytosolic helix are important for PLB localization (74) and phosphorylation-dependent relief of inhibition (21–24), respectively. We found that only interactions N34-G801 and N34-T805 were present in more than 80% of the simulation time in the trajectories, and intermolecular contacts between N34 and Q108 were only transiently present (Fig. S8; see also Fig. 2 B, with TM2 in blue and TM8 in orange). To determine how interactions N34-G801 and N34-T805 alter the geometry of the transport sites, we calculated the interresidue distance between residues E771 and D800/G801. We found that PLB binding resulted in an increase in the Cα-Cα distance between residues E771 and D800/G801, which was 0.2 nm longer than that found in E1⋅2K+ (64) (Fig. 5 C, red and blue lines). However, this relatively small change results in a 0.35 nm separation between the carboxylic groups of D800 and E771 (Fig. 5 C, green line). This structural change prevents D800 from participating in the occlusion of metal ions in site I (Fig. 5 A). Therefore, the local structural changes induced by PLB do not block metal-ion binding, but inhibit the formation of a competent transport site I.
It was recently proposed that PLB binding to SERCA facilitates the interaction between E771 and N796, which results in the protonation of the former (26). To test this hypothesis, we plotted the distance distributions of Cδ atoms between E771 and N796 (Fig. 6). In addition, we calculated the distance cutoff at which E771 and N796 interact through at least one hydrogen bond (dashed line, Fig. 6). We found that this plot captures both the actual spatial separation between these residues and the probability of hydrogen-bond formation between E771 and N796. We found that E771–N796 is in a dynamic equilibrium between two populations. The first population (R < 0.51 nm) corresponds to E771 and N796 interacting by at least one hydrogen bond. The second population (R > 0.51 nm) represents a spatial separation where E771 and N796 no longer form hydrogen bonds. In addition, pKa calculations showed that E771 remains protonated even in the absence of hydrogen bonds with N796 (Fig. 6). Additional calculations showed that transient binding of a K+ near E771 (e.g., position 3 in Fig. 5 A) does not decrease the pKa of this residue. To explain why E771 remains protonated in the SERCA-PLB complex, we calculated the pKa of E771 using a trajectory of E1⋅2K+ (64). In the absence of PLB, the pKa of E771 is 3.8 ± 0.3, indicating that this residue is deprotonated in E1⋅2K+. Closer inspection of the metal-ion interactions in the transport sites revealed that KI+ simultaneously coordinates oxygen atoms from D800 and E771 in E1⋅2K+, but not in E1⋅H+771. Binding of the first Ca2+ ion between E771 and D800 induces rotation of TM6 and a side-chain swivel of E309 on TM4, thereby forming the second Ca2+-binding site (N796, D800, and E309) and accounting for SERCA cooperative Ca2+ binding (48,49). These observations indicate that PLB-induced structural alterations in the transport sites inhibits a metal ion from simultaneously binding to both D800 and E771, thus maintaining protonation of E771and preventing TM6 rotation.
Figure 6.
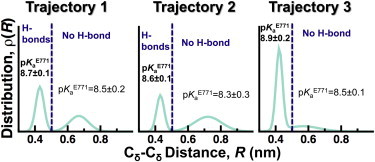
Distance distributions between E771 and N796 in E1⋅H+771-PLB. Cδ-Cδ distance distributions between residues E771 and N796 were calculated for each trajectory (MD1–MD3) of E1⋅H+771-PLB. Distance distributions were fitted to a two-Gaussian distribution. The dashed line indicates the distance cutoff at which E771 and N799 are bound by at least one hydrogen bond (R ≤ 0.5 nm). Distances longer than this cutoff value indicate the absence of a hydrogen bond. pKa values (mean ± SE) for E771 in the presence and absence of hydrogen bonds are shown in the plots. To see this figure in color, go online.
To further support the observation that E771 must be protonated in SERCA-PLB, we performed three additional 0.3 μs MD simulations: SERCA-PLB with E771 deprotonated, and SERCA without PLB with E771 protonated and deprotonated. The simulation of SERCA-PLB with E771 modeled as deprotonated showed that that proton removal from E771 indeed decreases the pKa of this residue to a mean value of 4.8, but ionization of E771 does not alter the spatial separation between Cδ and Cγ of E771 and D800, respectively (R = 0.9 ± 0.08 nm). Furthermore, we found that deprotonation of E771 facilitates the stabilization of two K+ ions bound to E771 and D800 (Fig. S9). These K+ ions have residence times in the range of hundreds of nanoseconds, and probably longer. This finding is in contrast to the electron density maps showing that the transports sites, and in particular the region around E771 and D800, are metal-ion free in SERCA-PLB. Upon removal of PLB, we found that the pKa of E771 decreases to a mean value of 5.9. An additional MD simulation starting from a structure at the end of this trajectory and with deprotonated E771 showed that the transport sites adopt a competent structure characteristic of apo E1 (RE771-D800 = 0.61 ± 0.05 nm; Fig. S9) (64) and that the pKa of E771 further decreases to a value of 3.7. Together, these sets of simulations strongly support the following observations: 1) E771 is protonated in SERCA-PLB, and 2) PLB is directly responsible for the structural changes in the transport sites that promote protonation of E771 in the E1 state.
Structural arrangement of the cytosolic headpiece of E1⋅H+771-PLB
We next sought to determine the cytosolic headpiece structure dynamics of E1⋅H+771. For this purpose, we plotted the interdomain distance distributions of E1⋅H+771 and compared them against the crystal structures of E1⋅2Ca2+, E1⋅2Ca2+⋅AMPPCP, and E1⋅Mg2+ (Fig. 7). Interdomain distance distributions of N-A, N-P, and A-P domains were calculated using Cα-Cα distances of the following amino acid pairs: K515-T171 (N-A domains), R489-E680 (N-P domains), and T171-E680 (A-P domains). We tested one- and two-Gaussian distribution models for each distance distribution, ρ(R), of each Cα-Cα distance pair. We found that all distances fit very well to either a one- or two-Gaussian distribution, with correlation coefficient values of ≥0.95 for all Gaussian distributions.
Figure 7.
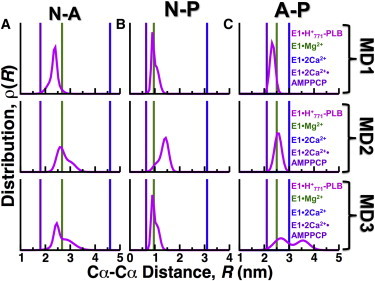
Distance distributions between the N, A, and P domains of E1⋅H+771-PLB. MD trajectories of E1⋅H+771-PLB (magenta) were used to calculate the Cα-Cα distance distribution between (A) residues K515 and T171 in N and A domains, (B) residues R489 and E680 in N and P domains, and (C) residues T171 and E680 in A and P domains (A-P). For comparison, discrete distances for the same pairs of residues were calculated from crystal structures of E1⋅Mg2+ (PDB: 3W5B in green), E1⋅2Ca2+ (PDB: 1SU4 in blue), and E1⋅2Ca2+⋅AMPPCP (PDB: 1VFP in purple). To see this figure in color, go online.
We found that interdomain distances K515-T171 (N-A domains) in only one trajectory the complex (MD1) fit to a single Gaussian distribution with mean = 2.4 nm, 0.2 nm shorter than the distance calculated from the crystal structure of E1⋅Mg2+ (Fig. 7 A, upper panel). The distance distributions in trajectories MD2 and MD3 fit to a two-Gaussian model, and the N-A domain distance of E1⋅Mg2+ falls within these boundaries of the distributions (Fig. 7 A, middle and lower panels). Except for trajectory 2 (Fig. 7 B, middle panel), the average geometry of the N-P interface in the trajectories of E1⋅H+771-PLB is very similar to the geometry observed in the crystal structure of E1⋅Mg2+ (Fig. 7 B, upper and lower panels). The distance T171-E680 (A-P domains) of E1⋅H+771-PLB shows the largest variability among the three independent trajectories (Fig. 7 C). These findings indicate that E1⋅H+771-PLB does not populate either a compact or an open headpiece conformation in solution. Instead, we found that the headpiece arrangement of E1⋅H+771-PLB populates a relatively closed structure, which is not suitable for either ATP hydrolysis or nucleotide exchange (64,66). These findings complement the cPCA analysis shown in Fig. 3, showing that PLB shifts SERCA equilibrium toward a closed headpiece structure.
Discussion
PLB populates an inhibited E1⋅H+771 state of SERCA
In this work, we used protein pKa calculations and microsecond MD simulations to show that PLB binding populates a protonated E1 state of SERCA, E1⋅H+771. Several studies have shown that PLB binds to SERCA throughout its catalytic cycle (34,36,75), and that SERCA inhibition depends on the structural shifts between the R and T states of PLB (21–24). Therefore, it is possible that E1⋅H+771-PLB does not represent the inhibited state of the complex, because the cytosolic region of PLB was not resolved in the crystal structure of the complex. However, we rule out this possibility for the following reasons: 1) The PLB mutant used to obtain crystals of the complex, PLB4, was shown to bind to SERCA with very high affinity, comparable to or even greater than that of thapsigargin (TG) (25). However, FRET experiments have shown that TG and Ca2+ decrease the binding affinity of PLB (34). Therefore, E1⋅H+771-PLB represents the high-affinity complex because the crystal structure of SERCA-PLB4 was obtained in the absence of TG and Ca2+ (26). 2) In a recent study (76), NMR residual dipolar couplings and restrained MD simulations showed that residues A24–Q29 are completely unfolded in the noninhibitory R state of PLB. However, the electron density maps in the crystal structure of SERCA-PLB4 and our MD simulations show that this segment folds as a stable α-helix. This evidence indicates that E1⋅H+771-PLB represents the structure of the inhibited complex.
PLB does not inhibit the E2-to-E1 transition of SERCA
One of the most controversial issues in SERCA regulation is whether PLB blocks the E2-to-E1 transition or traps the pump in a metal-ion-bound E1 intermediate. The first hypothesis suggests that PLB binds only to SERCA in E2 (17,77) or E2⋅ATP (16,25,26) states, and blocks the E2-to-E1 transition of the pump. The second hypothesis, based on crystallographic evidence, proposes that PLB binds to an Mg2+-bound E1 intermediate and blocks the progression of the cycle toward E1∼P⋅2Ca2+⋅ADP (28,29). Bidwell et al. (34) proposed a third scenario based on the results of FRET experiments on rabbit ventricular myocytes. These experiments showed that the SERCA-PLB complex is not disrupted by elevations of cytosolic calcium during cardiac contraction, suggesting that PLB remains bound to the metal-ion-bound E1 state of the pump, albeit with lower affinity (34). However, these experiments did not reveal which step along the catalytic cycle is affected by PLB. Our simulations showed that PLB does not inhibit the E2-E1 transition, because the crystal structure of SERCA-PLB represents an E1 state. This indicates that SERCA is not locked in a dead-end E2 state (e.g., E2 bound to the inhibitor TG), but has transitioned to E1. Our simulations demonstrate that PLB binding populates an inhibited E1⋅H+771 intermediate, indicating that PLB inhibition of SERCA occurs primarily in the E1 state. Kinetics experiments have shown that PLB interferes with binding of the first (47) and second (46) Ca2+ ions to E1. Although our simulations were performed in the absence of Ca2+, they provide an explanation for these experimental results: as cytosolic Ca2+concentration increases, it is possible that the first Ca2+ binds to E1⋅H+771 near D800 in the transport sites (K+ in position 1, Fig. 5 A). However, the structural distortion in the transport sites induced by PLB inhibits Ca2+ from interacting with both D800 and E771, which is required for the formation of a competent transport site I (64). The inability of transport site I to adopt a competent geometry will inevitably result in an altered geometry of the site II. Therefore, the PLB-induced structural alterations in the transport sites explain the altered rates of binding of the second Ca2+ ion (46). We propose that PLB populates an inhibited E1 state that interferes with the formation of a catalytically competent E1 state of the pump.
Functional role of E1⋅H+771 in the mechanism of SERCA regulation by PLB
Our findings suggest that PLB binding does not inhibit the E2-to-E1 transition of SERCA, but populates an inhibited E1⋅H+771 state of the pump. What is the functional importance of regulating SERCA activity through changes in protonation of the E1 state? E1 exists as a broad ensemble of structures separated by small energy barriers, and ligand binding (Ca2+, K+, and Mg2+) allosterically shifts this equilibrium toward an open or closed structure within the E1 continuum (28,29,64,66). Functional assays on native SERCA showed that activation of the pump by Ca2+ requires a pH-dependent, rate-limiting transition that is slower at acid than at alkaline pH (E2-to-E1 transition) (60). Based on these observations, it was proposed that activation by Ca2+ is easier if the enzyme resides in the E1 state rather than in a protonated E2 state (60). Indeed, coarse-grained simulations showed that the E2-to-E1 transition has a very large energy barrier (78). Alternatively, protonation/deprotonation of E1 offers other functional advantages. For instance, protonation can change the conformation and activity of a protein in a specific manner (79). In addition, protonation can impair regulation modes, including specificity, allostery, and cooperativity (79). Indeed, we found that PLB controls the headpiece dynamics of E1⋅H+771 and populates a headpiece structure that is not suitable for nucleotide binding and exchange (Fig. 7). Deprotonation of E771 after relief of SERCA inhibition should induce a rapid shift in the continuum of E1 structures to populate an open state, which is necessary for nucleotide exchange (64), and a productive closed state upon Ca2+ binding (66). Therefore, efficient and rapid regulation of SERCA activity is achieved by creating additional biochemical states in the E1 state, which serves as a kinetic trap in the structural transitions of E1. Hence, E1⋅H+771 ensures that the structural transitions associated with SERCA activation are temporarily depressed, but not abolished, at normal physiological conditions. We suggest that this mechanism of SERCA regulation by PLB results in both a rapid improvement of SR Ca2+ loading during diastole (relaxation) and higher Ca2+ availability during systole (contraction).
Conclusion
We have used protein pKa calculations and all-atom microsecond MD simulations to determine whether PLB inhibits the E1 or E2 states of SERCA. Our simulations not only showed that the inhibitory conformation of PLB binds to E1 SERCA but also revealed a novel (to our knowledge) protonated E1 state, designated as E1⋅H+771. We found that in the absence of Ca2+, K+ ions bind to the transport sites of E1⋅H+771, albeit transiently and nonspecifically. However, a PLB-induced structural deformation around residues D800/G801 renders SERCA incapable of producing competent transport sites. These findings indicate that PLB does not regulate SERCA by blocking the E2-to-E1 transition. Instead, PLB binding populates a novel (to our knowledge) E1⋅H+771 state that controls headpiece dynamics and depresses the structural transitions that are necessary for Ca2+-dependent activation of SERCA. Hence, PLB is an effector that introduces modifications in the energy landscape of the E1 state. We propose that E1⋅H+771 is an intermediate that is necessary for rapid Ca2+ transport after relief of SERCA inhibition. We conclude that the efficient PLB-mediated regulation of SERCA activity in the heart results from structural transitions that occur primarily in the E1 state of the pump.
Author Contributions
L.M.E.-F. and D.D.T. designed research. L.M.E.-F. and G.L.R.-S. performed research. G.L.R.-S. contributed analytic tools. L.M.E.-F. and J.M.A. analyzed data. L.M.E.-F., J.M.A., and D.D.T. wrote the paper.
Acknowledgments
We thank Bengt Svensson, Jesse E. McCaffrey, and Zach James for insightful discussions, and Octavian Cornea and Sarah Blakely for administrative assistance.
This work was supported by grants from the American Heart Association (12SDG12060656 to L.M.E.-F.) and the National Institutes of Health (R01GM27906 to D.D.T.). G.L.R.-S. is supported by a predoctoral fellowship from CONACYT (Mexico). This project made extensive use of the outstanding facilities at the University of Minnesota Supercomputing Institute.
Supporting Material
References
- 1.Palmgren M.G., Nissen P. P-type ATPases. Annu. Rev. Biophys. 2011;40:243–266. doi: 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- 2.Yu X., Carroll S., Inesi G. H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys. J. 1993;64:1232–1242. doi: 10.1016/S0006-3495(93)81489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zafar S., Hussain A., Inesi G. Specificity of ligand binding to transport sites: Ca2+ binding to the Ca2+ transport ATPase and its dependence on H+ and Mg2+ Arch. Biochem. Biophys. 2008;476:87–94. doi: 10.1016/j.abb.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantilina T., Sagara Y., Jones L.R. Comparative studies of cardiac and skeletal sarcoplasmic reticulum ATPases. Effect of a phospholamban antibody on enzyme activation by Ca2+ J. Biol. Chem. 1993;268:17018–17025. [PubMed] [Google Scholar]
- 5.Sahoo S.K., Shaikh S.A., Periasamy M. Sarcolipin protein interaction with sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) is distinct from phospholamban protein, and only sarcolipin can promote uncoupling of the SERCA pump. J. Biol. Chem. 2013;288:6881–6889. doi: 10.1074/jbc.M112.436915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asahi M., McKenna E., MacLennan D.H. Physical interactions between phospholamban and sarco(endo)plasmic reticulum Ca2+-ATPases are dissociated by elevated Ca2+, but not by phospholamban phosphorylation, vanadate, or thapsigargin, and are enhanced by ATP. J. Biol. Chem. 2000;275:15034–15038. doi: 10.1074/jbc.275.20.15034. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki T., Inui M., Tada M. Molecular mechanism of regulation of Ca2+ pump ATPase by phospholamban in cardiac sarcoplasmic reticulum. Effects of synthetic phospholamban peptides on Ca2+ pump ATPase. J. Biol. Chem. 1992;267:1674–1679. [PubMed] [Google Scholar]
- 8.MacLennan D.H., Kranias E.G. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 9.Minamisawa S., Hoshijima M., Chien K.R. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 10.del Monte F., Harding S.E., Hajjar R.J. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwanaga Y., Hoshijima M., Ross J., Jr. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J. Clin. Invest. 2004;113:727–736. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer M., Belke D.D., Dillmann W.H. A recombinant antibody increases cardiac contractility by mimicking phospholamban phosphorylation. FASEB J. 2004;18:1312–1314. doi: 10.1096/fj.03-1231fje. [DOI] [PubMed] [Google Scholar]
- 13.Hoshijima M., Ikeda Y., Chien K.R. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat. Med. 2002;8:864–871. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 14.Akin B.L., Jones L.R. Characterizing phospholamban to sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) protein binding interactions in human cardiac sarcoplasmic reticulum vesicles using chemical cross-linking. J. Biol. Chem. 2012;287:7582–7593. doi: 10.1074/jbc.M111.334987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z., Akin B.L., Jones L.R. Ca2+ binding to site I of the cardiac Ca2+ pump is sufficient to dissociate phospholamban. J. Biol. Chem. 2010;285:3253–3260. doi: 10.1074/jbc.M109.080820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Akin B.L., Jones L.R. Cross-linking of C-terminal residues of phospholamban to the Ca2+ pump of cardiac sarcoplasmic reticulum to probe spatial and functional interactions within the transmembrane domain. J. Biol. Chem. 2006;281:14163–14172. doi: 10.1074/jbc.M601338200. [DOI] [PubMed] [Google Scholar]
- 17.Toyoshima C., Asahi M., MacLennan D.H. Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. USA. 2003;100:467–472. doi: 10.1073/pnas.0237326100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zamoon J., Nitu F., Veglia G. Mapping the interaction surface of a membrane protein: unveiling the conformational switch of phospholamban in calcium pump regulation. Proc. Natl. Acad. Sci. USA. 2005;102:4747–4752. doi: 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asahi M., Kimura Y., MacLennan D.H. Transmembrane helix M6 in sarco(endo)plasmic reticulum Ca(2+)-ATPase forms a functional interaction site with phospholamban. Evidence for physical interactions at other sites. J. Biol. Chem. 1999;274:32855–32862. doi: 10.1074/jbc.274.46.32855. [DOI] [PubMed] [Google Scholar]
- 20.Morita T., Hussain D., Maclennan D.H. Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem. Biophys. Res. Commun. 2008;369:188–194. doi: 10.1016/j.bbrc.2007.11.098. [DOI] [PubMed] [Google Scholar]
- 21.Dong X., Thomas D.D. Time-resolved FRET reveals the structural mechanism of SERCA-PLB regulation. Biochem. Biophys. Res. Commun. 2014;449:196–201. doi: 10.1016/j.bbrc.2014.04.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gustavsson M., Verardi R., Veglia G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. USA. 2013;110:17338–17343. doi: 10.1073/pnas.1303006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha K.N., Traaseth N.J., Veglia G. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J. Biol. Chem. 2007;282:37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- 24.Karim C.B., Kirby T.L., Thomas D.D. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc. Natl. Acad. Sci. USA. 2004;101:14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akin B.L., Chen Z., Jones L.R. Superinhibitory phospholamban mutants compete with Ca2+ for binding to SERCA2a by stabilizing a unique nucleotide-dependent conformational state. J. Biol. Chem. 2010;285:28540–28552. doi: 10.1074/jbc.M110.151779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akin B.L., Hurley T.D., Jones L.R. The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum. J. Biol. Chem. 2013;288:30181–30191. doi: 10.1074/jbc.M113.501585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura Y., Asahi M., MacLennan D.H. Phospholamban domain I/cytochrome b5 transmembrane sequence chimeras do not inhibit SERCA2a. FEBS Lett. 1998;425:509–512. doi: 10.1016/s0014-5793(98)00151-3. [DOI] [PubMed] [Google Scholar]
- 28.Winther A.M., Bublitz M., Buch-Pedersen M.J. The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature. 2013;495:265–269. doi: 10.1038/nature11900. [DOI] [PubMed] [Google Scholar]
- 29.Toyoshima C., Iwasawa S., Inesi G. Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nature. 2013;495:260–264. doi: 10.1038/nature11899. [DOI] [PubMed] [Google Scholar]
- 30.Bublitz M., Musgaard M., Nissen P. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 2013;288:10759–10765. doi: 10.1074/jbc.R112.436550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clausen J.D., Bublitz M., le Maire M. SERCA mutant E309Q binds two Ca(2+) ions but adopts a catalytically incompetent conformation. EMBO J. 2013;32:3231–3243. doi: 10.1038/emboj.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorski P.A., Glaves J.P., Young H.S. Sarco(endo)plasmic reticulum calcium ATPase (SERCA) inhibition by sarcolipin is encoded in its luminal tail. J. Biol. Chem. 2013;288:8456–8467. doi: 10.1074/jbc.M112.446161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Meis L., Vianna A.L. Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 1979;48:275–292. doi: 10.1146/annurev.bi.48.070179.001423. [DOI] [PubMed] [Google Scholar]
- 34.Bidwell P., Blackwell D.J., Robia S.L. Phospholamban binds with differential affinity to calcium pump conformers. J. Biol. Chem. 2011;286:35044–35050. doi: 10.1074/jbc.M111.266759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.James Z.M., McCaffrey J.E., Thomas D.D. Protein-protein interactions in calcium transport regulation probed by saturation transfer electron paramagnetic resonance. Biophys. J. 2012;103:1370–1378. doi: 10.1016/j.bpj.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller B., Karim C.B., Thomas D.D. Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using fluorescence resonance energy transfer. Biochemistry. 2004;43:8754–8765. doi: 10.1021/bi049732k. [DOI] [PubMed] [Google Scholar]
- 37.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 38.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 39.Olsson M.H.M., Sondergaard C.R., Jensen J.H. PROPKA3: consistent treatment of internal and surface residues in empirical pK(a) predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 40.Sondergaard C.R., Olsson M.H.M., Jensen J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pK(a) values. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 41.Tanford C., Roxby R. Interpretation of protein titration curves. Application to lysozyme. Biochemistry. 1972;11:2192–2198. doi: 10.1021/bi00761a029. [DOI] [PubMed] [Google Scholar]
- 42.Cornelius F., Kanai R., Toyoshima C. A structural view on the functional importance of the sugar moiety and steroid hydroxyls of cardiotonic steroids in binding to Na,K-ATPase. J. Biol. Chem. 2013;288:6602–6616. doi: 10.1074/jbc.M112.442137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H., Ratheal I.M., Roux B. Protonation of key acidic residues is critical for the K+-selectivity of the Na/K pump. Nat. Struct. Mol. Biol. 2011;18:1159–1163. doi: 10.1038/nsmb.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiser A., Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 45.Traaseth N.J., Shi L., Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trieber C.A., Afara M., Young H.S. Effects of phospholamban transmembrane mutants on the calcium affinity, maximal activity, and cooperativity of the sarcoplasmic reticulum calcium pump. Biochemistry. 2009;48:9287–9296. doi: 10.1021/bi900852m. [DOI] [PubMed] [Google Scholar]
- 47.Mahaney J.E., Autry J.M., Jones L.R. Kinetics studies of the cardiac Ca-ATPase expressed in Sf21 cells: new insights on Ca-ATPase regulation by phospholamban. Biophys. J. 2000;78:1306–1323. doi: 10.1016/S0006-3495(00)76686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toyoshima C., Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 49.Obara K., Miyashita N., Toyoshima C. Structural role of countertransport revealed in Ca(2+) pump crystal structure in the absence of Ca(2+) Proc. Natl. Acad. Sci. USA. 2005;102:14489–14496. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reference deleted in proof.
- 51.Reference deleted in proof.
- 52.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber W., Hünenberger P.H., McCammon J.A. Molecular dynamics simulations of a polyalanine octapeptide under ewald boundary conditions: influence of artificial periodicity on peptide conformation. J. Phys. Chem. B. 2000;104:3668–3675. [Google Scholar]
- 54.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 55.Essmann U., Perera L., Berkowitz M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 56.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 57.Glykos N.M. Software news and updates. Carma: a molecular dynamics analysis program. J. Comput. Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 58.Clarke D.M., Loo T.W., MacLennan D.H. Location of high affinity Ca2+-binding sites within the predicted transmembrane domain of the sarcoplasmic reticulum Ca2+-ATPase. Nature. 1989;339:476–478. doi: 10.1038/339476a0. [DOI] [PubMed] [Google Scholar]
- 59.Inesi G., Lewis D., de Meis L. Conformational fluctuations of the Ca2+-ATPase in the native membrane environment. Effects of pH, temperature, catalytic substrates, and thapsigargin. J. Biol. Chem. 2008;283:1189–1196. doi: 10.1074/jbc.M707189200. [DOI] [PubMed] [Google Scholar]
- 60.Lewis D., Pilankatta R., Tadini-Buoninsegni F. Distinctive features of catalytic and transport mechanisms in mammalian sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and Cu+ (ATP7A/B) ATPases. J. Biol. Chem. 2012;287:32717–32727. doi: 10.1074/jbc.M112.373472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y., Pilankatta R., Moncelli M.R. High-yield heterologous expression of wild type and mutant Ca(2+) ATPase: characterization of Ca(2+) binding sites by charge transfer. J. Mol. Biol. 2009;391:858–871. doi: 10.1016/j.jmb.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steenbergen C., Deleeuw G., Williamson J.R. Effects of acidosis and ischemia on contractility and intracellular pH of rat heart. Circ. Res. 1977;41:849–858. doi: 10.1161/01.res.41.6.849. [DOI] [PubMed] [Google Scholar]
- 63.Musgaard M., Thøgersen L., Schiøtt B. Protonation states of important acidic residues in the central Ca2+ ion binding sites of the Ca2+-ATPase: a molecular modeling study. Biochemistry. 2011;50:11109–11120. doi: 10.1021/bi201164b. [DOI] [PubMed] [Google Scholar]
- 64.Espinoza-Fonseca L.M., Autry J.M., Thomas D.D. Microsecond molecular dynamics simulations of Mg2+- and K+-bound E1 intermediate states of the calcium pump. PLoS ONE. 2014;9:e95979. doi: 10.1371/journal.pone.0095979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Das A., Gur M., Roux B. Exploring the conformational transitions of biomolecular systems using a simple two-state anisotropic network model. PLOS Comput. Biol. 2014;10:e1003521. doi: 10.1371/journal.pcbi.1003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Espinoza-Fonseca L.M., Thomas D.D. Atomic-level characterization of the activation mechanism of SERCA by calcium. PLoS ONE. 2011;6:e26936. doi: 10.1371/journal.pone.0026936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winters D.L., Autry J.M., Thomas D.D. Interdomain fluorescence resonance energy transfer in SERCA probed by cyan-fluorescent protein fused to the actuator domain. Biochemistry. 2008;47:4246–4256. doi: 10.1021/bi702089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pallikkuth S., Blackwell D.J., Robia S.L. Phosphorylated phospholamban stabilizes a compact conformation of the cardiac calcium-ATPase. Biophys. J. 2013;105:1812–1821. doi: 10.1016/j.bpj.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou Z., Hu Z., Robia S.L. 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS ONE. 2012;7:e40369. doi: 10.1371/journal.pone.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sugita Y., Ikeguchi M., Toyoshima C. Relationship between Ca2+-affinity and shielding of bulk water in the Ca2+-pump from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2010;107:21465–21469. doi: 10.1073/pnas.1015819107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagle J.F., Morowitz H.J. Molecular mechanisms for proton transport in membranes. Proc. Natl. Acad. Sci. USA. 1978;75:298–302. doi: 10.1073/pnas.75.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimura Y., Kurzydlowski K., MacLennan D.H. Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 1997;272:15061–15064. doi: 10.1074/jbc.272.24.15061. [DOI] [PubMed] [Google Scholar]
- 73.Trieber C.A., Douglas J.L., Young H.S. The effects of mutation on the regulatory properties of phospholamban in co-reconstituted membranes. Biochemistry. 2005;44:3289–3297. doi: 10.1021/bi047878d. [DOI] [PubMed] [Google Scholar]
- 74.Abrol N., Smolin N., Robia S.L. Phospholamban C-terminal residues are critical determinants of the structure and function of the calcium ATPase regulatory complex. J. Biol. Chem. 2014;289:25855–25866. doi: 10.1074/jbc.M114.562579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Negash S., Yao Q., Squier T.C. Phospholamban remains associated with the Ca2+- and Mg2+-dependent ATPase following phosphorylation by cAMP-dependent protein kinase. Biochem. J. 2000;351:195–205. doi: 10.1042/0264-6021:3510195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Simone A., Gustavsson M., Vendruscolo M. Structures of the excited states of phospholamban and shifts in their populations upon phosphorylation. Biochemistry. 2013;52:6684–6694. doi: 10.1021/bi400517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Waggoner J.R., Huffman J., Mahaney J.E. Phospholamban inhibits Ca-ATPase conformational changes involving the E2 intermediate. Biochemistry. 2007;46:1999–2009. doi: 10.1021/bi061365k. [DOI] [PubMed] [Google Scholar]
- 78.Nagarajan A., Andersen J.P., Woolf T.B. Coarse-grained simulations of transitions in the E2-to-E1 conformations for Ca ATPase (SERCA) show entropy-enthalpy compensation. J. Mol. Biol. 2012;422:575–593. doi: 10.1016/j.jmb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Schönichen A., Webb B.A., Barber D.L. Considering protonation as a posttranslational modification regulating protein structure and function. Annu. Rev. Biophys. 2013;42:289–314. doi: 10.1146/annurev-biophys-050511-102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Best R.B., Zhu X., Mackerell A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allnér O., Nilsson L., Villa A. Magnesium ion–water coordination and exchange in biomolecular simulations. J. Chem. Theory Comput. 2012;8:1493–1502. doi: 10.1021/ct3000734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.